

Abstract
Fragment-based drug discovery has shown promise as an approach for challenging targets such as protein-protein interfaces. We developed and applied an activity-based fragment screen against dimeric Kaposi’s sarcoma-associated herpesvirus protease (KSHV Pr) using an optimized fluorogenic substrate. Dose response determination was performed as a confirmation screen and NMR spectroscopy was used to map fragment inhibitor binding to KSHV Pr. Kinetic assays demonstrated that several initial hits also inhibit human cytomegalovirus protease (HCMV Pr). Binding of these hits to HCMV Pr was also confirmed via NMR spectroscopy. Despite the use of a target-agnostic fragment library, more than 80% of confirmed hits disrupted dimerization and bound to a previously reported pocket at the dimer interface of KSHV Pr, not to the active site. One class of fragments, an aminothiazole scaffold, was further explored using commercially available analogs. These compounds demonstrated greater than 100-fold improvement of inhibition. This study illustrates the power of fragment-based screening for these challenging enzymatic targets and provides an example of the potential druggability of pockets at protein-protein interfaces.
Keywords: Fragment-based screening, dimer disruption, human herpesviruses, NMR, protease
Introduction
Fragment-based drug discovery shows great promise for the development of inhibitors, particularly of challenging targets such as protein-protein interfaces.[1–4] Over 80% of proteins are predicted to function in complexes, giving rise to an estimated 650,000 protein-protein interactions in the human interactome, and still more arising from host-pathogen interactions.[5–7] The ability to modulate even a fraction of these with small molecules derived from fragments holds enormous potential to both better understand biology and address clinical unmet need.[8] The human herpesviruses provide a case study for this approach.
All human herpesviruses rely on an essential conserved protease for capsid maturation and viral replication.[9–11] Since the discovery of these proteases and their genetic validation as potential drug targets, numerous attempts have been made to develop active-site inhibitors.[12–16] However, despite considerable efforts and some in vitro success, no inhibitors targeting the active-site of the human herpesvirus proteases (HHV Pr) have advanced to the clinic.[16, 17]
All HHV proteases are homodimeric and require dimerization for activity due to an allosteric link between the dimer interface and the active site of each monomer (Figure 1a).[10, 18] The HHV Pr dimer interface consists of two conformationally dynamic helices, one from each monomer, located ~ 15 – 20 Å from their two independent active sites.[19, 20] As with most protein-protein interactions, the interfacial helices of the HHV proteases bury a large hydrophobic surface area containing a surface-exposed aromatic residue.[21] In the case of one model member of the HHV Pr family, Kaposi's sarcoma-associated herpesvirus protease (KSHV Pr), loss of these interfacial helices in the monomeric state perturbs the conformation of the substrate binding pocket into an inactive conformation.[21] From an inhibitor development perspective this opens up the possibility of developing dimer disruptors: protein-protein interaction antagonists that prevent dimerization, thereby allosterically inhibiting the enzyme active site of not only KSHV Pr, but the other homologous HHV proteases as well. This approach was previously demonstrated with helical mimetics, which were designated the DD2 analogs.[21–23] With the goal of increasing the chemical diversity of HHV Pr dimerization inhibitors, we focused our attention on fragment-based screening (FBS) approaches. Compared to traditional high-throughput screening methods, where libraries consist of larger "drug-like" molecules, the smaller compound libraries of a FBS campaign could allow the exploration of potential dimer interface binders in a more efficient manner.[24]
Figure 1.

(a) The structure of dimeric wild-type KSHV Pr (PDB: 2PBK) with the interfacial helices (black dots), Met197 (red, orange) and the individual active sites (cyan, magenta dots) as indicated on each monomeric unit. (b) The Met197 ε-methyl dimer-to-monomer peak height ratios in the presence of the Table 1 fragments 13C/1H-HSQC spectral overlays of selectively [13C-methyl] methionine labeled wild-type KSHV Pr in the absence (black) and presence (red) of Fragments (c) 1 and (d) 6.
In order to identify new scaffolds for the development of HHV Pr dimer disruptors, we developed a high-throughput screen for use in fragment-based screening of KSHV Pr. Inhibition was directly measured by monitoring proteolytic activity, allowing for detection of any mode of inhibition (e.g. competitive, uncompetitive, dimer disruptor). The confirmation screen consisted of dose response curves based on enzymatic activity using an optimized fluorescently labeled substrate.[25] NMR spectroscopy was used to confirm mechanism of inhibition, as well as map fragment binding sites on the protease and rule out non-specific binders. Remarkably, all but one of the confirmed primary hits from an unbiased library disrupted dimerization and did so by binding to a cryptic binding site at the dimer interface. These screens identified two scaffold classes (indoles and aminothiazoles) that exhibited modest micromolar IC50 values ranging from 51 to 557 µM. Further evaluation of these scaffolds identified compounds with enhanced inhibitory activity against KSHV Pr, relative to the primary hits.
