

Abstract
Type 1 diabetes (T1D) is caused by autoreactive T cells that recognize pancreatic islet antigens and destroy insulin-producing β-cells. This attack results from a breakdown in tolerance for self-antigens, which is controlled by ectopic antigen expression in the thymus and pancreatic lymph nodes (PLNs). The autoantigens known to be involved include a set of islet proteins, such as insulin, GAD65, IA-2, and ZnT8. In an attempt to identify additional antigenic proteins, we performed an expression-based genome-wide association study using microarray data from 118 arrays of the thymus and PLNs of T1D mice. We ranked all 16,089 protein-coding genes by the likelihood of finding repeated differential expression and the degree of tissue specificity for pancreatic islets. The top autoantigen candidate was vitamin D–binding protein (VDBP). T-cell proliferation assays showed stronger T-cell reactivity to VDBP compared with control stimulations. Higher levels and frequencies of serum anti-VDBP autoantibodies (VDBP-Abs) were identified in patients with T1D (n = 331) than in healthy control subjects (n = 77). Serum vitamin D levels were negatively correlated with VDBP-Ab levels in patients in whom T1D developed during the winter. Immunohistochemical localization revealed that VDBP was specifically expressed in α-cells of pancreatic islets. We propose that VDBP could be an autoantigen in T1D.
Introduction
Type 1 diabetes (T1D) is a multifactorial polygenic disease caused by self-reactive T-cells that recognize pancreatic islet antigens and destroy their own pancreatic islet insulin-producing β-cells. This process leads to severe hyperglycemia, lifelong dependence on exogenous insulin, and potentially severe chronic complications that increase the risk of premature mortality (1,2). Autoimmune attack may result from a failure in central and/or peripheral tolerance that is controlled by expression of peripheral tissue antigen (PTA) in the thymus and pancreatic lymph nodes (PLNs). Central tolerance is enforced by medullary thymic epithelial cells that ectopically express a range of PTAs under the transcriptional control of Aire, an autoimmune regulator (3).
We and others have shown that peripheral tolerance is mediated by the ectopic expression of PTAs in stromal cells of peripheral lymph nodes, including the PLN. PTA expression is controlled by a transcription regulator called deformed epidermal autoregulatory factor 1 (Deaf1). During the progression of T1D, inflammation and hyperglycemia lead to alternative splicing of Deaf1 and reduce PTA expression in the PLN (4–6). These PTAs include a set of islet-specific autoantigens critical for disease development, such as insulin (INS), GAD 65 kDa (GAD65), islet antigen-2 (IA-2), zinc transporter 8 (ZnT8), and chromogranin A (CHGA) (3,5,7–9).
Studies of T1D genetics have long supported a model in which the major risk factor for T1D resides in the HLA region. Candidate gene association studies and genome-wide association studies (GWAS) have revealed additional loci, including insulin (INS); cytotoxic T-lymphocyte antigen 4 (CTLA4); protein tyrosine phosphatase, nonreceptor type 22 (PTPN22); interleukin 2 receptor α (IL2RA); and interleukin 7 receptor (IL7R) (10). These genes have substantially expanded our knowledge of the genetic architecture and functional aspects of adaptive immunity in T1D. However, they do not fully explain the molecular mechanisms underlying pathogenesis and have provided little insight into other possible autoantigens.
We have described a new meta-analytic methodology, gene expression–based GWAS (eGWAS), that is a computational approach for calculating the likelihood of repeated differential expression for every gene across a large number of case-controlled gene-expression microarray experiments (11). Genes that are most repeatedly implicated across a large set of experimental representations of polygenic diseases serve as data-driven causal disease genes and are candidates for further analysis. We used eGWAS to study type 2 diabetes, integrating 130 independent, publicly available microarray experiments totaling 1,175 samples (11). We identified the immune receptor CD44 as the top functional candidate and by using mouse models and human samples, verified that it plays a novel causative role in the development of type 2 diabetes (11–13). These findings were later reproduced and verified in the same mouse strain by other researchers (14,15).
In T1D, autoimmune reactivity spreads from one protein to another during the induction and progression of disease; this process is called epitope spreading (16,17). Immune recognition of islet-specific autoantigens is one of the principal pathogenic factors. Of note, some of these antigens are present not only in β-cells but also in other endocrine cells in the pancreas, such as α-cells adjacent to β-cells (7).
Autoantibodies against islet cell antigens have served as diagnostic biomarkers for islet autoimmunity in patients with diabetes and prediabetes. They are also useful as predictive markers for subjects with prediabetes. Four antibodies that react to INS, GAD65, IA-2, or ZnT8 have been identified and tested as predictive markers of T1D in at-risk children (18). The chance of progression to T1D within 10 years after islet autoantibody seroconversion with a single-islet autoantibody was found to be 14.5%. Subjects with two or more autoantibodies had a 69.7% chance of T1D developing within 10 years (19).
