

Abstract
The anti-parasitic benzimidazole flubendazole has been used for many years to treat intestinal infections in humans and animals. Previous genotoxicity studies have shown that the compound is not a bacterial mutagen and a bone marrow micronucleus test, using a formulation that limited systemic absorption, was negative. The purpose of this study is to explore the genotoxicity of flubendazole and its main metabolites in in vitro micronucleus studies and to test a new oral formulation that improves systemic absorption in an in vivo micronucleus test. The isolated metabolites were also screened using the Ames test for bacterial mutagenicity. It was found that flubendazole, like other chemically related benzimidazoles used in anti-parasitic therapies, is a potent aneugen in vitro. The hydrolysed metabolite of flubendazole is negative in these tests, but the reduced metabolite (R- and S-forms) shows both aneugenic and clastogenic activity. However, in vitro micronucleus tests of flubendazole in the presence of rat liver S9 gave almost identical signals for aneugenicity as they did in the absence of S9, suggesting that any clastogenicity from the reduced metabolite is not sufficient to change the overall profile. Like flubendazole itself, both metabolites are negative in the Ames test. Analysis of dose–response curves from the in vitro tests, using recently developed point of departure approaches, demonstrate that the aneugenic potency of flubendazole is very similar to related anti-parasitic benzimidazoles, including albendazole, which is used in mass drug administration programmes to combat endemic filarial diseases. The in vivo micronucleus test of the new formulation of flubendazole also showed evidence of induced aneugenicity. Analysis of the in vivo data allowed a reference dose for aneugenicity to be established which can be compared with therapeutic exposures of flubendazole when this has been established. Analysis of the plasma from the animals used in the in vivo micronucleus test showed that there is increased exposure to flubendazole compared with previously tested formulations, as well as significant formation of the non-genotoxic hydrolysed metabolite of flubendazole and small levels of the reduced metabolite. In conclusion, this study shows that flubendazole is a potent aneugen in vitro with similar potency to chemically related benzimidazoles currently used as anti-parasitic therapies. The reduced metabolite also has aneugenic properties as well as clastogenic properties. Treatment with a new formulation of flubendazole that allows increased systemic exposure, compared with previously used formulations, also results in detectable aneugenicity in vivo. Based on the lack of carcinogenicity of this class of benzimidazoles and the intended short-term dosing, it is unlikely that flubendazole treatment will pose a carcinogenic risk to patients.
Introduction
Filarial worm parasitic diseases are highly prevalent in the poorest regions of the world and are transmitted by specific insect vectors, the river blackfly for onchocerciasis and the mosquito for lymphatic filariasis (LF). Approximately 1.3 billion people are at risk and 120 million are infected with LF caused by Wuchereria bancrofti and Brugia spp. (1). For onchocerciasis, at least 25 million people are infected with Onchocerca volvulus and 130 million people are at risk of infection (2,3). Many millions are also at high risk of infection with a third form of filariasis, loiasis, caused by the Loa loa parasite (4).
Filarial diseases are treated in mass drug administration (MDA) programmes, currently with the benzimidazole, albendazole in combination with either ivermectin or diethylcarbamazine. The aims of the MDA programmes are to reduce the immature microfilarial load in patients and to halt disease transmission. As reproductively active adult macrofilariae can live for 12 years, MDA treatments are prolonged and so far have been running for more than 12 years in some territories (5).
Over 14 million people are co-infected with Onchocerca and L. loa and are treated using ivermectin alone or in combination with albendazole, which can result in undesirable side-effects of encephalopathy and brain dysfunction, potentially leading to coma and death (4). Thus, there is an urgent need for a new, safe and effective treatment.
Flubendazole, an antihelmintic benzimidazole carbamate, is effective for treating gastrointestinal (GI) parasites in humans and animals (6) and is a highly active agent against macrofilariae. However, flubendazole, as currently formulated for this purpose, has poor systemic availability when given orally (6). In order to improve systemic bioavailability it has been reformulated as an amorphous solid dispersion (ASD) oral formulation. If clinical development of such a new formulation is successful, flubendazole would be valuable for case management in areas of high risk of co-infection. Since flubendazole has macrofilaricidal activity, it is hoped that the dosing period could be significantly reduced to a few days (7).
