

Abstract
It is still poorly understood why α-synuclein, the intrinsically disordered protein involved in Parkinson’s and other neurodegenerative diseases, is so prone to aggregation. Recent work has shown a correlation between the aggregation rate and the rate of diffusional reconfiguration by varying temperature and pH. Here we examine the effects of several point mutations in the sequence on the conformational ensemble and reconfiguration rate. We find that at lower temperatures the PD causing aggregation enhancing mutations slow down and aggregation reducing mutations drastically speed up intramolecular diffusion, as compared to the wild type sequence. However, at higher temperatures, one of three familial mutations that enhance aggregation slows intramolecular diffusion while non-natural mutations that inhibit aggregation speed up intramolecular diffusion. These results support the hypothesis that the first step of aggregation is kinetically controlled by reconfiguration in which the protein chain cannot reconfigure rapidly enough to escape oligomerization. Finally we provide physical and chemical insights into why small point mutations cause these dramatic changes in the conformational ensemble and dynamics.
Graphical abstract
INTRODUCTION
A number of neurological diseases, most notably Parkinson’s disease (PD), seem to result from the aggregation of the 140 residue, intrinsically disordered protein α-synuclein.1 Recent work has shown that α-synuclein is mostly unstructured in vitro, though not a true random coil, and highly diffusive.2–9 However, it is still not very well understood why α-synuclein and some of its naturally occurring mutant forms are so very aggregation prone. The process is likely initiated by aggregation prone segments becoming exposed to solvent and enabling them to form intermolecular interactions.10 Ultimately, the determinant of whether a sequence will aggregate is embedded within the ensemble of unfolded states.11 This ensemble samples different conformations by intramolecular diffusion and it has been shown by a simple model that when such conformational sampling slows down, aggregation competent monomer conformations can easily associate with each other, thus initiating the process of bimolecular association.12 On the other hand, when reconfiguration is extremely fast, these aggregation competent conformations do not have time to make stabilizing interactions and aggregation is prevented.
In agreement with this model, Ahmad et al. showed that the average size of disordered α-synuclein compacts and intramolecular diffusion slows with increasing temperature.13 This nonintuitive result correlates with the propensity to aggregate. Therefore, it is the nature of the conformational ensemble and dynamics of reconfiguration that are in crucial control of aggregation speed. This observation has been used to find small molecule inhibitors that are able to prevent early aggregation of α-synuclein.14,15 It is possible that point mutations in the amino acid sequence would also affect reconfiguration in a manner that correlates with aggregation propensity.
There have been numerous attempts in vitro to study the effects of aggregation-enhancing, PD-causing, familial mutations16,17 on wild type α-synuclein by measuring and characterizing the aggregates that appear at later stages of aggregation using conventional methods such as ThT fluorescence, atomic force microscopy, electron microscopy, size exclusion chromatography, and light scattering.18–22 Each of these mutations is unique in the lag time, formation rate, and/or morphology of fibrils and protofibrils, yet they all accelerate disease. Therefore, understanding how these mutations affect how an oligomeric dimer may form from an unstructured monomer could be useful in exploring the early stages in aggregation, which is difficult to probe using existing methods. A potential application of this technique is to screen natural mutations to identify those that encourage early stage aggregation and increase the risk of PD.23,24
In this work we attempt to understand the sequence-dependent effect in more detail by examining the effects of point mutations on monomeric α-synuclein. The PD causing A30P mutant was reported to slow down reconfiguration by a factor of 2 at physiological temperatures,13 but here we attempt to investigate if the previous measurement reflected a localized effect on the sequence or if it was indeed a global phenomenon. We also test the effect of the other PD causing mutants A53T and E46K on the wildtype sequence of alpha-synuclein. Our measurements show that all the three mutants slow down diffusion of alpha-synuclein at lower temperatures. At higher temperatures, both A53T and E46K have intramolecular diffusion similar to the wild type, which lies in the dangerous regime where aggregation is extremely likely. However, A30P slows down diffusion more than wildtype at higher temperatures, making bimolecular contacts more likely compared to the wildtype and the other two mutants A53T and E46K. This correlates with previously reported studies where A30P was reported to form early stage oligomers faster than wild-type,25 while E46K and A53T were found to accelerate aggregation by enhancing fibrillation.20,26 To test if early stage aggregation can be correlated with slower diffusion in sequences, we further created two non-natural mutants in different parts of the alpha-synuclein sequence. Both these mutants reportedly inhibit the aggregation of the protein.23,24 Our measurements show that both these mutants drastically speed up the diffusion of alpha-synuclein. This supports a model in which formation of early stage oligomers such as dimers is a competition between intramolecular reconfiguration and bimolecular diffusion, where faster diffusion enables the sequence to escape from forming bimolecular contacts.
MATERIALS AND METHODS
α-Synuclein Mutation, Expression, and Purification
The α-synuclein plasmid was a kind gift from Gary Pielak (University of North Carolina, Chapel Hill, NC). The α-synuclein mutant containing the tryptophan/cysteine (Trp/Cys) pairs Y39W/A69C (hereafter named WT39–69) and A69C/F94W (hereafter named WT69–94) was created using the QuikChange site-directed mutagenesis kit (Stratagene). For mutation studies, a third mutation was then introduced in either the WT39–69 or WT69–94, and five such triple mutants were created, namely A30P, A53T, E46K, V74E, and T72P. Each of the mutations was confirmed by DNA sequencing. The mutants were then expressed in E. coli BL21 (DE3) cells and purified by a procedure described previously.1,2 The monomeric α-synuclein peak obtained using size exclusion was found to be >95% pure as assessed by SDS-PAGE and Coomassie blue staining.