Identifying additional autoantigens could expand our understanding of epitope spreading and improve the risk assessment and prediction of disease progression in T1D. We have attempted to identify potential candidates by applying our eGWAS methodology to data obtained from the thymus and PLN of NOD mice. The NOD mouse has been established as a genetically susceptible animal model of autoimmune diabetes in which progression of the disease closely resembles that of T1D in humans (20). We hypothesized that genes that are most repeatedly dysregulated across a large set of experimental representations of T1D in the thymus and PLN may serve as data-driven T1D autoantigens and are candidates for validation. This approach is aided by the increasing amounts of publicly available raw microarray experimental results. We meta-analyzed 17 independent genome-wide gene-expression experiments, totaling 118 case and control samples from the thymus and PLN. We identified a top candidate and performed confirmatory studies using samples obtained from the NOD mouse model of T1D and human subjects (Supplementary Fig. 1).
Research Design and Methods
eGWAS
An eGWAS was performed as previously described (11). All T1D-related genome-wide microarray experiments used for this study were collected from the National Center for Biotechnology Information Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo). Studies were found using the following key words: (“type 1 diabetes” OR “T1D”) AND (“thymus” OR “pancreatic lymph nodes” OR “PLN”). We identified 118 array samples (58 subjects with T1D and 60 control subjects) in 17 independent data sets (Supplementary Table 1). To estimate differences between samples from subjects with diabetes and control subjects, raw postquantitation microarray data were reanalyzed using Significance Analysis of Microarrays software (21). For each gene in every microarray experiment, we calculated a D score (di), which denotes the standardized change in gene expression (Eq. 1):
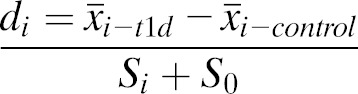 |
(Eq. 1) |
Here, 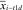 is the mean expression level of gene i in the T1D group,
is the mean expression level of gene i in the T1D group, 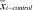 is the mean expression level of gene i in the control group, Si is the SD for the numerator calculation, and S0 is a small positive constant. We considered genes to be significantly dysregulated with either an absolute value of the di ≥2 or a fold change ≥2 between control and case samples. We then converted all probe identifiers across the various mouse microarray platforms to the latest human Entrez Gene identifiers by using our Array Information Library Universal Navigator system (22). Gene expression profiles were assigned in our eGWAS database according to the standardized human Entrez Gene identifier. We counted the observed number of microarray experiments in which each gene was significantly dysregulated. We then calculated P values from the number of positive/negative experiments for each gene and the sum of the number of positive/negative experiments for all other genes by using a 2 × 2 χ2 analysis. A tissue specificity index (TSI) for pancreatic islets (= log10 [signal intensity of pancreatic islets/mean signal intensity of all other tissues]) was calculated for every gene by using multitissue transcriptome data downloaded from BioGPS (http://biogps.org/dataset).
is the mean expression level of gene i in the control group, Si is the SD for the numerator calculation, and S0 is a small positive constant. We considered genes to be significantly dysregulated with either an absolute value of the di ≥2 or a fold change ≥2 between control and case samples. We then converted all probe identifiers across the various mouse microarray platforms to the latest human Entrez Gene identifiers by using our Array Information Library Universal Navigator system (22). Gene expression profiles were assigned in our eGWAS database according to the standardized human Entrez Gene identifier. We counted the observed number of microarray experiments in which each gene was significantly dysregulated. We then calculated P values from the number of positive/negative experiments for each gene and the sum of the number of positive/negative experiments for all other genes by using a 2 × 2 χ2 analysis. A tissue specificity index (TSI) for pancreatic islets (= log10 [signal intensity of pancreatic islets/mean signal intensity of all other tissues]) was calculated for every gene by using multitissue transcriptome data downloaded from BioGPS (http://biogps.org/dataset).
Mice
Female NOD/ShiJcl (NOD) mice were purchased from CLEA Japan, Inc. (Tokyo, Japan). They were housed in a barrier facility under specific pathogen-free conditions. The Animal Care and Use Committee of Kitasato University (Tokyo, Japan) approved all animal experiments.
T-Cell Proliferation Assay
Antigen-induced blastogenesis was measured in spleen cells from 4-, 8-, or 12-week-old female NOD mice (n = 5). Recombinant vitamin D–binding protein (VDBP) produced in yeast (GenScript, Piscataway, NJ), recombinant GAD65 produced in yeast (RSR, Cardiff, U.K.), or regular insulin (Novo Nordisk) was used for this assay. Splenocytes (5 × 106 cells/mL) were plated into a 96-well microtiter plate with 10 μg/mL individual islet antigens and cultured at 37°C, 5% CO2, and 95% humidity for 48 h. BrdU was added to the cell suspension to a concentration of 10 μmol/L, and incubation was continued for an additional 8 h. Supernatants were removed, and a peroxidase-labeled anti-BrdU reagent was added. After 60 min, tetramethylbenzidine substrate was added for color development. Results were read on an ELISA reader. The stimulation index (SI) was calculated by dividing antigen-induced proliferation by no-antigen control. Interferon-γ (IFN-γ) release was measured by ELISA in supernatants (R&D Systems, Minneapolis, MN).