Due to its poor bioavailability and solubility, the original oral formulation for GI infections induces limited systemic or GI toxicities. In addition, bone marrow micronucleus tests to detect chromosome damage are negative, as are tests to measure the induction of dominant lethal mutations in treated mice. Flubendazole is negative in the Ames bacterial mutation test and also tests to measure the induction of DNA repair in Bacillus subtilis (8). Mutation tests in the yeast Saccharomyces cerevisiae and the fruit fly Drosophila melanogaster are also negative (8). Many benzimidazoles, including flubendazole, bind to the same site on the tubulin protein as colchicine but distinct from Vinca alkaloids such as vinblastine, inhibiting the polymerisation of tubulin and thus disrupting the mitotic spindle (9). Spindle poisons all have the potential to induce polyploidy (multiple sets of the chromosome complement) and aneugenicity (loss and gain of small numbers of chromosomes). Flubendazole has been shown to induce polyploidy in CHL cells and morphological transformation in C3H/10T1/2 Cl 8 mouse embryo fibroblasts (9).
As the new oral ASD formulation allows improved systemic absorption, it was necessary to re-evaluate the toxicity of flubendazole including potential genotoxicity.
Non-DNA reactive genotoxic substances are widely accepted as having threshold dose responses (10–15). Aneugens were the first substances within this category, to have clear non-linear dose responses accepted with a clear mode of action (MOA) (10,11). In fact, benzimidazoles were included in this first set of studies that initiated the paradigm shift from the default that all genotoxic substances had a linear dose response. The aneugenic MOA is either mitotic spindle depolymerisation or inhibition of polymerisation, and both mechanisms lead to chromosome loss and non-disjunction. Chromosome loss is readily quantified using the micronucleus assay, and numerous aneugens have well characterised threshold responses for this endpoint in vitro and in vivo (10–18). A recent review presents clearly defined no observed genotoxic effect levels (NOGELs) in vitro and in vivo, for the aneugenic substances colchicine, carbendazim, nocodazole, mebendazole, benomyl, nitrobenzene, benzonitrile, paclitaxel, bisphenol-A, rotenone, vincristine and vinblastine (19).
The UK Department of Health’s Committee on Mutagenicity (COM) have stated that ‘it is reasonable to assume that aneuploidy inducing chemicals (particularly those that function by interfering with the spindle apparatus of cell division) may have a threshold mode of action’ (20). In a more recent report the COM advised that ‘there is sound scientific basis to assume that benzimidazoles have a threshold of action in both somatic and germ cells’ (11). ‘It was not considered essential for a benzimidazole to have been studied in vivo for it to be included in the common mechanism group, since the available data indicated that in vitro aneugenicity is a good predictor of in vivo aneugenicity for the benzimidazoles’. Mebendazole has also been well characterised as having a NOGEL and threshold dose response for chromosome loss (12–14,18). It is therefore key to note that benzimidazoles are used as an exemplar case to show that similar acting aneugens can be assessed together for hazard and risk assessment (12–22). This, together with the fact that they are accepted as having threshold dose responses for aneuploidy, means it is likely that the benzimidazole flubendazole, will have similar points of departure (PoD) in the form of NOGEL, threshold (Td) and/or benchmark dose (BMD) as previously studied compounds of this class (22).
The purpose of this study is to investigate the aneugenicity of flubendazole and its significant metabolites, the hydrolysed and reduced (R- and S-derivatives) using in vitro and in vivo micronucleus tests. Micronucleus tests are established assays to measure both chromosome structural damage (clastogenicity) and chromosome number changes (aneugenicity) when used in conjunction with centromeric staining. The in vivo test was carried out to determine if the new oral ASD formulation, with improved bioavailability of flubendazole, increases systemic levels sufficiently to induce aneugenicity. This is in contrast to the previous oral GI formulation, which gave a negative response in the bone marrow micronucleus test. In addition, the consequence of in vivo metabolism could also be evaluated. Recently developed methods to explore PoD of response were used to help establish reference doses from the in vivo micronucleus test dose–response data and to allow in vitro data comparisons with established aneugens of the same chemical class, to aid understanding of the hazards posed by flubendazole treatment of patients. Thus, this work aimed to establish the aneugenicty potency of flubendazole and its metabolites in vitro and in vivo for the first time and to provide a valuable example of how genetic toxicity dose response data from aneugens can be used for human health risk assessment purposes.
