

Abstract
The present work analyzed 120 high-resolution X-ray crystal structures and identified 335 RNA–protein π-interactions (154 nonredundant) between a nucleobase and aromatic (W, H, F, or Y) or acyclic (R, E, or D) π-containing amino acid. Each contact was critically analyzed (including using a visual inspection protocol) to determine the most prevalent composition, structure, and strength of π-interactions at RNA–protein interfaces. These contacts most commonly involve F and U, with U:F interactions comprising one-fifth of the total number of contacts found. Furthermore, the RNA and protein π-systems adopt many different relative orientations, although there is a preference for more parallel (stacked) arrangements. Due to the variation in structure, the strength of the intermolecular forces between the RNA and protein components (as determined from accurate quantum chemical calculations) exhibits a significant range, with most of the contacts providing significant stability to the associated RNA–protein complex (up to −65 kJ mol−1). Comparison to the analogous DNA–protein π-interactions emphasizes differences in RNA– and DNA–protein π-interactions at the molecular level, including the greater abundance of RNA contacts and the involvement of different nucleobase/amino acid residues. Overall, our results provide a clearer picture of the molecular basis of nucleic acid–protein binding and underscore the important role of these contacts in biology, including the significant contribution of π–π interactions to the stability of nucleic acid–protein complexes. Nevertheless, more work is still needed in this area in order to further appreciate the properties and roles of RNA nucleobase–amino acid π-interactions in nature.
Keywords: RNA–protein interactions, X-ray crystal structure analysis, π-interactions, DFT calculations, π-containing amino acids
INTRODUCTION
Although DNA predominantly adopts a B-type double-helical structure in living cells, RNA commonly appears in several forms, including double strands, single strands, hairpins, loops, bulges, and pseudoknots (Sweeney et al. 2015). As a result, DNA nucleotides interact in Watson-Crick hydrogen-bonded pairs, while RNA nucleotides adopt different relative configurations, leading to Watson-Crick and non-Watson-Crick hydrogen-bonded pairs, unpaired bases, base triples, or even higher-order interactions (Sweeney et al. 2015). Furthermore, RNA nucleotide interactions exploit the 2′-hydroxyl group that distinguishes RNA from DNA (Sweeney et al. 2015). In addition to the greater structural versatility of RNA over DNA, RNA has more diverse biological roles, which include catalytic functions (Neugebauer 2015). Nevertheless, most essential cellular tasks of nucleic acids require interactions with proteins. Indeed, DNA–protein interactions are known to be critical for replication and repair, whereas RNA–protein interactions are vital for the post-transcriptional regulation of gene expression, protein synthesis, and viral assembly and replication.
Due to the importance of nucleic acid–protein interactions, a number of studies have analyzed experimental X-ray crystal structures to understand the types of atomic level interactions between DNA (Luscombe et al. 2001; Luscombe and Thornton 2002; Gromiha et al. 2004a,b, 2005; Mao et al. 2004; Lejeune et al. 2005; Prabakaran et al. 2006; Baker and Grant 2007; Sathyapriya et al. 2008; Wilson et al. 2014, 2015) or RNA (Allers and Shamoo 2001; Jones et al. 2001; Treger and Westhof 2001; Cheng et al. 2003; Jeong et al. 2003; Lejeune et al. 2005; Morozova et al. 2006; Baker and Grant 2007; Ellis et al. 2007; Bahadur et al. 2008; Barik et al. 2015) nucleotides and amino acids that govern nucleic acid recognition and binding. These studies have determined that nucleic acid–protein contacts span hydrogen bonding (direct or water mediated), ionic (salt-bridge or phosphate backbone), van der Waals, and hydrophobic interactions. Importantly, key differences between RNA and DNA recognition have become apparent (Allers and Shamoo 2001; Jones et al. 2001; Lejeune et al. 2005; Morozova et al. 2006; Baker and Grant 2007; Bahadur et al. 2008; Barik et al. 2015). Indeed, the 2′-hydroxyl group plays a role in RNA recognition (Bahadur et al. 2008; Barik et al. 2015), with estimates that this moiety is involved in approximately one-quarter of all RNA–protein hydrogen bonds (Treger and Westhof 2001). Furthermore, although the double-helical structure of DNA necessitates that most DNA–protein interactions involve the phosphate backbone, interactions with the nucleobase and ribose moieties are more important for RNA–protein binding since protein interactions typically occur with loops, bulges, kinks, and other irregular structures (Allers and Shamoo 2001; Lejeune et al. 2005; Bahadur et al. 2008).
Previous analyses of nucleic acid–protein crystal structures reveal small distances between the nucleobases and π-containing amino acids (Jones et al. 2001; Luscombe et al. 2001; Treger and Westhof 2001; Lejeune et al. 2005; Morozova et al. 2006; Baker and Grant 2007; Ellis et al. 2007; Wilson et al. 2014; Barik et al. 2015; Wilson et al. 2015). Indeed, despite the involvement of the DNA nucleobases in stable stacking interactions within the double helix, an abundance of nucleobase–amino acid π-interactions (π-contacts) have been identified in DNA–protein complexes (Luscombe et al. 2001; Lejeune et al. 2005; Baker and Grant 2007; Wilson et al. 2014; Wilson et al. 2015). Since most RNA nucleobases are not stacked within duplexes, nucleobase–amino acid π-interactions can occur without requiring unfavorable conformational changes and with the added benefit of sequestering the bases from solvent. Indeed, hydrogen bonds have been determined to be less dominating in RNA than DNA–protein complexes (Jones et al. 2001; Treger and Westhof 2001), and van der Waals contacts play a more prevalent role than hydrogen bonding in RNA–protein interactions (Jones et al. 2001; Ellis et al. 2007), comprising 72% of the contacts at RNA–protein interfaces (Treger and Westhof 2001). Furthermore, with an increase in the number of resolved structures of RNA binding proteins, more RNA–protein nucleobase–amino acid π-interactions have been shown to be critical for cellular functions (Supplemental Fig. S1). For example, three nucleobase–amino acid π-interactions have been reported to be important for binding RNA to the U1 small nuclease ribonucleoprotein particle that is involved in pre-mRNA splicing (Shiels et al. 2002; Guzman et al. 2015). Additionally, a conserved His–nucleobase π-interaction has been implicated in controlling gene expression by the Pumilo and FBF homology family of RNA-binding proteins (Nahalka et al. 2015), while the S1 protein involved in translocation initiation primarily uses π-stacking interactions to bind to the ribosome (Byrgazov et al. 2015).
Despite the prevalence and biological relevance of RNA–protein π-contacts, little definitive information is available about these nucleobase–amino acid interactions. Jones et al. (2001) concluded based on 32 RNA–protein complexes that van der Waals contacts are prevalent in RNA binding sites, with a base preference of G and U and an amino acid preference of phenylalanine (F) and tyrosine (Y). Alternatively, analysis of van der Waals interactions in 89 RNA–protein complexes revealed that the most favored nucleotide–amino acid pairings include U:Y, A:F, and G:tryptophan (W) (Ellis et al. 2007). Although three studies specifically considered nucleobase–amino acid π–π stacking interactions and consistently concluded that all aromatic (cyclic) amino acids are involved in this type of nucleobase contact, variations occur in the amino acid and nucleobase reported to most commonly participate in stacking contacts (Morozova et al. 2006; Baker and Grant 2007; Barik et al. 2015). Besides the aromatic amino acids, Morozova et al. (2006) and Barik et al. (2015) both report significant involvement of R in nucleobase π–π stacking interactions. Furthermore, previous work has illustrated that other relative orientations of nucleobases and π-containing amino acids besides (planar) stacked arrangements (e.g., perpendicular T-shaped orientations) may also contribute significantly to the stability and function of nucleic acid–protein complexes (Rutledge et al. 2009; Wilson et al. 2014, 2015).
The scarcity and diversity in the information available about RNA–protein π-interactions contrasts detailed bioinformatics studies that characterized the abundance, composition, structure, and stability of DNA–protein π-interactions between cyclic (W, histidine [H], F, and Y) or acyclic (arginine [R], glutamate [E], and aspartate [D]) π-containing amino acid side chains and the DNA nucleobases (Supplemental Fig. S2; Wilson et al. 2014, 2015). In this previous study, 1765 contacts were identified in 672 X-ray crystal structures published prior to January 2, 2014 with a resolution better than 2.0 Å. To unequivocally analyze the importance of nucleobase–amino acid π-interactions, each π-containing amino acid that was within 5 Å of a nucleobase was classified as a pair, and each pair was visually inspected to ensure the relative orientation of the DNA nucleobase and protein residues was consistent with a π-interaction and did not represent a hydrogen-bonding interaction or noninteracting amino acid and nucleobase (Supplemental Fig. S3). This approach avoids errors due to technical challenges defining π-interactions in automated search routines and ensures all contacts included in our data set represent π–π interactions. Indeed, the use of automated search routines, which typically classify contacts based on distance and/or angle cutoffs, have erroneously classified hydrogen bonding, van der Waals, and noninteracting amino acid–nucleotides pairs as nucleobase–amino acid π–π contacts (see, for example, Supplemental Fig. S4; Baker and Grant 2007). Furthermore, although the recently released beta-r06-2015oct23 version of 3DNA-SNAP (Lu and Olson 2008) is able to distinguish between such errors, and accurately detects stacking interactions between nucleobases and amino acids, it unfortunately is currently unable to identify T-shaped interactions (see, for example, Supplemental Table S1). Additionally, through the use of visual inspection, interactions can be unambiguously classified based on the angle between the planes of the two π-systems (tilt angle or ω) as π–π stacked (parallel or ω = 0−20°), inclined (20° < ω < 70°), or T-shaped (perpendicular or ω = 70–90°; Supplemental Fig. S1). By using visual inspection, the relative arrangements of the DNA and protein components spanning the full range of possible interplanar angles were distinctly identified, reflecting the structural diversity of DNA–protein π-interactions that can occur in nature. In addition to verifying the relative abundance of each nucleobase–amino acid pairing, key information about the significant energetic contribution of each π-contact to DNA–protein binding was obtained from accurate quantum chemical calculations. However, due to the unique structures adopted by DNA and RNA, and differences already identified in their noncovalent interactions with proteins (discussed above) (Allers and Shamoo 2001; Jones et al. 2001; Lejeune et al. 2005; Morozova et al. 2006; Baker and Grant 2007; Bahadur et al. 2008; Barik et al. 2015), the known features of DNA–protein π-interactions cannot be confidently extrapolated to RNA–protein π-interactions.
