

ABSTRACT
Immunotherapy is emerging as a major treatment for patients with cancer, predominantly via blocking immune inhibitory pathways and through adoptive T cell therapy. However, only a subset of patients shows clinical responses to these interventions. Emerging data indicates a correlation between clinical response and a pre-existing T cell-inflamed tumor microenvironment. Tumor-intrinsic β-catenin activation has been identified as mediating exclusion of T cells from the tumor microenvironment and other oncogene pathways are being explored similarly. Understanding the molecular mechanisms underlying immune avoidance should identify new therapeutic targets for expanding efficacy of immunotherapies.
Keywords: Cancer immunotherapy, checkpoint blockade, non-T cell inflamed tumor, T cell infiltration, T cell-inflamed tumor, tumor immune evasion
Introduction
Recent developments in immunotherapy approaches for cancer are having significant clinical impact. In particular, monoclonal antibodies (mAbs) targeting the inhibitory receptors CTLA-4 and PD-1 have shown marked efficacy and have been FDA-approved for the treatment of patients with metastatic melanoma, and for non-small cell lung cancer in the case of anti-PD-11-3. Significant clinical activity of anti-PD-1 or anti-PD-L1 mAbs has also been observed in multiple additional cancer types, including triple-negative breast cancer, head and neck cancer, bladder cancer, renal cell carcinoma, and others4-6. However, despite these exciting advances, only a subset of patients experiences clinical benefit in each of these tumor types. As such, understanding molecular mechanisms of primary resistance to immunotherapies has become paramount.
Gene expression profiling of melanoma metastases has revealed that a subset of patients shows evidence of a T cell-inflamed tumor microenvironment at baseline7. This phenotype includes evidence for expression of T cell-specific transcripts, chemokines, and a type I IFN gene signature3,7. The T cell-inflamed phenotype, in addition to demonstrating presence of CD8+ T cells, also shows the highest expression of immune-inhibitory pathways, including expression of PD-L1 and indoleamine-2,3-dioxygenase (IDO) as well as presence of FoxP3+ regulatory T cells8. Thus, antitumor immune responses in these cases appear to be held in check by immune-intrinsic negative feedback mechanisms. In contrast, tumors lacking T cells within the tumor microenvironment (non-T cell-inflamed phenotype) lack these inhibitory factors but instead seems to escape immune destruction through exclusion of T cells from the tumor site (Fig. 1). Thus, the mechanisms of immune escape appear to be distinct in these two major subsets of tumors. Perhaps not surprisingly based on this biology, the majority of clinical responses to immunotherapies appear to be restricted to tumors displaying the T cell-inflamed tumor microenvironment. This is the case for therapeutic cancer vaccines9, but also has been observed with the anti-CTLA-4 mAb ipilimumab and recently with anti-PD-110. Interestingly, post-treatment biopsies in melanoma patients treated with anti-PD-1 have demonstrated a marked increase in Ki67+ proliferating CD8+ T cells penetrating deep into the tumor microenvironment in response to therapy10. These observations are consistent with preclinical data indicating that immunotherapies targeting immune-inhibitory pathways predominantly function through re-activation of CD8+ T cells already present within the tumor microenvironment11.
Figure 1.

(T)cell-inflamed and non-(T)cell-inflamed phenotypes. (A) T cell-inflamed phenotype. CD8α+and/or CD103+ dendritic cells (DCs) are sensing the tumor and upon stimulation through STING pathway activation, type I interferons are produced. These activated DCs migrate to lymph nodes to prime tumor antigen-specific T cells. Activated T cells are recruited back into the tumor microenvironment via CXCL9 and CXCL10 chemokines. Upon tumor infiltration and encounter with antigen, T cells produce IFNγ which leads to the upregulation of immune inhibitory mechanisms including PD-L1 and IDO, and also to produce chemokines that recruit regulatory T cells. (B) Non-T cell-inflamed phenotype. In this scenario the tumor microenvironment is lacking activated T cells, which could potentially be caused by 4 independent mechanisms (indicated by the indicated STOPs): (1) Lack of DC recruitment, (2) Lack of innate immune activation within DCs (3) Lack of adequate priming of antitumor T cells, or (4) Lack of trafficking of activated T cells into the tumor site. Despite lacking T cells, this phenotype still contains tumor-associated macrophages.