Human Subjects
We analyzed serum samples from 331 randomly selected patients (mean age ± SD 12.2 ± 6.8 years, male/female sex 163/168) who had been given a diagnosis of T1D in the previous 6 months at the Barbara Davis Center for Childhood Diabetes (Aurora, CO). All patients had at least one islet autoantibody present (INS, GAD65, IA-2, and ZnT8). Of the 331 patients, 34 were positive for transglutaminase autoantibodies (related to celiac disease) and 4 were positive for 21-hydroxylase autoantibodies (Addison disease). Patients taking vitamin D medication were not included in this study. In addition, 77 healthy control subjects negative for all islet autoantibodies were also included (mean age 16.9 ± 6.0 years, male/female sex 30/47). Signed informed consent was obtained from all subjects, and the study was approved by the institutional review board of the University of Colorado (Aurora, CO).
VDBP Autoantibody-Autoantigen Complex Detection
Serum diluted five times (20 μL) was incubated with 20 μL of biotinylated goat anti-human IgG monoclonal antibody in PBS for 1 h at room temperature followed by plate capture with a streptavidin-coated plate (Meso Scale Discovery, Gaithersburg, MD). The plate was incubated for 1 h at room temperature and then washed three times with PBS containing 0.1% Tween-20. Finally, 30 μL mouse anti-human VDBP monoclonal antibody (ab23484 [species reactivity: human]; Abcam, Cambridge, MA) was added to each well and incubated for 2 h at room temperature. The plate was then washed three times with PBS containing 0.1% Tween-20 and counted on a SECTOR Imager 2400 (Meso Scale Discovery). An assay cutoff index of 0.020 was based on the 100th percentile and mean ± 3 SD of the sample set from 77 healthy control subjects after removing 11 outliers. The interassay coefficient of variation was 9.8% (n = 6).
25-Hydroxyvitamin D Assay
Serum concentrations of 25-hydroxyvitamin D (25OHD) were measured in a Clinical Laboratory Improvement Amendments–certified clinical laboratory of Children’s Hospital Colorado by using a commercial kit with an automated chemiluminescence immunoassay (Immunodiagnostic Systems, Gaithersburg, MD). All samples were coded, and the assay was performed in a blinded fashion.
Immunohistochemistry
Immunohistochemical staining of frozen sections (humans) or paraffin sections (mice) was performed as previously described (11,23). Human pancreas samples from islet autoantibody–positive donors were obtained through the Network for Pancreatic Organ Donors with Diabetes (nPOD) of JDRF. Mouse pancreatic tissues were harvested from 8-week-old prediabetic NOD mice. Primary and secondary antibodies used for multiple immunofluorescence labeling are listed in Supplementary Table 2.
Statistics
For verification studies in mice and human subjects, comparisons between two groups were performed using a two-tailed Welch t test, a Wilcoxon rank sum test, or a Fisher exact test. P < 0.05 was considered significant. All experimental data are represented as mean ± SE unless otherwise noted.
Results
eGWAS Identifies VDBP as a T1D Autoantigen Candidate
We carried out an eGWAS for T1D by using 17 independent microarray experiments, totaling 118 case and control samples from the thymus and PLN (research design and methods). Sample data were collected from a public repository (Supplementary Table 1). For all 16,089 protein-coding genes, we calculated the likelihood that repeated differential expression for every gene was due to chance (Supplementary Fig. 2). We filtered the list for pancreatic islet–specific genes by using a public human multitissue transcriptome database (BioGPS), yielding 246 genes (24). We then ranked these genes by the TSI for human pancreatic islets and combined this value with eGWAS data (Fig. 1 and research design and methods). The set of genes with high TSI values that were also at the top of the eGWAS list includes several known T1D autoantigens, such as INS, GAD65, and CHGA. VDBP was our top autoantigen candidate (eGWAS P = 2.8 × 10−11, TSI = 1.75) (Fig. 1). This protein was most frequently downregulated in the thymus and PLN across multiple microarray experiments in NOD mice and highly expressed in human pancreatic islet tissues (Fig. 1 and Supplementary Fig. 3).
Figure 1.

Identification of possible autoantigenic proteins in T1D. Pancreatic islet specificity is plotted against the likelihood of frequent differential expression in thymus and PLNs.
VDBP, also known as group-specific component (GC), is located on chromosome 4q13 and codes for a glycosylated α-globulin found in blood plasma. It binds to vitamin D and its plasma metabolites and transports them to target tissues (25).
T-Cell Reactivity Against VDBP Is Increased
To examine whether VDBP exhibits antigenicity during T1D progression in rodent models, we measured antigen-induced blastogenesis for VDBP and known islet autoantigens (INS and GAD65) in spleen cells from 4-, 8-, or 12-week-old female NOD mice (five per group). At 8 weeks of age, we found striking T-cell reactivity against VDBP (SI 3.38 ± 0.41) that was stronger than INS (SI 1.40 ± 0.14), GAD65 (SI 1.36 ± 0.13), and control samples (Fig. 2A). At 4 and 12 weeks of age, there also was stronger reactivity against VDBP compared with control samples. Reactivity against INS and GAD65 was higher than in control samples in all age-groups. In addition, we measured the release of IFN-γ in the culture supernatant of the T-cell proliferation assay by ELISA. The levels of IFN-γ were higher in the VDBP- and INS-stimulated groups compared with the control group (Fig. 2B).
Figure 2.