To complement previous studies of nucleic acid–protein interactions, the present study systematically investigates contacts between the RNA nucleobases and π-containing amino acid side chains in RNA–protein complexes. Critical information is obtained regarding the nucleobase and amino acid residues involved in RNA–protein π-interactions and the relative arrangement of their π-systems. Due to previous successes using computational methods to gain information about the energetic contributions of discrete nucleic acid–protein π-interactions (Mao et al. 2004; Cauët et al. 2005; Baker and Grant 2007; Copeland et al. 2008, 2013; Wilson et al. 2014, 2015), quantum mechanical techniques are used to assess the stability of each discrete RNA–protein π-contact identified. Detailed comparison to analogous studies on DNA–protein π-interactions (Wilson et al. 2014, 2015) reveals both similarities and differences between the π-interactions used by RNA and DNA binding proteins. The information obtained about nucleic acid–protein interactions will have broad implications for understanding key cellular processes, such as splicing, protein synthesis, and viral replication, as well as for developing improved computational routines (including automated identification of RNA–protein interactions, force fields, and docking procedures) that will afford additional atomic level information about nucleic acid structure in the future.
RESULTS
Nucleobase–amino acid π–π interactions frequently occur when a variety of RNA types bind to proteins
The set of 120 high-resolution RNA–protein crystal structures were chosen based on availability and our desire to study the largest possible data set based on accurate atomic positions, but were not restricted according to sequence similarities. Therefore, in some instances, structures with up to 100% sequence similarity were included in our analysis. Furthermore, crystallographic copies of RNA–protein interactions were included in the data set. To ensure that this redundancy does not bias our conclusions, the same analysis procedure was applied to a set of 75 crystal structures with ≤30% sequence identity and no crystallographic copies were included. The full and nonredundant data sets of RNA–protein crystal structures were determined to unambiguously contain 335 and 154 nucleobase–amino acid π–π interactions, respectively. To avoid potential bias from analyzing structures with a high sequence similarity, the results of the 154 contacts identified in the nonredundant data set are presented from this point forward in the main text, while the results for the full data set are provided in the Supplemental Material.
The 154 π–π contacts occur with a variety of different RNA types (Fig. 1A), with most contacts involving mRNA (29%), rRNA (13%), viral RNA (12%), and dsRNA (10%). Nevertheless, this distribution across RNA types is an artifact of the structures searched. Specifically, there is a <3% difference in the distribution of the RNA types searched and the distribution in the RNA types forming π–π contacts with protein π-systems (Fig. 1A). Among the 75 crystal structures considered, 69% (52) contain at least one nucleobase–amino acid π–π interaction (Fig. 1B). Although up to 22 contacts were identified in a single structure, this case arose for a multimeric protein in which two contacts were found with each polypeptide chain, although discrete structural deviations exist between the contacts formed with each chain. For structures containing a single polypeptide chain, a maximum of nine RNA–protein π–π contacts were identified.
FIGURE 1.
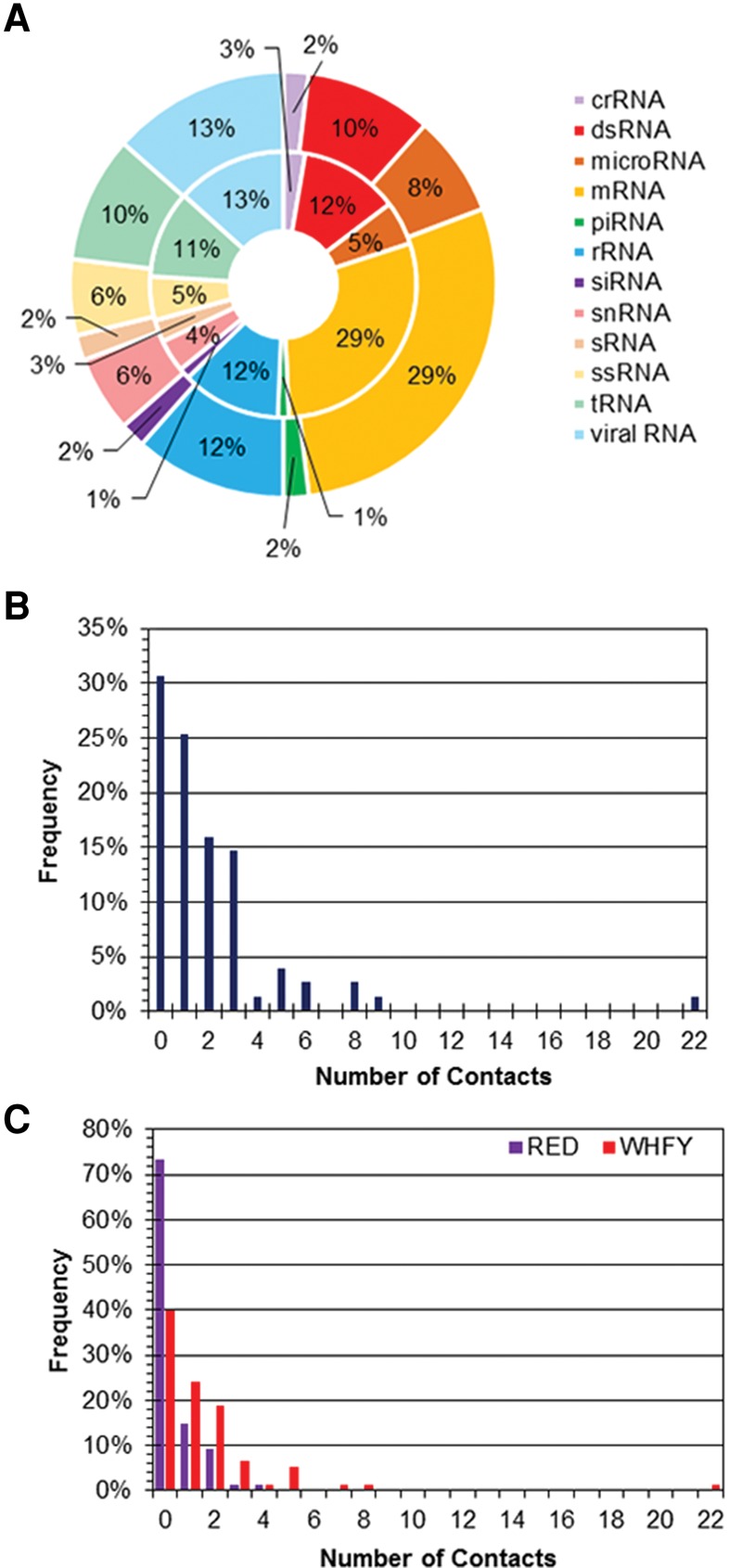
(A) Distribution in the RNA types searched (inner circle) and the RNA types that form at least one protein π–π contact (outer circle) in the nonredundant data set. (B) Overall number of RNA–protein π–π contacts found in each crystal structure searched in the nonredundant data set. (C) Number of RNA–protein π–π contacts found in each crystal structure searched as a function of the amino acid (cyclic versus acyclic) classification in the nonredundant data set.
RNA nucleobase–amino acid π–π interactions most commonly involve the aromatic amino acids, specifically phenylalanine, while uracil is the most frequent nucleobase
Within the 75 RNA–protein complexes considered, 60% (45) of the structures contain a close contact between a nucleobase and an aromatic (cyclic) amino acid (W, H, F, or Y), yielding a total of 122 interactions (Fig. 1C). Additionally, most complexes with a nucleobase–aromatic amino acid π–π interaction contain one (24%) or two (19%) of such contacts. In contrast, only 27% (20) of structures searched contain an interaction with an acyclic amino acid (Fig. 1C), which corresponds to a total of 32 interactions (or 21% of the total number of contacts). Furthermore, most structures with a nucleobase–acyclic amino acid interaction have only one such contact (56%). In terms of the amino acid (Fig. 2A), nearly half of the interactions occur with F (45%), and one-fifth of the interactions occur with Y (22%). The prevalence of F and Y interactions is consistent with their previously predicted role in RNA binding sites (Jones et al. 2001), and evidence that Y forms the greatest number, while W forms the least number of stacking interactions (defined as <30° between the planes of the DNA and protein π-systems) (Barik et al. 2015). Among the acyclic counterparts, R forms the majority of π–π interactions with the RNA nucleobases (19%), which agrees with previous studies identifying R contacts as being prevalent at RNA–protein binding sites (Jones et al. 2001; Morozova et al. 2006; Ellis et al. 2007). Each of W, H, D, and E are involved in less than ∼5% of the identified nucleobase–amino acid π–π interactions. In terms of the nucleobase (Fig. 2B), most contacts occur with uracil (36%), which along with guanine was previously predicted to exhibit the most prevalent role in RNA–protein van der Waals contacts (Jones et al. 2001). Although this contrasts previous reports that guanine and adenine occur overall most frequently (Ellis et al. 2007), our results suggest that the prevalent U contacts are followed by the purines (25%–26%), while significantly fewer contacts occur with C (12%). As a result, overall slightly more RNA–protein π–π contacts are found with the purines (51%) than pyrimidines (49%). This distribution is in good agreement with a previous study (54% purine; 46% pyrimidines), which only considered stacked RNA–protein arrangements (defined as <30° between the planes of the π-systems) (Barik et al. 2015).