Viewed from the perspective of immunotherapy resistance, therefore, absence of a T cell-inflamed tumor microenvironment at baseline appears to be an important biomarker. As such, the problem of resistance can, at first approximation, be reduced to a question of identifying molecular mechanisms that explain why a subset of patients have metastatic lesions that disallow accumulation of T cells within the tumor microenvironment. A major source of inter-patient heterogeneity is likely derived from differences in somatic mutations between individual patients' cancers12. Collectively, these concepts have led to the hypothesis that differential activation of specific oncogene pathways might explain the phenomenon of immune exclusion in a subset of cancers. Successful identification of such pathways should lead to new therapeutic approaches that may enable T cell entry into non-inflamed tumors and expand the fraction of patients capable of responding to novel immunotherapies.
Immunological mechanisms leading to spontaneous T cell priming and tumor infiltration when it does occur
One approach toward understanding why some tumors lack a T cell infiltrate is to gain insights from the mechanisms required for a spontaneous antitumor T cell response and T cell entry into the tumor microenvironment when it does occur. At minimum, tumors capable of priming a spontaneous antitumor T cell response must have antigens capable of being recognized by specific T cells. Although it has been suggested that non-T cell-infiltrated tumors might have far fewer antigens13,14, preliminary data from our own laboratory has indicated that this is not likely the case. Analysis of the TCGA metastatic melanoma dataset has indicated comparable expression of differentiation antigens, cancer-germline antigens, as well as mutated self-proteins generating peptides presented by HLA-A*020115. Thus, non-T cell-inflamed melanomas appear to have at least as many antigens as the T cell-inflamed tumors. However, the relevant innate immune pathways required to trigger a productive adaptive immune response against those antigens might not be engaged. Preclinical data have indicated that type I IFN signaling on host cells is necessary upstream from spontaneous T cell priming against tumor-associated antigens3,16. The mechanism of this effect is mediated via the Batf3-dependent subset of dendritic cells (DCs), which in the mouse express CD8α or CD103 and are superior at cross-presentation of antigens to CD8+ T cells3,17,18. Interestingly, while the T cell-inflamed subset of human tumors shows evidence for a type I IFN gene signature, the non-T cell-inflamed tumors lack this type I IFN-driven gene expression profile3,7,19. Thus, the required innate immune pathways involved in this process might not be engaged. Recent work has indicated that the innate immune signals involved in triggering type I IFN production by host DCs in the tumor context occur predominantly via the STING pathway of cytosolic DNA sensing 16,20. As such, interventions aimed at stimulating this pathway directly, for example using pharmacologic agonists of STING, are impressively therapeutic in preclinical tumor models21 and attractive to consider for clinical development as a means to promote innate immune activation and an endogenous antitumor T cell response as a novel therapeutic. Some key steps in induction of antitumor immunity are depicted in Fig. 1A.
In the effector phase of the antitumor T cell response, expression of appropriate chemokines in the tumor microenvironment is critical for effector T cell trafficking into tumor sites. Gene expression profiling and confirmatory protein-based assays have confirmed that T cell-inflamed tumors show expression of a wide array of chemokines capable of recruiting CD8+ effector T cells, whereas the non-T cell-inflamed subset lacks these chemokines7,22,23. Recent work has indicated that the mandatory chemokines for T cell entry into tumors are those that engage CXCR3, including CXCL9 and CXCL1024. Inasmuch as in vitro activation of the STING pathway in DCs triggers both production of type I IFNs and also the chemokines CXCL9 and CXCL1016, it is plausible to consider that the minimal defect in non-T cell-inflamed tumors might be attributed to poor recruitment and/or activation of Batf3-lineage DCs into the tumor microenvironment. These concepts have formulated a reasonable working model as molecular explanations responsible for the non-T cell-inflamed tumor microenvironment phenotype are being pursued (illustrated in Fig. 1). Escape mechanisms from the immune system via exclusion could be at the level of recruitment of DCs and other innate immune cells (Stop 1, Fig. 1B), poor activation of DCs (Stop 2, Fig. 1B), inefficient priming of antigen-specific T cells (Stop 3, Fig. 1B), or failure to efficiently recruit primed T cells into the tumor microenvironment (Stop 4, Fig. 1B). All of these candidate arrests in the induction or execution of a tumor antigen-specific T cell response can be influenced by the tumor microenvironment, and could in principle be impacted by specific oncogene pathways activated within the tumor cells.
