

Abstract
To drive lymphocyte proliferation and differentiation, common γ-chain (γc) cytokine receptors require hours to days of sustained stimulation. While JAK1 and JAK3 kinases are found together in all γc-receptor complexes, it is not known how their respective catalytic activities contribute to signaling over time. Here, we dissect the temporal requirements for JAK3 kinase activity with a selective covalent inhibitor (JAK3i). By monitoring STAT5 phosphorylation over 20 hours in IL-2-stimulated CD4+ T cells, we document a previously unappreciated second wave of signaling that is much more sensitive to JAK3i than the first wave. Selective inhibition of this second wave is sufficient to block cyclin expression and S-phase entry. An inhibitor-resistant JAK3 mutant (Cys905Ser) rescued all effects of JAK3i in isolated T cells and in mice. Our chemical genetic toolkit elucidates a biphasic requirement for JAK3 kinase activity in IL-2-driven T-cell proliferation and will find broad utility in studies of γc-receptor signaling.
Introduction
The common gamma chain (γc) receptor cytokines (IL-2, 4, 7, 9, 15 and 21) are essential for lymphocyte development, survival, differentiation and proliferation1. All six of these cytokines signal by engaging a receptor complex comprising the common gamma chain (IL-2Rγ, CD132) and one or two additional chains. These receptors have no intrinsic kinase activity. Hence, the tightly associated cytoplasmic kinases, JAK1 (bound to IL-2Rβ and others) and JAK3 (bound to IL-2Rγ)2, are required for signal transduction and are clinically validated targets for the treatment of autoimmune diseases1,3. In the prototypical case of IL-2, ligand binding rapidly triggers the phosphorylation of 3 tyrosines within the IL-2Rβ cytoplasmic tail (Y341, Y395, Y498) and the subsequent recruitment, phosphorylation, and activation of the transcription factor STAT5. In addition, IL-2 receptor ligation activates the PI3K/AKT and MEK/ERK pathways via the recruited adapter protein Shc4. Genetic studies indicate that among these downstream signaling events, STAT5 activation is especially critical, enabling full lymphocyte expansion in vitro and in vivo as well as immune regulation by CD4+ regulatory T-cells5–8.
Loss of JAK3 in mice and humans results in severely compromised T-cell function9,10. Nevertheless, the requirement for JAK3 kinase activity downstream of IL-2R and other γc cytokine receptors remains controversial. Critical tyrosines on IL-2Rβ and STAT5 are likely phosphorylated by JAK1 and/or JAK3, but none of these phosphorylation events have been uniquely attributed to either kinase11. Interestingly, a recent study concluded that JAK1 kinase activity is necessary and sufficient for IL-2-stimulated STAT5 phosphorylation, whereas JAK3 kinase activity is dispensable12. Instead, JAK3 was proposed to play an essential scaffolding role. These conclusions were based primarily on experiments in cell lines expressing combinations of kinase-dead and analog-sensitive JAK1/3 mutants. Moreover, and of particular relevance to the work presented here, signaling events were only followed for one hour after IL-2 stimulation.
The temporal requirements for JAK3 kinase activity in driving cytokine-induced cell proliferation are completely unknown. Is a transient pulse of JAK3 activity sufficient to drive quiescent T cells into S-phase (DNA synthesis), or is sustained JAK3 signaling required? As is true for other γc cytokine-driven processes13–16, T-cell proliferation requires at least 6 hours of continuous exposure to IL-217. Yet, receptor-proximal signaling events (e.g., IL-2Rβ and STAT5 phosphorylation) are most often assessed at early time points (< 1 hr) after receptor stimulation. The relative contributions of JAK1 and JAK3 kinase activity to cell proliferation, and how they evolve over time, is a central unanswered question in JAK-STAT signaling.
To define the temporal requirements for JAK3 kinase activity in a manner not achievable with genetic knockout approaches, we employed a highly selective inhibitor, JAK3i. Like other recently reported JAK3-selective inhibitors18–20, JAK3i forms a covalent bond with a cysteine found in JAK3, but not the closely related kinase domains in JAK1, JAK2, or TYK2. We exploited the nonessential nature of this cysteine to generate an inhibitor-resistant JAK3 mutant (Cys905Ser). This combined chemical and genetic toolkit revealed new insights into JAK3 signaling requirements in the context of IL-2-stimulated primary CD4+ T cells. Contrary to the report described above12, we find that JAK3 kinase activity is absolutely essential for STAT5 phosphorylation. Through detailed time-course experiments, we characterize a previously unreported second wave of STAT5 phosphorylation that is sustained for at least 10 hours after IL-2 addition. This second, more prolonged wave of signaling is exquisitely sensitive to JAK3 inhibition and is essential for T-cell proliferation. Finally, we demonstrate that JAK3i abolishes IL-2-driven T-cell proliferation in mice, a phenotype that is completely rescued by C905S JAK3.
Results
JAK3i is selective for JAK3 over closely related kinases
To dissect the role of JAK3 in γc-receptor signaling, we sought a highly selective chemical probe that would allow us to study signaling with temporal control and without perturbing lymphocyte development. However, the discovery of JAK3-selective inhibitors has proven challenging due to the nearly identical ATP binding sites shared between all JAK-family kinase domains21. Although we recently identified reversible covalent JAK3 inhibitors with selectivity over other JAK-family kinases by targeting a cysteine unique to JAK3 (Supplementary Results, Supplementary Fig. 1)18, these compounds were not sufficiently potent for cellular studies and lacked selectivity toward other cysteine-containing kinases such as ITK, a critical T-cell kinase22. As an alternative, we identified a series of acrylamide-containing pyrrolopyrimidines, exemplified by JAK3i (1, Fig. 1), in a patent application23. Although compounds related to JAK3i were reported to inhibit JAK3 with subnanomolar potency in biochemical assays, no data were provided on the activity of these compounds in cells or animals.
Figure 1.
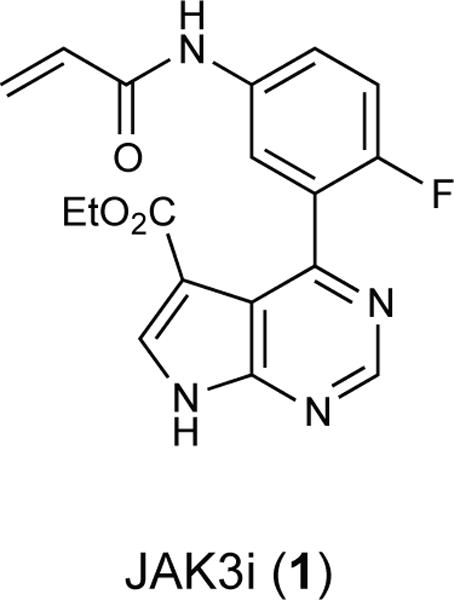
Chemical Structure of JAK3i.
JAK3i was highly selective in biochemical kinase assays, with >3,000-fold selectivity for JAK3 over the closely related kinases JAK1, JAK2, and TYK2 (Supplementary Table 1). In addition to JAK3, 9 other kinases share a structurally equivalent cysteine. They include the TEC-family kinases, which play an important role in antigen receptor signaling22. When tested against a small panel of these kinases in the presence of physiological ATP, JAK3i was also highly selective for JAK3: 1,300-fold vs. EGFR, 600-fold vs. ITK and 50-fold vs. BTK (Supplementary Table 2).