Trp-Cys Contact Quenching Experiments
For each measurement, the respective α-synuclein mutant fractions were thawed and diluted 10× in 25 mM sodium phosphate buffer (pH 7.5), 10 mM TCEP (to prevent disulfide bond formation), 10 mM NaCl and various sucrose concentrations (0, 10, 20, and 30% w/w). The A30P39–69 was completed with 0 mM NaCl and measured by Ahmad et al.13 The final concentration of the mutant proteins was kept fixed at 30 μM for each set of temperature and viscosity measurements. The buffer, sucrose, and TCEP solutions were bubbled with N2O for 1 h to eliminate oxygen and scavenge solvated electrons created in the UV laser pulse. Triplet lifetime decay kinetics of tryptophan as a result of cysteine quenching was measured with an instrument described previously.27 Briefly, the tryptophan triplet state was excited by a 10 ns laser pulse at 289 nm created from the fourth harmonic of an Nd:YAG laser (Continuum) and a 1-m Raman cell filled with 450 PSI of D2 gas. The triplet population was probed at 441 nm by a HeCd laser (Kimmon). The probe and the reference beam were measured with silicon detectors and then combined in a differential amplifier (DA 1853A, LeCroy) with an additional stage of a 350-MHz preamplifier (SR445A, Stanford Research Systems). The total gain was 50-fold. All measurements were completed within 30 min, much less than the observed lag times for α-synuclein (~60 h). Variation of solution viscosity was achieved by varying the sucrose concentrations. Each sucrose concentration was measured at six temperatures (0, 10, 15, 20, 30, and 40 C). The viscosity of each sucrose concentrations at each different temperature was measured independently using a cone-cup viscometer (Brookfield engineering).
RESULTS
Choice of Mutations
To employ the Trp-Cys quenching method in this work, the sequence of α-synuclein was modified to contain a Tyr(Y) → Trp(W) mutation at position 39 and an Ala(A) → Cys(C) mutation at position 69, creating a W39/C69 loop on which the Trp-Cys quenching could be measured. Ahmad et al. showed that these mutations did not change the aggregation rate significantly as measured by ThT fluorescence or the CD spectrum.13 Thus, we will call the Y39W/A69C mutation hereafter WT39–69. Two sets of further mutations were chosen in order to investigate the effects on the polypeptide chain dynamics of α-synuclein using the Trp-Cys quenching method on the W39/C69 loop. The first set is the well-known familial mutations, A53T, A30P (previously measured in ref 13), and E46K, which have been found to accelerate PD in humans. In order to investigate the effect of aggregation reducing mutations on α-synuclein, we chose two previously reported fibrillation-defective mutants of α-synuclein, V74E and T72P.23,24
Trp-Cys Measurements and Data Analysis
The schematic representation of Trp-Cys contact quenching measurement is shown in Figure 1a. To measure intramolecular diffusion, we excite the W39 to a long-lived triplet state, which is quenched upon close contact with C69, almost 400-times more efficiently than with any other amino acid. The observed rate of triplet decay consists of two processes, intramolecular diffusion and irreversible quenching of the triplet by Cys on close contact. In equilibrium, intramolecular diffusion brings the Trp and Cys within the same polypeptide together with a diffusion-limited forward rate kD+, where it may be quenched with rate q or diffuse away with rate kD−. The observed rate is given by28
| (1) |
Equation 1 can be rewritten as29
| (2) |
We assume the reaction-limited rate kR depends only on temperature (T) but kD+ depends on both temperature and viscosity of the solvent (η). Therefore, by making measurements at different viscosities for a constant temperature we can extract both kR and kD+ by fitting a plot of 1/kobs vs η at a given temperature to a line in which the intercept is 1/kR and the slope is 1/ηkD+.
Figure 1.
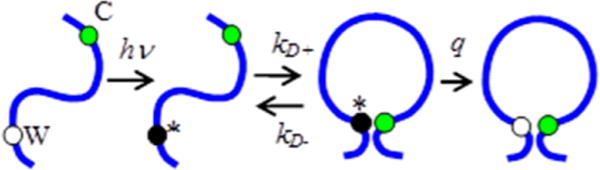
Schematic of Trp-Cys quenching measurement.
Figure S1 shows the plots of 1/kobs vs η for all the 39–69 sequences. These times are all ~1–10 μs, indicating that the chains are freely diffusing monomers. Stable oligomers would not be freely diffusing, and the lifetimes would be ~40 μs, the natural lifetime of the Trp triplet. For the wildtype and aggregation enhancing mutations, the intercept decreases and slope increases drastically with temperature. The reaction-limited and diffusion-limited rates are plotted in Figure 2a,b. At low temperatures, the reaction-limited rates for A53T and E46K are ~2× lower than those for the wildtype and the A30P is within error, but the diffusion-limited rates are fairly similar to the wildtype. At 30 and 40 °C the intercept approaches zero, 1/kR ≈ 0 (for some measurements, the fitted value is actually negative but within error of 0). Therefore, in these cases the observed rate is diffusion-limited, kobs = kD+. The triangles in Figure 2b plot kobs for the wildtype and aggregation enhancing mutants at 30 and 40 C. Only A30P is significantly different from the wildtype. For the aggregation-inhibiting mutations (T72P and V74E) the intercepts do not approach zero for any temperature, and the slopes are fairly constant with temperature, displaying behavior seen previously in the cases of unstructured peptides and denatured proteins.11,27,29–34 The reaction-limited and diffusion-limited rates for the aggregation-inhibiting mutations are plotted in Figure 3a,b and diverge significantly from the wildtype at T = 30 and 40 °C.
Figure 2.