Tumor-intrinsic Wnt/β-catenin pathway activation as a direct cause of T cell exclusion in melanoma
To begin to investigate the possibility that somatic differences at the level of the tumor cells themselves might explain the lack of a T cell-inflamed tumor microenvironment in a subset of cases, gene expression profiling of 266 individual melanoma metastases was analyzed in concert with exome sequencing of the same tumors25. Interestingly, exome sequence data revealed that seven tumors (14%) in the non-T cell-inflamed group showed gain of function mutations in the β-catenin gene. On closer examination, loss of function mutations in negative regulators of the β-catenin pathway (APC, Axin1, TCF1) were identified in an additional ten of the non-T cell-inflamed tumors (23%). Based on gene expression profiling of six defined β-catenin target genes, 48% of the non-T cell-inflamed tumors showed evidence for activation of the WNT/β-catenin pathway. The remainder of the tumors with evidence for β-catenin activation showed increased expression of either a Wnt-ligand family member (Wnt7b, 29.5%; 13 patients) or a receptor family member (Fzd3, 20.5%; 9 patients) or β-catenin itself (11%; 5 patients). Analysis of individual β-catenin target genes showed a negative correlation with CD8α expression in the tumor, whereas PD-L1 expression showed a positive correlation with CD8α as expected based on published results8. Immunohistochemistry confirmed high β-catenin protein expression predominantly in tumors that lacked CD8+ T cells. These data therefore indicate a significant inverse correlation between β-catenin pathway activation and a T cell-inflamed tumor microenvironment25.
To investigate the functional relevance of tumor-intrinsic β-catenin signaling in controlling the host immune response to melanoma, genetically engineered mice were constructed using a tamoxifen-regulated Cre driven by the tyrosinase promoter as developed by Marcus Bosenberg utilizing active Braf (BrafV600E) and conditional PTEN deletion (PTEN−/−), with or without a conditional active β-catenin mutant (CAT-STA)26-28. Tamoxifen-induced melanomas arising from BrafV600E/PTEN−/− mice did have a modest T cell infiltrate as analyzed by flow cytometry and immunohistochemistry. However, melanomas induced by mutated Braf combined with active β-catenin (BrafV600E/CT-STA) completely lacked a T cell infiltrate. Moreover, when PTEN deletion and stabilization of β-catenin (BrafV600E/PTEN−/−/CAT-STA) were combined, the melanomas that arose also lacked a T cell infiltrate. These results directly demonstrate that activation the β-catenin pathway within melanoma tumor cells can dominantly exclude immune cell activation and result in a non-T cell-inflamed tumor microenvironment.
Using this model system and based on fundamental knowledge of the mechanisms involved in spontaneous antitumor T cell responses, the mechanism by which tumor-intrinsic β-catenin activation antagonized antitumor immune responses was pursued. By combining the Cre-inducible expression of the model antigen SIY (SIYRYYGL)29 with adoptive transfer of SIY-specific 2C TCR-transgenic T cells the extent of endogenous T cell activation could be determined. In fact, mice with SIY+ tumors driven by BrafV600E mutation and PTEN deletion (BrafV600E/PTEN−/−) indeed showed spontaneous activation of 2C T cells as measured by CFSE dilution. However, no activation of 2C T cells was observed in mice bearing tumors driven by mutated Braf, PTEN deletion and active β-catenin (BrafV600E/PTEN−/−/CT-STA). These results indicated an early defect in immune priming, likely at the level of DC activation. Closer interrogation revealed that tumors expressing β-catenin showed a complete lack of recruitment of the Batf3-lineage DCs expressing the surface markers CD103 or CD8α. The mechanism of this defect was mapped to failed production of the critical chemokine CCL4 by the melanoma cells, which was downregulated by β-catenin via the transcriptional repressor ATF3 (see Fig. 2, zoom in). Importantly, this difference in baseline immune phenotype impacted on the ability of the mice to respond in vivo to immunotherapy. Whereas the combination of anti-CTLA-4 + anti-PD-L1 mAbs slowed tumor growth in inducible BrafV600E/PTEN−/− mice, there was no therapeutic effect in BrafV600E/PTEN−/−/CAT-STA mice. Thus, these data collectively have identified the Wnt/β-catenin pathway as the first defined tumor-intrinsic oncogene pathway that can abort the induction of antitumor T cell responses, prevent the T cell-inflamed tumor microenvironment, and generate resistance to checkpoint blockade therapy25. A diagram representing this mechanism in the context of a developing antitumor immune response is depicted in Fig. 2.
Figure 2.
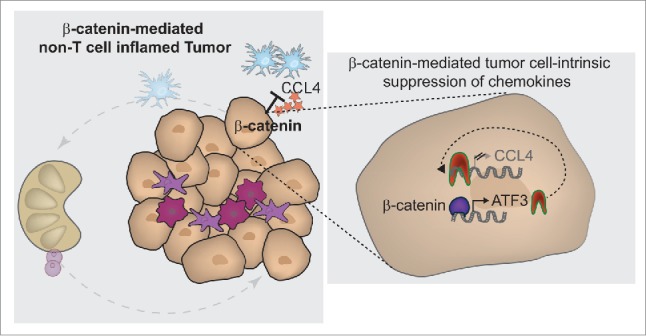
Molecular mechanism of β-catenin-driven immune escape. β-catenin-mediated immune avoidance occurs via inhibition of CCL4 production by the tumor cells themselves, as a result of induction of the transcriptional repressor ATF3, which blocks CCL4 gene transcription (see right zoom-in panel). This lack of CCL4-secretion results in failed recruitment of CD103+ dendritic cells, thereby preventing cross-priming of antitumor T cells (indicated on left overview panel).