JAK3i differentially affects IL-2 signaling events
The high selectivity of JAK3i within the JAK family enabled us to elucidate the specific requirements for JAK3 catalytic activity in γc-receptor signaling. For these experiments, we isolated primary murine CD4+ T cells and activated them via plate-bound αCD3 and αCD28 to induce expression of the high-affinity IL-2 receptor. We then monitored IL-2-stimulated STAT5 phosphorylation by FACS. Contrary to an earlier chemical genetic study12, we found that JAK3 kinase activity was absolutely required for STAT5 phosphorylation (pSTAT5) after 15 minutes of IL-2 stimulation (IC50 47 nM, Fig. 2a). Our finding is consistent with a recently published report in which a structurally distinct, irreversible JAK3 inhibitor was found to block STAT5 phosphorylation at early time points19. The effect of JAK3-selective inhibitors on later IL-2-driven signaling events has not been reported and is the primary focus of our study.
Figure 2. Early and late signaling events are differentially affected by JAK3i.
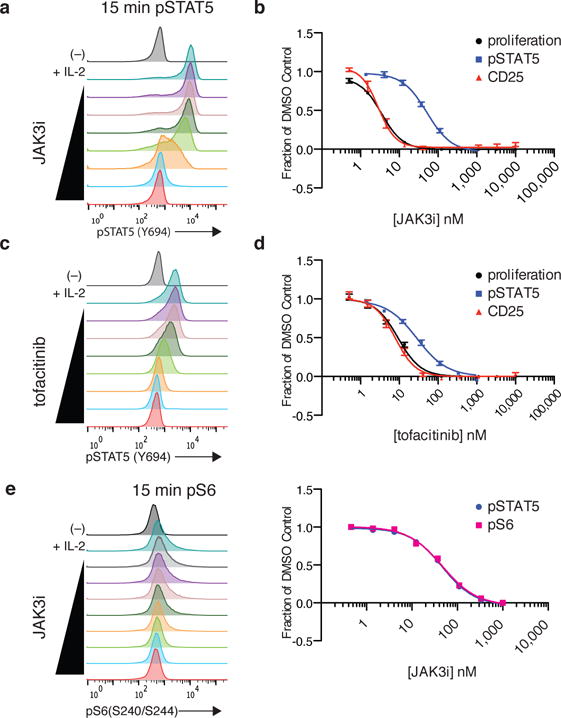
CD4+ T-cell blasts were pretreated for 2 hrs with indicated dose of JAK3i (a–b, e) or tofacitinib (c–d) and stimulated with IL-2 (50 u/mL). (a & c) STAT5 phosphorylation was monitored by phosphoflow 15 min after IL-2 stimulation. (c & d) Dose-response curves comparing effects on pSTAT5, proliferation, and CD25 upregulation. Proliferation was assessed by 3H-thymidine uptake after 24–48 hrs. CD25 (IL-2Rα) upregulation was quantified by FACS after 24 hrs (see Supplementary Fig. 2a for FACS histograms). e) S6 phosphorylation (Ser240/244) was monitored by phosphoflow in parallel with pSTAT5 after 15 minutes of IL-2 stimulation. See Supplementary Fig. 4c for corresponding pSTAT5 histograms. Data in a,c & e are representative of 5, 8 and 2 independent experiments. Data in b are cumulative of 6 (proliferation) and 4 (pSTAT5 & CD25) independent experiments, plotted as mean ± SEM. Data in c are cumulative of 3 (proliferation), 7 (pSTAT5) and 5 (CD25) independent experiments, plotted as mean ± SEM. See Supplementary Table 3 for average IC50 values from all experiments.
To our surprise, three downstream readouts of IL-2 signaling, all measured at 24–48 hours after stimulation, were much more sensitive to JAK3i (IC50 3–7 nM), with significant inhibition occurring at concentrations that had no effect on pSTAT5: (1) upregulation of IL-2Rα (CD25) at 24 hours (IC50 3.1 nM, Supplementary Fig. 2a and Fig. 2b); (2) proliferation, quantified by 3H-thymidine uptake from 24–48 hours (IC50 3.7 nM, Fig. 2b); (3) IFN-γ production at 24 hours (IC50 6.8 nM, Supplementary Fig. 2b). This disconnect between early and late signaling readouts was less pronounced with the pan-JAK inhibitor tofacitinib21. Rather, tofacitinib’s concentration-dependent effects on early pSTAT5 more closely mirrored its effects on CD25 upregulation, T-cell proliferation, and IFN-γ production (Fig. 2c–d and Supplementary Fig. 2a–b), as expected given that all three processes require STAT5-mediated transcription. Thus, in contrast to tofacitinib, a JAK3-selective inhibitor can prevent critical STAT5-dependent processes (e.g., T-cell proliferation) in the absence of any effect on early STAT5 phosphorylation. This suggests that the requirements for JAK3 (and possibly, JAK1) catalytic activity change over time, a possibility we address in greater detail below.
The apparent disconnect between JAK3i effects on early pSTAT5 and downstream readouts could potentially be explained by inhibition of MEK/ERK or PI3K/mTOR pathways, both of which are canonically activated by IL-24. In our primary CD4+ T-cell blasts, we detected very little phospho-ERK (Supplementary Fig. 3a), and MEK/ERK signaling was dispensable for proliferation (Supplementary Fig. 3b). By contrast, inhibition of PI3K, AKT or mTOR was sufficient to abolish IL-2-stimulated proliferation (Supplementary Fig. 4a), consistent with prior studies in human T-cell lines24. Upon stimulation with IL-2 for 15 minutes, we consistently detected a small induction of phosphorylated ribosomal protein S6 (a p70S6K substrate and proxy for PI3K and mTOR signaling25) by phosphoflow (Supplementary Fig. 4b). This induction was blocked by JAK3i, but with an equivalent IC50 to pSTAT5 (Fig. 2e), suggesting that these two processes have similar requirements for JAK3 catalytic activity.
C905S JAK3 mutant rescues JAK3i effects
Before pursuing additional mechanistic studies, we sought to ensure that all phenotypes induced by JAK3i were indeed the result of JAK3 kinase inhibition. We established this through multiple independent approaches. Compound 2, which lacks cysteine reactivity but is otherwise structurally identical to JAK3i, is >200-fold less potent in all biochemical and cellular assays (Supplementary Fig. 5). To address the possibility that JAK3i acts by targeting an alternative cysteine-containing kinase (e.g., ITK or RLK, both expressed in T cells) or any other protein besides JAK3, we retrovirally overexpressed a JAK3 mutant lacking the key cysteine (C905S) (Supplementary Fig. 6a). Relative to the empty vector, overexpression of wild-type JAK3 in mouse CD4+ T-cell blasts caused a slight reduction in JAK3i sensitivity in the IL-2 proliferation assay (IC50 23 nM, Fig. 3a), consistent with the large increase in cellular JAK3 levels (Supplementary Fig. 6b). In contrast to WT JAK3, overexpression of C905S JAK3 completely prevented JAK3i effects on T-cell proliferation at relevant concentrations, resulting in an apparent 1,000-fold shift in potency (IC50 > 4,000 nM). STAT5 phosphorylation and CD25 upregulation were similarly rescued by C905S JAK3 (Fig. 3b and Supplementary Fig. 6c). These genetic perturbations had no effect on tofacitinib sensitivity (Supplementary Fig. 6d–e). JAK3i thus provides a powerful tool for inactivating JAK3 catalytic activity in an acute, selective, and graded manner. The C905S JAK3 mutant, which is resistant to JAK3i, completes the chemical genetic toolkit by enabling genetic rescue experiments to rigorously control for potential off-target effects.