Proliferative T-cell responses to VDBP and other islet antigens. A: Antigen-induced blastogenesis was measured in spleen cells from 4-, 8-, or 12-week-old female NOD mice (five per group). Islet antigens examined were GAD65, insulin, and VDBP. The SI was calculated by dividing antigen-induced proliferation by no-antigen control (-). B: The release of IFN-γ was measured by ELISA in supernatants. Representative data are from one of two independent experiments for each age. *P < 0.05, **P < 0.01, ***P < 0.001 vs. control (-). w, weeks.
VDBP Autoantibodies in Patients With T1D
We designed an assay to identify the autoantibody-autoantigen (Ab-Ag) complex of VDBP in serum (Supplementary Fig. 4). A standard assay designed to detect VDBP antibodies failed to detect any signal and showed only background levels (data not shown). Because excess VDBP protein in circulating blood (26) could saturate VDBP autoantibodies (VDBP-Abs), we designed a new assay to capture VDBP Ab-Ag complexes. This assay found high levels of VDBP Ab-Ag complexes in the blood of patients with T1D. VDBP Ab-Ag complex levels in 331 patients with newly diagnosed T1D were higher than levels in 77 healthy control subjects (Wilcoxon rank sum test P < 0.0001) (Fig. 3). With the assay cutoff set at an index of 0.020, 27.5% (91 of 331) of subjects were positive in the T1D group versus only 14.3% (11 of 77) in the control group (Fisher exact test P = 0.004). Furthermore, eight patients with T1D had dramatically high signals (>0.220), which was not seen in control subjects. We found no correlation of VDBP-Ab positivity with autoantibody positivity in each of the six autoantibodies (INS, GAD65, IA-2, ZnT8, transglutaminase, and 21-hydroxylase).
Figure 3.

VDBP-Ab assay in humans. The levels of VDBP-Ab (index) in 331 patients with newly diagnosed T1D and 77 healthy control subjects are shown. The dotted line represents an assay cutoff value of index 0.020. In patients with T1D, assays were positive in 27.5% (91 of 331) compared with 14.3% (11 of 77) in healthy control subjects.
VDBP-Ab Levels Are Inversely Correlated With Serum Vitamin D Levels in Patients With T1D During Winter
Vitamin D has a wide spectrum of activity, including calcium and bone homeostasis, cardiovascular function, and skin and muscle cell proliferation. It is derived from a limited number of foods, and its primary source comes from skin conversion of 7-dehydrocholesterol induced by exposure to solar ultraviolet B radiation. Low exposure to sunlight during winter can lead to vitamin D insufficiency. It has been reported that in northern Canadians, serum 25OHD concentrations are lower in winter and higher in summer, and in contrast, transporter VDBP concentrations are higher in winter and lower in summer (27). We therefore assumed that VDBP-Ab may have different interactions seasonally with vitamin D metabolism. The level of serum 25OHD is considered a reliable indicator of vitamin D levels because of its long serum half-life and lack of hormonal control by hepatic 25-hydroxylase (28). To assess the seasonal association of VDBP-Ab with vitamin D status, we measured serum 25OHD levels in patients with T1D who were positive for VDBP-Ab (index >0.020) and performed a correlational analysis between serum concentrations of 25OHD and levels of VDBP-Ab. The concentration of 25OHD was inversely correlated with levels of VDBP-Ab in blood samples collected during the wintertime (November–April) (n = 29, r2 = 0.14, P = 0.049). No correlation was observed during the summertime (n = 34, r2 = 0.0021, P = 0.80) (Fig. 4).
Figure 4.

Correlation between serum levels of 25OHD and VDBP-Ab in patients with T1D. Concentration of 25OHD in serum was analyzed in patients with T1D and positive for VDBP-Ab (index >0.020). Correlation analysis was performed between the concentration of 25OHD and levels of VDBP-Ab in patients with blood samples collected during the winter or summer season.
VDBP Is Specifically Expressed in α-Cells of Pancreatic Islets
VDBP is synthesized and secreted by the liver. It is also expressed by pancreatic islet cells (29,30). Thus, we investigated its localization in pancreatic islets. Immunohistochemistry was performed to detect VDBP, α-cells (glucagon), and β-cells (insulin) in pancreatic tissue sections of human subjects with islet autoantibody positivity (Fig. 5 and Supplementary Figs. 5 and 6) and prediabetic NOD mice (Supplementary Fig. 7). VDBP was highly and specifically expressed in α-cells of pancreatic islets in both species.
Figure 5.

Immunohistochemical localization of VDBP in human pancreatic islets. Immunofluorescent double staining was performed to detect VDBP and glucagon (in α-cells) or insulin (in β-cells) in human pancreas sections obtained from an islet autoantibody-positive individual (nPOD ID #6090 [VDBP + insulin] and #6170 [VDBP + glucagon]). Frozen sections were incubated with anti-VDBP and antiglucagon or anti-insulin antibodies followed by incubation in secondary antisera conjugated to Alexa Fluor 594 (red) or Alexa Fluor 488 (green). Colocalization is shown in yellow. Scale bar = 25 μm.