A model is emerging in which it is hypothesized that β-catenin can mediate direct immune evasion from an antitumor immune response through direct tumor immune avoidance. This process of immune avoidance can theoretically occur at any given time in tumor development, although data obtained to date have been generated with activation of β-catenin at the initial stage of tumorigenesis (Fig. 3 lower left). In concordance with the concept of immune evasion (3 E hypothesis, Elimination, Equilibrium and Escape, illustrated in Fig. 3 upper panel)30, in which the tumor escapes from the immune system by elimination of tumor cells expressing immunogenic antigens followed by upregulation of immune inhibitory mechanisms that suppress the function of residual T cells of borderline avidity (leading to the T cell-inflamed phenotype, Fig. 3 upper right, Local immune suppression), we propose that tumor escape also can emerge through selection of tumor cells that possess activation of the Wnt/β-catenin pathway. This latter mechanism would lead to immune exclusion from the tumor microenvironment, corresponding to the second major phenotype observed clinically (non-T cell-inflamed, Fig. 3 lower right). A prediction of this model is that patients who develop secondary resistance to immunotherapies also may show acquisition of β-catenin pathway activation, a hypothesis that that is being investigated clinically.
Figure 3.

Oncogene-mediated immune avoidance in contrast to immunogenic escape through selection. Upper panel: Illustrated tumorigenesis of melanoma in the absence of β-catenin signaling. From left to right: tumor initiation is accompanied by recruitment of innate immune cells including CD103+ dendritic cells via CCL4. Those dendritic cells become activated by danger signals, foremost through tumor-derived DNA sensing via the STING pathway. DC activation results in production of type I interferons and facilitates antigen-specific T cell activation (See Fig. 1 for details). Following T cell activation, tumor-specific T cells infiltrate the tumor and result in tumor cell killing, leading either to an equilibrium state in which the immune system can control the tumor growth, or to complete tumor elimination. If the tumor is not eliminated in its entirety, less-immunogenic tumor cells which survived the elimination grow out and an immune-suppressive tumor microenvironment becomes established (characterized by upregulated PD-L1, IDO, and Treg recruitment). Lower panel: Illustrated tumorigenesis in the presence of tumor-intrinsic β-catenin activation. If the initial tumor cells show upregulation of the Wnt/β-catenin pathway, an immediate block in recruitment of DCs cells results due to the lack of CCL4 secretion. It is conceivable that this block can also occur later in the tumor development if activating mutations triggering β-catenin activation are acquired over time, leading to aborted DC recruitment and blunted T cell activation and accumulation. Eventually in this scenario, T cell-negative tumors would grow out with the tumor cells “avoiding” T cell encounter.
Other candidate ancillary oncogene pathways active in subsets of cancers that could contribute to immune exclusion
Inasmuch as the Wnt/β-catenin pathway only explains around 48% of non-T cell-inflamed melanomas, it seems likely that additional molecular perturbations might function to limit host immunity in the remaining non-T cell-inflamed melanomas and in other cancer types as well. One potential candidate is activation of the STAT3 signaling pathway. Constitutively active STAT3 signaling in transplantable tumor cell lines has been reported to lead to decreased expression of proinflammatory mediators, while expression of a dominant negative STAT3 variant resulted in augmented expression of proinflammatory molecules31,32. These factors included the chemokines CCL5 and CXCL10, making them functionally relevant for immune cell recruitment. Additional evidence for this mechanism has been provided through more recent studies using a carcinogen-induced lung cancer model as well as a genetically-induced prostate cancer model33,34. Using a conditional knockout model for STAT3, Ihara and colleagues showed an increased antitumor immune response in the absence of STAT3 signaling. This was associated with increased expression of CCL5 and CXCL10. This phenotype was associated with increased T cell accumulation and T cell function within the tumor microenvironment. Thus, STAT3 may represent another viable mechanistic pathway for diminishing immune cell recruitment into tumor sites and based on the currently available data this might interfere with recruitment of both DCs as well as T cells (Fig. 1B, Stop1 and 4).