Figure 3. C905S JAK3 mutant rescues JAK3i effects in primary CD4+ T cells.
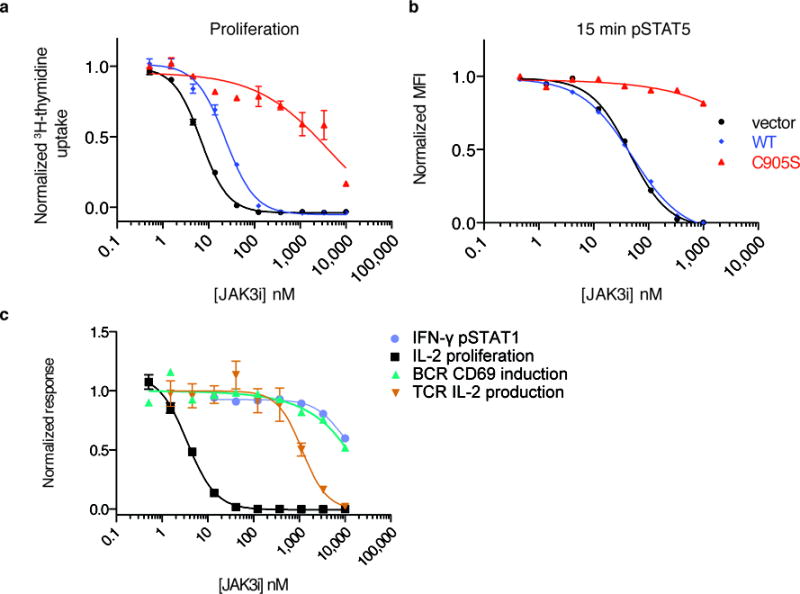
Rescue of JAK3i-mediated inhibition of (a) proliferation in unsorted CD4+ cells and (b) pSTAT5 (t = 15 min) in CD4+GFP+ cells. Cells were retrovirally transduced with empty vector, WT JAK3, or C905S JAK3, treated with JAK3i for 2 hrs, and stimulated with 50 units/mL rhIL-2. See Supplementary Fig. 6a for transduction efficiency. (a) and (b) are representative of two independent experiments. (c) Cellular selectivity of JAK3i in primary murine immune cells. IFN-γ-stimulated pSTAT1 (JAK1/JAK2 dependent), measured by phosphoflow, and IL-2-driven proliferation (JAK1/JAK3 dependent), measured by 3H-thymidine uptake, were assessed in CD4+ T-cell blasts. T-cell receptor (TCR)-driven IL-2 production (ITK/RLK dependent) was assessed by ELISA in naïve CD4+ T cells. Activation of naïve B cells (BTK dependent) by B-cell receptor (BCR) stimulation (α-IgM) was determined by FACS measurement of CD69 upregulation. Data are representative of 2 (IFN-γ, BCR), 9 (IL-2), or 3 (TCR) independent experiments. See Supplementary Table 4 for IC50 values from all cellular selectivity experiments. Proliferation (a, c) and IL-2 ELISA (c) data are plotted as mean ± SEM of triplicates.
We obtained additional evidence supporting the selectivity of JAK3i from cellular assays dependent on JAK or TEC-family kinases: IFN-γ-stimulated STAT1 phosphorylation (JAK1 and JAK2), TCR-stimulated IL-2 production (ITK and TXK/RLK27), and BCR-stimulated CD69 expression (BTK). Consistent with the results from biochemical kinase assays, JAK3i had no significant effect on these potentially confounding pathways (IC50 > 1,100 nM, Fig. 3c, Supplementary Table 4) at concentrations that abolished IL-2-driven proliferation and STAT5 phosphorylation (Fig. 2b and Fig. 3a,b).
Two waves of IL-2 signaling
The inability of JAK3i to inhibit STAT5 phosphorylation at concentrations that blocked proliferation, CD25 induction, and IFN-γ production was initially puzzling, since these processes are known to be STAT5-dependent5. However, by monitoring the dynamics of STAT5 phosphorylation over 20 hours, we discovered a second wave of signaling that, to our knowledge, has not been characterized previously. STAT5 phosphorylation peaked initially at 15 minutes and then declined to an intermediate level by one hour. In a majority of cells, pSTAT5 intensity rapidly returned to the original peak level and remained at this level for at least 10 hours (Fig. 4a,b). Signaling through the PI3K/mTOR pathway, while detected at early time points, was most prominent 2–6 hours after stimulation (Supplementary Fig. 7).
Figure 4. A second wave of STAT5 signaling is highly sensitive to JAK3 inhibition.
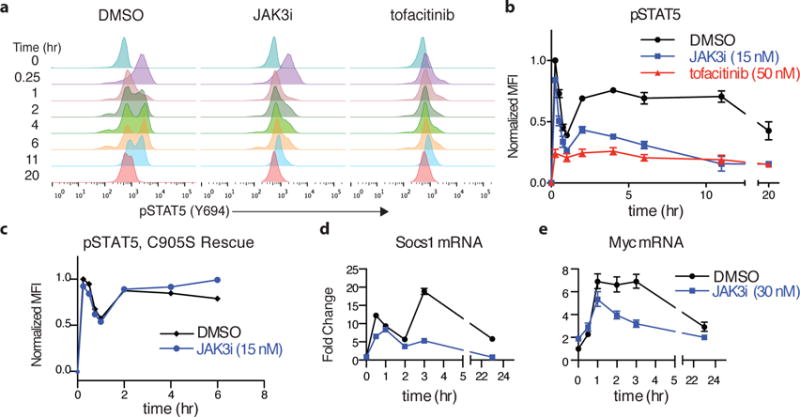
(a) CD4+ T-cell blasts were pretreated with inhibitors at the EC90 dose for blocking T-cell proliferation (JAK3i 15 nM, tofacitinib 50 nM) for 2 hrs and then stimulated with IL-2 (50 u/mL). pSTAT5 was monitored by phosphoflow at the indicated times. (b) pSTAT5 time course from (a), quantified by mean fluorescence intensity (MFI), normalized to the maximum value (t = 15 min, DMSO) and plotted ± SEM of 10 independent experiments. (c) Time course showing C905S JAK3 rescue of pSTAT5 inhibition by JAK3i. (d–e) Cells were pretreated with JAK3i (30 nM) and stimulated with IL-2. mRNA levels were monitored by qPCR at the indicated times. Plotted as mean fold-induction with 95% confidence intervals. Data are representative of 10 (a), 2 (c) or 3 (d–e) independent experiments.