FIGURE 2.
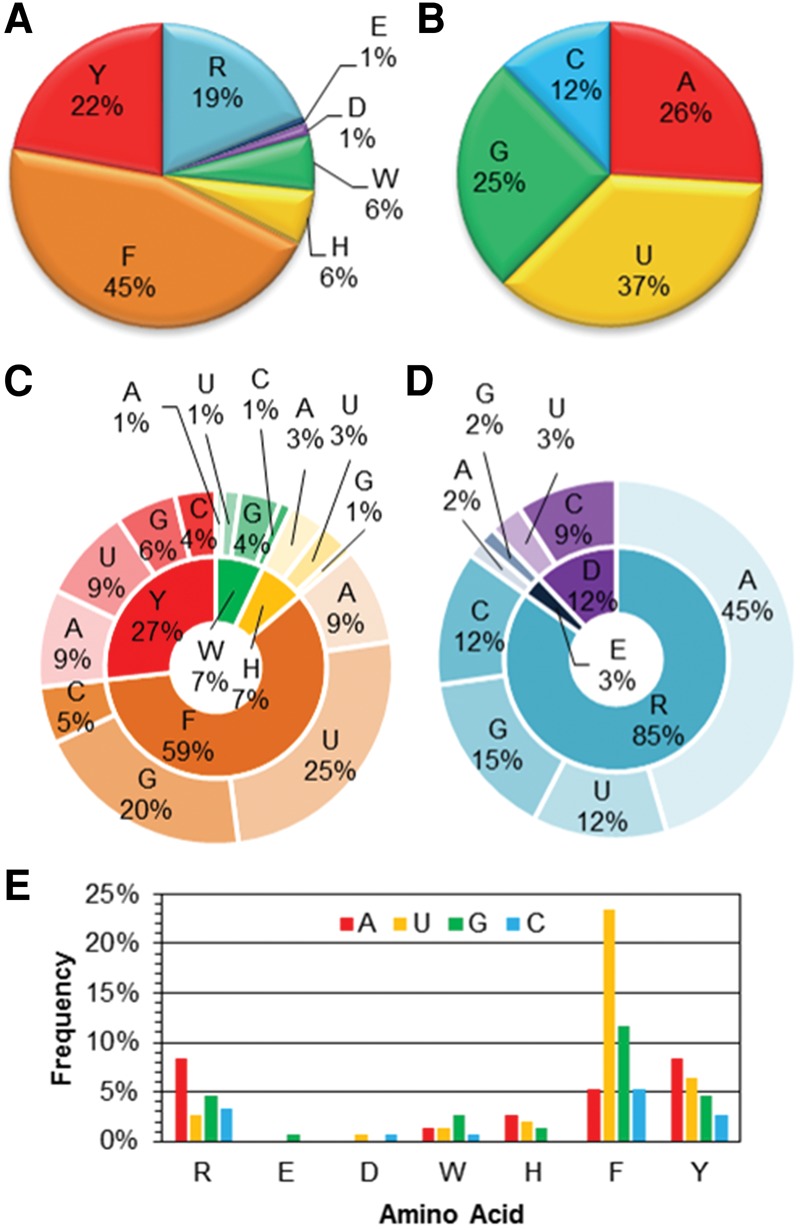
Distribution in the composition of the RNA–protein π–π contacts in the nonredundant data set as a function of (A) amino acid, (B) nucleobase, (C) the aromatic (cyclic) amino acids, (D) the acyclic amino acids, and (E) both (cyclic and acyclic) amino acid classes.
Most common RNA nucleobase–amino acid π–π pairings are phenylalanine with uracil or guanine, and arginine with adenine
When the RNA–protein π–π interactions are dissected into the nucleobase and amino acids involved, the majority of contacts involving the aromatic amino acids occur with U (33%) and G (20%; Fig. 2C). This trend is caused by the most abundant F interactions, where the U:F and G:F pairs comprise 30% and 15% of the total π–π interactions with the aromatic amino acids, respectively. Y forms the most interactions with A (38% of the Y interactions or 10% of the total interactions with the cyclic residues) and U (29% of the Y interactions or 8% of the total interactions with the cyclic residues). These results are at least in part consistent with previous reports that F is the most common aromatic amino acid engaged in stacking at RNA–protein interfaces (Morozova et al. 2006), and that RNA binding sites exhibit a base preference of G and U and an amino acid preference of F and Y (Jones et al. 2001). W forms the most interactions with G, with these contacts comprising <4% of the total number of interactions with the aromatic amino acids. This finding does not support previous claims that W is the most common aromatic amino acid in RNA–protein recognition (Baker and Grant 2007) and G:W is among the most prevalent pairings at RNA binding sites (Ellis et al. 2007), although it supports suggestions that W forms the least number of stacking interactions (defined as <30° between the planes of the π-systems) (Barik et al. 2015). Interestingly, no interactions were identified between H and C. In contrast to the W, H, F, and Y set, 41% of the total number of interactions with the acyclic π-containing amino acids involve A (Fig. 2D) and all of these contacts occur between R and A. R interactions also involve the three other RNA nucleobases with a frequency >10% of the total number of acyclic contacts. The abundance of nucleobase contacts with R is consistent with previous reports in the literature (Morozova et al. 2006; Barik et al. 2015). Notably, there were only one and two interactions identified with E and D, respectively, in the nonredundant data set. Among all of the RNA–protein nucleobase–amino acid π–π contacts identified (Fig. 2E), there is a clear preference for U:F and G:F interactions, which comprise 23 and 12% of the total number of nucleobase–amino acid π–π interactions, respectively. The next most abundant RNA–protein pair is A:R, which comprises only 8% of the total number of contacts.
Although the RNA and protein π-systems adopt many different relative arrangements, both cyclic and acyclic amino acids generally prefer a (parallel) stacked orientation relative to the RNA nucleobases
Consistent with previous literature, the structure of each interacting pair was classified based on the interplanar angle between the two π-systems (tilt or ω) as stacked (ω = 0–20°), inclined (20° < ω < 70°), or T-shaped (ω = 70–90°; Supplemental Fig. S1; Wilson et al. 2014, 2015). The majority of RNA–protein nucleobase–amino acid interactions adopt a stacked π–π orientation regardless of whether the contact involves an aromatic (64%) or acyclic (56%) amino acid (Fig. 3A,B). Furthermore, both classes of amino acids are more likely to adopt an inclined than a T-shaped orientation. However, the proportions of inclined and T-shaped π-arrangements are similar for the cyclic amino acids, while the acyclic amino acids rarely adopt a tilt angle (ω) >50°. H, R, and Y most commonly adopt a tilt angle between 5 and 10°, while F prefers ω = 0–5° and W prefers ω = 10–15°. Interestingly, π-containing amino acids that can potentially adopt a cationic charge strongly prefer stacking arrangements relative to the RNA nucleobases, with H never adopting ω > 30° and 79% of R interactions having ω < 30°. Regardless, F adopts the full range of tilt angles relative to the RNA nucleobase π-systems.
FIGURE 3.

Frequency of the tilt angle (degrees) between the ring planes for all π–π interactions in the nonredundant data set as a function of the protein (A,B) or RNA (C,D) component for the cyclic (A,C) and acyclic (B,D) amino acids.
When the preferred relative arrangement of the RNA and aromatic amino acid π-systems is considered as a function of the nucleobase (Fig. 3C), the preferred angle between the π-systems increases as C (ω = 0–5°; 31%) < A (ω = 5–10°; 41%) < U (ω = 10–15°; 11%) < G (ω = 20–25°; 26%). Nevertheless, C adopts a range of tilt angles, while G rarely adopts a tilt >40°. Furthermore, a third of the U contacts with the aromatic amino acids involve a T-shaped arrangement of the π-systems. When the preferred structure of contacts involving an acyclic amino acid π-system is considered as a function of the nucleobase (Fig. 3D), G and A most commonly adopt stacked structures. In fact, although A adopts a range of tilt angles, G interactions rarely acquire a tilt angle >15°. In contrast, 60% of the U interactions are slightly tilted (ω = 20–25°), while C adopts a variety of orientations with respect to the acyclic amino acids.
T-shaped interactions between RNA and protein components can involve the edge of the amino acid interacting with the face (π-system) of the nucleobase or an edge of the nucleobase interacting with the face (π-system) of the amino acid. Furthermore, nucleobase or amino acid edges with different chemical properties can be involved in the interactions, including a single proton, a lone pair or bridged structures, which direct more than one atom toward the π-system. The majority (74%) of RNA–protein π–π interactions with a tilt angle >45° involve an amino acid edge directed toward a nucleobase π-system, with 65% of the contacts involving an F edge (Supplemental Table S2). Furthermore, among the F interactions identified, 77% are bridged contacts, which direct two protons toward the nucleobase π-system, while 14% direct a single F proton toward the nucleobase.