Another molecular aberration in cancer cells that may be associated with immune modulatory effects is mutant p53. Intact p53 signaling has been associated with increased recruitment and activation of innate immune cells35. In a murine liver carcinoma model, reactivation of the p53 pathway resulted in tumor regression, which was associated with increased expression of proinflammatory chemokines. In a related study, tumor regression associated with re-expression of wildtype p53 was strongly dependent on the activation and recruitment of Natural Killer (NK) cells into the tumor microenvironment36. That recruitment was dependent on the p53-dependent production of the chemokine CCL2. Consistent with these data, a recent study analyzing triple negative breast cancer identified a correlation between wildtype p53 and the presence of T cells in the tumor microenvironment37. Cumulatively, these data suggest that steady-state p53 signaling could contribute to enhanced recruitment of innate immune cells as well as their activation (Fig. 1B, Stop1 and 2).
Another candidate oncogenic pathway that has potential impact on host immune responses is the NFκB signaling pathway. Activation of this pathway in cancer cells has been associated with tumor progression38,39. In a hepatocellular carcinoma model, increased immune-derived TNF signaling augmented NFκB signaling in liver cells and promoted tumor progression40. Constitutive activation of NFκB has also been shown to increase expression of tumor cell-derived chemokines, which could have positive immune effects41. Further, hyperactivation of NFκB within the tumor microenvironment has been shown to enhance the production of chemokines that recruit activated T cells 42. Therefore, the impact of tumor-intrinsic NFκB activation on host immunity might depend on the cellular context, and whether tumor-promoting inflammatory cells are involved versus antitumor adaptive immunity. Additional cancer types-specific studies will be needed to determine if an NFκB driven tumor microenvironment is enhancing or dampening the antitumor immune response.
The PI3K/PTEN/AKT pathway is another interesting candidate to consider that could impact on host immune responses. Several studies focusing on inflammation-induced cancer progression have identified that active PI3K signaling, either through activating mutations in PIK3CA or loss of function mutations in PTEN, results in increased accumulation of tumor-associated macrophages, which in turn induce an immune suppressive microenvironment43,44. This phenomenon was associated with increased production of TNF, IL-6, CSF-1 VEGF-A and IL-8 by the tumor cells, which contributed to recruitment of macrophages and the induction of an M2 macrophage phenotype45. In contrast, recent reports in triple negative breast cancer have indicated that expression of PTEN was associated with the absence of T cells as well as low PD-L1 expression in the tumor microenvironment46, arguing that loss of PTEN expression (and constitutive PI3K activation) is associated with presence of T cells in the tumor microenvironment. Similarly to intra-tumoral NFkB signaling, the impacts of active PI3K signaling need to be further studied in a tumor-specific context to draw definitive conclusions on its effect on T cell infiltration and those might be different between tumor types.
Host factors that may contribute to regulation of the T cell-inflamed phenotype
Besides differential activation of ancillary oncogene pathways within the tumor cells themselves, several host-derived factors could contribute to robustness of a spontaneous antitumor adaptive immune response. One consideration is the immunologic history of the patient and exposure to chronic viral infections. Life-long latent infection with cytomegalovirus (CMV) has been reported to consume the memory T cell pool and result in fewer T cell clones available to respond to new antigens47. Reduction in T cell repertoire diversity might have severe effects on the ability to recognize tumor-derived antigens in the context of cancer and in the face of immunotherapies. Additionally, emerging data have suggested that environmental factors, including the composition of the intestinal microbiota, might influence the magnitude of an antitumor immune response in the context of certain therapeutic interventions48. Thus, it is conceivable that the baseline composition of intestinal microbiota can influence the endogenous adaptive immune response toward the tumor. Another critical host factor is the constellation of germline polymorphisms in immune regulatory genes. Like predisposition toward autoimmune diseases, it is plausible that specific polymorphisms might favor the ability of the host to mount an adaptive immune response against their tumor49-51.