Results And Discussion
Primary high-throughput FBS against KSHV Pr
We screened a 16,225 compound fragment library at 500 µ;M against KSHV Pr. This resulted in 317 hits (2.0% hit rate), defined as fragments that resulted in greater than 50% inhibition based on endpoint fluorescence intensity. We repurchased 67 fragments based on availability and diversity of chemical structure, and tested these for dose response and mode of binding. Across the repurchased fragments, 17 out of 67 (Table 1, Table S1) had mean IC50 values less than or approximately equal to 500 µ;M while the other 50 could not be confirmed (not shown). IC50 values spanned approximately ten-fold, ranging from 51.2 to 557 µ;M with ligand efficiencies ranging from 0.23 to 0.49. Interestingly, common scaffolds were observed in the list of the 17 primary confirmed hits, in particular, the aminothiazoles (1–4, 24%) and the indoles (5–7, 18%).
Table 1.
Confirmed Primary Hits against KSHV Pr
| Structure | Fragment ID |
KSHV Pr IC50 (µM) |
HCMV Pr IC50 (µM) |
KSHV Pr DD?[a] |
|---|---|---|---|---|
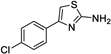 |
1 | 51.2 | 154 | Yes |
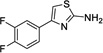 |
2 | 404 | 199 | Yes |
| 3 | 495 | > 500 | Yes | |
| 4 | 187 | ---[b] | Yes | |
| 5 | 337 | ---[b] | Yes | |
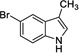 |
6 | 421 | 954 | Yes |
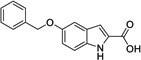 |
7 | > 500 | > 500 | Yes |
| 8 | 81.7 | 179 | Yes | |
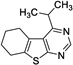 |
9 | 89.5 | 261 | Yes |
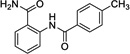 |
10 | 197 | ---[b] | Data not acquired |
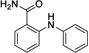 |
11 | > 500 | > 500 | Data not acquired |
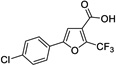 |
12 | 445 | > 500 | Yes |
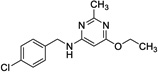 |
13 | 338 | 199 | No |
| 14 | 387 | > 500 | Yes | |
| 15 | 337 | 300 | Yes | |
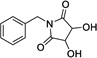 |
16 | 367 | > 500 | Yes |
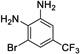 |
17 | 235 | > 500 | Yes |
| 18 | 405 | > 500 | Data not acquired | |
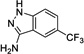 |
19 Negative control |
> 1000 | ---[b] | No |
Dimer Disruption: determined by reduced 13C-Met Dimer-to-Monomer HSQC peak ratios.
No quantifiable inhibition.
Validation of primary hit fragments by protein-based NMR
Having confirmed enzymatic activity in the dose response confirmation screen, we next evaluated the fragment inhibitors by 13C/1H-HSQC NMR spectroscopy using selectively [13C-methyl]-methionine labeled KSHV Pr, with the goal of determining whether dimerization is perturbed. The monomer-dimer equilibrium exhibits slow exchange on the NMR time scale, resulting in distinct monomer and dimer peaks for the interfacial Met197 ε-methyl group, as previously described (Figure 1c–d, Figure S1).[21–23] The hallmark of dimer disruption in this experiment is the loss of Met197 dimer peak intensity with a concomitant chemical shift perturbation (CSP) and increase of peak intensity observed for its monomeric species. Conversely, enhancement of the dimer peak with a concomitant loss of the monomer peak reflects competitive active-site inhibition.[26] Such stabilization of the KSHV Pr dimeric state was observed for a non-cleavable peptidic active-site inhibitor, which covalently binds to the catalytic Ser114.[25] Our group previously established a strong correlation between the 13C-Met spectra and gel filtration data. Specifically, in its apo form, populations of the dimeric and monomeric forms of KSHV Pr are observed in the analytical size exclusion chromatograms. While addition of the covalent active-site inhibitor enhances the peak corresponding to the dimer,[26] incubation of KSHV Pr with our known dimer disruptor DD2 shifts the population towards the monomeric state.[22] Examination of the 13C-Met NMR data indicated that fifteen of the seventeen inhibitors not only bind to KSHV Pr, but also disrupt dimerization (Table 1, Figure 1b).
To further investigate the mode of binding at KSHV Pr, we acquired 13C/1H- and 15N/1H-HSQC spectra using selectively [13C-methyl]-isoleucine and uniform 15N/1H labeled protein. A truncated monomeric construct of the enzyme, KSHV Pr Δ196 (Figure 2a),[21] was used to map fragment binding sites for these experiments. Examination of the 13C/1H-HSQC spectra revealed that, with the exception of 10 and 13, the majority of the primary hits from Table 1 bind near the putative aromatic hotspot of the dimer interface, Trp109. This was reflected in significant CSPs (> 0.025 ppm) observed for the neighboring Ile44 and Ile105 δ1-methyl groups (Figure 2b–d, Figure S2). This pattern of CSPs, as illustrated for 1 and 6 is similar to that previously reported for helical mimetics that disrupt KSHV Pr dimerization, suggesting these fragments are binding the same site.[21–23]
Figure 2.