Discussion
T1D is a polygenic autoimmune disease in which effector T cells mistakenly attack and destroy insulin-producing β-cells in pancreatic islets (cellular immune responses). The immune system is believed to lose tolerance to >10 islet self-antigens, including INS, GAD65, IA-2, ZnT8, and CHGA (7). T-cell reactivity to islet antigens is followed by activation of additional T cells with various islet antigen specificities (antigen/epitope spreading) (16,17). Additional autoreactive T cells may contribute to β-cell destruction (pathogenic activation) or represent nondestructive islet autoimmunity (bystander activation). The list of target antigens has gradually increased since the 1970s, and it is conceivable that additional islet autoantigens will be discovered (7). Many of these antigens are preferentially expressed not only in β-cells but also in multiple non–β-cells and other tissues. For instance, GAD65 and IA-2 are neuroenzymes expressed in β- and non–β-cells of islets and in the CNS, neurons, testis, and ovary; CHGA is expressed in neurons and non–β-endocrine cells (α-cells) (7,31,32). Although why loss of tolerance to certain proteins expressed in β- and non–β-cells and other tissues is associated only with β-cell–specific pathology and disease is still a mystery, these proteins could confer autoantigenicity during the early to midstages of β-cell destruction and may be involved in the progression of disease, with intermolecular antigen recognition occurring as a consequence of this autoantigenicity. In T1D, immune tolerance to self-proteins is broken, and cohorts of autoreactive T cells are recruited around pancreatic islets. This process is intricately involved in T1D pathogenesis, and any information regarding additional self-antigens is crucial to understanding the mechanism of antigen spreading in T1D. This information could be useful for developing therapies and predictive/diagnostic biomarkers.
A number of islet autoantigens have been identified based on the recognition of islet cell proteins (α-, β-, and δ-cells) by human serum autoantibodies (33–37). Other approaches, including T-cell reactivity, have also led to the discovery of islet autoantigens (9,38–40). The breakdown of immune tolerance in T1D may result from the loss of ectopic PTA expression in thymic and/or peripheral lymphoid tissues (i.e., thymus, PLN). We have identified additional islet autoantigens by using an in silico approach, eGWAS. We calculated the likelihood of finding repeated differential expression of a gene in disease-related tissues by using a large number of case-control genome-wide gene-expression arrays. We performed an eGWAS analysis on data from >100 publicly available microarrays from the thymus and PLN of the NOD mouse model of T1D. We identified VDBP, a transporter of vitamin D in the circulation, as the top candidate for an islet autoantigen. Across microarrays, VDBP was most frequently downregulated in the thymus and PLN of NOD mice and is specifically and highly expressed in human pancreatic islets based on multitissue transcriptome database data. We performed additional experiments to determine whether VDBP possesses antigenicity in T1D and to examine the cellular localization of VDBP in pancreatic islets.
T-cell reactivity assays performed using splenocytes from the same strain of mice as those used in the meta-analyzed microarray experiments showed a strong cellular autoimmune response to VDBP, which was comparable with other known antigens, including INS and GAD65. For the present analysis, we used prediabetic NOD mice at stages when nondestructive peri-insulitis occurs and progresses (4 and 8 weeks of age) and invasive insulitis develops (12 weeks of age). The data suggest that VDBP may already possess antigenicity during the early stage of disease progression in NOD mice and that VDBP could be part of the early antigen spreading cascade during the progression of T1D.
We have previously developed and extensively validated nonradioactive islet autoantibody assays for T1D. The assays use electrochemiluminescence detection and have excellent sensitivity and specificity compared with the current standard radioimmunoassays for this purpose (18,41–43). In the current study, we developed an electrochemiluminescence VDBP-Ab assay by using a format similar to that for islet autoantibody measurement but with some modifications to detect VDBP Ab-Ag complexes in patient sera. The assay showed that VDBP-Ab was present in higher levels and at higher frequencies in patients with newly diagnosed T1D compared with healthy control subjects. This result demonstrates that autoimmunity against VDBP is present in a substantial number of patients with T1D and that VDBP could be an autoantigen contributing to the progression of T1D. On the other hand, autoimmunity to VDBP could be a newly discovered independent autoimmune phenomenon in humans that occurs with higher frequency in subjects with existing autoimmune diseases, like T1D. This possibility would explain why some healthy control subjects were also positive for VDBP-Ab. A series of large studies in cohorts of patients with T1D and cohorts of patients with other autoimmune diseases is needed to validate VDBP autoimmunity and to further explore its etiology, clinical pathology, and relationship with T1D and other autoimmune diseases.