Conclusions and future directions
The presence of a T cell-inflamed tumor microenvironment is indicative of an endogenous adaptive immune response against a given tumor, and is emerging as a useful predictive biomarker for response to immunotherapies. The molecular mechanisms that mediate the presence or absence of a T cell-infiltrated tumor are just beginning to be understood. Tumor-intrinsic β-catenin pathway activation has been identified as the first oncogene pathway mechanistically confirmed to mediate exclusion of immune cells from the melanoma tumor microenvironment. This pathway also may be relevant for other cancers beyond melanoma, and alternative oncogene pathways also could explain immune exclusion for some of the remaining melanoma cases and also for other cancer types. In addition, it is conceivable that activation of oncogenic pathways during the elimination phase can result in an alternative mechanism of immune escape through immune avoidance (Fig. 3, lower panel). Even more importantly, upregulation of these immune-inhibitory oncogene pathways could be predicted to result in secondary resistance after immunotherapies such as checkpoint inhibitors. Therefore, it will be of critical importance to analyze such tumor-intrinsic pathways in patients with disease progression/recurrence after initial response. Host-derived elements are also being considered as contributory factors, including germline polymorphisms in immune regulatory genes, history of persistent viruses and a contracting T cell repertoire, as well as the specific composition of the intestinal microbiota. All of these factors are measurable, and should be analyzed prospectively from patients being treated with immunotherapeutic agents such as anti-PD-1 or anti-PD-L1 mAbs.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
Work represented in this review was supported by a Team Science Award from the Melanoma Research Alliance and a Cancer Research Institute Translational Research Award; SS is supported by the Cancer Research Institute Irvington Postdoctoral Fellowship
References
- 1.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB et al.. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012; 366:2443-54; PMID:22658127; http://dx.doi.org/ 10.1056/NEJMoa1200690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC et al.. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010; 363:711-23; PMID:20525992; http://dx.doi.org/ 10.1056/NEJMoa1003466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{α}+ dendritic cells. J Exp Med. 2011; 208:2005-16; PMID:21930765; http://dx.doi.org/ 10.1084/jem.20101159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hamid O, Carvajal RD. Anti-programmed death-1 and anti-programmed death-ligand 1 antibodies in cancer therapy. Expert Opin Biol Ther. 2013; 13:847-61; PMID:23421934; http://dx.doi.org/ 10.1517/14712598.2013.770836 [DOI] [PubMed] [Google Scholar]
- 5.Stagg J, Allard B. Immunotherapeutic approaches in triple-negative breast cancer: latest research and clinical prospects. Ther Adv Med Oncol. 2013; 5:169-81; PMID:23634195; http://dx.doi.org/ 10.1177/1758834012475152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swanson MS, Sinha UK. Rationale for combined blockade of PD-1 and CTLA-4 in advanced head and neck squamous cell cancer-review of current data. Oral Oncol. 2015; 51:12-5; PMID:25459157; http://dx.doi.org/ 10.1016/j.oraloncology.2014.10.010 [DOI] [PubMed] [Google Scholar]
- 7.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009; 69:3077-85; PMID:19293190; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, Gajewski TF. Up-regulation of PD-L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med. 2013; 5:200ra116; PMID:23986400; http://dx.doi.org/ 10.1126/scitranslmed.3006504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajewski TF, Louahed J, Brichard VG. Gene signature in melanoma associated with clinical activity: a potential clue to unlock cancer immunotherapy. Cancer J. 2010; 16:399-403; PMID:20693853; http://dx.doi.org/ 10.1097/PPO.0b013e3181eacbd8 [DOI] [PubMed] [Google Scholar]
- 10.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spranger S, Koblish H, Horton B, Scherle P, Newton R, Gajewski T. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8+ T cells directly within the tumor microenvironment. J Immunother Cancer. 2014; 2; PMID:24829760; shttp://dx.doi.org/ 10.1186/2051-1426-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015; 348:69-74; PMID:25838375; http://dx.doi.org/ 10.1126/science.aaa4971 [DOI] [PubMed] [Google Scholar]
- 13.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS et al.. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189-99; PMID:25409260; http://dx.doi.org/ 10.1056/NEJMoa1406498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS et al.. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015; PMID:25765070; http://dx.doi.org/ 10.1126/science.aaa1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gajewski T, Zha Y, Hernandez K, Li Y, Bao R, Alexieff P, Andrade J, Luke J, Spranger S. Density of immunogenic antigens and presence or absence of the T cell-inflamed tumor microenvironment in metastatic melanoma. Journal of Clinical Oncology, 2015 ASCO Annual Meeting (May 29 - June 2, 2015). Vol 33, No 15_suppl (May 20 Supplement), 2015; 3002 [Google Scholar]