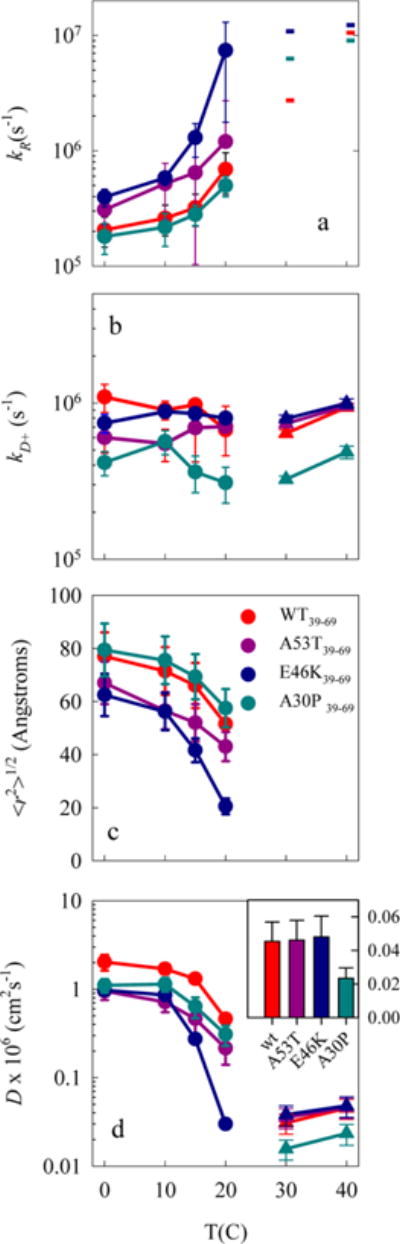
Effect of aggregation-enhancing, PD-causing, familial mutations (A53T, E46K, and A30P). (a) Reaction-limited rates. The dashes at 30 and 40 °C represent the lower limit of the rates. (b) (circles) Diffusion-limited rates, normalized to the viscosity at water at that temperature. Error bars are determined from the linear fits. (triangles) At T = 30 and 40 °C the observed rates are diffusion limited (kobs ~ kD+) and the error bars are the standard deviation of multiple measurements. The subscripts in the legend indicate the location of the Trp and Cys. (c) Average root-mean-square distance between the Trp and Cys determined for each reaction-limited rate using eqs 4 and 6. Error bars are determined as the change in distance required to change kR by the measured error. (d) (circles) Intramolecular diffusion coefficients determined for each diffusion-limited rate using eq 5. The error bars are determined as proportional to the error in kD+. (triangles) D determined from values in (b) using eq 3 and ⟨r2⟩ = 400 ± 100 Å2. The error bars are the propagated error of kD+ and ⟨r2⟩. Inset: The diffusion coefficients of the different mutants at 30 °C.
Figure 3.
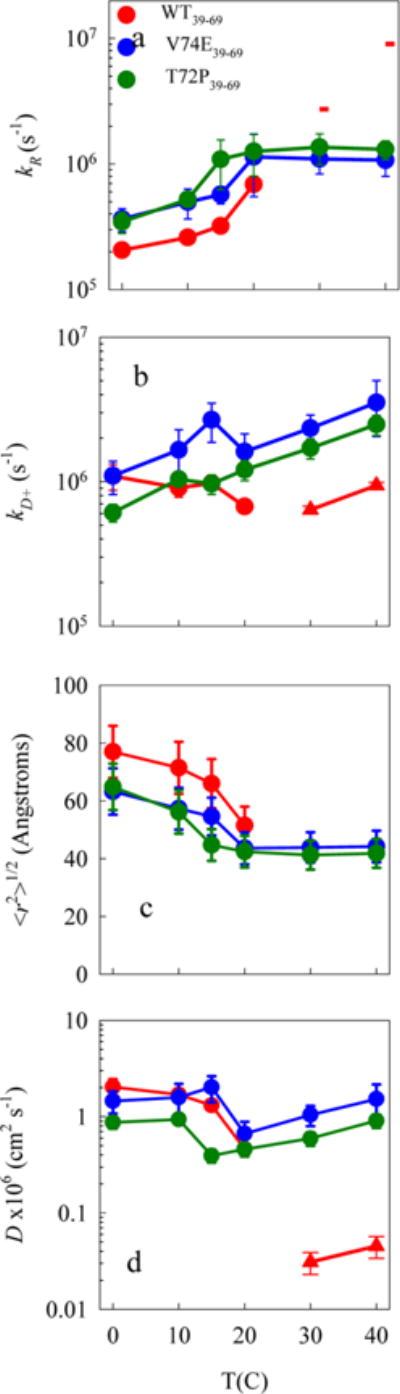
Effect of aggregation reducing mutations (V74E and T72P). (a) Reaction-limited rates. The dashes represent the lower limit of this rate. (b) (circles) Diffusion-limited rates, normalized to the viscosity of water at that temperature. Error bars are determined from the linear fits. (triangles) The observed rates are diffusion limited (kobs ≈ kD+), and the error bars are the standard deviation of multiple measurements. The subscripts in the legend indicate the location of the Trp and Cys. (c) Average root-mean-square distance between the Trp and Cys determined for each reaction-limited rate using eqs 4 and 6. Error bars are determined as the change in distance required to change kR by the measured error. (d) Intramolecular diffusion coefficients determined for each diffusion-limited rate using eq 5. The error bars are determined as proportional to the error in kD+. (triangles) D determined from values in (b) using eq 3 and ⟨r2⟩ = 400 ± 100 Å2. The error bars are the propagated error of kD+ and ⟨r2⟩.
To interpret these rates we use a theory by Szabo, Schulten, and Schulten (SSS), which models intramolecular diffusion as diffusion on a 1-dimensional potential of mean force determined by the probability of intrachain distances (P(r)). If the observed rate is diffusion-limited and we assume the intrachain distances are approximated by a Gaussian distribution, the diffusion-limited rate is given by28
| (3) |
where dα = 4 Å, the distance of closest approach, ⟨r2⟩ is the average Trp-Cys distance, squared, and D is the effective intramolecular diffusion coefficient. For the aggregation-enhancing mutations plus wildtype at T ≤ 20 C and the aggregation-inhibiting mutations at all temperatures, the observed rate is not diffusion-limited because diffusion is much faster. In this case we use a more detailed form of SSS theory. The measured reaction-limited (kR) and diffusion-limited (kD+) rates are given by29
| (4) |
| (5) |
where the integration limits are dα = 4 Å, and lc, the contour length of the protein. r is the distance between the Trp and Cys, and q(r) = 4.2 × 109 exp(4.0(r − 4.0)) s−1 is the experimentally determined distance-dependent quenching rate.35 This rate for the Trp-Cys system drops off very rapidly beyond 4.0 Å, so the reaction-limited rate is mostly determined by the probability of the shortest distances.35 Very generally, kR and kD+ are both inversely proportional to the average volume of the chain and kD+ is directly proportional to D.