Washout experiments have shown that continuous exposure to IL-2 over a period at least 6 hours is required to drive T-cell blasts into S phase17. Based on this precedent and our observation of a second wave of signaling, we surmised that JAK3 catalytic activity might be especially critical during the second wave, potentially accounting for the potent effects of JAK3i on T-cell proliferation. To test this, we treated cells with a concentration of JAK3i sufficient to block IL-2-driven proliferation by 90% (EC90 15 nM) and monitored pSTAT5 levels over time. Although it had little effect on the first wave, JAK3i at 15 nM knocked down pSTAT5 and pS6 levels by >50% in a sustained manner throughout the second wave (Fig. 4a,b, Supplementary Fig. 7). As expected, this inhibitory effect was completely prevented in cells expressing the JAK3i-resistant allele, C905S JAK3 (Fig. 4c and Supplementary Fig. 8). The pan-JAK inhibitor tofacitinib had a completely different profile from JAK3i, reducing pSTAT5 to a similar extent during the first and second waves when tested at its EC90 for T-cell proliferation (50 nM). Importantly, the remaining levels of second-wave pSTAT5 were nearly identical in cells treated with JAK3i and tofacitinib (Fig. 4a,b). Hence, with both JAK3i and tofacitinib, a sustained yet partial reduction (>50%) in pSTAT5 levels during the second wave correlates with a nearly complete block in T-cell proliferation. Moreover, this second wave of STAT5 phosphorylation is much more sensitive to selective JAK3 inhibition than the first wave, whereas both waves are equally sensitive to pan-JAK inhibition.
To test whether the effects of JAK3i on pSTAT5 correlate with effects on transcription, we analyzed a panel of STAT5-dependent genes implicated in T-cell proliferation, survival, and cytokine production. Based on their temporal expression patterns, these genes can be divided into three groups: first, a biphasic pattern exemplified by Socs1 (a negative regulator of JAK-STAT signaling) and Fos (Fig. 4d and Supplementary Fig. 9a); second, early and sustained induction, exemplified by Myc and Ifng (Fig. 4e and Supplementary Fig. 9b); and third, delayed induction, roughly coinciding with the second wave of pSTAT5 and exemplified by Bcl2 and Il2ra (Supplementary Fig. 9c). In all cases, pretreatment with JAK3i had little effect during the first 1–2 hours following IL-2 stimulation, yet profoundly reduced mRNA levels at later time points, consistent with more potent effects on pSTAT5 during the second wave.
Cell cycle progression requires continuous JAK3 activity
The above results demonstrate a correlation between JAK3i’s effects on late IL-2 signaling events and T-cell proliferation. To test directly whether JAK3 inhibition during the second wave is sufficient to prevent cell cycle progression, we added JAK3i to IL-2-stimulated cells after the initial peak of pSTAT5 had subsided. Treatment with JAK3i at the onset of the second wave induced rapid (within 30 min) and sustained reduction of both pSTAT5 and pS6 levels, similar to pretreating cells before IL-2 stimulation (Fig. 5a, Supplementary Fig. 10a). Moreover, adding JAK3i with or 2 hours after IL-2 stimulation similarly inhibited S-phase entry (Fig. 5b), as assessed by labeling cells with 5-ethynyl-2′-deoxyuridine (EdU) for one hour at the onset of S phase (t = 24 hrs, Supplementary Fig. 10b). Addition of 15 nM JAK3i at later time points during the second wave led to a progressive increase in the fraction of cells incorporating EdU. However, even an 8-hour delay in JAK3i treatment prevented more than half the cells from entering S phase (Fig. 5b). Strikingly, late addition of 100 nM JAK3i, a higher but still selective dose (Fig. 3), up to 8 hours after IL-2 stimulation inhibited S-phase entry by 95% (Fig. 5c); a minority of cells (~30%) managed to escape when this higher dose was delayed for 12 hours. These experiments establish a minimal requirement of 8–12 hours of sustained JAK3 catalytic activity for T cells to progress from G1 to S phase in response to IL-2.
Figure 5. Sustained JAK3 activity is required for S-phase entry.
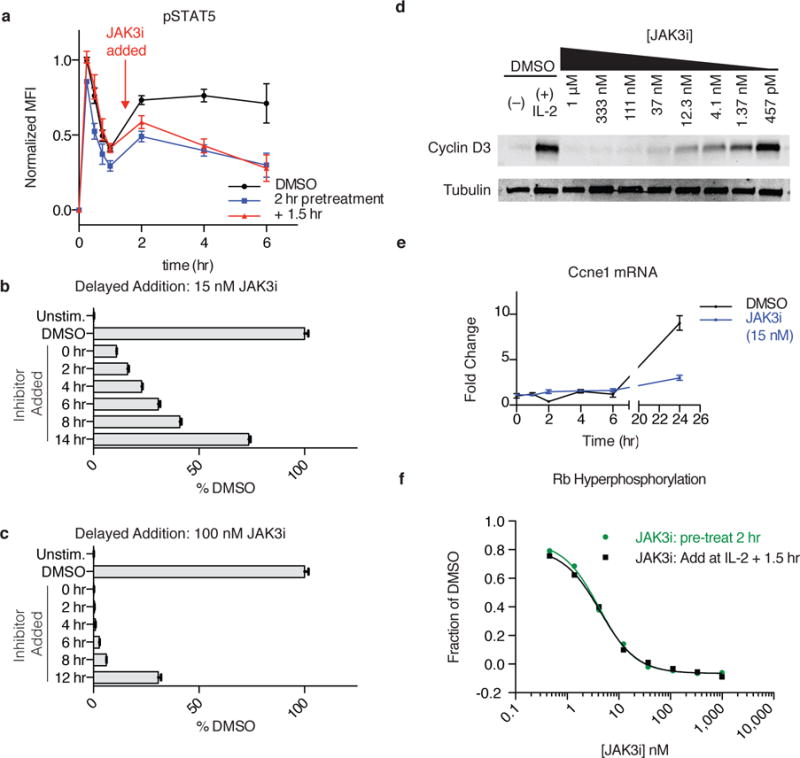
(a) pSTAT5 levels were monitored over time in T cells treated with JAK3i (15 nM) either 2 hrs before or 90 min after IL-2 stimulation. Data are normalized the maximum value (t=15 min DMSO) and plotted as mean ± SEM of 4 independent experiments. (b–c) JAK3i was added at a concentration of 15 nM (b) or 100 nM (d) at the indicated times after IL-2 stimulation. After 24 hrs, cells were labeled with EdU for 1 hr, and the percentage of EdU+ cells was measured by FACS and normalized to DMSO (± SEM of replicates, n = 3). Typically, 35–45% of cells were in S-phase 24–25 hours after stimulation. (d) Cyclin D3 was monitored by immunoblot 24 hrs after IL-2 stimulation in the presence of indicated concentrations of JAK3i. For full gel, see Supplementary Fig. 12. (e) Cells were pretreated with 15 nM JAK3i, and cyclin E1 mRNA was quantified by qPCR at the indicated times after IL-2 stimulation. (f) T cells were treated with JAK3i 2 hrs before or 1.5 hrs after IL-2 stimulation. After 24 hrs, phosphorylation of the retinoblastoma protein (Rb) at S800/S804 was quantified by FACS. Original histograms are shown in Supplementary Figure 10d. Data are representative of 4 (d), 3 (b, e) or 2 (c, f) independent experiments.