(a) The structure of monomeric KSHV Pr Δ196 (PDB: 3NJQ), with the catalytic (cyan), hot spot Trp109 (red), and surface exposed Ile44 and Ile105 (blue) residues. Helix 1, Ile71 (orange) and the remaining isoleucine residues (purple) also indicated. (b) The chemical shift perturbations (CSPs) of the isoleucine δ1-methyl groups in the presence of the Table 1 fragments. The gray dotted line represents a CSP threshold of 0.025 ppm. Representative 13C/1H-HSQC spectral overlays of selectively [13C-methyl] isoleucine labeled KSHV Pr Δ196 in the absence (black) and presence (red) of Fragments (c) 1 and (d) 6.
15N/1H-HSQC spectra strongly support this conclusion, with the most significantly perturbed residues being a part of the surface of the previously reported cryptic binding site at the dimer interface (Figure S3). Indeed, the significant CSP values (> 0.05 ppm) and conformational exchange broadening are observed for backbone amides in four distinct regions of the enzyme: (i) near the dimer interface hot spot (Trp109), (ii) the loop containing the oxyanion hole residues (Arg142 and Arg143), (iii) helix 1 (residues 75–86), and (iv) the C-terminal residues (residues 190–196) that would normally constitute the first turn of the interfacial helix in the wild-type enzyme (Figure 3). Importantly, these results indicate that the fragments do not inhibit KSHV Pr via aggregation. Had they done so, significant global shift perturbations and resonance peak broadening would have been observed in all the HSQC spectra.
Figure 3.

Backbone 15N/1HN amide chemical shift perturbations (CSPs) of KSHV Pr Δ196 in the presence of 20–25× molar excess of (a) all Table 1 Fragments and (b) Fragments 1 and 6. The most perturbed backbone amides are highlighted in dotted boxes, and include residues at dimer interface near the hot spot W109, the oxyanion hole, helix 1, and the C-terminus. The largest CSP values for those Fragments which demonstrate binding to KSHV Pr are consistently observed for Leu110.
Mapping the CSP data onto the KSHV Pr Δ196 structure indicates that all the fragments bind to a single face of the protease (Figure 4, Figure S4). The pocket into which these fragments bind is formed when Trp109 changes its rotameric state: instead of being flush against the core of the protein in a "closed" conformation, the Trp109 indole sidechain adopts an “open” conformation, exposing a hydrophobic binding pocket (Figure 2a, Figure 4, Figure S4).[21] This may be the basis for enrichment in indole-containing fragments (5–7) and other multi-cyclic aromatic planar structures (3, 8, 9). These fragments, such as 6 (Figure S4), could stack on top of the tryptophan indole or take its place in the “closed” position.
Figure 4.

The structure of monomeric KSHV Pr Δ196 (PDB: 3NJQ) with the 15N/1HN-HSQC chemical shift perturbations for Fragment 1 indicated by color. Backbone amide resonances which displayed peak broadening upon addition of fragments are indicated in dark gray. Amide backbone nitrogen atoms are shown as colored spheres in (a), while surfaces are displayed in (b). The catalytic triad (His46, Ser114, and His134) and oxyanion hole (Arg142 and Arg143) residues are highlighted in cyan. Left and right structures are rotated 180° about the vertical axis. CSP mapping results for Fragment 6 are displayed in Figure S4.
Rotation of the Trp109 indole is reflected in the observation that the Leu110 amide peak consistently displays the largest CSP values upon binding to fragments (Figure 3, Figure S5). Contrary to the Ile44 and Ile105 δ1-methyl peaks, the shift perturbations for the Leu110 amide peak appear to progress in a linear fashion. While the CSPs of these three residues are a reflection of the indole rotameric state, only the Ile44 and Ile105 methyl groups are surface exposed when the Trp109 adopts the "open" conformation (Figure 2a). As a result, CSPs observed for the Ile44 and Ile105 δ1-methyl groups also reflect direct fragment binding events. Interestingly, some correlations are observed between the CSPs and the molecular volume of the fragments, in which the "smaller" scaffolds, regardless of their chemical moieties or their measured IC50 values, induce larger chemical shift perturbations (Figure S6).
Primary hit fragments also inhibit HCMV Pr
Several of the primary validated hits also showed inhibition of a homologous protease, human cytomegalovirus protease (HCMV Pr), with IC50 values ranging from 154 to > 500 µM and ligand efficiencies ranging from 0.21 to 0.46. (Table 1, Table S1). Saturation-transfer difference NMR (STD-NMR) was used to further evaluate binding of selected Table 1 fragments to HCMV Pr. These data (Figure S7) displayed NOEs with the enzyme, indicating binding for selected aminothiazoles (1 and 3) and indoles (5–7) that were inhibitory against HCMV Pr. Interestingly, 5 showed binding in the STD data, but did not demonstrate robust inhibition of HCMV Pr. Taken together, these results suggest that although the fragments bind to HCMV Pr, they may target a different position on the protease, warranting further exploration of the binding mode for these compounds.