Charged π-containing amino acids generally appear closer to the nucleobase π-system than neutral amino acids
Unsurprisingly, as a result of electrostatics, the shortest heavy atom distances between the RNA and protein π-components generally occur for the potentially charged amino acids (H, R, and D) regardless of their cyclic or acyclic nature (Supplemental Fig. S4A,B). Indeed, the most frequent shortest heavy atom separation distance for Y, F, and W ranges from 3.3–3.5 Å (Supplemental Fig. S5A), with average distances of 3.4 ± 0.2, 3.5 ± 0.4, and 3.4 ± 0.1 Å, respectively. In comparison, the most commonly adopted interspatial separation between the RNA and protein π-systems are 3.2–3.3 Å for R, D, and H (Supplemental Fig. S5B), with average distances of 3.3 ± 0.2, 3.4 ± 0.1, and 3.2 ± 0.1 Å, respectively. Nevertheless, F adopts the largest range of distances (2.7–4.3 Å), which may in part reflect the greater number of contacts identified for this amino acid in general. The only exception to this general trend is E interactions, which occupy the largest distances (3.8–3.9 Å) of the acyclic (potentially charged) amino acids. There is no clear trend in the interseparation distances according to the nucleobase, with all four nucleobases adopting a range of distances and an average separation distance of 3.3–3.5 Å (Supplemental Fig. S5C,D).
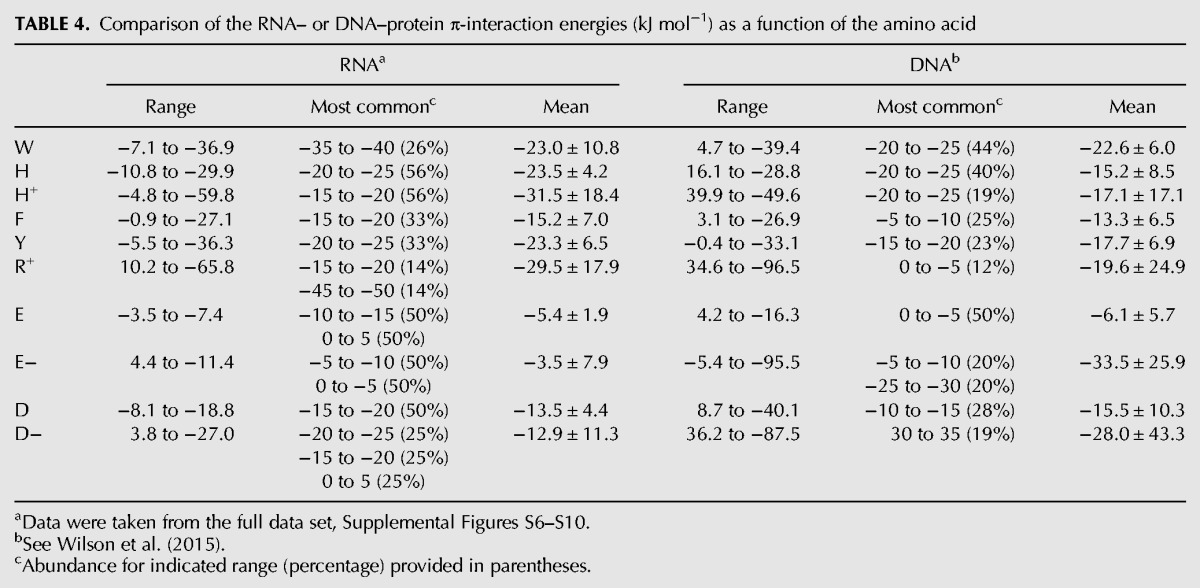
RNA–protein nucleobase–amino acid π–π intermolecular forces adopt a range of strengths, which are primarily dictated by the amino acid charge
Figure 4 summarizes the gas-phase binding strengths calculated using quantum chemical methods for each RNA–protein contact identified. In terms of the neutral amino acids (Fig. 4A), the aromatic amino acids typically exhibit a binding strength on average of −10 to −25 kJ mol−1, while neutral E and D contacts have interaction energies of −3 to −20 kJ mol−1, respectively. Consistent with this trend, the most common interaction energies for Y fall between −20 and −25 kJ mol−1, while the most common H binding strengths range from −15 to −20 kJ mol−1 and the most common F and D binding strengths range from −10 to −15 kJ mol−1. However, the most common W intermolecular forces are stronger (more negative), falling between −35 and −40 kJ mol−1. Nevertheless, the contacts involving neutral amino acids collectively exhibit a significant range of binding strengths, corresponding to both very weak (approximately −1 kJ mol−1) and very strong (approximately −37 kJ mol−1) interactions.
FIGURE 4.

Frequency of the binding energy (kJ mol−1) for nucleobase–amino acid π–π interactions in the nonredundant data set as a function of the protein (A,B) or RNA (C,D) component for the neutral (A,C) and charged (B,D) π-containing amino acids.
The average anionic interactions with E and D have similar strengths to the corresponding neutral contacts (3.8 to −15 kJ mol−1). Since previous computational studies suggest that anionic nucleobase–aromatic amino acid interactions can be very stable (Wells et al. 2013), the equivalent strength found in the present study suggests that the relative orientations of the RNA and protein components considered in the present work are not conducive for these interactions and this may in part be due to the small number of interactions found involving E and D. In contrast, the cationic interactions are significantly more stable than the corresponding neutral interactions, with average interaction energies of 33.4 ± 17.2 and 31.8 ± 16.8 kJ mol−1 for H and R, respectively (Fig. 4B). Indeed, the cationic interactions are the overall strongest contacts, contributing to the stability of RNA–protein complexes by up to approximately −65 kJ mol−1. This agrees with previous computational studies highlighting the significant strength of cationic nucleobase–aromatic amino acid contacts (Churchill and Wetmore 2009; Rutledge et al. 2010; Leavens et al. 2011). It should be noted that some of the charged interactions were determined to be repulsive (positive binding energy; Fig. 4B), which may simply reflect the fact that the amino acids are neutral in the corresponding binding arrangements.
When the intermolecular forces involving the neutral amino acids are considered as a function of the nucleobase (Fig. 4C), the majority of interactions fall between −15 and −25 kJ mol−1 regardless of the nucleobase considered. The average interaction energy decreases as G (−22.2 ± 8.4 kJ mol−1) > A (−19.5 ± 6.8 kJ mol−1) > U (−16.3 ± 8.4 kJ mol−1) > C (−13.6 ± 5.7 kJ mol−1). However, U is involved in the strongest interaction (−36.9 kJ mol−1), and adopts the largest range of interaction strengths, which fall between approximately −1 and −37 kJ mol−1. Nevertheless, G interactions display a similar range (−4 and −37 kJ mol−1) and strongest (−36.5 kJ mol−1) interaction energy. When the dependence of the binding strength on the nucleobase is considered for the charged amino acids, there is greater variance (Fig. 4D). Specifically, about half of the interactions with U or C fall between −10 and −20 kJ mol−1 or +15 and −5 kJ mol−1, respectively. Therefore, the average binding strengths between the charged amino acids and U (−19.5 ± 4.3 kJ mol−1) or C (−7.7 ± 18.5 kJ mol−1) are similar or even less than the corresponding nucleobase interaction energies for the neutral amino acids. In contrast, the most common A and G binding strengths (−30 to −35, and −35 to −40 kJ mol−1, respectively), as well as averages (−37.6 ± 10.4 and −40.5 ± 16.5 kJ mol−1, respectively) are stronger for the charged amino acids.
Sequence similarities within the RNA–protein data set do not bias the reported conclusions about RNA–protein π–π interactions
Although the results discussed to this point in the main text correspond to the nonredundant data set of 75 RNA–protein crystal structures, a detailed analysis was also completed on the larger data set of 120 RNA–protein crystal structures, which does not have restrictions on sequence similarity (Supplemental Figs. S6–S10). Consistent with previous literature on nucleic acid–protein π-interactions (Baker and Grant 2007), our global conclusions were not influenced by redundancies in the data set. Specifically, the overall relative trends in the number of RNA–protein π–π contacts found in each crystal structure searched (Fig. 1; Supplemental Fig. S6), as well as the distribution in the composition (Fig. 2; Supplemental Fig. S7), the frequency of the tilt angle (Fig. 3; Supplemental Fig. S8), or distance (Supplemental Figs. S5, S9) and the frequency of the binding energy (Fig. 4; Supplemental Fig. S10) for RNA–protein π–π contacts, do not significantly deviate between the two data sets. Thus, it can be concluded that the results of our full data set are not biased by the overrepresentation of certain types of structures. We therefore encourage researchers to review the full data set provided in the Supplemental Material since this represents the most complete and accurate database of RNA–protein π–π interactions generated to date. This large data set can, for example, aid the future development of automated search programs, and allow researchers to gain further information about discrete π–π interactions in many different systems of interest.
DISCUSSION
The present work analyzed the abundance, composition, structure, and strength of RNA–protein π-interactions between the nucleobases and cyclic (W, H, F, or Y) or acyclic (R, E, or D) π-containing amino acids found in 120 high-resolution X-ray crystal structures published in the PDB. Each structure was critically analyzed (including visual inspection) to unambiguously verify that the associated contact reflects a π-interaction, and eliminate pairings that represent hydrogen-bonding contacts or noninteracting RNA nucleobase and protein components. This work is critical due to previous suggestions that van der Waals contacts are prevalent in RNA–protein binding sites (Jones et al. 2001; Treger and Westhof 2001; Ellis et al. 2007), but conflicting reports of the relative abundance of different nucleobase–amino acid pairings (Morozova et al. 2006; Baker and Grant 2007; Barik et al. 2015), which may in part arise due to difficulties definitively characterizing π-contacts using predefined automated search algorithms (Supplemental Fig. S4; Supplemental Table S1). Furthermore, previous work has identified a large number of analogous DNA–protein π-interactions (Wilson et al. 2014, 2015), suggesting these noncovalent contacts are important throughout biology even though the DNA nucleobases are somewhat sequestered within the double helix. In contrast, RNA nucleobases may more readily form π-interactions with proteins due to the unique and varied structures known to be adopted by RNA. In this section, the RNA–protein π-interactions investigated in the present work are compared to the DNA–protein π-contacts previously identified using the same search protocol (Wilson et al. 2014, 2015). Since previous work on DNA–protein interactions did not restrict the sequence identity of the structures considered and it has been shown in the present work that the conclusions for RNA–protein interactions are not affected by the sequence identity considered, RNA– and DNA–protein interactions are compared using the full data sets. Together, this work affords the most accurate comparison of these nucleic acid–protein π-interactions in nature to date.