To determine D we assume the probability distribution, P(r), for each mutant and temperature is given by a Gaussian chain model
| (6) |
where ⟨r2⟩, the average Trp-Cys distance, is an adjustable parameter, and N is a normalization constant such that ∫P(r) = 1. For each mutant and temperature, ⟨r2⟩ was found such that the calculated kR from eq 3 matched the measured rate. These distances are plotted in Figures 2c and 3c. In the absence of any mutations the WT39–69 compacts significantly and its diffusion slows down significantly with temperature.13 These values are likely unrealistic because a Gaussian curve is a much broader distribution than expected for a compact chain, but they serve as a rough guide for comparison of the chain size at various temperatures and sequences.
Alternatively, an energy reweighted wormlike chain (WLC) is a more realistic model of the unfolded chain. This model begins with 10 million randomly generated wormlike chains with a persistence length of 4 Å and a backbone excluded diameter of 4 Å. Each of these chains is given an energy determined by the number of close range interactions between residues of similar hydrophobicity.11
| (7) |
where the hydropathy, h, for each residue in the sequence is given using the Miyazawa-Jernigan scale. We assume that two residues with h values within 30% of each other contribute to hydrophobic or hydrophilic interactions, reducing the free energy of the system and therefore making conformations containing such interactions favorable. We also apply a distance cutoff to EH such that EH = 0 for |ri − rj| > 6.5 Å. Using the interaction energy EH, we can reweight the probability distribution P(r) to yield a new distribution Z(r)11
| (8) |
The reaction-limited rate is calculated using eq 4 and substituting Z(r) for P(r). The only adjustable parameter in this model is σ, which can be tuned from 0 to ~2.5. This model is sequence specific so, in principle, the different mutants should produce the measured differences in kR for the same tuning parameter, σ. The model fails on this account and therefore σ is assigned individually for each measurement. Table S1 shows the best values of σ that can be computed and the resulting ⟨r⟩ at 20 and 30 °C.
Using either polymeric model, the intramolecular diffusion constant, D, is determined from eq 5 using the appropriate calculated distribution and the measured kD+. Figure 4 shows the computed values of D for both models at 20 °C. Although the average Trp-Cys distance is significantly smaller for the reweighted WLC than for the Gaussian chain, the values of D are fairly close. Thus, for purposes of comparing D between different mutants, the Gaussian chain and the reweighted WLC are equivalent. The values of D for the Gaussian chain are plotted in Figures 2d and 3d. For those sequences (WT and aggregation-enhancing mutations) that are diffusion-limited at 30 and 40 °C, we assume ⟨r2⟩1/2 ≈ 20 Å for all mutants and use eq 3 to calculate D. This value is approximately what is required to achieve k ≈ 107s−1 using eq 4. It is also close to the value of a maximally compact polymer given by Flory, R ≈ lkN1/3 ≈ 19 Å, where lk = 3.8 Å is the single amino acid length and N = 140 is the peptide length.
Figure 4.
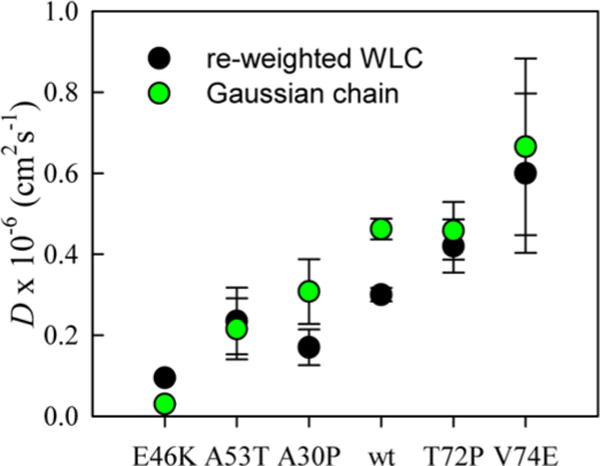
Average intramolecular diffusion coefficient of the W39–C69 loop for each mutant calculated using the Gaussian chain and energy reweighted wormlike chain (WLC) at 20 °C.
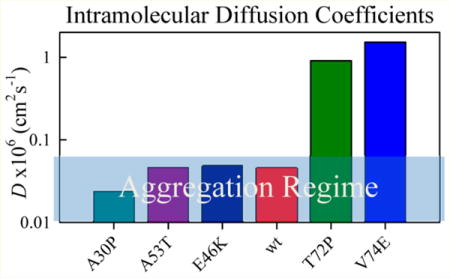
At low temperatures (0–15 °C), diffusion is about 2× slower for A30P, A53T, and E46K compared to wildtype, as shown in Figure 2d. Near 20 C, diffusion begins to slow significantly for these sequences, though it appears to occur at a lower temperature for E46K than for the others, resulting in a diffusion coefficient that is ~15× lower than the wildtype. Above 30 C the observed rates for all these sequences become diffusion-limited and we can no longer distinguish differences in chain volume. If we assume ⟨r2⟩ = 400 ± 100 Å2 for all these sequences, then diffusion coefficients for A53T and E46K are within error of the wildtype. Only A30P is significantly slower, as illustrated by the inset to Figure 2d.
Figure 3d compares the aggregation-inhibiting mutations. Both V74E and T72P mutations behaved similarly by opening up the polypeptide chain and making it diffuse faster at higher temperatures. At 30 °C the V74E mutation was observed to increase diffusion by at least 25-fold while the T72P mutation by at least 14-fold compared to WT39–69.
Ahmad et al. also demonstrated that other Trp-Cys loops throughout the sequence showed the same dynamics at pH 7.5. Therefore, we assume the 39W-69C loop is sensitive to the global dynamics of the chain.13 To check this assumption we made the A30P and V74E mutations to the WT69–94 sequence containing the 69C-94W loop. These mutations are plotted in Figure S2 and show similar changes as the mutations discussed above, although the reduction in diffusion coefficient by A30P at 30 °C is smaller in the 69W-94C loop than the 39W-69C loop.
DISCUSSION
α-Synuclein is an intrinsically disordered protein in vitro that adopts an ensemble of conformations that are stabilized by long-range interactions involving the N- and C-termini. A number of SAXS and NMR studies have looked at how these long-range interactions change with the mutants E46K, A30P, and A53T and have produced conflicting reports on whether these mutations compact the chain compared to the wildtype.19,36–40 CD and other NMR studies show minor differences in the preference for secondary structure between the mutants and the wildtype, but no difference represents a significant population within the broad ensemble that can explain the difference in aggregation behavior of the familial mutants with that of the wild type.36,38,39 Molecular dynamics simulations have produced similarly conflicting reports.37,40,41 Therefore, the key factor that drives these mutants to be so much more aggregation prone and pathogenic in nature than the WT and causes early onset of PD is still unknown.