SAR by Catalogue
We next pursued SAR by catalogue[27] based on these novel dimer disruptor fragments (Table 2, Tables S2–S7). Given the large CSP values in the KSHV Pr HSQC spectra, we initially examined analogs of the indole-containing compounds (Table S3). However, these scaffolds showed limited SAR and little improvement from the original hits (5–7). Addition of functional groups at the 1- and/or 2-positions (37–40) of 5 resulted in diminished inhibition. Despite this reduction in potency, the KSHV Pr NMR data clearly indicates disruption of dimerization and binding to the same pocket at the dimer interface (Figures 1–3, Figures S1–S3). The only indole-containing compound that showed improved activity in the SAR by catalogue efforts was 62 (Table S7) with an IC50 of 76 µM, a 4.4-fold improvement over 5. However, this compound is more closely related to the tricyclic analogs 8 and 9 (Tables S5–S6). Similarly to the indole scaffold, the initial set of trifluoromethylbenzene-containing analogs (Table S4), as well as the tricyclic fragments (Tables S5–S6), displayed no significant improvement in inhibitory activity over their initial primary hits, and were not further pursued.
Table 2.
Phenylaminothiazoles
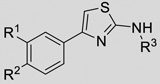 | |||||
|---|---|---|---|---|---|
| Fragment ID |
R1 | R2 | R3 | KSHV Pr IC50 (µM) |
HCMV Pr IC50 (µM) |
| 20 | H | H | H | > 500 | > 500 |
| 21 | H | Br | H | > 500 | 414 |
| 22 | H | CH3 | H | > 500 | > 500 |
| 23 | Cl | Cl | H | 38.4 | 67.6 |
| 24 | Br | H | H | 151.0 | 381 |
| 25 | H | OCH3 | 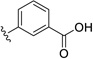 |
29.9 | ---[a] |
| 26 | H | Br | 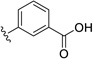 |
21.5 | ---[a] |
| 27 | H | Cl | 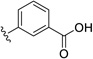 |
50.0 | ---[a] |
| 28 | H | Cl | 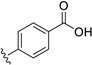 |
12.4 | 42.3 |
| 29 | H | Br | 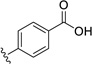 |
20.8 | ---[a] |
| 30 | Cl | Cl | 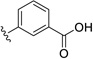 |
6.72 | ---[a] |
| 31 | Cl | Cl | 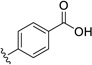 |
6.75 | 47.5 |
| 32 | H | H | 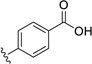 |
265.0 | ---[a] |
| 33 | H | Br | 211.0 | ---[a] | |
| 34 | H | Br | 350.0 | ---[a] | |
No quantifiable inhibition.
The aminothiazoles showed the most promising SAR by catalogue with IC50 values ranging from > 500 µ;M to 6.75 µ;M and exhibiting clear chemical trends (Table 2, Table S2). Previous publications have expressed concern about the aminothiazole scaffold, particularly as a common hit in fragment-based screening.[28] Specifically, aminothiazoles have been reported as promiscuous binders and have exhibited flat SAR against other diverse targets. While these properties are cause for concern, the data herein provide evidence of specific binding as well as clear chemical trends that allow for optimization of the 2-aminothiazole scaffold as inhibitors of KSHV Pr dimerization and activity. SAR is also apparent for those aminothiazoles that inhibit HCMV Pr and differs from that of KSHV Pr, ranging from no inhibition to 42.3 µ;M — further support of specific inhibition by this class of molecules.
The substitution pattern on the phenyl ring of the 4-phenyl-2-aminothiazole scaffold appears to have a significant effect on KSHV Pr inhibition. The original hit compound with a para-chloro substitution (1) resulted in an inhibitor with an IC50 of 51 µ;M. Conversely, the unsubstituted 4-phenyl-2-aminothiazole (20) exhibited an IC50 against KSHV Pr greater than 1 mM. Both the para-bromo (21) and para-methyl (22) substitutions exhibited slight improvement of potency relative to 20. Meta-bromo substitution (24) resulted in an IC50 of 151 µ;M. Di-chloro substitution in the para and meta positions (23) further improved inhibition, yielding an IC50 of 38.4 µ;M, while the di-fluoro substitution (2) did not improve potency.
Further improvements in potency were also observed upon substitution of the amine (Figure 5a, Table 2). Addition of 3- or 4-benzoic acid gave rise to fragments that inhibited KSHV Pr with IC50 values less than 50 µM (25–31). Consistent with the unsubstituted amines (32, IC50 = 265 µM), mono- (28, IC50 = 12.4 µM) and di-halogenation (31, IC50 = 6.75 µM) of the phenyl ring at the meta- or para-positions significantly improves inhibitory activity. Substitution of the benzoic acid with either a 1,3-benzodioxole (34) or a 3-methoxyphenyl group (33) adversely affected inhibition, reducing potency by greater than ten-fold relative to (26).
Figure 5.