Since treatment with low-dose JAK3i blocked proliferation without inducing apoptosis (Supplementary Fig. 10c), we next focused on the mechanism of the observed G0/G1 arrest. In T cells, progression into S phase requires phosphorylation of the retinoblastoma protein (Rb) by CDK4/6 and CDK2 kinases28. IL-2 regulates these kinases, in part, via STAT5-mediated induction of cyclin D3 and E128. We asked whether this pathway was particularly sensitive to JAK3 kinase inactivation. Indeed, JAK3i potently blocked IL-2-stimulated induction of cyclin D3 (Fig. 5d) and cyclin E1 (Fig. 5e, detected by qPCR due to a lack of suitable antibodies). Consistent with its effects on cyclin induction, JAK3i abolished Rb phosphorylation at Ser800 and Ser804, assessed by flow cytometry 24 hours after IL-2 stimulation. Importantly, pRb inhibition occurred with identical potency whether JAK3i was added 2 hours before or 90 minutes after IL-2 stimulation (Fig. 5f). Thus, JAK3 kinase activity is required during a critical window – between 2 and 8 hours after IL-2 stimulation – to upregulate G1 and S-phase cyclins, which in turn promote Rb hyperphosphorylation and DNA replication. It is likely that additional JAK3- and STAT5-dependent processes (e.g. mTOR signaling, Supplementary Fig. 7, Myc induction, Fig. 4e) contribute to timely cell cycle progression. Indeed, addition of the PI3K inhibitor GDC0941 – even 12 hours after IL-2 stimulation – prevented the majority of cells from entering S-phase, suggesting a similar requirement for sustained PI3K activity in cell cycle progression (Supplementary Fig. 10e). Thus, JAK3i’s antiproliferative effects likely reflect partial suppression of both STAT5 and PI3K activities during this crucial second wave of IL-2 signaling.
JAK3i blocks IL-2-driven T-cell proliferation in vivo
Whereas in vitro experiments enabled careful dissection of JAK3-dependent signaling dynamics in primary T cells, the cells were necessarily isolated from the milieu of cytokines, growth factors, and other cell types that contribute to T-cell functions in vivo. To test whether JAK3 activity is required for IL-2 proliferation in this environment and assess the utility of JAK3i as an in vivo tool compound, we devised a proliferation assay similar to our in vitro system using adoptively transferred T-cell blasts. Ex vivo-activated CD4+ T-cell blasts were labeled with carboxyfluorescein succinimidyl ester (CFSE) and injected into congenically marked (CD45.1+) hosts. The mice were then dosed with JAK3i twice daily and stimulated over two days with thrice-daily injections of IL-2 (Fig. 6a). Cell division was monitored by CFSE dilution 50 hours after the first IL-2 injection, which yielded 2–4 divisions for most cells in the control mice (Fig. 6b). JAK3i strongly inhibited T-cell proliferation in vivo, with a nearly complete block at 40 mg/kg and substantial reduction at 10 mg/kg (Fig. 6c).
Figure 6. JAK3 inhibition blocks IL-2-driven proliferation in vivo.
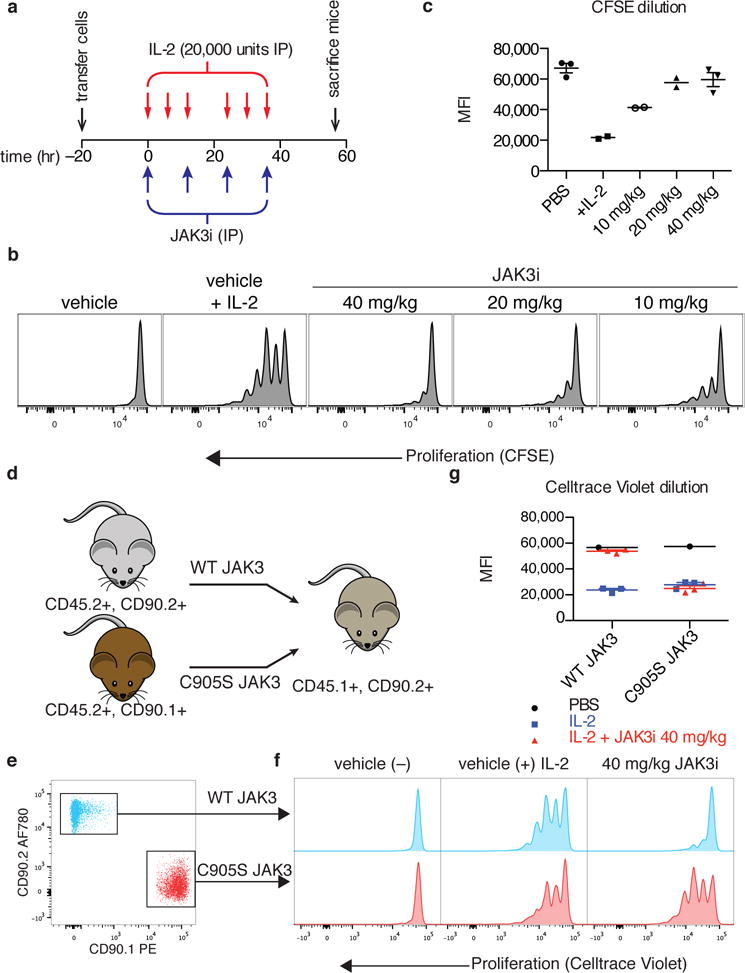
(a) Experimental timeline, showing adoptive transfer of CD4+ T-cell blasts (5 × 106) labeled with carboxyfluorescein succinimidyl ester (CFSE), twice-daily dosing with JAK3i, and thrice-daily injections of IL-2 (20,000 units). (b) Representative CFSE dilution histograms for the indicated treatments. (c) Median CFSE intensity (plotted ± SEM for n>2) for the indicated treatments, 2–3 mice per group as indicated. (d) Congenic marking and transfer scheme for in vivo proliferation and rescue experiment. A 1:1 mixture of WT and C905S T-cells (total 5 × 106) was transferred to each mouse. (e) FACS identification of WT and C905S JAK3-transduced cells from the same mouse (see complete gating in Supplementary Figure 11). (f) Representative Celltrace Violet dilution profiles. Each column depicts cells from the same mouse. (g) Median Celltrace Violet intensity for the indicated treatments and JAK3 overexpression construct (n = 3 mice per IL-2-stimulated treatment condition, n = 1 mouse for the unstimulated control). Data in a-c are pooled from two independenet experiments and data in d-g are from one experiment with indicated numbers of mice.
To rule out off-target effects, we developed an assay that allowed the simultaneous assessment of T cells expressing either WT or C905S JAK3 in the same mouse (Fig. 6d). Two congenic markers were used to distinguish host cells (CD45.1/CD90.2) from donor cells transduced ex vivo with WT JAK3 (CD45.2/CD90.2) or C905S JAK3 (CD45.2/CD90.1) (Fig. 6e). Upon IL-2 stimulation as described above, T cells transduced with WT or C905S JAK3 proliferated to the same extent in vehicle-treated mice (Fig. 6f,g). By contrast, in mice dosed with JAK3i, T-cells expressing WT JAK3 failed to proliferate, whereas T-cells expressing C905S JAK3 proliferated normally in the exact same animals. These results strongly implicate JAK3 as the relevant in vivo target of JAK3i, even at relatively high doses (40 mg/kg, twice daily).