- 16.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, Duggan R, Wang Y, Barber GN, Fitzgerald KA et al.. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014; 41:830-42; PMID:25517615; http://dx.doi.org/ 10.1016/j.immuni.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Edelson BT, Kc W, Juang R, Kohyama M, Benoit LA, Klekotka PA, Moon C, Albring JC, Ise W, Michael DG et al.. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010; 207:823-36; PMID:20351058; http://dx.doi.org/ 10.1084/jem.20091627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, Krummel MF. Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer Cell. 2012; 21:402-17; PMID:22439936; http://dx.doi.org/ 10.1016/j.ccr.2012.01.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013; 14:1014-22; PMID:24048123; http://dx.doi.org/ 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, Fu YX. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014; 124:687-95; PMID:24382348; http://dx.doi.org/ 10.1172/JCI67313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corrales L, Glickman LH, McWhirter SM, Kanne DB, Sivick KE, Katibah GE, Woo SR, Lemmens E, Banda T, Leong JJ et al.. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015; 11:1018-30; PMID:25959818; http://dx.doi.org/ 10.1016/j.celrep.2015.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salerno EP, Olson WC, McSkimming C, Shea S, Slingluff CL Jr. T cells in the human metastatic melanoma microenvironment express site-specific homing receptors and retention integrins. Int J Cancer. 2014; 134:563-74; PMID:23873187; http://dx.doi.org/ 10.1002/ijc.28391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erdag G, Schaefer JT, Smolkin ME, Deacon DH, Shea SM, Dengel LT, Patterson JW, Slingluff CL Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012; 72:1070-80; PMID:22266112; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-3218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mikucki MFD; Matsuzaki J.; Skitzki J.; Gaulin N.; Muhitch J.; Ku A.; Frelinger J.; Odunsi K.; Gajewski T.; Luster A.; Evans S.. Non-redundant Requirement for CXCR3 Signaling during Tumoricidal T Cell Trafficking across Tumor Vascular Checkpoints. Nat Commun. 2015; in press; PMID:26109379; http://dx.doi.org/ 10.1038/ncomms8458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015; PMID:25970248; http://dx.doi.org/ 10.1038/nature14404 [DOI] [PubMed] [Google Scholar]
- 26.Bosenberg M, Muthusamy V, Curley DP, Wang Z, Hobbs C, Nelson B, Nogueira C, Horner JW 2nd, Depinho R, Chin L. Characterization of melanocyte-specific inducible Cre recombinase transgenic mice. Genesis 2006; 44:262-7; PMID:16676322; http://dx.doi.org/ 10.1002/dvg.20205 [DOI] [PubMed] [Google Scholar]
- 27.Damsky WE, Curley DP, Santhanakrishnan M, Rosenbaum LE, Platt JT, Gould Rothberg BE, Taketo MM, Dankort D, Rimm DL, McMahon M et al.. β-catenin signaling controls metastasis in Braf-activated Pten-deficient melanomas. Cancer Cell. 2011; 20:741-54; PMID:22172720; http://dx.doi.org/ 10.1016/j.ccr.2011.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE Jr., You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009; 41:544-52; PMID:19282848; http://dx.doi.org/ 10.1038/ng.356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cheung AF, Dupage MJ, Dong HK, Chen J, Jacks T. Regulated expression of a tumor-associated antigen reveals multiple levels of T-cell tolerance in a mouse model of lung cancer. Cancer Res. 2008; 68:9459-68; PMID:19010921; http://dx.doi.org/ 10.1158/0008-5472.CAN-08-2634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002; 3:991-8; PMID:12407406; http://dx.doi.org/ 10.1038/ni1102-991 [DOI] [PubMed] [Google Scholar]
- 31.Wang T, Niu G, Kortylewski M, Burdelya L, Shain K, Zhang S, Bhattacharya R, Gabrilovich D, Heller R, Coppola D et al.. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med. 2004; 10:48-54; PMID:14702634; http://dx.doi.org/ 10.1038/nm976 [DOI] [PubMed] [Google Scholar]
- 32.Burdelya L, Kujawski M, Niu G, Zhong B, Wang T, Zhang S, Kortylewski M, Shain K, Kay H, Djeu J et al.. Stat3 activity in melanoma cells affects migration of immune effector cells and nitric oxide-mediated antitumor effects. J Immunol. 2005; 174:3925-31; PMID:15778348; http://dx.doi.org/ 10.4049/jimmunol.174.7.3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toso A, Revandkar A, Di Mitri D, Guccini I, Proietti M, Sarti M, Pinton S, Zhang J, Kalathur M, Civenni G et al.. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014; 9:75-89; PMID:25263564; http://dx.doi.org/ 10.1016/j.celrep.2014.08.044 [DOI] [PubMed] [Google Scholar]