VDBP is the primary carrier of vitamin D in the bloodstream and plays a role in maintaining total levels of vitamin D in the body (25). Vitamin D is a key regulator of bone and calcium homeostasis. It is mainly (>90%) synthesized in the skin by sunlight (44). 25OHD levels can vary naturally throughout the seasons in some climates, with lower levels in winter due to lack of sun exposure. The number of newly diagnosed cases of T1D is higher in autumn or winter compared with spring or summer, and the incidence of T1D is much higher in northern Europe where winter sun exposure is minimal compared with southern Europe (45,46). A large number of studies have linked/associated serum 25OHD levels or vitamin D metabolism–related genes (e.g., CYP27B1, CYP2R1, VDR) with T1D risk (29,47–53). These studies have demonstrated that vitamin D insufficiency may increase the risk of T1D onset or autoimmunity in infancy, early childhood, and young adulthood. They have also suggested a direct immunomodulatory role for vitamin D on the autoimmune response in T1D. In the current study, we found that serum VDBP-Ab levels were inversely correlated with 25OHD levels in patients with T1D. This result enabled us to speculate that vitamin D insufficiency could induce the increased VDBP autoimmunity. Although islet autoantibodies (humoral immune responses) are not believed to be directly pathogenic in T1D, they are important markers of islet autoimmunity. Further studies are needed to explore the mechanisms underlying the appearance of VDBP autoreactivity in this disease. The potential relationship between vitamin D status and VDBP autoimmunity may shed light on the role of vitamin D in the etiology and pathology of T1D and its seasonal/geographical differences.
Finally, immunohistochemistry localized VDBP in the pancreas of islet autoantibody–positive humans and NOD mice. High levels of VDBP expression were confirmed in the target tissue (pancreatic islets) as previously described by other researchers (29,30). Double-staining experiments showed that like another known antigen, CHGA, VDBP is expressed in α-cells rather than in β-cells. In addition, similar to GAD65 and IA-2, VDBP is synthesized in nontarget tissue (liver). Future studies are needed to determine the detailed molecular and cellular mechanisms that lead to the emergence of VDBP autoimmunity in T1D. These studies may provide hints about how self-antigens in non–β-cells and/or other tissues can contribute to β-cell (INS)–specific autoimmune destruction in T1D. VDBP autoimmunity possibly occurs only in a subgroup of patients with T1D or occurs as a result of bystander activation during antigen spreading, given that VDBP-Ab positivity occurs at a lower frequency (27.5%) than IA-2, GAD, INS, and ZnT8 autoantibodies (55–72%) in the same study cohort (8).
In conclusion, by using a data-driven candidate gene approach (eGWAS), we discovered that VDBP is expressed in pancreatic islet α-cells and acquires autoantigenicity during the development of T1D. VDBP-Abs were more frequently detected in patients with T1D than in control subjects and could be an additional T1D biomarker. This would aid in the prediction of T1D risk and development. The data also contribute to ongoing research on the role of vitamin D in the development of T1D and, possibly, in other autoimmune diseases.
Supplementary Material
Article Information
Acknowledgments. The authors thank Osamu Hiraku and Ken Sasaki, of the Experimental Animal Center, Kitasato Institute for Life Sciences, Kitasato University, Tokyo, Japan, for support of animal experiments. The authors also thank Valerie Natale, Forgotten Diseases Research Foundation, Santa Clara, CA, for copyediting and manuscript preparation.
Funding. This work was supported by the JDRF Innovative Grant Program (1-INO-2015-124-A-V) and the National Institute for Diabetes and Digestive and Kidney Diseases (R01-DK-32083). This work was also performed with support from the Howard Hughes Medical Institute, the National Institute of General Medical Sciences (R01-GM-079719), the National Library of Medicine (R01-LM-009719), and the Lucile Packard Foundation for Children’s Health. This research was performed with the support of nPOD, a collaborative T1D research project sponsored by JDRF. Organ procurement organizations partnering with nPOD to provide research resources are listed at www.jdrfnpod.org/for-partners/npod-partners.
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. K.K., A.J.B., and L.Yu designed research. K.K., Z.Z., K.T., L.Yi., R.F., D.M., S.Y., and L.Yu performed research. K.K., Z.Z., L.Yi., R.F., D.M., C.G.F., S.Y., and L.Yu analyzed data. All authors participated in data interpretation. K.K. and L.Yu wrote the manuscript. All authors provided critical review of the draft and approved the final version. K.K. and L.Yu are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db15-1308/-/DC1.