Materials and methods
Test materials
The chemical structure of flubendazole and its two major metabolites, the reduced form and the hydrolysed form, are shown in Figure 1. The chemical structures of mebendazole, albendazole and albendazole sulfoxide are also presented. Flubendazole batch number EPL-BS1065 Batch 3, purity 99.2% (Epichem, Murdoch University Campus, Murdoch, Australia) was used for the initial in vitro experiments. Flubendazole Amorphous Solid Dispersion batch number 10035642-0060, purity 99.2% (Abbot Laboratories, Abbott Park, IL, USA) was used for the in vivo experiment. Flubendazole (batch number SZBB157XV, purity 99.2%, supplied by Sigma–Aldrich) was used as a comparator in the in vitro experiments with the metabolites. For the in vivo experiment with flubendazole, an ASD formulation (Flubendazole: vitamin E TPGS: Copovidone (10:5:85) %w/w) was used. For this study doses are expressed in terms of mg flubendazole/kg following a correction factor for purity of ×11.35 (88.1mg flubendazole/g solid dispersion). Formulations were freshly prepared in purified water prior to each dosing occasion. To ensure homogeneity, dose bottles were stirred continuously (on a magnetic stirrer) immediately before and throughout dosing. Samples were assessed for achieved concentration and homogeneity (Day 1 preparation) and achieved concentration (Day 2 preparation).
Figure 1.

Chemical structures of flubendazole, its metabolites and related benzimidazole anti-parasitic compounds.
For the in vitro micronucleus tests, hydrolysed flubendazole (JNJ-114699) batch number EPL BS1151 was used. Its purity was 98.4%. S-reduced flubendazole (JNJ-60281156), batch number 42417796 was used. Purity was >95%. R-reduced flubendazole (JNJ-60774643), batch number 42414540 was used. Purity was >95%. These compounds were kindly supplied by Janssen Pharmaceutica N.V, Beerse, Belgium (a Johnson and Johnson company). The metabolites were dissolved in anhydrous, analytical grade DMSO.
For the Ames tests, hydrolysed flubendazole, EPL-BS 1151, batch number 1 and reduced flubendazole (racemate), batch number 1, both manufactured by Epichem Pty Ltd were used. Both had a purity of 98.4%.
For in vitro studies flubendazole and its metabolites, were dissolved in anhydrous analytical grade DMSO.
Albendazole (Sigma–Aldrich, batch number SLBD9170V, purity 99%) and albendazole sulfoxide (Sigma-Aldrich, batch number SZBD049XV, purity 99.9%) were used as comparators in the in vitro experiments with the metabolites. These compounds were dissolved in anhydrous, analytical grade DMSO.
The positive control compounds mitomycin C and vinblastine sulfate were obtained from Sigma–Aldrich Chemical Co, Poole, Dorset, UK. Cyclophosphamide was obtained from Arcos Organics, Loughborough, Leicestershire, UK.
Test methods
In vitro micronucleus test
Blood was taken from two healthy non-smoking, healthy male volunteers, who were not taking any medication. Whole blood cultures were prepared by pipetting 0.8ml of pooled heparinised blood into hydroxyethylpiperazine-N’-2-ethanesulfonic acid-buffered RPMI media containing 10% (v/v) heat-inactivated foetal calf serum plus penicillin and streptomycin. The mitogen phytohaemagglutinin was included in the media at a concentration of approximately 2% (v/v) and the cultures (9.4ml) incubated at 37 C for 48h prior to treatment.
Test chemicals were dissolved in reagent grade DMSO. Stock solutions of test chemicals were prepared at 100× the required final concentrations approximately 2–3h prior to treatment and diluted 100-fold directly in the blood cultures with mixing (0.1ml additions).