In this work we have investigated how point mutations of the α-synuclein sequence affect the dynamics of the intrinsically disordered protein chain. The aggregation-inhibiting mutations, T72P and V74E, each make the chain more expanded and diffusion faster at T > 20 °C (see Figure 3d), while the aggregation-inducing mutations, A53T and E46K, make the chain slightly more compact and diffusion slower at T ≤ 20 °C but have no measurable effect at T > 20 °C compared to wildtype possibly due to our inability to assess differences in chain size at these temperatures. Ahmad et al.13 showed that WT α-synuclein compacts significantly and enters a dangerous regime of aggregation at T > 30 °C wherein it is much more likely to make stable bimolecular contacts, making it significantly aggregation prone at those temperatures.13 Our measurements here show that both A53T and E46K preserves the same kind of behavior of the sequence at higher temperatures, while further lowering the diffusion at lower temperatures and making bimolecular contact formation much more likely than the wildtype. The aggregation-inducing mutation A30P does not compact the chain significantly but slows down reconfiguration dynamics at all temperatures, enabling the sequence to make bimolecular contacts more easily than the wildtype under all conditions measured.
It should be noted that the intramolecular diffusion coefficient, D, is not the same as that obtained from classical Brownian diffusion which is entirely governed by the viscosity of the solution. If the chain was freely diffusing, each residue would randomly move as if it was a free amino acid. In reality, the chain constrains the movement of each residue both sterically and by adding some drag to the motion. This produces diffusion on a potential of mean force which is related to P(r). Furthermore, a real chain in water has many attractive and repulsive intramolecular interactions that further constrain diffusion. D as calculated in eq 5 reflects all of these contributions. The effect of these mutations is to change the pattern of intramolecular interactions to bias D compared to the wildtype pattern.
The correlation of diffusion constant with aggregation propensity can be reconciled with a model of aggregation in which the first step of dimerization depends on the speed of reconfiguration dynamics. The details of the model are described in the SI. Briefly, if the rate of diffusive reconfiguration of the monomer chain is much faster than bimolecular diffusion, when two monomers come into contact in an encounter complex, stabilizing interactions cannot be made because each chain reconfigures too quickly. However, if reconfiguration slows to about the rate of bimolecular association, stabilizing interactions can be made to form a stable oligomer. Therefore, if intramolecular diffusion (D) is slower than WT, as seen for A30P, oligomerization will be more rapid, and if diffusion is faster than WT, as observed for T72P and V74E, oligomerization is slower. As shown in the SI, assuming all other rates are the same, the oligomer formation rate scales with D. Note that later steps of oligomerization, fibril conversion and fibril elongation may not depend on reconfiguration because stable intermolecular contacts slow down intramolecular diffusion and may instead be more sensitive to structure of these states, which could also be altered with point mutations. For example, A30P is well-known to form fibrils significantly slower than the wildtype, even though it appears to accelerate Parkinson’s disease.42,43 It is likely that oligomers are formed faster but the rate of fibril conversion is slower. Conversely, A53T and E46K form fibrils faster than wildtype but do not change reconfiguration significantly compared to that of the wildtype under physiological temperatures.
Dimer formation rates were recently measured directly using single molecule methods. Lv et al.44 observed the residence times of fluorescently labeled α-synuclein interacting with unlabeled surface-immobilized α-synuclein. For the wildtype, Lv et al. measured a residence time of ~200 ms, much longer than the reconfiguration times measured here, indicating the encounter complex is quite stable. Furthermore, the aggregation enhancing mutations, A53T and E46K showed only modest (15% and 50%, respectively) increases in residence times, while A30P was 3 times longer than the wildtype, commensurate with our measurements of D. These lifetimes therefore support the model in which the aggregation-enhancing mutations increase time spent in the encounter complex by slowing diffusion. Although we could not measure slower diffusion for A53T and E46K outside of error, these measurements suggest diffusion is indeed slightly slower for these mutants compared to wildtype. Intriguingly Lv et al. also observe a longer lifetime for 10–20% of the dimers, ~3 s. This population could correspond to the final oligomers described in the model.
Our measurements show that a single-point mutation in a 140 residue long chain can affect the largely disordered monomeric ensemble quite significantly by either increasing or decreasing diffusion coefficients by up to 1 order of magnitude. Such differences may very well be the underlying cause of drastic differences between aggregation behaviors of the mutants with that of the wildtype. Hoffmann et al. showed that proteins of different sequences followed different polymer scaling laws and that the scaling exponent varied by mean net charge and mean hydrophobicity.45 Unfortunately no coarse sequence determinants such as these precisely capture the observed effects of the single-point mutations measured in this work. Furthermore, wildtype α-synuclein is not a typical unstructured polypeptide. Other unstructured sequences that have been measured typically show only a small increase in kR and actually an increase in kD+ with temperature.11,27,46 Wildtype α-synuclein shows opposite behavior, with a drastic increase in kR with temperature, wherein the chain compacts significantly, and a decrease in kD+, implying a dramatic slowdown in diffusion. The temperature-dependent behavior of kD+ for α-synuclein may indicate that this sequence is on the verge of an anomalous glass transition near physiological temperatures.13 It appears E46K is shifting this glass-transition temperature down while T72P and V74E are shifting it up or eliminating the transition entirely.
Most of the mutations have a change in hydrophobicity which could affect the temperature dependence as well. The seminal work by Brandts showed that the free energy of hydration for different residues had different temperature dependencies.47 Theoretically, Huang and Chandler showed the temperature of maximum chemical potential depended on the size of the hydrophobic solute.48 More recently, Li and Walker directly measured the compaction of hydrophobic homopolymers by single molecule force measurements and showed that different types of hydrophobic groups shifted the temperature dependence of free energy of extension significantly.49 Data compiled by Wuttke et al. of the free energy of solvation of all amino acids vs temperature showed that Ala and Pro have similar positive free energies and little temperature dependence, while Thr has a negative free energy and a strong temperature dependence,50 which could explain the A53T and T72P results. However, there is no well-developed sequence-specific theory to predict the ensemble changes that we observe here. As we showed above, the reweighted WLC model cannot account for these sequence changes using the same tuning parameter, σ. This makes it a challenge to predict changes in intramolecular diffusion quantitatively.