(a) SAR by catalogue results for selected Table 2 aminothiazole analogues. Notes for R3 moieties: a = 1,3-benzodioxole; b = 3-methoxyphenyl; c = 4-benzoic acid; d = 3-benzoic acid. (b) The induced-fit docking pose of 30 shown in complex with monomeric KSHV Pr Δ196 (PDB: 4P2T). Residues within the putative binding site that exhibit strong chemical shift perturbations in the 13C/1H-HSQC spectra (Ile44 and Ile105, violet) are shown. The putative binding pocket consisting of the hot spot (Trp109, red) and the surrounding polar (Ser87, green; Arg82, yellow) and hydrophobic residues (Pro192, Phe76, Leu137, Ala80, Leu83 and Val89, pale green) are also indicated.
The position of the acid on the benzoic acid moiety did not change the degree of inhibition (25–32). For example, no change in inhibitory activity was observed for our most potent fragments, 30 and 31, both exhibiting IC50 values of 6.7 µM. Although both are di-chloro substituted on one phenyl ring, 30 contains a 3-benzoic acid, while 31 contains a 4-benzoic acid moiety. Interestingly, 30 and 31 demonstrate selectivity against different members of the HHV Pr family. Both fragments inhibit KSHV Pr with nearly equal potency, but only 31 inhibits HCMV Pr with measurable efficacy.
To further verify the mode of action for Fragment 30, we employed size exclusion chromatography (Figure S8). Similar to our known dimer disruptor DD2,[22] addition of 30 shifts the KSHV Pr from the dimer-monomer equilibrium to a population that is predominantly monomeric. The area under the curve for both chromatograms is nearly identical, indicating no apparent aggregation of the enzyme. Consistent with this result, no peaks corresponding to higher molecular weight species were observed. Examination of the 13C-Met HSQC data (Figure S8) also verifies this transition to the monomeric state. In addition, the 13C-Ile and 15N-HSQC data (Figure S9), display CSPs that map to residues located at the dimer interface, with the Ile44 and Ile105 δ1-methyl and Leu110 amide peak resonances the most perturbed (Figure S10, S13). Dynamic light scattering performed on 30 at a concentration of 20 µM also indicated no aggregate formation (Figure S8). Examination of Michaelis-Menten kinetics with KSHV Pr and 30 in the presence of varying concentrations of our optimized substrate (Figure S8) indicate mixed inhibition, as previously observed for dimer disruptor DD2.[22] The decrease in Vmax with increasing inhibitor concentration (Table S8) rules out strictly competitive inhibition. Taken together, these data suggest that interactions between this elaborated aminothiazole scaffold and KSHV Pr are specific, cause mixed inhibition, and are not due to aggregation.
Docking of Selected Fragments to KSHV Pr
In light of both the 13C/1H and 15N/1H-HSQC results from addition of the mono-chlorinated 4-phenyl-2-aminothiazole (1) to KSHV Pr we performed in silico induced fit docking of the benzoic acid analogs 30 and 31. As illustrated for Fragment 30, the results from this study suggest that these fragments adopt an orientation within the binding pocket to allow for an interaction between the benzoic acid groups and the Arg82 sidechain (Figure 5b). This residue has previously been identified by our group to be important in the binding of the DD2 class of helical mimetic compounds.[23] That class of compounds also contains a carboxylic acid moiety and this binding model could help explain the robust improvement in potency when the carboxylic acid is present. These results are consistent with the HSQC data (Figures S8–S13) and SAR, confirming there is sufficient volume for the ligands within the allosteric pocket, but, in the absence of any crystallographic data, fall short of a definitive binding mode.
Conclusions
Utilizing a fragment library screen and SAR by catalogue, we have identified low micromolar inhibitors of KSHV Pr dimerization, as well as less potent scaffolds that, with further chemistry, may yield additional potent inhibitors. Of the 117 fragments tested in our confirmation screen, NMR assays, and SAR by catalogue efforts, the bromoindole and aminothiazole scaffolds gave rise to the most promising initial compounds with modest micromolar IC50 values. Evaluation of these fragments demonstrated that they also inhibit activity of the related HCMV Pr. Given this, and the potential to target other homologous members of the HHV Pr family, further exploration of these scaffolds is warranted.
The result that the overwhelming majority of fragments in a target-agnostic library cause dimer disruption is, at face value, highly counterintuitive. Typically protein-protein interfaces are considered less “druggable” than enzyme active sites due to their large relatively featureless surfaces.[24, 29] But, in the case of some viral proteases, the opposite might be true. Viral protease active sites are usually shallow and solvent exposed, and preferentially lend themselves towards inhibition by peptidomimetics.[10] The most successful cases of fragment-based drug design against viral proteases have taken advantage of allosteric mechanisms, as in the case of the hepatitis C virus NS3 protein[30] and the NS2B-NS3 proteins of Dengue, West Nile, and Chikungunya viruses,[31] and herpesvirus proteases have a well-understood allosteric mechanism to exploit.[10, 21, 23] It was previously shown that substrates and active site competitive inhibitors promote dimerization of KSHV Pr.[25, 26] This suggests that in order for the identified fragments to disrupt dimerization they must either bind at the interface or a site distant from the interface that allosterically causes dimer disruption. Attempts to co-crystallize fragments with the enzyme were unsuccessful. In instances such as this, where crystallography is not feasible, NMR provides a powerful way to map potential fragment binding sites and confirm the hypothesis that dimer-disrupting fragments indeed bind an allosteric site at the dimer interface.