- 34.Ihara S, Kida H, Arase H, Tripathi LP, Chen YA, Kimura T, Yoshida M, Kashiwa Y, Hirata H, Fukamizu R et al.. Inhibitory roles of signal transducer and activator of transcription 3 in antitumor immunity during carcinogen-induced lung tumorigenesis. Cancer Res. 2012; 72:2990-9; PMID:22659452; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-4062 [DOI] [PubMed] [Google Scholar]
- 35.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656-60; PMID:17251933; http://dx.doi.org/ 10.1038/nature05529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. J Exp Med. 2013; 210:2057-69; PMID:24043758; http://dx.doi.org/ 10.1084/jem.20130783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Quigley D, Silwal-Pandit L, Dannenfelser R, Langerod A, Vollan HK, Vaske C, Siegel JU, Troyanskaya O, Chin SF, Caldas C et al.. Lymphocyte Invasion in IC10/Basal-Like Breast Tumors Is Associated with Wild-Type TP53. Mol Cancer Res. 2015; 13:493-501; PMID:25351767; http://dx.doi.org/ 10.1158/1541-7786.MCR-14-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basseres DS, Baldwin AS. Nuclear factor-kappaB and inhibitor of kappaB kinase pathways in oncogenic initiation and progression. Oncogene. 2006; 25:6817-30; PMID:17072330; http://dx.doi.org/ 10.1038/sj.onc.1209942 [DOI] [PubMed] [Google Scholar]
- 39.Baldwin AS. Regulation of cell death and autophagy by IKK and NF-kappaB: critical mechanisms in immune function and cancer. Immunol Rev. 2012; 246:327-45; PMID:22435564; http://dx.doi.org/ 10.1111/j.1600-065X.2012.01095.x [DOI] [PubMed] [Google Scholar]
- 40.Pikarsky E, Porat RM, Stein I, Abramovitch R, Amit S, Kasem S, Gutkovich-Pyest E, Urieli-Shoval S, Galun E, Ben-Neriah Y. NF-kappaB functions as a tumour promoter in inflammation-associated cancer. Nature 2004; 431:461-6; PMID:15329734; http://dx.doi.org/ 10.1038/nature02924 [DOI] [PubMed] [Google Scholar]
- 41.Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. 2004; 118:285-96; PMID:15294155; http://dx.doi.org/ 10.1016/j.cell.2004.07.013 [DOI] [PubMed] [Google Scholar]
- 42.Muthuswamy R, Berk E, Junecko BF, Zeh HJ, Zureikat AH, Normolle D, Luong TM, Reinhart TA, Bartlett DL, Kalinski P. NF-kappaB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012; 72:3735-43; PMID:22593190; http://dx.doi.org/ 10.1158/0008-5472.CAN-11-4136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schmid MC, Avraamides CJ, Dippold HC, Franco I, Foubert P, Ellies LG, Acevedo LM, Manglicmot JR, Song X, Wrasidlo W et al.. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3kgamma, a single convergent point promoting tumor inflammation and progression. Cancer Cell. 2011; 19:715-27; PMID:21665146; http://dx.doi.org/ 10.1016/j.ccr.2011.04.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coussens LM, Zitvogel L, Palucka AK. Neutralizing tumor-promoting chronic inflammation: a magic bullet?. Science 2013; 339:286-91; PMID:23329041; http://dx.doi.org/ 10.1126/science.1232227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bronte V, Murray PJ. Understanding local macrophage phenotypes in disease: modulating macrophage function to treat cancer. Nat Med. 2015; 21:117-9; PMID:25654601; http://dx.doi.org/ 10.1038/nm.3794 [DOI] [PubMed] [Google Scholar]
- 46.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM, Akcakanat A et al.. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014; 2:361-70; PMID:24764583; http://dx.doi.org/ 10.1158/2326-6066.CIR-13-0127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Almanzar G, Schwaiger S, Jenewein B, Keller M, Herndler-Brandstetter D, Wurzner R, Schonitzer D, Grubeck-Loebenstein B. Long-term cytomegalovirus infection leads to significant changes in the composition of the CD8+ T-cell repertoire, which may be the basis for an imbalance in the cytokine production profile in elderly persons. J Virol. 2005; 79:3675-83; PMID:15731261; http://dx.doi.org/ 10.1128/JVI.79.6.3675-3683.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zitvogel L, Galluzzi L, Viaud S, Vetizou M, Daillere R, Merad M, Kroemer G. Cancer and the gut microbiota: an unexpected link. Sci Transl Med. 2015; 7:271ps1; PMID:25609166; http://dx.doi.org/ 10.1126/scitranslmed.3010473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang E, Uccellini L, Marincola FM. A genetic inference on cancer immune responsiveness. Oncoimmunology 2012; 1:520-5; PMID:22754772; http://dx.doi.org/ 10.4161/onci.19531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Uccellini L, De Giorgi V, Zhao Y, Tumaini B, Erdenebileg N, Dudley ME, Tomei S, Bedognetti D, Ascierto ML, Liu Q et al.. IRF5 gene polymorphisms in melanoma. J Transl Med. 2012; 10:170; PMID:22909381; http://dx.doi.org/ 10.1186/1479-5876-10-170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cescon DW, Haibe-Kains B, Mak TW. APOBEC3B expression in breast cancer reflects cellular proliferation, while a deletion polymorphism is associated with immune activation. Proc Natl Acad Sci U S A. 2015; 112:2841-6; PMID:25730878; http://dx.doi.org/ 10.1073/pnas.1424869112 [DOI] [PMC free article] [PubMed] [Google Scholar]