Two mutations, V74E and E46K involve changes in charge. The net charge of the wildtype sequence at physiological pH is ~−9, so the aggregation-inhibiting mutation makes the net charge more negative (−10) and the aggregation-enhancing mutation makes it less negative (−7). Using the theory of Higgs and Joanny,51 we calculate that the chain radius of E46K should be 18% smaller than the wildtype, similar to the difference in chain size at 0 C. The radius of V74E should be 8% larger, within our error at 0 °C but much smaller than we estimate at 30–40 °C. Pappu and co-workers have further postulated that there exists a coil-to-globule transition for most intrinsically disordered sequences at high net positive or negative charge, though the charge pattern and hydrophobicity affect where this transition occurs.52,53 Commensurate with the discussion above, the wildtype sequence may sit on the edge of this transition and simply increasing or decreasing the net charge will push the ensemble into coil or globule states, with bigger changes than estimated by net charge alone. As with hydrophobicity, there is still no sequence-specific theory to predict these effects.
The only mutation that does not significantly alter the conformational ensemble (as detected by kR) at any temperature is A30P, in agreement with the similar free energies of solvation for Ala and Pro found by Wuttke et al.50 However, A30P does have about a 2-fold lower diffusion coefficient than the WT suggesting that proline acts as a stiffer link in the chain than alanine. This may also explain why the effect is bigger for the 39W-69C loop than the 69C-94W loop since the stiffness is essentially a local sequence effect. The same effect can also be seen for T72P. This mutant has practically the same reaction-limited rates at all temperatures as V74E but the diffusion coefficient is ~2-fold slower than V74E. That proline adds stiffness to the chain is not at all surprising. Lapidus et al. showed that polyproline chains as short as 3 residues had no measurable end-to-end contact.28
In conclusion, we have shown that the reconfiguration rates of various mutants of α-synuclein correlates strongly with the propensity to make early bimolecular contacts both in vitro and in vivo, supporting the hypothesis that aggregation is kinetically controlled by intramolecular diffusion. Experimentally, this work is unique in directly measuring reconfiguration dynamics but is relatively poor at describing the entire conformational ensemble, especially at high temperatures when the ensemble is quite compact. Thus, future measurements need to be coupled to other temperature-dependent measurements of specific intramolecular distances, such as single-molecule FRET. A fundamental question that remains incompletely answered is how these small changes to sequence have such a huge effect on dynamics. Future work is needed to produce predictive models of conformation ensembles based on the sequence that is sensitive to single-point mutations.
Supplementary Material
Acknowledgments
We thank Gary Pielak for the gift of the α-synuclein plasmid and Terry Ball with assistance in protein mutation, expression, and purification. This work is supported by the National Institutes of Health, R01 GM100908.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.5b10136.
Description of the model of oligomer formation, Table S1 and Figures S1 and S2. (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Cookson MR. The Biochemistry of Parkinson’s Disease. Annu Rev Biochem. 2005;74:29–52. doi: 10.1146/annurev.biochem.74.082803.133400. [DOI] [PubMed] [Google Scholar]
- 2.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Griesinger C, Jovin TM, Zweckstetter M. Release of Long-Range Tertiary Interactions Potentiates Aggregation of Natively Unstructured Alpha-Synuclein. Proc Natl Acad Sci U S A. 2005;102(5):1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cho M-K, Nodet G, Kim H-Y, Jensen MR, Bernado P, Fernandez CO, Becker S, Blackledge M, Zweckstetter M. Structural Characterization of A-Synuclein in an Aggregation Prone State. Protein Sci. 2009;18(9):1840–1846. doi: 10.1002/pro.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping Long-Range Interactions in A-Synuclein Using Spin-Label Nmr and Ensemble Molecular Dynamics Simulations. J Am Chem Soc. 2005;127(2):476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 5.Eliezer D, Kutluay E, Bussell R, Browne G. Conformational Properties of Alpha-Synuclein in Its Free and Lipid-Associated States. J Mol Biol. 2001;307(4):1061–1073. doi: 10.1006/jmbi.2001.4538. [DOI] [PubMed] [Google Scholar]
- 6.Grupi A, Haas E. Segmental Conformational Disorder and Dynamics in the Intrinsically Disordered Protein [Alpha]-Synuclein and Its Chain Length Dependence. J Mol Biol. 2011;405(5):1267–1283. doi: 10.1016/j.jmb.2010.11.011. [DOI] [PubMed] [Google Scholar]
- 7.Lee JC, Gray HB, Winkler JR. Tertiary Contact Formation in Alpha-Synuclein Probed by Electron Transfer. J Am Chem Soc. 2005;127(47):16388–16389. doi: 10.1021/ja0561901. [DOI] [PubMed] [Google Scholar]