Discussion
In this study, we elucidated a previously unknown requirement for JAK3 catalytic activity in sustaining a second wave of STAT5 phosphorylation in IL-2-stimulated CD4+ T cells. Although JAK3 activity is also required for the initial wave of pSTAT5 that peaks within 15 minutes of IL-2 stimulation, the second, more prolonged wave shows greater sensitivity to acute inhibition by JAK3i. This increased sensitivity accounts for the potent effects of JAK3i on STAT5-dependent processes occurring many hours after IL-2 stimulation, which include transcriptional activation of genes required for cell cycle progression. Sustained activation of the PI3K/mTOR pathway was also more sensitive to JAK3i and essential for cell cycle progression, suggesting a general requirement for second-wave JAK3 activity in multiple signaling pathways downstream of the IL-2 receptor.
What determines this biphasic pattern of STAT5 phosphorylation? In IL-6-stimulated macrophages, STAT3 phosphorylation shows a similar biphasic pattern. Here, the initial decrease in pSTAT3 is ablated in cells lacking the Suppressor of Cytokine Signaling-3 (Socs3)29. Based on this precedent, we speculate that induction of one or more SOCS proteins could promote the rapid decrease in pSTAT5 that occurs within one hour of IL-2 stimulation (Fig. 4). The SOCS-family genes, Socs1, Socs3, and Cish, are all transcriptionally induced by IL-25. Moreover, Socs1-deficient T-cells are hyper-responsive to IL-2 at the level of both STAT5 phosphorylation and proliferation30. Finally, in IL-2-stimulated T cells lacking Cish, the temporal pattern of STAT5 phosphorylation and dephosphorylation is altered31. Hence, SOCS-family proteins likely play complex and partially overlapping roles in tuning the dynamics of IL-2 signaling. Induction of SOCS1 or SOCS3 is expected to shift the balance of JAK kinase dependency toward JAK3, since both proteins bind and directly inhibit JAK1 but not JAK3 kinase domains32. Induction of SOCS1 or SOCS3 could therefore lead to diminished JAK1 activity during the second wave, and hence, a greater reliance on JAK3 activity.
High levels of pSTAT5 were sustained for at least 12 hours after IL-2 stimulation, suggesting that acute JAK3 inhibition during the second wave might be sufficient to block IL-2-stimulated T-cell proliferation. Consistent with this prediction, delayed addition of 100 nM JAK3i 8 hours after IL-2 stimulation completely prevented almost all cells from entering S phase. In patients, tofacitinib inhibits JAK1/3 activity for less than half the time between each dose and yet still exerts a clinically beneficial effect33. Extrapolating from our in vitro studies, tofacitinib may work by preventing the sustained activation of STATs required for committed cellular responses. An inhibitor that selectively blocks JAK3 activity for short periods of time may show similar efficacy to pan-JAK or JAK1-selective inhibitors, but without the side effects resulting from inhibition of JAK1/2-dependent cytokines.
The combination of JAK3i and the C905S JAK3 mutant provides a powerful toolkit to decipher the function of JAK3. Tofacitinib sensitivity is often cited as evidence for JAK3 dependence (see, for example, recent work on alopecia areata34). However, many of tofacitinib’s effects result from blockade of JAK3-independent cytokines35. It remains unclear whether blocking γc cytokines is sufficient to achieve therapeutic efficacy in the setting of autoimmune disease, or whether it is instead necessary to block additional cytokines (e.g., IL-6), as observed with tofacitinib. Together with the inhibitor-resistant C905S JAK3 mutant, JAK3i will enable future preclinical studies to address this important question.
Online Methods
Mice
C57BL/6J mice, B6.Cg-Gpi1aThy1aIgha/J and B6.SJL-Ptprca Pepcb/BoyJ (Jackson) used in this study were housed in the specific pathogen–free facilities at the University of California, San Francisco, and were treated according to protocols approved by the Institutional Animal Care and Use Committee in accordance with US National Institutes of Health guidelines. Both male and female mice were used as a source of primary CD4+ T-cells, all aged 6–12 weeks.
Reagents
pan-Jak inhibitor tofacitinib (CP-690,550), MEK inhibitor PD0325901 and AKT inhibitor MK2206 were purchased from Selleck Chemicals and MEK inhibitor U0126 was purchased from Cell Signaling Technologies. JAK3i 1 and inactive analogue 2 were synthesized as described in the Supplementary Note. pan-PI3K inhibitor GDC-0941 and mTOR inhibitor INK128 were a gift of K. Shokat (UCSF). Recombinant human IL-2 was from the NIH AIDS Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH: Maurice Gately (Hoffmann-La Roche). Recombinant mouse IFN-γ was a gift of R. Locksley.
In vitro kinase assays
Purified JAK1 (9.4 nM), JAK2 (1.15 nM) or JAK3 (3.13 nM) kinase (all Invitrogen) were preincubated with compound and 100 μM ATP for 30 minutes in manufacturer prescribed buffer. Reactions were run for 30 minutes at 23 °C with γ-32P ATP (16.7 nM, Perkin Elmer) and the appropriate substrate: JAK1 - IRS1 (0.06 mg/mL, Enzo Life Sciences); JAK2 - PDKTide (0.19 mg/mL, EMD Millipore); JAK3 - JAK3tide (0.0325 mg/mL, EMD-Millipore). ITK (3.13 nM, Invitrogen) was performed similarly except using 1 mM ATP and Myelin Basic Protein substrate (Sigma). Kinase assays with BTK, EGFR, JAK3 (all with 1 mM ATP), and TYK2 (100 μM ATP) were performed by Nanosyn.
Primary T-Cell Culture
T cells were purified from single-cell suspensions of spleen and lymph nodes from male and female mice aged 6–12 weeks by negative selection with biotinylated antibodies (CD8, CD19, B220, CD11b, CD11c, DX5, Ter119 and CD24, UCSF Monoclonal Antibody Core) and magnetic anti-Biotin beads (MACSi Beads, Miltenyi Biotec). For IL-2 stimulation, purified T cells were pre-activated on 96-well plates coated with anti-CD3 (2C11) and anti-CD28 (37.51) for 72 hours, removed and cultured with rhIL-2 (100 u/mL, Roche) for 36 hours, and then cultured without rhIL-2 for the 36 hours prior to all experiments.
T-Cell Proliferation
Pre-activated T cells were cultured in 96-well plates with titrations of inhibitor or DMSO (0.1%) for 2 hours prior to stimulation with rhIL-2 (50 u/mL). After 18 hours, cells were pulsed with [Methyl-3H]-thymidine (1 μCi/well, Perkin Elmer) and incubated for 24 hours. Cells were harvested onto glass fiber filtermats, dried, and counted on a Microbeta 1450 Trilux Liquid Scintillation Counter (Wallac). Alternatively, T cells were grown in 24-well plates for 10–30 hours and pulsed with 10 μM 5-ethynyl-2′-deoxyuridine for 1 hour and assayed per the manufacturer’s procedure (Click-IT Plus EdU, Life Technologies).
IFN-γ production
CD4+ T cells, purified and pre-activated as described above, were cultured in 96-well plates with titrations of inhibitor or DMSO for 2 hours and then stimulated with rhIL-2 (50 u/mL). After 24 hours, the plate was spun down and the media was carefully removed. Media was diluted 1:1 in assay diluent and assayed for IFN-γ content as per the manufacture’s procedure (BD OptEIA Set Mouse IFN-γ, Cat# 555138).