References
- 1.Lind M, Svensson AM, Kosiborod M, et al. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med 2014;371:1972–1982 [DOI] [PubMed] [Google Scholar]
- 2.Harjutsalo V, Forsblom C, Groop PH. Time trends in mortality in patients with type 1 diabetes: nationwide population based cohort study. BMJ 2011;343:d5364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson MS, Venanzi ES, Klein L, et al. Projection of an immunological self shadow within the thymus by the aire protein. Science 2002;298:1395–1401 [DOI] [PubMed] [Google Scholar]
- 4.Kodama K, Butte AJ, Creusot RJ, et al. Tissue- and age-specific changes in gene expression during disease induction and progression in NOD mice. Clin Immunol 2008;129:195–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yip L, Su L, Sheng D, et al. Deaf1 isoforms control the expression of genes encoding peripheral tissue antigens in the pancreatic lymph nodes during type 1 diabetes. Nat Immunol 2009;10:1026–1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yip L, Fuhlbrigge R, Taylor C, et al. Inflammation and hyperglycemia mediate Deaf1 splicing in the pancreatic lymph nodes via distinct pathways during type 1 diabetes. Diabetes 2015;64:604–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roep BO, Peakman M. Antigen targets of type 1 diabetes autoimmunity. Cold Spring Harb Perspect Med 2012;2:a007781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wenzlau JM, Juhl K, Yu L, et al. The cation efflux transporter ZnT8 (Slc30A8) is a major autoantigen in human type 1 diabetes. Proc Natl Acad Sci U S A 2007;104:17040–17045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stadinski BD, Delong T, Reisdorph N, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol 2010;11:225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barrett JC, Clayton DG, Concannon P, et al.; Type 1 Diabetes Genetics Consortium . Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet 2009;41:703–707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kodama K, Horikoshi M, Toda K, et al. Expression-based genome-wide association study links the receptor CD44 in adipose tissue with type 2 diabetes. Proc Natl Acad Sci U S A 2012;109:7049–7054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kodama K, Toda K, Morinaga S, Yamada S, Butte AJ. Anti-CD44 antibody treatment lowers hyperglycemia and improves insulin resistance, adipose inflammation, and hepatic steatosis in diet-induced obese mice. Diabetes 2015;64:867–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu LF, Kodama K, Wei K, et al. The receptor CD44 is associated with systemic insulin resistance and proinflammatory macrophages in human adipose tissue. Diabetologia 2015;58:1579–1586 [DOI] [PubMed] [Google Scholar]
- 14.Kang HS, Liao G, DeGraff LM, et al. CD44 plays a critical role in regulating diet-induced adipose inflammation, hepatic steatosis, and insulin resistance. PLoS One 2013;8:e58417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Egan CE, Daugherity EK, Rogers AB, Abi Abdallah DS, Denkers EY, Maurer KJ. CCR2 and CD44 promote inflammatory cell recruitment during fatty liver formation in a lithogenic diet fed mouse model. PLoS One 2013;8:e65247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks-Worrell B, Gersuk VH, Greenbaum C, Palmer JP. Intermolecular antigen spreading occurs during the preclinical period of human type 1 diabetes. J Immunol 2001;166:5265–5270 [DOI] [PubMed] [Google Scholar]
- 17.Tisch R, Yang XD, Singer SM, Liblau RS, Fugger L, McDevitt HO. Immune response to glutamic acid decarboxylase correlates with insulitis in non-obese diabetic mice. Nature 1993;366:72–75 [DOI] [PubMed] [Google Scholar]
- 18.Yu L, Dong F, Miao D, Fouts AR, Wenzlau JM, Steck AK. Proinsulin/insulin autoantibodies measured with electrochemiluminescent assay are the earliest indicator of prediabetic islet autoimmunity. Diabetes Care 2013;36:2266–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ziegler AG, Rewers M, Simell O, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA 2013;309:2473–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yip L, Fathman CG. Type 1 diabetes in mice and men: gene expression profiling to investigate disease pathogenesis. Immunol Res 2014;58:340–350 [DOI] [PubMed] [Google Scholar]
- 21.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A 2001;98:5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen R, Li L, Butte AJ. AILUN: reannotating gene expression data automatically. Nat Methods 2007;4:879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yip L, Taylor C, Whiting CC, Fathman CG. Diminished adenosine A1 receptor expression in pancreatic α-cells may contribute to the pathology of type 1 diabetes. Diabetes 2013;62:4208–4219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Su AI, Cooke MP, Ching KA, et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A 2002;99:4465–4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chun RF. New perspectives on the vitamin D binding protein. Cell Biochem Funct 2012;30:445–456 [DOI] [PubMed] [Google Scholar]
- 26.Speeckaert M, Huang G, Delanghe JR, Taes YE. Biological and clinical aspects of the vitamin D binding protein (Gc-globulin) and its polymorphism. Clin Chim Acta 2006;372:33–42 [DOI] [PubMed] [Google Scholar]
- 27.Larcombe L, Mookherjee N, Slater J, et al. Vitamin D in a northern Canadian first nation population: dietary intake, serum concentrations and functional gene polymorphisms. PLoS One 2012;7:e49872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holick MF. Resurrection of vitamin D deficiency and rickets. J Clin Invest 2006;116:2062–2072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolden-Kirk H, Rondas D, Bugliani M, et al. Discovery of molecular pathways mediating 1,25-dihydroxyvitamin D3 protection against cytokine-induced inflammation and damage of human and male mouse islets of Langerhans. Endocrinology 2014;155:736–747 [DOI] [PubMed] [Google Scholar]