- 8.Lee JC, Lai BT, Kozak JJ, Gray HB, Winkler JR. A-Synuclein Tertiary Contact Dynamics. J Phys Chem B. 2007;111(8):2107–2112. doi: 10.1021/jp068604y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trexler AJ, Rhoades E. Single Molecule Characterization of [Alpha]-Synuclein in Aggregation-Prone States. Biophys J. 2010;99(9):3048–3055. doi: 10.1016/j.bpj.2010.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bemporad F, Chiti F. Protein Misfolded Oligomers: Experimental Approaches, Mechanism of Formation, and Structure-Toxicity Relationships. Chem Biol. 2012;19(3):315–327. doi: 10.1016/j.chembiol.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Wedemeyer WJ, Lapidus LJ. A General Polymer Model of Unfolded Proteins under Folding Conditions. J Phys Chem B. 2010;114(48):15969–15975. doi: 10.1021/jp104746g. [DOI] [PubMed] [Google Scholar]
- 12.Lapidus LJ. Understanding Protein Aggregation from the View of Monomer Dynamics. Mol BioSyst. 2013;9(1):29–35. doi: 10.1039/c2mb25334h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmad B, Chen Y, Lapidus LJ. Aggregation of A-Synuclein Is Kinetically Controlled by Intramolecular Diffusion. Proc Natl Acad Sci U S A. 2012;109(7):2336–2341. doi: 10.1073/pnas.1109526109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acharya S, Safaie BM, Wongkongkathep P, Ivanova MI, Attar A, Klärner F-G, Schrader T, Loo JA, Bitan G, Lapidus LJ. Molecular Basis for Preventing A-Synuclein Aggregation by a Molecular Tweezer. J Biol Chem. 2014;289:10727. doi: 10.1074/jbc.M113.524520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmad B, Lapidus LJ. Curcumin Prevents Aggregation in A-Synuclein by Increasing Reconfiguration Rate. J Biol Chem. 2012;287(12):9193–9199. doi: 10.1074/jbc.M111.325548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Alasopro Mutation in the Gene Encoding [Alpha]-Synuclein in Parkinson’s Disease. Nat Genet. 1998;18(2):106–108. doi: 10.1038/ng0298-106. [DOI] [PubMed] [Google Scholar]
- 17.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, et al. Mutation in the A-Synuclein Gene Identified in Families with Parkinson’s Disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 18.Conway KA, Harper JD, Lansbury PT. Accelerated in Vitro Fibril Formation by a Mutant [Alpha]-Synuclein Linked to Early-Onset Parkinson Disease. Nat Med. 1998;4(11):1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 19.Li J, Uversky VN, Fink AL. Effect of Familial Parkinson’s Disease Point Mutations A30p and A53t on the Structural Properties, Aggregation, and Fibrillation of Human A-Synuclein†. Biochemistry. 2001;40(38):11604–11613. doi: 10.1021/bi010616g. [DOI] [PubMed] [Google Scholar]
- 20.Greenbaum EA, Graves CL, Mishizen-Eberz AJ, Lupoli MA, Lynch DR, Englander SW, Axelsen PH, Giasson BI. The E46k Mutation in A-Synuclein Increases Amyloid Fibril Formation. J Biol Chem. 2005;280(9):7800–7807. doi: 10.1074/jbc.M411638200. [DOI] [PubMed] [Google Scholar]
- 21.Pandey N, Schmidt RE, Galvin JE. The Alpha-Synuclein Mutation E46k Promotes Aggregation in Cultured Cells. Exp Neurol. 2006;197(2):515–520. doi: 10.1016/j.expneurol.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 22.Fredenburg RA, Rospigliosi C, Meray RK, Kessler JC, Lashuel HA, Eliezer D, Lansbury PT. The Impact of the E46k Mutation on the Properties of A-Synuclein in Its Monomeric and Oligomeric States†. Biochemistry. 2007;46(24):7107–7118. doi: 10.1021/bi7000246. [DOI] [PubMed] [Google Scholar]
- 23.Koo HJ, Choi MY, Im H. Aggregation-Defective Alpha-Synuclein Mutants Inhibit the Fibrillation of Parkinson’s Disease-Linked Alpha-Synuclein Variants. Biochem Biophys Res Commun. 2009;386(1):165–169. doi: 10.1016/j.bbrc.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 24.Koo HJ, Lee HJ, Im H. Sequence Determinants Regulating Fibrillation of Human Alpha-Synuclein. Biochem Biophys Res Commun. 2008;368(3):772–778. doi: 10.1016/j.bbrc.2008.01.140. [DOI] [PubMed] [Google Scholar]
- 25.Conway KA, Lee S-J, Rochet J-C, Ding TT, Williamson RE, Lansbury PT. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both A-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc Natl Acad Sci U S A. 2000;97(2):571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narhi L, Wood SJ, Steavenson S, Jiang YJ, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, et al. Both Familial Parkinson’s Disease Mutations Accelerate Alpha-Synuclein Aggregation. J Biol Chem. 1999;274(14):9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 27.Singh VR, Kopka M, Chen Y, Wedemeyer WJ, Lapidus LJ. Dynamic Similarity of the Unfolded States of Proteins L and G. Biochemistry. 2007;46:10046–10054. doi: 10.1021/bi700270j. [DOI] [PubMed] [Google Scholar]
- 28.Lapidus LJ, Eaton WA, Hofrichter J. Measuring the Rate of Intramolecular Contact Formation in Polypeptides. Proc Natl Acad Sci U S A. 2000;97(13):7220–7225. doi: 10.1073/pnas.97.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lapidus LJ, Steinbach PJ, Eaton WA, Szabo A, Hofrichter J. Effects of Chain Stiffness on the Dynamics of Loop Formation in Polypeptides. Appendix: Testing a 1-Dimensional Diffusion Model for Peptide Dynamics. J Phys Chem B. 2002;106(44):11628–11640. [Google Scholar]
- 30.Neuweiler H, Löllmann M, Doose S, Sauer M. Dynamics of Unfolded Polypeptide Chains in Crowded Environment Studied by Fluorescence Correlation Spectroscopy. J Mol Biol. 2007;365(3):856–869. doi: 10.1016/j.jmb.2006.10.021. [DOI] [PubMed] [Google Scholar]
- 31.Bieri O, Kiefhaber T. Elementary Steps in Protein Folding. Biol Chem. 1999;380(7–8):923–929. doi: 10.1515/BC.1999.114. [DOI] [PubMed] [Google Scholar]
- 32.Fierz B, Kiefhaber T. End-to-End Vs Interior Loop Formation Kinetics in Unfolded Polypeptide Chains. J Am Chem Soc. 2007;129(3):672–679. doi: 10.1021/ja0666396. [DOI] [PubMed] [Google Scholar]