These results not only provide novel chemical entities from which to develop dimer disruptors, they suggest that the dimer interface may be more “druggable” than the active site. Out of a library of 16K fragments, not one was identified as a competitive active site-directed inhibitor. These results lend credence to the importance of hotspots (Trp109 in the case of KSHV Pr) in binding to protein-protein interfaces and suggest fragments may preferentially bind these regions and thus provide efficient starting points for development of protein-protein interaction antagonists.
Materials and methods
Materials
Protein expression and purification was performed as previously described.[21–23] SAR by catalogue purchases were made from ChemBridge (Hit2lead.com) or www.emolecules.com, and used without further purification. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
FBS primary screen
A biochemical fluorescence intensity-based assay was used to screen 16,225 fragments at 500 µ;M. 0.25 µ;L compound (50 mM stock solution in DMSO) was incubated with 12.5 µ;L of KSHV Pr in 384-well black microtiter plates (Cat No. 95040020, Thermo Electron) and incubated for 1 hour. 12.5 µ;L Ac-Pro-Val-Tyr-tBG-Leu-Gln-Ala-(rhodamine110)-dPro substrate (P6R substrate, Biosyntan) was added to each plate to initiate the reaction. Fluorescence intensity was measured after 1 hour using a Tecan Safire2 monochromator-based fluorescence reader (excitation/emission: 485/535 nm). Final assay conditions were 500 µ;M compound, 1.5 µ;M KSHV Pr, and 2 µ;M substrate in 25 mM potassium phosphate pH 8.0, 150 mM KCl, 0.1 mM EDTA, 0.9 % DMSO, 0.1% prionex, 1 mM DTT. Z’ values were between 0.6–0.8 across the 46 plates, and a control IC50 plate was run after every 15 plates to assure assay robustness with KSHV Pr inhibitor DD2. Assay was run in duplicate; correlation between the two replicates was good and the percent inhibition was scored as an average of n1 and n2.
R-group analysis
R-group analysis and chemical database preparation and manipulation were performed using Instant JChem (ChemAxon). Ligand efficiency (LE) was calculated as LE = (1.4)*[−log(IC50)]*N−1, where IC50 is in molarity and N is the heavy atom count. Calculated distribution coefficient (logD) values were predicted assuming pH 7.4. Lipophilic efficiency (LipE) was computed as LipE = −log(IC50) − logD. Total polar surface area (TPSA) and molecular volumes of the fragments were estimated using the Molinspiration Property Calculation Service (www.molinspiration.com/cgi-bin/properties).
Dose-Response Kinetic Assays
All manual kinetic assays for KSHV Pr dose-response determination were performed with the P6R substrate using a Synergy H4 multimode plate reader (BioTek Instruments, Inc.) with excitation/emission wavelengths of 485/530 nm. Similar kinetic assays for HCMV Pr used a four residue substrate, Ac-tBG-tBG-Asn(NMe2)-Ala modified from[32], containing an ACC instead of an AMC fluorophore. Excitation/emission wavelengths were set to 380/460 nm. Sample conditions and IC50 determination were as previously described.[23]
Michaelis-Menten binding curves were performed as previously described.[22, 23]. In this case, serial dilutions from a 25 µM stock of Fragment 30 were pre-incubated with 1 µM of KSHV Pr for 30 minutes at 30 °C prior to addition of substrate. Concentrations of the rhodamine110-labeled substrate used in this assay were 20, 15, 10, and 5 µM.
1D-STD and 2D-HSQC NMR Assays
All 2D-NMR data were acquired at 27 °C on a Bruker Avance DRX 500 MHz spectrometer equipped with a QCI CryoProbe and a B-ACS 60-slot autosampler. For the 2D-HSQC experiments, two constructs were used: (i) selectively [13C-methyl]-methionine labeled wild-type KSHV Pr; and (ii) uniform 15N/1H and selectively [13C-methyl]-isoleucine labeled KSHV Pr Δ196. Nominal protein and fragment concentrations were 20 – 25 µM and 490 – 500 µM, respectively. All other protein sample conditions, data acquisition parameters and analysis were as previously described.[21–23]
1D-STD NMR experiments were acquired at 27 °C on a Bruker Avance AV 800 MHz Spectrometer equipped with a TXI CryoProbe. Unlabeled wild-type HCMV Pr was diluted to 10 µ;M in deuterated buffer containing 25 mM potassium phosphate pH 8.0, 150 mM KCl, 0.1 mM EDTA, and 1 mM TCEP. Fragments were added to a final concentration of 500 µ;M from 50 mM stocks diluted in d6-DMSO (Cambridge Isotope Laboratories, Inc.). Final sample volumes were 0.45 mL. Data were acquired using a saturation-transfer difference program equipped with a Watergate solvent suppression pulse sequence.[33] On-resonance experiments were set to 0.9 ppm, while off-resonance experiments were set to 30 kHz from the spectral offset. 1D-STD NMR data was analyzed using Mnova NMR (Mestrelab Research) or ACD NMR Processor (ACD/Labs).