Blood cultures were treated with test substance or controls for 3 or 24h. Solutions of 150mM KCl or rat liver S9-mix (0.5ml) were added as appropriate (final culture volume was 10ml). Cultures were incubated at 37°C.
Following short (3h) or extended (24h) treatments the compound was removed (via centrifugation), washed in sterile saline and resuspended in pre-warmed media containing foetal calf serum and streptomycin/penicillin. Cytochalasin B was added to give a final concentration of 6 μg/ml and the cell cultures incubated for an additional 21 or 24h (3 + 21h or 24 + 24h treatments). Total time from initiation of blood culture to cell harvest was either 72 or 96h.
Initial flubendazole experiments were performed using short (3h), plus +/−S9 and 24h treatment with 24h recovery −S9. For the studies with flubendazole metabolites plus comparator compounds, 24h treatment with 24h recovery alone was used.
Following treatment, at the time of cell harvest, cultures were then centrifuged, the supernatant discarded and the pellets resuspended in 4ml of 0.075M KCl (hypotonic solution) to swell the cells, for 4min. The cells were then then fixed in ice-cold methanol/glacial acetic acid (7:1 v/v). This procedure was repeated as deemed necessary. Cells were pelleted after fixation and resuspended in a small amount of fixative. Drops of this cell suspension were used to prepare several slides per culture. The slides were air-dried and stained with 125 µg/ml acridine orange. Slides were protected from light before scoring.
Where possible, 1000 binucleate cells from each culture (2000 per concentration of test substance and the positive controls and 4000 per concentration for vehicle controls) were analysed for micronuclei. After completion of scoring and decoding of slides, the numbers of binucleate cells with micronuclei (MNBN cells) in each culture were obtained.
The proportions of MNBN cells in each replicate were used to establish acceptable heterogeneity between replicates by means of a binomial dispersion test.
For each study acceptance criteria were met as follows:
The binomial dispersion test demonstrated acceptable heterogeneity (in terms of MNBN cell frequency) between replicate cultures, particularly where no positive responses were seen.
The frequency of MNBN cells in vehicle controls fell within the laboratory’s historical vehicle control (normal) ranges.
The positive control chemicals induced statistically significant increases in the proportion of cells with micronuclei. Both replicate cultures at the positive control concentration analysed under each treatment condition demonstrated MNBN cell frequencies that clearly exceeded the laboratory’s historical vehicle control ranges.
A minimum of 50% of cells had gone through at least one cell division (as measured by binucleate + multinucleate cell counts) in negative control cultures at the time of harvest.
The proportion of MNBN cells for each treatment condition was compared with the proportion in negative controls by using Fisher’s exact test. Probability values of P ≤ 0.05 were accepted as significant. Additionally, the number of micronuclei per binucleate cell were obtained and recorded.
The replication index (RI), which indicates the relative number of nuclei compared with controls was used as a measure of cytotoxicity and determined using the formulae below (23):
Relative RI (expressed in terms of percentage) for each treated culture was calculated as follows:
Cytotoxicity (%) is expressed as (100 − Relative RI).
Mechanism of action studies (FISH analysis)
In order to determine if the micronuclei observed were generated primarily as a result of clastogenicity or aneugenicity, fluorescence in situ hybridization (FISH) with pan-centromeric human DNA probes (PlaninumBright™550 (Ex/Em 550/580), Filter: TRITC (pan-centromere probe and chromosome specific centromere probes) was used to highlight if micronuclei contained centromeres or not. Fluorophores and filters for probes were obtained from Leica Microsytems, Ltd, UK.
Those micronuclei containing a centromere were deemed to contain a whole chromosome as a result of spindle malfunction (aneugenicity), whereas those that did not contain a centromere were deemed to result from chromosome breakage (clastogenicity).
Freshly prepared slides from stored cell pellets were processed from cultures exposed to flubendazole concentrations of 7.5 and 9.0 μg/ml (3 + 21h −S-9); at 16 and 25 μg/ml (3 + 21h +S-9) and 3.0 and 5.0 μg/ml (24 + 24h −S-9).