RNA–protein π–π interactions are more abundant than the analogous DNA–protein contacts
A large number of interactions between the nucleobases and aromatic or acyclic amino acids are found in structures of both RNA– and DNA–protein interactions. These interactions involve many different types of RNA (Fig. 1A) and DNA-binding proteins. In the 120 X-ray structures considered in the present work, 335 RNA–protein nucleobase–amino acid π-interactions were unambiguously identified. In contrast, 962 DNA–protein interactions were previously found between a nucleobase and π-containing amino acid in 672 crystal structures (Wilson et al. 2015). Although the absolute number of RNA contacts is significantly less than reported for DNA, this is an artifact of the greater abundance of structural data for DNA systems. In fact, when the number of contacts is scaled according to the number of structures considered, on average 2.79 interactions are found per RNA-binding protein compared to 1.43 contacts per DNA-binding protein. Furthermore, 69% of all RNA–protein complexes considered include at least one nucleobase–amino acid π-interaction (Fig. 1B), while 61% of DNA–protein structures contain analogous contacts (Supplemental Fig. S11). Thus, RNA nucleobases are overall more likely to participate in π-interactions with cyclic or acyclic amino acids than the DNA counterparts. This agrees with previous conclusions that stacking interactions (ω ≤ 30°) involving the side chains of aromatic residues are more prevalent in RNA than DNA–protein interfaces (Barik et al. 2015). Overall, while the frequency of π–π interactions in DNA and RNA will be affected by the available structures, the higher occurrence of RNA–protein interactions likely also reflects the unique biology of each nucleic acid class, where RNA adopts a greater variety of tertiary structures and therefore RNA nucleobases can more readily form π–π contacts with the protein.
The most common amino acid and nucleobase involved in nucleic acid–protein π-interactions differ between RNA and DNA
Table 1 summarizes the involvement of the nucleobases and amino acids involved in RNA– or DNA–protein π-interactions in nature. For both types of nucleic acids, there is a much greater abundance of nucleobase π–contacts with the aromatic amino acids (80% RNA; 76% DNA) than the acyclic π-containing protein residues. Overall, F is the most prevalent amino acid involved in these noncovalent interactions. However, this preference is greater for RNA (F contacts comprise 48% of the total number of interactions) than DNA (F contacts account for 33% of all interactions identified). In contrast, Y has a similar role in nucleobase–protein π-interactions in RNA or DNA systems, being the second most abundant amino acid (21%–22%), while contacts with H or W are less common in RNA (5%–6% each) than DNA (9%–11%). Thus, the DNA–protein π-interactions are more varied in terms of the aromatic amino acid involved. Among the acyclic π-containing amino acids, the majority of interactions with both RNA and DNA nucleobases occur with R (17% and 21% of the total contacts found, respectively).
TABLE 1.
Comparison of the relative abundance of different nucleobase–amino acid pairings in RNA– or DNA–protein π-interactions found in nature
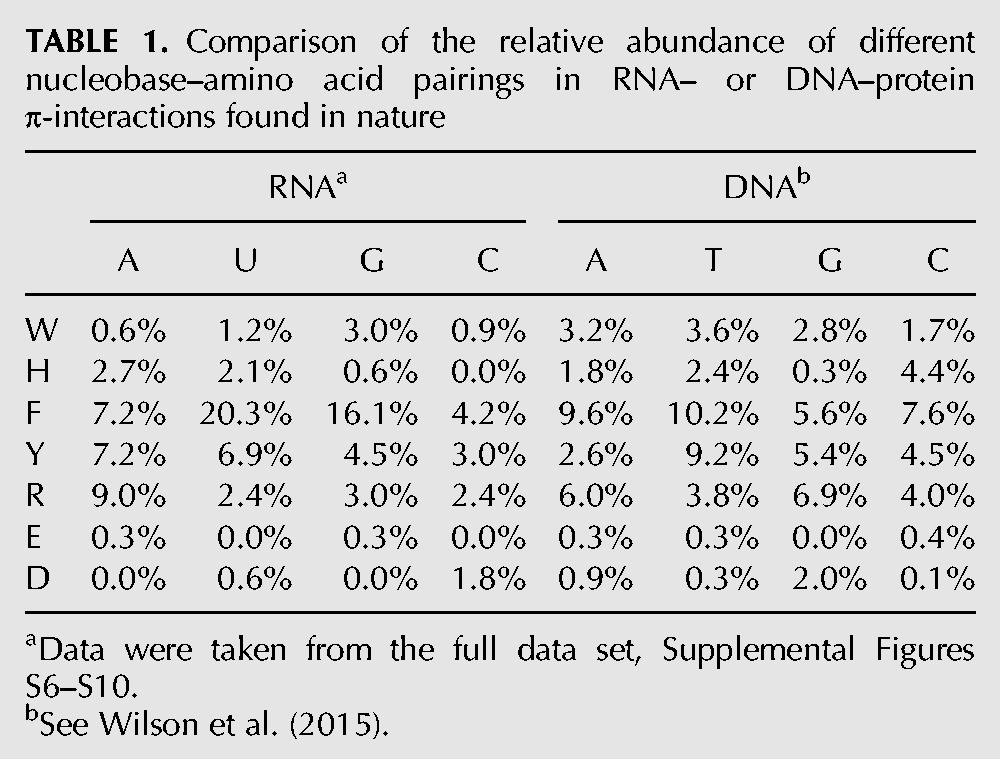
In terms of the nucleobase involved in the nucleic acid–protein π-interactions, U/T are most abundant for RNA/DNA (Table 1). Nevertheless, the preference for a given nucleobase varies more significantly in RNA–protein complexes, with the abundance ranging from 33% for U to 12% for C. In contrast, while T comprises 29% of all DNA–protein nucleobase–amino acid π-contacts, the remaining interactions are nearly equally distributed across the other three bases (23%–24%). Furthermore, although the RNA purines participate in more interactions with protein components than the RNA pyrimidines (purine:pyrimidine ratio of 54:46), the DNA pyrimidines are slightly more favored (purine:pyrimidine ratio of 48:52).
When the most common nucleobase–amino acid pairings are considered (Table 1), it becomes evident that there is a significantly greater preference in the π-contact composition for RNA compared to DNA. Specifically, the most common pairings in RNA–protein complexes are U:F (20%) and G:F (16%). Furthermore, the next most common RNA–protein pairing occurs for A:R, which is considerably less prevalent (9%). In contrast, the most abundant DNA–protein pairs, namely A:F and T:F, are appreciably less prevalent (9%–10%) than the most common RNA pairs, and many other DNA–protein combinations have relative abundances in the range of 4%–9%. Regardless, the strong prevalence of F in both RNA– and DNA–protein π-interactions disputes previous claims that differences in the relative abundances of the aromatic amino acids in RNA and DNA binding sites may provide a means to differentiate between the two nucleic acids (Baker and Grant 2007).
Both RNA and DNA nucleobases adopt many orientations relative to π-containing amino acids
The nucleobase–amino acid contacts identified in both RNA– and DNA–binding proteins adopt a wide variety of structures with various interplanar (tilt) angles (ω) between the nucleic acid and protein components. Table 2 summarizes the range in the tilt angles between the nucleobase and protein components, as well as the most common and average values. Consistent with previous literature indicating that other relative orientations of nucleobases and π-containing amino acids besides (planar) stacked arrangements may contribute as much or more significantly to the stability and function of nucleic acid–protein complexes (Rutledge et al. 2009), relative orientations of the biological π-systems that can be classified as stacked (ω = 0–20°), inclined (20° < ω < 70°), or T-shaped (ω = 70–90°; Supplemental Fig. S1) were identified in nature for both nucleic acid types. Interestingly, both RNA and DNA nucleobases preferentially adopt a stacked orientation with respect to the cyclic (aromatic) amino acids (W, H, F, or Y), comprising 59% of all RNA π-contacts (Fig. 5) and 64% of DNA π-interactions. The remaining contacts are equally distributed among inclined (25% for both RNA and DNA) and T-shaped (16% RNA; 11% DNA) orientations. Although the most common interplanar angle between a DNA nucleobase and aromatic amino acid consistently falls between 5–10°, larger angles are sometimes more populated when an RNA base binds with F or W. For nucleobase interactions with the acyclic amino acids (R, E, and D), RNA more strongly prefers a stacked orientation (57%, Fig. 5) than DNA (39%). Although there is approximately the same relative abundance of inclined structures (37% RNA; 33% DNA), RNA nucleobases rarely adopt a T-shaped orientation with respect to the protein (6% abundance), while DNA nucleobases adopt a T-shaped orientation in almost one-third of the contacts in nature (28%). For both RNA and DNA systems, the neutral and cationic π-containing amino acids preferentially adopt a stacked orientation relative to the nucleobase, while the anionic amino acids preferentially adopt T-shaped structures. The distances between the RNA and protein components are typically shorter for the neutral and cationic amino acids, but larger for the anionic counterparts, than the corresponding DNA contacts (Table 3). Nevertheless, the ranges are larger for DNA than RNA, while the most common distances are very similar between the two nucleic acids.