Size Exclusion Chromatography
Relative dimer-to-monomer populations of wild-type KSHV Pr was determined using a Superdex 75 10/300 GL analytical size exclusion column (GE Healthcare Life Sciences) attached to an ÄKTA Explorer (GE Healthcare Life Sciences), as previously described.[22, 26] Briefly, KSHV Pr was diluted to a final concentration of 5 µM in the filtered enzyme assay buffer described above and incubated with either 2% DMSO (negative control) or 30 µM compound for 30 minutes at 30 °C. A positive control experiment was performed using 30 µM DD2 (not shown).[22] Reaction and injection volumes were 500 µL. The column was pre-equilibrated with 30 µM compound in assay buffer prior to sample injection, and all data was acquired at room temperature. All data analysis was performed using Unicorn (version 5.31, GE Healthcare Life Sciences).
Dynamic Light Scattering
Fragment 30 was diluted from a 5 mM DMSO stock solution into filtered enzyme assay buffer (described above) to a final concentration of 20 µM. The solution was evaluated using a DynaPro Dynamic Light Scattering System (Protein Solutions/Wyatt). Data were acquired and processed using Dynamics (version 6).
Docking of Aminothiazole Fragments
Docking of elaborated aminothiazole fragments was performed using Small Molecule Drug Discovery Suite (Release 2015-1, Schrödinger, LLC.), including Glide (version 6.6), LigPrep (version 3.3), Epik (version 3.1), and Maestro (version 10.1). Briefly, ligands were prepared with LigPrep using the OPLS_2005 force field[34] and charge states predicted at pH 7.0 using Epik. The compounds were desalted and all possible chiralities were created to a total of 32 per ligand. Compound conformations used for induced-fit docking[35, 36] were generated using conformational search with the OPLS_2005 force field. Charge state was taken from the structure and all other settings were default for multi-ligand preparation. The protein structure (PDB: 4P2T) was prepared using the protein preparation wizard per the default settings. The binding site centroid (defined with residues 76, 87,109, and 193) in an attempt to encapsulate the entire putative binding site. All other parameters were left to default settings.
Supplementary Material
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (grant R01-AI090592 to C.S.C.; grant 1F32GM111012-01 to T.M.A.). J.E.G. was supported by NIH Structural Biology Training Grant GM008284 and the National Science Foundation Graduate Research Fellowship Program (1144247).This work was also supported by the National Center for Research Resources and the National Center for Advancing Translational Sciences (NIH), through the UCSF Clinical and Translational Science Institute (CTSI) Grants UL1 TR000004 and UL1 RR024131.
The authors also wish to thank Prof. Adam Renslo, and Drs. Mark Kelly, Da Duan, and Markus Bohn for helpful discussions and technical support.
Abbreviations
- CSP
chemical shift perturbation
- FBS
fragment-based screen
- HCMV Pr
human cytomegalovirus protease
- HHV Pr
human herpesvirus protease
- HSQC
heteronuclear single quantum coherence
- KSHV Pr
Kaposi's sarcoma-associated herpesvirus protease
- NOE
nuclear Overhauser effect
- SAR
structure-activity relationship
- STD
saturation-transfer difference.
Footnotes
Supporting information, which includes Tables S1–S8 and Figures S1–S13, is available with the on-line version of this article.