- 30.Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin D and diabetes. Endocrinol Metab Clin North Am 2010;39:419–446 [DOI] [PubMed] [Google Scholar]
- 31.Schmid KW, Brink M, Freytag G, et al. Expression of chromogranin A and B and secretoneurin immunoreactivity in neoplastic and nonneoplastic pancreatic alpha cells. Virchows Arch 1994;425:127–132 [DOI] [PubMed]
- 32.Lukinius A, Stridsberg M, Wilander E. Cellular expression and specific intragranular localization of chromogranin A, chromogranin B, and synaptophysin during ontogeny of pancreatic islet cells: an ultrastructural study. Pancreas 2003;27:38–46 [DOI] [PubMed] [Google Scholar]
- 33.Bottazzo GF, Florin-Christensen A, Doniach D. Islet-cell antibodies in diabetes mellitus with autoimmune polyendocrine deficiencies. Lancet 1974;2:1279–1283 [DOI] [PubMed] [Google Scholar]
- 34.Palmer JP, Asplin CM, Clemons P, et al. Insulin antibodies in insulin-dependent diabetics before insulin treatment. Science 1983;222:1337–1339 [DOI] [PubMed] [Google Scholar]
- 35.Baekkeskov S, Aanstoot HJ, Christgau S, et al. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase [published correction appears in Nature 1990;347:782]. Nature 1990;347:151–156 [DOI] [PubMed] [Google Scholar]
- 36.Martin S, Kardorf J, Schulte B, et al. Autoantibodies to the islet antigen ICA69 occur in IDDM and in rheumatoid arthritis. Diabetologia 1995;38:351–355 [DOI] [PubMed] [Google Scholar]
- 37.Payton MA, Hawkes CJ, Christie MR. Relationship of the 37,000- and 40,000-M(r) tryptic fragments of islet antigens in insulin-dependent diabetes to the protein tyrosine phosphatase-like molecule IA-2 (ICA512). J Clin Invest 1995;96:1506–1511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roep BO, Arden SD, de Vries RR, Hutton JC. T-cell clones from a type-1 diabetes patient respond to insulin secretory granule proteins. Nature 1990;345:632–634 [DOI] [PubMed] [Google Scholar]
- 39.Arden SD, Roep BO, Neophytou PI, et al. Imogen 38: a novel 38-kD islet mitochondrial autoantigen recognized by T cells from a newly diagnosed type 1 diabetic patient. J Clin Invest 1996;97:551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Han B, Serra P, Amrani A, et al. Prevention of diabetes by manipulation of anti-IGRP autoimmunity: high efficiency of a low-affinity peptide. Nat Med 2005;11:645–652 [DOI] [PubMed] [Google Scholar]
- 41.Yu L, Miao D, Scrimgeour L, Johnson K, Rewers M, Eisenbarth GS. Distinguishing persistent insulin autoantibodies with differential risk: nonradioactive bivalent proinsulin/insulin autoantibody assay. Diabetes 2012;61:179–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Miao D, Guyer KM, Dong F, et al. GAD65 autoantibodies detected by electrochemiluminescence assay identify high risk for type 1 diabetes. Diabetes 2013;62:4174–4178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miao D, Steck AK, Zhang L, et al.; Type 1 Diabetes TrialNet Study Group . Electrochemiluminescence assays for insulin and glutamic acid decarboxylase autoantibodies improve prediction of type 1 diabetes risk. Diabetes Technol Ther 2015;17:119–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Norris JM. Can the sunshine vitamin shed light on type 1 diabetes? Lancet 2001;358:1476–1478 [DOI] [PubMed] [Google Scholar]
- 45.Soltesz G, Patterson CC, Dahlquist G; EURODIAB Study Group . Worldwide childhood type 1 diabetes incidence–what can we learn from epidemiology? Pediatr Diabetes 2007;8(Suppl. 6):6–14 [DOI] [PubMed] [Google Scholar]
- 46.Patterson C, Gyürüs E, Rosenbauer J, et al. Seasonal variation in month of diagnosis in children with type 1 diabetes registered in 23 European centers during 1989-2008: little short-term influence of sunshine hours or average temperature. Pediatr Diabetes 2015;16:573–580 [DOI] [PubMed] [Google Scholar]
- 47.Hyppönen E, Läärä E, Reunanen A, Järvelin MR, Virtanen SM. Intake of vitamin D and risk of type 1 diabetes: a birth-cohort study. Lancet 2001;358:1500–1503 [DOI] [PubMed] [Google Scholar]
- 48.The EURODIAB Substudy 2 Study Group. Vitamin D supplement in early childhood and risk for type I (insulin-dependent) diabetes mellitus. Diabetologia 1999;42:51–54 [DOI] [PubMed] [Google Scholar]
- 49.Dong JY, Zhang WG, Chen JJ, Zhang ZL, Han SF, Qin LQ. Vitamin D intake and risk of type 1 diabetes: a meta-analysis of observational studies. Nutrients 2013;5:3551–3562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gorham ED, Garland CF, Burgi AA, et al. Lower prediagnostic serum 25-hydroxyvitamin D concentration is associated with higher risk of insulin-requiring diabetes: a nested case-control study. Diabetologia 2012;55:3224–3227 [DOI] [PubMed] [Google Scholar]
- 51.Littorin B, Blom P, Schölin A, et al. Lower levels of plasma 25-hydroxyvitamin D among young adults at diagnosis of autoimmune type 1 diabetes compared with control subjects: results from the nationwide Diabetes Incidence Study in Sweden (DISS). Diabetologia 2006;49:2847–2852 [DOI] [PubMed] [Google Scholar]
- 52.Cooper JD, Smyth DJ, Walker NM, et al. Inherited variation in vitamin D genes is associated with predisposition to autoimmune disease type 1 diabetes. Diabetes 2011;60:1624–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Israni N, Goswami R, Kumar A, Rani R. Interaction of vitamin D receptor with HLA DRB1 0301 in type 1 diabetes patients from North India. PLoS One 2009;4:e8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.