TABLE 2.
Comparison of the relative orientation (tilt angle or ω, degrees) of different nucleobase–amino acid π-systems in RNA– or DNA–protein π-interactions found in nature as a function of the amino acid
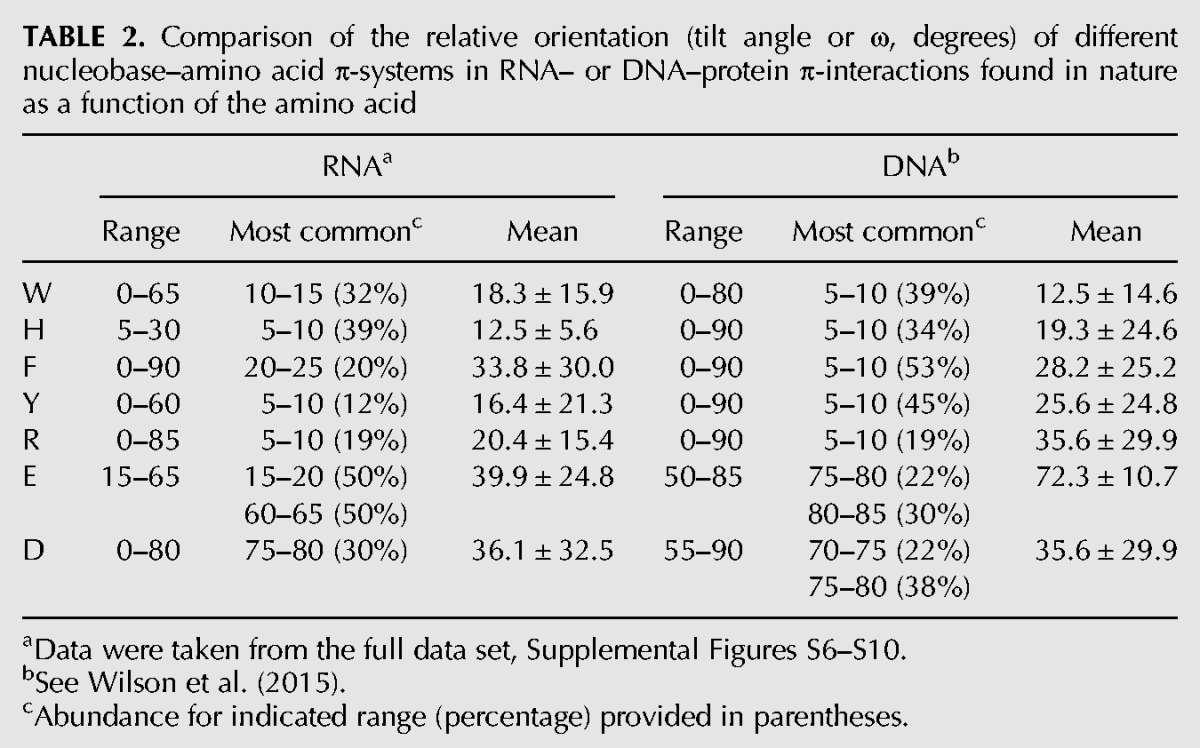
TABLE 3.
Comparison of the distance (Å) between nucleobase–amino acid π-systems in RNA– or DNA–protein π-interactions found in nature as a function of the amino acid
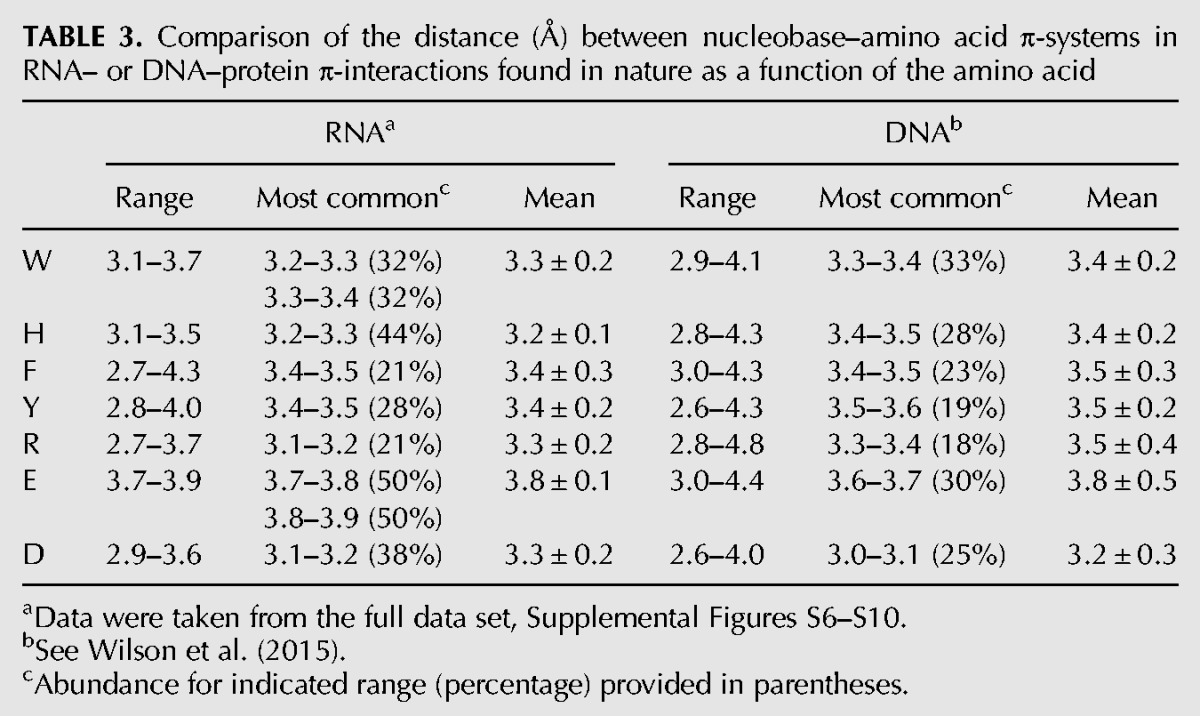
In contrast to the trends as a function of the amino acid involved in the RNA/DNA–protein interaction discussed above, few trends are evident when the geometry of the π-contacts are considered as a function of the nucleobase (Supplemental Tables S3, S4). Specifically, most nucleobases form interactions that span the full range of tilt angles, but most commonly adopt a stacked orientation. The exceptions include cyclic amino acid interactions with G or acyclic protein residue contacts with U in RNA–protein complexes, or acyclic C contacts in DNA–protein complexes, which all preferentially form inclined interactions. Additionally, the acyclic amino acids preferentially form T-shaped interactions with C in RNA–protein complexes. Overall, the general structures of nucleobase–amino acid π-interactions are the same for DNA and RNA. The small deviations in the preferences of the relative arrangement of the nucleic acid and protein components discussed could arise due to the more limited sampling in the case of RNA or differences in RNA and DNA structures.
Strength of intermolecular forces between RNA nucleobases and π-containing amino acids in nature are comparable to the corresponding DNA interactions
A full range of interaction energies are exhibited when either RNA or DNA nucleobases are bound to π-containing amino acids (Tables 4, 5). Neutral DNA interactions with the aromatic amino acids adopt a wider range of binding strengths than neutral RNA interactions, which in part arises due to a greater number of repulsive interactions for the DNA contacts. Interestingly, no repulsive contacts between the RNA bases and cyclic amino acids were found, which may reflect the less constrained nucleobase orientations in RNA compared to DNA. Additionally, the most common and average RNA interaction energies are up to ∼10 kJ mol−1 stronger than the corresponding DNA values (Table 4). This finding again correlates with a greater flexibility in the nucleobase orientation in RNA–protein complexes. Although the strength of neutral contacts between E/D and the RNA/DNA nucleobases are similar, the cationic interactions exhibit significant deviation between the two nucleic acids. Specifically, although the most stable contact between R and DNA is nearly 30 kJ mol−1 stronger than the most stable RNA interaction, the mean binding strength is ∼10 kJ mol−1 more stable for RNA than DNA. In contrast, the anionic E/D contacts with DNA are significantly more stable than with RNA (by up to 30 kJ mol−1 for the mean values). Thus, there is more variation in the strength of the RNA and DNA nucleobase–amino acid π-interactions when the amino acids are charged. Nevertheless, since the data set of charged interactions is much smaller than for the neutral contacts, this discrepancy may simply reflect the specific geometries of the isolated contacts identified in nature to date.
TABLE 4.
Comparison of the RNA– or DNA–protein π-interaction energies (kJ mol−1) as a function of the amino acid
When the DNA–protein π-binding strengths are considered as a function of the nucleobase (Table 5), similar ranges in the stability of contacts with neutral amino acids are seen for RNA and DNA. However, the most common and average binding strengths are greater for the RNA purines than the DNA purines. Although C interactions exhibit similar stability in RNA– and DNA–protein complexes, interactions with T are on average slightly (∼3 kJ mol−1) stronger for T in DNA than U in RNA, which may reflect differences in the relative orientations of the nucleic acid and protein components and/or weak C–H···π contacts due to the additional methyl group of T (Rutledge et al. 2009). Nevertheless, the reverse trend holds when charged amino acids are considered, such that U contacts involving RNA are up to ∼3 kJ mol−1 on average more stable than T contacts involving DNA. The remaining trends discussed for the neutral contacts prevail when charged amino acids are considered, with the RNA purine interactions being more stable than the DNA purine contacts and little variation existing in the RNA versus DNA contacts with C.
TABLE 5.