Cellular Selectivity
ITK and RLK inhibition:IL-2 production was assessed by culturing purified naive CD4+ T cells on anti-CD3 anti-CD28-coated 96-well plates in the presence of inhibitors and measuring IL-2 in the media after 24 hours by ELISA (BD OptEIA Mouse IL-2 ELISA Set #555148). JAK1/JAK2: CD4+ T-cell blasts prepared as above were stimulated with 100 u/mL IFN-γ for 30 minutes, fixed and analyzed by FACS as described below. BTK: Freshly isolated mouse splenocytes were stimulated and assayed for CD69 expression as previously described33.
Flow Cytometry
Cells were live/dead stained using the Live/Dead Fixable Near IR Dead Cell Stain Kit (Life Technologies). For surface stains, cells were stained for 30 minutes on ice with indicated antibodies. Clones, sources & dilutions can be found in the Supplementary Table 5. For intracellular stains, samples were fixed at the indicated time after stimulation in 2% paraformaldehyde, surface stained with CD25-biotin and CD4-BUV395, fixed again and permeabilized with ice cold 90% methanol at −20 °C overnight. Samples were then barcoded using Pacific Orange-NHS ester (0.33 or 5 μg/mL), Pacific Blue-NHS Ester (0.67 or 10 μg/mL), and AlexaFluor (AF) 488-NHS Ester (0.26 or 2 μg/mL) (Life Technologies), as previously described36. Intracellular antigens were then stained for 30 minutes at 23 °C with antibodies indicated in Supplementary Table 5. Samples were acquired on a BD LSR Fortessa and analyzed in FlowJo (Tree Star).
Apoptosis Assay
CD4+ T-cells, pre-activated and rested as described above, were initially stained with Live/Dead Fixable Near IR (Life Tech) to identify any dead cells at beginning of the assay, and then cultured with either 15 or 100 nM JAK3i in the presence of 50 u/mL IL-2 for 24 hours. Cells were then stained with Annexin V FITC and PI as per manufacture’s protocol (BD) and acquired on a BD LSR Fortessa. Using FlowJo (Tree Star), results were first gated on cells that were live at the beginning of the assay (~90%) and then as live (Annexin V−PI−), undergoing apoptosis (Annexin V+PI−) or dead (Annexin V+PI+). These gates were determined by reference to cells cultured in PBS or in media without IL-2 for 24 hours.
Retroviral Overexpression
Murine JAK3 (plasmid provided by L. Berg) was cloned into a pMIG-derived vector (MSCV-IRES-GFP) via Gibson cloning and the C905S mutant was generated by site-directed mutagenesis. Retrovirus was generated in Phoenix cells co-transfected with pCL-Eco. CD4+ T-cells were isolated and activated as described above. After 24 hours of T-cell receptor stimulation, cells were spin transduced for 1 hr with fresh viral supernatant in the presence of Lipofectamine 2000 (Life Technologies) and 100 u/mL rhIL-2. Cells were left on coated plates for 48 hours and then expanded and rested as described above. Transduction efficiency was >75% and cells were used unsorted for proliferation assays. In CD25 and pSTAT5 assays, cells were gated on GFP expression.
qPCR
RNA was isolated from 1–2 × 106 CD4+ T-cell blasts per condition using RNAeasy kit (Qiagen) and cDNA was synthesized using qScript (Quanta Biosciences). mRNA was detected by Primetime (IDT) or Taqman (Life Technologies) predesigned qPCR assays. Primer & probe sequences can be found in Supplementary Table 6. Data are from 3 replicates collected on a QuantStudio 12k (Life Technologies), plotted with 95% confidence intervals as calculated by Quantstudio (Life Technologies).
Western Blots
5–10 × 106 cells were lysed in 50 μL RIPA buffer with 500 mM NaCl, clarified, and protein normalized by bicinchoninic acid assay (Pierce). Proteins (20–30 μg) were resolved by 7.5% or 10% SDS-PAGE, transferred to a polyvinylidene fluoride membrane and blocked with Odyssey Block (LI-COR Biosciences). Blots were probed for Cyclin D3 (DCS22, 1:1000) or JAK3 (D7B12, 1:1000) (Cell Signaling Technologies), normalized to tubulin (DM1A, 1:5000, Sigma-Aldrich) and detected with fluorescent secondary antibodies on an Odyssey Scanner (LI-COR Biosciences). Alternatively, blots were probed with HRP-linked goat-anti-rabbit secondary (Southern Biotech, Birmingham AL), developed with Western Lightening Plus ECL (Pierce) and detected on a ChemiDocMP (Biorad).
In vivo proliferation
Prior to transfer, mice were marked by ear punch, assigned a number and then randomized to treatment group without blinding. Pilot experiments demonstrated that the effect size was large enough to use 2 mice per dose group. CD4+ T-cell blasts from C57BL/6J mice were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE, Life Technologies), and 5 × 106 cells were adoptively transferred into 8-week old B6.SJL-Ptprca Pepcb/BoyJ male mice via lateral tail vein injection. After 12 hours, mice were treated with JAK3i or vehicle by intraperitoneal injection twice a day and stimulated 3 times a day with 20,000 units rhIL-2 IP for 2 days. On day 3, mice were sacrificed, spleens isolated, and proliferation of adoptively transferred cells was assessed by FACS as described above. For in vivo proliferation with cells overexpressing either WT or C905S JAK3, isolated CD4+ T cells from C57BL/6J were transduced with WT JAK3 and CD4+ T-cells from B6.Cg-Gpi1aThy1aIgha/J mice were transduced with C950S JAK3, then mixed 1:1 and labeled with Celltrace Violet (Life Technologies) prior to transfer of 5 × 106 cells (2.5 × 106 each of WT and C905S) into 8-week old B6.SJL-Ptprca Pepcb/BoyJ male mice via lateral tail vein injection. Mice were treated with vehicle + IL-2 (3) or JAK3i + IL-2 (3) or vehicle only (1) and proliferation assessed as described above.
Statistical Analysis
Prism (Graphpad Software) was used to calculate mean and standard error of the mean for all graphs except qPCR data. Relative gene expression is plotted as mean with 95% confidence intervals calculated by Quantstudio software.
Supplementary Material
Acknowledgments
We thank B. Au-Yeung, J. Carelli, H. Wang and K. Shokat for helpful discussion and suggestions and A. Roque for animal husbandry. This work was funded by Rosalind Russell and Ephraim P. Engleman Rheumatology Research Center (AW) and HHMI (AW and JT). GS was supported by the UCSF MSTP and NIAID (F30AI120517-01).
Footnotes
Author Contributions
G.A.S. designed experiments, performed chemical synthesis and biological experiments and wrote the manuscript. K.U. performed chemical synthesis. A.W. designed experiments and wrote the manuscript. JT designed experiments and wrote the manuscript.
Competing Financial Interests
None.