- 33.Buscaglia M, Schuler B, Lapidus LJ, Eaton WA, Hofrichter J. Kinetics of Intramolecular Contact Formation in a Denatured Protein. J Mol Biol. 2003;332:9–12. doi: 10.1016/s0022-2836(03)00891-x. [DOI] [PubMed] [Google Scholar]
- 34.Chen YJ, Parrini C, Taddei N, Lapidus LJ. Conformational Properties of Unfolded Hypf-N. J Phys Chem B. 2009;113(50):16209–16213. doi: 10.1021/jp904189b. [DOI] [PubMed] [Google Scholar]
- 35.Lapidus LJ, Eaton WA, Hofrichter J. Dynamics of Intramolecular Contact Formation in Polypeptides: Distance Dependence of Quenching Rates in a Room-Temperature Glass. Phys Rev Lett. 2001;87(25):4. doi: 10.1103/PhysRevLett.87.258101. [DOI] [PubMed] [Google Scholar]
- 36.Bussell R, Eliezer D. Residual Structure and Dynamics in Parkinson’s Disease-Associated Mutants of A-Synuclein. J Biol Chem. 2001;276(49):45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 37.Coskuner O, Wise-Scira O. Structures and Free Energy Landscapes of the A53t Mutant-Type A-Synuclein Protein and Impact of A53t Mutation on the Structures of the Wild-Type A-Synuclein Protein with Dynamics. ACS Chem Neurosci. 2013;4(7):1101–1113. doi: 10.1021/cn400041j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferreon ACM, Moran CR, Ferreon JC, Deniz AA. Alteration of the A-Synuclein Folding Landscape by a Mutation Related to Parkinson’s Disease. Angew Chem Int Ed. 2010;49(20):3469–3472. doi: 10.1002/anie.201000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rospigliosi CC, McClendon S, Schmid AW, Ramlall TF, Barré P, Lashuel HA, Eliezer D. E46k Parkinson’s-Linked Mutation Enhances C-Terminal-to-N-Terminal Contacts in A-Synuclein. J Mol Biol. 2009;388(5):1022–1032. doi: 10.1016/j.jmb.2009.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wise-Scira O, Dunn A, Aloglu AK, Sakallioglu IT, Coskuner O. Structures of the E46k Mutant-Type A-Synuclein Protein and Impact of E46k Mutation on the Structures of the Wild-Type A-Synuclein Protein. ACS Chem Neurosci. 2013;4(3):498–508. doi: 10.1021/cn3002027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wise-Scira O, Aloglu AK, Dunn A, Sakallioglu IT, Coskuner O. Structures and Free Energy Landscapes of the Wild-Type and A30p Mutant-Type A-Synuclein Proteins with Dynamics. ACS Chem Neurosci. 2013;4(3):486–497. doi: 10.1021/cn300198q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Conway KA, Lee S-J, Rochet J-C, Ding TT, Williamson RE, Lansbury PT. Acceleration of Oligomerization, Not Fibrillization, Is a Shared Property of Both A-Synuclein Mutations Linked to Early-Onset Parkinson’s Disease: Implications for Pathogenesis and Therapy. Proc Natl Acad Sci U S A. 2000;97(2):571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Narhi L, Wood SJ, Steavenson S, Jiang Y, Wu GM, Anafi D, Kaufman SA, Martin F, Sitney K, Denis P, et al. Both Familial Parkinson’s Disease Mutations Accelerate A-Synuclein Aggregation. J Biol Chem. 1999;274(14):9843–9846. doi: 10.1074/jbc.274.14.9843. [DOI] [PubMed] [Google Scholar]
- 44.Lv Z, Krasnoslobodtsev AV, Zhang Y, Ysselstein D, Rochet J-C, Blanchard SC, Lyubchenko YL. Direct Detection of A-Synuclein Dimerization Dynamics: Single-Molecule Fluorescence Analysis. Biophys J. 2015;108(8):2038–2047. doi: 10.1016/j.bpj.2015.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hofmann H, Soranno A, Borgia A, Gast K, Nettels D, Schuler B. Polymer Scaling Laws of Unfolded and Intrinsically Disordered Proteins Quantified with Single-Molecule Spectroscopy. Proc Natl Acad Sci U S A. 2012;109(40):16155–16160. doi: 10.1073/pnas.1207719109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Voelz VA, Jäger M, Yao S, Chen Y, Zhu L, Waldauer SA, Bowman GR, Friedrichs M, Bakajin O, Lapidus LJ, et al. Slow Unfolded-State Structuring in Acyl-Coa Binding Protein Folding Revealed by Simulation and Experiment. J Am Chem Soc. 2012;134(30):12565–12577. doi: 10.1021/ja302528z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brandts JF. The Thermodynamics of Protein Denaturation. Ii. A Model of Reversible Denaturation and Interpretations Regarding the Stability of Chymotrypsinogen. J Am Chem Soc. 1964;86(20):4302–4314. [Google Scholar]
- 48.Huang DM, Chandler D. Temperature and Length Scale Dependence of Hydrophobic Effects and Their Possible Implications for Protein Folding. Proc Natl Acad Sci U S A. 2000;97(15):8324–8327. doi: 10.1073/pnas.120176397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li ITS, Walker GC. Signature of Hydrophobic Hydration in a Single Polymer. Proc Natl Acad Sci U S A. 2011;108(40):16527–16532. doi: 10.1073/pnas.1105450108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wuttke R, Hofmann H, Nettels D, Borgia MB, Mittal J, Best RB, Schuler B. Temperature-Dependent Solvation Modulates the Dimensions of Disordered Proteins. Proc Natl Acad Sci U S A. 2014;111(14):5213–5218. doi: 10.1073/pnas.1313006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Higgs PG, Joanny JF. Theory of Polyampholyte Solutions. J Chem Phys. 1991;94(2):1543–1554. [Google Scholar]
- 52.Mao AH, Crick SL, Vitalis A, Chicoine CL, Pappu RV. Net Charge Per Residue Modulates Conformational Ensembles of Intrinsically Disordered Proteins. Proc Natl Acad Sci U S A. 2010;107(18):8183–8188. doi: 10.1073/pnas.0911107107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng W, Lyle N, Luan B, Raleigh DP, Pappu RV. Experiments and Simulations Show How Long-Range Contacts Can Form in Expanded Unfolded Proteins with Negligible Secondary Structure. Proc Natl Acad Sci U S A. 2013;110(6):2123–2128. doi: 10.1073/pnas.1216979110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.