Comparison of the RNA– or DNA–protein π-interaction energies (kJ mol−1) as a function of the nucleobase
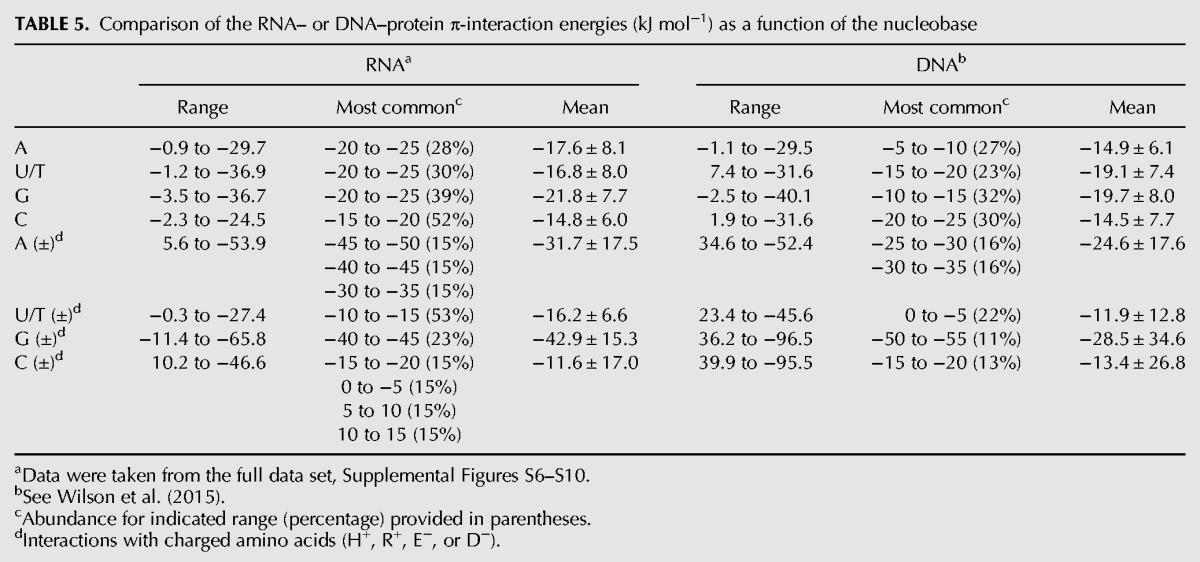
In summary, the intermolecular forces between the RNA or DNA nucleobases and cyclic or acyclic π-containing amino acid side chains can adopt a range of strengths regardless of the nucleic acid or protein components involved. There is no clear correlation between the maximum stability for a given nucleobase–amino acid pair and the relative abundance of that pair in nature. There is also no clear correlation between the strength of an interaction and the RNA/DNA base or amino acid involved in the contact. Nevertheless, since these noncovalent π-interactions provide significant stability to RNA/DNA–protein complexes, it is likely that their important role in biology will continue to be unveiled as more experimental structures of nucleic acid–protein complexes become available in the future.
Conclusions
A key problem in modern structural biology is understanding how proteins interact with nucleic acids. However, much less is known about RNA–protein than DNA–protein interactions since fewer structures have been resolved of RNA-binding proteins and RNA exhibits greater structural diversity. Nevertheless, with a growing number of crystal structures available for RNA–protein complexes, it is now possible to obtain accurate information about discrete RNA–protein interactions. In this light, the present study analyzed π-interactions between nucleobases and aromatic (cyclic) or acyclic π-containing amino acids. It was determined that RNA–protein π-interactions occur with many different RNA types. Contacts were identified for almost all combinations of nucleobases and aromatic amino acids, with F interactions comprising nearly half of all RNA contacts, while R interactions are most prevalent among those involving the acyclic protein components. In general, despite many different observed orientations of the nucleic acid and protein components, a nearly planar (stacked) relative arrangement of the RNA nucleobases and amino acids is preferred. Furthermore, regardless of the nucleobase or amino acid involved, a range of stabilizing intermolecular forces was determined for RNA–protein interactions in nature. Compared to DNA–protein interactions, RNA–protein π-contacts are relatively more abundant, exhibit different nucleobase and amino acid prevalence, and show a stronger preference with respect to the amino acid, which emphasizes differences in RNA– and DNA–protein π-interactions at the molecular level. However, RNA– and DNA–protein π-contacts exhibit similar relative nucleobase and amino acid arrangements in space, and similar ranges in the strength of the associated intermolecular forces. Overall, our results clarify the current picture of the molecular basis of nucleic acid–protein binding. Most importantly, our data emphasize that nucleobase–amino acid π-interactions can greatly contribute to the stability of nucleic acid–protein complexes and therefore have an important role to play in biology. Nevertheless, more work is still needed in this area, including further investigation of the properties of RNA nucleobase–amino acid π-interactions, the role and catalytic effects of specific RNA–protein nucleobase–amino acid π–π interaction, and the relation between RNA type and the role of specific π–π contacts. Furthermore, since other DNA components, specifically the phosphate (Luscombe et al. 2001; Lejeune et al. 2005) and sugar (Wilson et al. 2014, 2015) moieties, have been shown to commonly form strong interactions with π-containing amino acids, future work should investigate the prevalence, structure, and strength of the analogous RNA interactions.
MATERIALS AND METHODS
Data set
Experimental X-ray structures were selected and searched for RNA–protein nucleobase–amino acid π-interactions using the detailed protocol developed and implemented by our group to study the analogous DNA–protein contacts (Wilson et al. 2014, 2015). Specifically, experimental X-ray structures of RNA–protein complexes available in the protein data bank (PDB) were chosen for investigation using the predefined search algorithms present in the PDB, and the requirements that the resolution is better than 2.0 Å, and the publication date is prior to June 30, 2014. As a result, 120 high-resolution crystal structures were considered that contain a wide range in RNA types, but do not include hybrid RNA/DNA. This data set has a high level of sequence identity, which is consistent with a previous study of 61 RNA–protein complexes (Baker and Grant 2007), and permits study of a larger data set with accurate atomic positions. Nevertheless, using the Swiss Institute of Bioinformatics ExPASy server (Decrease Redundancy algorithm), a nonredundant data set that contains structures with a sequence identity of ≤30% and no crystallographic copies of interactions was extracted. This data set confirmed that our results are not biased by overrepresenting particular types of structures in our full data set, which is consistent with previous literature (Baker and Grant 2007). Additionally, we strongly believe that including all identified interactions allows us to generate a database of RNA–protein π–π interactions that can aid the development of automated search programs and allow researchers to gain further information about discrete π–π interactions in many different systems of interest.
In the crystal structures searched, each RNA–protein pair between an aromatic (W, H, F, and Y) or acyclic (R, E, and D) π-containing amino acid and a nucleobase (A, C, G, and U, Supplemental Fig. S2) with a separation distance <5 Å was selected using PyMOL (The PyMOL molecular graphics system, version 1.3r1, Schrodinger LLC). This distance criteria was confidently used based on optimum separation distances predicted for isolated pairs using high-level quantum mechanical studies (Rutledge et al. 2009; Wells et al. 2013) and previous successes using the same cutoff to study DNA–protein π-interactions in experimental structures (Wilson et al. 2014, 2015). To reduce ambiguity in the coordinates of all RNA–protein pairings, only residues that were fully resolved (contained no missing atoms) were included in the analysis. To unequivocally ensure that all contacts included in our analysis represent π-contacts, each nucleobase–amino acid pair identified was subsequently visually inspected, and structures that represent hydrogen-bonding interactions or noninteracting pairs were removed from the data set. To classify the identified π–π interactions, the closest heavy atom distance and the angle between the planes of the two π-systems (ω) were measured using Mercury (Macrae et al. 2008). Full details of the crystal structures searched, the nucleobase–amino acid pairs identified, and the classification of each π-interaction are provided in the Supplemental Material.
Quantum mechanical interaction energies
The strength of the intermolecular forces between each nucleobase–amino acid pair (binding or interaction energy) was calculated using models that neglect the RNA backbone and solely include the π-system in the protein side chain. Specifically, RNA nucleobases were considered by replacing the sugar–phosphate backbone with a hydrogen atom, and the protein backbone was replaced by a hydrogen atom at Cβ for the aromatic amino acids, or at Cα for D, Cβ for E, and Cγ for R. To consider different biologically relevant environments, H was considered in cationic and two neutral (δ and ε) forms, and D and E were modeled as neutral and anionic (Supplemental Fig. S2). Additionally, the Y and (neutral) D/E hydroxyl group was orientated in two directions that differ by mirror flipping the amino acid prior to stacking with the nucleobase. However, since the effects of the hydroxyl orientation are minimal, only the energy of the orientation resulting in the strongest energy is discussed. Each truncated model was optimized in a planar (Cs symmetric) conformation using MP2/6-31G(d), and overlaid onto the crystal structure coordinates using root-mean-square fitting of the heavy atoms according to algorithms available in Hyperchem 8.0.8 (Hypercube Inc.). Overlaying of the optimized truncated monomers onto the crystal structure orientations circumvents problems caused by deviations from the optimal nucleobase or amino acid structure results from high B-values. The gas-phase binding energy was calculated as the electronic energy difference between the nucleobase–amino acid pair and each monomer at the M06-2X/6-31+G(d,p) level using the Gaussian 09 program suite (revision A.02) (Frisch et al. 2009). The M06-2X methodology was chosen due to the computational speed required to consider the large number of interactions in the present work and is justified based on previous successes predicting the stability of π-interactions in nucleic acid–protein systems (Wilson et al. 2014, 2015). Although the calculated gas-phase interaction energies represent the more frequent RNA–protein binding environments of low polarity, binding strengths are anticipated to decrease in environments corresponding to more polar active sites. In these cases, the effects of amino acid charge will likely be significantly diminished. Nevertheless, previous work has shown that neutral and charged π–π interactions maintain significant strength even in more polar environments (Cauët et al. 2005; Rutledge et al. 2008; Churchill and Wetmore 2009). Regardless, future work should consider the effects of solvation in order to generalize our conclusions to many different RNA–protein binding environments.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC, 249598-07), Canada Research Chain Program (950-228175), and the Canada Foundation of Innovation (22770). K.A.W. also thanks NSERC (Vanier), Alberta Innovates-Technology Futures, and the University of Lethbridge for student scholarships. Computational resources from the Upscale and Robust Abacus for Chemistry in Lethbridge (URACIL), and those provided by Westgrid and Compute/Calcul Canada, are greatly appreciated.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.054924.115.