References
- 1.Rochman Y, Spolski R, Leonard WJ. New insights into the regulation of T cells by γc family cytokines. Nat Rev Immunol. 2009;9:480–490. doi: 10.1038/nri2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyazaki T, et al. Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits. Science. 1994;266:1045–1047. doi: 10.1126/science.7973659. [DOI] [PubMed] [Google Scholar]
- 3.Ghoreschi K, Laurence A, O’shea JJ. Janus kinases in immune cell signaling. Immunol Rev. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liao W, Lin JX, Leonard WJ. Interleukin-2 at the Crossroads of Effector Responses, Tolerance, and Immunotherapy. Immunity. 2013;38:13–25. doi: 10.1016/j.immuni.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin JX, et al. Critical Role of STAT5 transcription factor tetramerization for cytokine responses and normal immune function. Immunity. 2012;36:586–599. doi: 10.1016/j.immuni.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laurence A, et al. Interleukin-2 Signaling via STAT5 Constrains T Helper 17 Cell Generation. Immunity. 2007;26:371–381. doi: 10.1016/j.immuni.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 7.Lockyer HM, Tran E, Nelson BH. STAT5 is essential for Akt/p70S6 kinase activity during IL-2-induced lymphocyte proliferation. J Immunol. 2007;179:5301–5308. doi: 10.4049/jimmunol.179.8.5301. [DOI] [PubMed] [Google Scholar]
- 8.Yao Z, et al. Stat5a/b are essential for normal lymphoid development and differentiation. Proc Natl Acad Sci USA. 2006;103:1000–1005. doi: 10.1073/pnas.0507350103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thomis DC, Gurniak CB, Tivol E, Sharpe AH, Berg LJ. Defects in B lymphocyte maturation and T lymphocyte activation in mice lacking Jak3. Science. 1995;270:794–797. doi: 10.1126/science.270.5237.794. [DOI] [PubMed] [Google Scholar]
- 10.Thomis DC, Berg LJ. Peripheral expression of Jak3 is required to maintain T lymphocyte function. J Exp Med. 1997;185:197–206. doi: 10.1084/jem.185.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Witthuhn BA, Williams MD, Kerawalla H, Uckun FM. Differential substrate recognition capabilities of Janus family protein tyrosine kinases within the interleukin 2 receptor (IL2R) system. Leuk Lymphoma. 1999;32:289–297. doi: 10.3109/10428199909167389. [DOI] [PubMed] [Google Scholar]
- 12.Haan C, et al. Jak1 Has a Dominant Role over Jak3 in Signal Transduction through γc-Containing Cytokine Receptors. Chem Biol. 2011;18:314–323. doi: 10.1016/j.chembiol.2011.01.012. [DOI] [PubMed] [Google Scholar]
- 13.Perona-Wright G, Mohrs K, Mohrs M. Sustained signaling by canonical helper T cell cytokines throughout the reactive lymph node. Nature Immunol. 2010;11:520–526. doi: 10.1038/ni.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Swainson L, et al. IL-7-induced proliferation of recent thymic emigrants requires activation of the PI3K pathway. Blood. 2006;109:1034–1042. doi: 10.1182/blood-2006-06-027912. [DOI] [PubMed] [Google Scholar]
- 15.Liao W, et al. Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc Natl Acad Sci USA. 2014;111:3508–3513. doi: 10.1073/pnas.1301138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Arneja A, Johnson H, Gabrovsek L, Lauffenburger DA, White FM. Qualitatively Different T Cell Phenotypic Responses to IL-2 versus IL-15 Are Unified by Identical Dependences on Receptor Signal Strength and Duration. J Immunol. 2014;192:123–135. doi: 10.4049/jimmunol.1302291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cantrell DA, Smith KA. The interleukin-2 T-cell system: a new cell growth model. Science. 1984;224:1312–1316. doi: 10.1126/science.6427923. [DOI] [PubMed] [Google Scholar]
- 18.London N, et al. Covalent docking of large libraries for the discovery of chemical probes. Nat Chem Bio. 2014;10:1066–1072. doi: 10.1038/nchembio.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Goedken ER, et al. Tricyclic covalent inhibitors selectively target Jak3 through an active site thiol. J Biol Chem. 2015;290:4573–4589. doi: 10.1074/jbc.M114.595181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tan L, et al. Development of Selective Covalent Janus Kinase 3 Inhibitors. J Med Chem. 2015;58:6589–6606. doi: 10.1021/acs.jmedchem.5b00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Clark JD, Flanagan ME, Telliez JB. Discovery and development of Janus kinase (JAK) inhibitors for inflammatory diseases. J Med Chem. 2014;57:5023–5038. doi: 10.1021/jm401490p. [DOI] [PubMed] [Google Scholar]
- 22.Readinger JA, Mueller KL, Venegas AM, Horai R, Schwartzberg PL. Tec kinases regulate T-lymphocyte development and function: new insights into the roles of Itk and Rlk/Txk. Immunol Rev. 2009;228:93–114. doi: 10.1111/j.1600-065X.2008.00757.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ahearn SP, et al. Pyrrolopyrimidines as Janus Kinase Inhibitors. 2013 WO085802 A1. [Google Scholar]
- 24.Brennan P, et al. Phosphatidylinositol 3-Kinase Couples the Interleukin-2 Receptor to the Cell Cycle Regulator E2F. Immunity. 1997;7:679–689. doi: 10.1016/s1074-7613(00)80388-x. [DOI] [PubMed] [Google Scholar]
- 25.Cornish GH, Sinclair LV, Cantrell DA. Differential regulation of T-cell growth by IL-2 and IL-15. Blood. 2006;108:600–608. doi: 10.1182/blood-2005-12-4827. [DOI] [PubMed] [Google Scholar]
- 26.Lanning BR, et al. A road map to evaluate the proteome-wide selectivity of covalent kinase inhibitors. Nat Chem Bio. 2014;10:760–767. doi: 10.1038/nchembio.1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schaeffer EM, et al. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science. 1999;284:638–641. doi: 10.1126/science.284.5414.638. [DOI] [PubMed] [Google Scholar]
- 28.Rowell EA, Wells AD. The role of cyclin-dependent kinases in T-cell development, proliferation, and function. Crit Rev Immunol. 2006;26:189–212. doi: 10.1615/critrevimmunol.v26.i3.10. [DOI] [PubMed] [Google Scholar]
- 29.Wormald S, et al. The Comparative Roles of Suppressor of Cytokine Signaling-1 and -3 in the Inhibition and Desensitization of Cytokine Signaling. J Biol Chem. 2006;281:11135–11143. doi: 10.1074/jbc.M509595200. [DOI] [PubMed] [Google Scholar]
- 30.Cornish AL, et al. Suppressor of Cytokine Signaling-1 Regulates Signaling in Response to Interleukin-2 and Other γc-dependent Cytokines in Peripheral T Cells. J Biol Chem. 2003;278:22755–22761. doi: 10.1074/jbc.M303021200. [DOI] [PubMed] [Google Scholar]
- 31.Yang XO, et al. The signaling suppressor CIS controls proallergic T cell development and allergic airway inflammation. Nature Immunol. 2013;14:732–740. doi: 10.1038/ni.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Babon JJ, et al. Suppression of Cytokine Signaling by SOCS3: Characterization of the Mode of Inhibition and the Basis of Its Specificity. Immunity. 2012;36:239–250. doi: 10.1016/j.immuni.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dowty ME, et al. Preclinical to Clinical Translation of Tofacitinib, a Janus Kinase Inhibitor, in Rheumatoid Arthritis. J Pharmacol Exp Ther. 2014;348:165–173. doi: 10.1124/jpet.113.209304. [DOI] [PubMed] [Google Scholar]
- 34.Xing L, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014 doi: 10.1038/nm.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ghoreschi K, et al. Modulation of innate and adaptive immune responses by tofacitinib (CP-690,550) J Immunol. 2011;186:4234–4243. doi: 10.4049/jimmunol.1003668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krutzik PO, Nolan GP. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat Meth. 2006;3:361–368. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.