References
- 1.Surade S, Blundell TL. Chem Biol. 2012;19(1):42–50. doi: 10.1016/j.chembiol.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 2.Jin L, Wang W, Fang G. Annu Rev Pharmacol Toxicol. 2014;54:435–456. doi: 10.1146/annurev-pharmtox-011613-140028. [DOI] [PubMed] [Google Scholar]
- 3.Magee TV. Bioorganic & Medicinal Chemistry Letters. 2015;25(12):2461–2468. doi: 10.1016/j.bmcl.2015.04.089. [DOI] [PubMed] [Google Scholar]
- 4.Sheng C, Dong G, Miao Z, Zhang W, Wang W. Chem Soc Rev. 2015 [Google Scholar]
- 5.Berggard T, Linse S, James P. Proteomics. 2007;7(16):2833–2842. doi: 10.1002/pmic.200700131. [DOI] [PubMed] [Google Scholar]
- 6.Stumpf MP, Thorne T, de Silva E, Stewart R, An HJ, Lappe M, Wiuf C. Proc Natl Acad Sci U S A. 2008;105(19):6959–6964. doi: 10.1073/pnas.0708078105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shah PS, Wojcechowskyj JA, Eckhardt M, Krogan NJ. Curr Opin Microbiol. 2015;27:62–68. doi: 10.1016/j.mib.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Nature. 2001;411(6833):41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 9.Gao M, Matusick-Kumar L, Hurlburt W, DiTusa SF, Newcomb WW, Brown JC, McCann PJ, Deckman I, Colonno RJ. Journal of Virology. 1994;68(6):3702–3712. doi: 10.1128/jvi.68.6.3702-3712.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tong L. Chemical Reviews. 2002;102(12):4609–4626. doi: 10.1021/cr010184f. [DOI] [PubMed] [Google Scholar]
- 11.Jiang X, Gong H, Chen YC, Vu GP, Trang P, Zhang CY, Lu S, Liu F. Proc Natl Acad Sci U S A. 2012;109(32):13070–13075. doi: 10.1073/pnas.1201620109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teshima T, Griffin JC, Powers JC. J Biol Chem. 1982;257(9):5085–5091. [PubMed] [Google Scholar]
- 13.Holwerda BC. Antiviral Res. 1997;35(1):1–21. doi: 10.1016/s0166-3542(97)00012-0. [DOI] [PubMed] [Google Scholar]
- 14.Waxman L, Darke PL. Antivir Chem Chemother. 2000;11(1):1–22. doi: 10.1177/095632020001100101. [DOI] [PubMed] [Google Scholar]
- 15.Borthwick AD. Med Res Rev. 2005;25(4):427–452. doi: 10.1002/med.20030. [DOI] [PubMed] [Google Scholar]
- 16.Gable JE, Acker TM, Craik CS. Chemical Reviews. 2014;114(22):11382–11412. doi: 10.1021/cr500255e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Skorenski M, Sienczyk M. Expert Opin Ther Pat. 2014;24(8):925–941. doi: 10.1517/13543776.2014.927442. [DOI] [PubMed] [Google Scholar]
- 18.Reiling KK, Pray TR, Craik CS, Stroud RM. Biochemistry. 2000;39(42):12796–12803. doi: 10.1021/bi001019h. [DOI] [PubMed] [Google Scholar]
- 19.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. Nat Struct Mol Biol. 2005;12(11):1019–1020. doi: 10.1038/nsmb1006. [DOI] [PubMed] [Google Scholar]
- 20.Nomura AM, Marnett AB, Shimba N, Dotsch V, Craik CS. Biochemistry. 2006;45(11):3572–3579. doi: 10.1021/bi0523658. [DOI] [PubMed] [Google Scholar]
- 21.Lee GM, Shahian T, Baharuddin A, Gable JE, Craik CS. J Mol Biol. 2011;411(5):999–1016. doi: 10.1016/j.jmb.2011.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shahian T, Lee GM, Lazic A, Arnold LA, Velusamy P, Roels CM, Guy RK, Craik CS. Nat Chem Biol. 2009;5(9):640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gable JE, Lee GM, Jaishankar P, Hearn BR, Waddling CA, Renslo AR, Craik CS. Biochemistry. 2014;53(28):4648–4660. doi: 10.1021/bi5003234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arkin MR, Tang Y, Wells JA. Chem Biol. 2014;21(9):1102–1114. doi: 10.1016/j.chembiol.2014.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lazic A, Goetz DH, Nomura AM, Marnett AB, Craik CS. J Mol Biol. 2007;373(4):913–923. doi: 10.1016/j.jmb.2007.07.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marnett AB, Nomura AM, Shimba N, Ortiz de Montellano PR, Craik CS. Proc Natl Acad Sci U S A. 2004;101(18):6870–6875. doi: 10.1073/pnas.0401613101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubbard RE, Murray JB. Methods Enzymol. 2011;493:509–531. doi: 10.1016/B978-0-12-381274-2.00020-0. [DOI] [PubMed] [Google Scholar]
- 28.Devine SM, Mulcair MD, Debono CO, Leung EW, Nissink JW, Lim SS, Chandrashekaran IR, Vazirani M, Mohanty B, Simpson JS, Baell JB, Scammells PJ, Norton RS, Scanlon MJ. J Med Chem. 2015;58(3):1205–1214. doi: 10.1021/jm501402x. [DOI] [PubMed] [Google Scholar]
- 29.Wells JA, McClendon CL. Nature. 2007;450(7172):1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 30.Saalau-Bethell SM, Woodhead AJ, Chessari G, Carr MG, Coyle J, Graham B, Hiscock SD, Murray CW, Pathuri P, Rich SJ, Richardson CJ, Williams PA, Jhoti H. Nat Chem Biol. 2012;8(11):920–925. doi: 10.1038/nchembio.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bhakat S, Karubiu W, Jayaprakash V, Soliman ME. Eur J Med Chem. 2014;87:677–702. doi: 10.1016/j.ejmech.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 32.Bonneau PR, Plouffe C, Pelletier A, Wernic D, Poupart M-A. Analytical Biochemistry. 1998;255(1):59–65. doi: 10.1006/abio.1997.2445. [DOI] [PubMed] [Google Scholar]
- 33.Mayer M, Meyer B. Journal of the American Chemical Society. 2001;123(25):6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 34.Jorgensen WL, Tirado-Rives J. Journal of the American Chemical Society. 1988;110(6):1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 35.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 36.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. J Med Chem. 2006;49(21):6177–6196. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.