REFERENCES
- Allers J, Shamoo Y. 2001. Structure-based analysis of protein–RNA interactions using the program entangle. J Mol Biol 311: 75–86. [DOI] [PubMed] [Google Scholar]
- Bahadur RP, Zacharias M, Janin J. 2008. Dissecting protein–RNA recognition sites. Nucleic Acids Res 36: 2705–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CM, Grant GH. 2007. Role of aromatic amino acids in protein-nucleic acid recognition. Biopolymers 85: 456–470. [DOI] [PubMed] [Google Scholar]
- Barik A, C N, Pilla SP, Bahadur RP. 2015. Molecular architecture of protein–RNA recognition sites. J Biomol Struct Dyn 33: 2738–2751. [DOI] [PubMed] [Google Scholar]
- Byrgazov K, Grishkovskaya I, Arenz S, Coudevylle N, Temmel H, Wilson DN, Djinovic-Carugo K, Moll I. 2015. Structural basis for the interaction of protein S1 with the Escherichia coli ribosome. Nucleic Acids Res 43: 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauët E, Rooman M, Wintjens R, Liévin J, Biot C. 2005. Histidine-aromatic interactions in proteins and protein-ligand complexes: quantum chemical study of X-ray and model structures. J Chem Theory Comput 1: 472–483. [DOI] [PubMed] [Google Scholar]
- Cheng AC, Chen WW, Fuhrmann CN, Frankel AD. 2003. Recognition of nucleic acid bases and base-pairs by hydrogen bonding to amino acid side-chains. J Mol Biol 327: 781–796. [DOI] [PubMed] [Google Scholar]
- Churchill CDM, Wetmore SD. 2009. Noncovalent interactions involving histidine: the effect of charge on π-π stacking and T-shaped interactions with the DNA nucleobases. J Phys Chem B 113: 16046–16058. [DOI] [PubMed] [Google Scholar]
- Copeland KL, Anderson JA, Farley AR, Cox JR, Tschumper GS. 2008. Probing phenylalanine/adenine π-stacking interactions in protein complexes with explicitly correlated and CCSD(T) computations. J Phys Chem B 112: 14291–14295. [DOI] [PubMed] [Google Scholar]
- Copeland KL, Pellock SJ, Cox JR, Cafiero ML, Tschumper GS. 2013. Examination of tyrosine/adenine stacking interactions in protein complexes. J Phys Chem B 117: 14001–14008. [DOI] [PubMed] [Google Scholar]
- Ellis JJ, Broom M, Jones S. 2007. Protein–RNA interactions: structural analysis and functional classes. Proteins 66: 903–911. [DOI] [PubMed] [Google Scholar]
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, et al. 2009. Gaussian 09. Gaussian Inc, Wallingford, CT. [Google Scholar]
- Gromiha MM, Santhosh C, Ahmad S. 2004a. Structural analysis of cation-π interactions in DNA binding proteins. Int J Biol Macromol 34: 203–211. [DOI] [PubMed] [Google Scholar]
- Gromiha MM, Santhosh C, Suwa M. 2004b. Influence of cation-π interactions in protein–DNA complexes. Polymer 45: 633–639. [Google Scholar]
- Gromiha MM, Siebers JG, Selvaraj S, Kono H, Sarai A. 2005. Role of inter- and intramolecular interactions in protein–DNA recognition. Gene 364: 108–113. [DOI] [PubMed] [Google Scholar]
- Guzman I, Ghaemi Z, Baranger A, Luthey-Schulten Z, Gruebele M. 2015. Native conformational dynamics of the spliceosomal U1A protein. J Phys Chem B 119: 3651–3661. [DOI] [PubMed] [Google Scholar]
- Jeong E, Kim H, Lee S-W, Han K. 2003. Discovering the interaction propensities of amino acids and nucleotides from protein–RNA complexes. Mol Cells 16: 161–167. [PubMed] [Google Scholar]
- Jones S, Daley DTA, Luscombe NM, Berman HM, Thornton JM. 2001. Protein–RNA interactions: a structural analysis. Nucleic Acids Res 29: 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leavens FMV, Churchill CDM, Wang S, Wetmore SD. 2011. Evaluating how discrete water molecules affect protein–DNA π–π and π+–π stacking and T-shaped interactions: the case of histidine-adenine dimers. J Phys Chem B 115: 10990–11003. [DOI] [PubMed] [Google Scholar]
- Lejeune D, Delsaux N, Charloteaux B, Thomas A, Brasseur R. 2005. Protein-nucleic acid recognition: statistical analysis of atomic interactions and influence of DNA structure. Proteins 61: 258–271. [DOI] [PubMed] [Google Scholar]
- Lu XJ, Olson WK. 2008. 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat Protoc 3: 1213–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscombe NM, Thornton JM. 2002. Protein–DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J Mol Biol 320: 991–1009. [DOI] [PubMed] [Google Scholar]
- Luscombe NM, Laskowski RA, Thornton JM. 2001. Amino acid-base interactions: a three-dimensional analysis of protein–DNA interactions at an atomic level. Nucleic Acids Res 29: 2860–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae CF, Bruno IJ, Chisholm JA, Edgington PR, McCabe P, Pidcock E, Rodriguez-Monge L, Taylor R, van de Streek J, Wood PA. 2008. Mercury CSD 2.0–new features for the visualization and investigation of crystal structures. J Appl Crystallogr 41: 466–470. [Google Scholar]
- Mao L, Wang Y, Liu Y, Hu X. 2004. Molecular determinants for ATP-binding in proteins: a data mining and quantum chemical analysis. J Mol Biol 336: 787–807. [DOI] [PubMed] [Google Scholar]
- Morozova N, Allers J, Myers J, Shamoo Y. 2006. Protein–RNA interactions: exploring binding patterns with a three-dimensional superposition analysis of high resolution structures. Bioinformatics 22: 2746–2752. [DOI] [PubMed] [Google Scholar]
- Nahalka J, Hrabarova E, Talafova K. 2015. Protein–RNA and protein–glycan recognitions in light of amino acid codes. Biochim Biophys Acta 1850: 1942–1952. [DOI] [PubMed] [Google Scholar]
- Neugebauer KM. 2015. RNA: master or servant? RNA 21: 701–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabakaran P, Siebers JG, Ahmad S, Gromiha MM, Singarayan MG, Sarai A. 2006. Classification of protein–DNA complexes based on structural descriptors. Structure 14: 1355–1367. [DOI] [PubMed] [Google Scholar]
- Rutledge LR, Durst HF, Wetmore SD. 2008. Computational comparison of the stacking interactions between the aromatic amino acids and the natural or (Cationic) methylated nucleobases. Phys Chem Chem Phys 10: 2801–2812. [DOI] [PubMed] [Google Scholar]
- Rutledge LR, Durst HF, Wetmore SD. 2009. Evidence for stabilization of DNA/RNA-protein complexes arising from nucleobase-amino acid stacking and T-shaped interactions. J Chem Theory Comput 5: 1400–1410. [DOI] [PubMed] [Google Scholar]
- Rutledge LR, Churchill CDM, Wetmore SD. 2010. A preliminary investigation of the additivity of π−π or π+−π stacking and T-shaped interactions between natural or damaged DNA nucleobases and histidine. J Phys Chem B 114: 3355–3367. [DOI] [PubMed] [Google Scholar]
- Sathyapriya R, Vijayabaskar M, Vishveshwara S. 2008. Insights into protein–DNA interactions through structure network analysis. PLoS Comput Biol 4: e1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels JC, Tuite JB, Nolan SJ, Baranger AM. 2002. Investigation of a conserved stacking interaction in target site recognition by the U1A protein. Nucleic Acids Res 30: 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney BA, Roy P, Leontis NB. 2015. An introduction to recurrent nucleotide interactions in RNA. Wiley Interdiscip Rev RNA 6: 17–45. [DOI] [PubMed] [Google Scholar]
- Treger M, Westhof E. 2001. Statistical analysis of atomic contacts at RNA–protein interfaces. J Mol Recognit 14: 199–214. [DOI] [PubMed] [Google Scholar]
- Wells RA, Kellie JL, Wetmore SD. 2013. Significant strength of charged DNA-protein π-π interactions: a preliminary study of cytosine. J Phys Chem B 117: 10462–10474. [DOI] [PubMed] [Google Scholar]
- Wilson KA, Kellie JL, Wetmore SD. 2014. DNA–protein π-interactions in nature: abundance, structure, composition and strength of contacts between aromatic amino acids and DNA nucleobases or deoxyribose sugar. Nucleic Acids Res 42: 6726–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson KA, Wells RA, Abendong MN, Anderson CB, Kung RW, Wetmore SD. 2015. Landscape of π–π and sugar–π contacts in DNA–protein interactions. J Biomol Struct Dyn 34: 184–200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.