Prior to hybridisation, the sample slides were pre-treated in 2 × SSC/0.5% IGEPAL®CA630 (Sigma–Aldrich, UK), a non-ionic, non-denaturing detergent, octylphenoxypolyethoxyethanol, pH 7.0±0.1, at approximately 37°C for 15min. The slides were dehydrated by placing them through a series of ethanol washes (70%, 85% and 100% ethanol) for 1min each prior to drying at room temperature (15–30°C). The samples and FISH probe (pan-centromere) were co-denatured using the StatSpin®ThermoBriteTM Denaturation and Hybridisation System, as follows: 20 µl of FISH probe (per slide) was added with the application of a glass coverslip and sealed by rubber cement. The sample and FISH probe were denatured at 75±1°C for 5min using the StatSpin®ThermoBriteTM. The slides were hybridised overnight in a humidified chamber at 37±1°C using the StatSpi®ThermoBriteTM, containing humidity strips saturated with distilled water. Following overnight hybridisation, the rubber cement was removed and the slides washed with 2 × SSC/0.1% igepal for 2min at room temperature. The coverslips were carefully removed and the slides washed in 0.4 × SSC/0.3% igepal for 2min at 72±1°C followed by 2 × SSC/0.1% igepal for 1min at room temperature. The slides were dehydrated by placing them through a series of ethanol washes (70%, 85% and 100% ethanol) for 1min each. Slides were allowed to air dry prior to mounting in DAPI antifade and stored at −20°C until analysis.
Slide analysis was conducted using fluorescence microscopy using appropriate filters. Slides from the selected treatments and controls were coded (analysis was conducted ‘blind’). Where possible, one hundred MNBN cells per concentration were analysed. Micronuclei were classified as centromere-positive if one or more fluorescent signals were present and centromere-negative if no fluorescent signals were observed. After completion of scoring and decoding of slides, the proportions of centromere positive and centromere negative micronuclei (per concentration) were obtained and tabulated.
Bone marrow micronucleus test
Groups of seven young, out-bred female Han Wistar rats [Cr1 WI(Han)], were obtained from Charles River (UK) Ltd, UK. Animals were housed in groups of no more than seven per cage in wire topped, solid bottomed cages. Animals were housed in rooms air-conditioned to provide 15–20 air changes/hour. Temperature and humidity ranges were 20–24°C and 45–65%, respectively. Fluorescent lighting was controlled automatically to give a cycle of 12h light (0600 to 1800) and 12h dark. Animals were acclimatised for 7 days prior to dosing. Throughout the study, animals had access ad libitum to SQC Rat and Mouse Maintenance Diet No 1, Expanded (Special Diets Services Ltd. Witham). Mains water was provided ad libitum via water bottles. Bedding used was European softwood bedding (Datesand Ltd, Manchester, UK). Aspen chew block and rodent retreats were provided to enrich the environment and welfare of the animals. The rats were approximately 9 weeks old with a weight range of 169–216g on the first day of dosing.
Animals were given flubendazole formulations, or solutions of the controls, by oral gavage. Doses were given at 0 and 24h and the animals killed 24h after the second dose. The doses of flubendazole selected were 65, 130 and 400mg/kg/day based on previous dose range finding studies. The top dose was limited by the viscosity of the formulation. Female animals were chosen for the study based on previous toxicology studies showing that slightly higher exposures could be achieved in this gender compared to male animals. Satellite animals were included to allow estimation of the exposure of flubendazole and its reduced and hydrolysed major metabolites.
Bone marrow smears were prepared from washed and filtered femoral bone marrow samples obtained from the animals on test. After fixing and staining with acridine orange, slides were scored using fluorescence microscopy. Two thousand polychromatic erythrocytes (PCE) were scored for the presence of micronuclei and the relative proportion of PCE were determined among a total of 500 erythrocytes [PCE and normochromatic erythrocytes (NCE)] scored.
The percentage of PCE’s for each animal and the mean percentage of PCE’s of each group were used to estimate if any bone marrow toxicity was induced in the treated animals compared with the vehicle controls. For each group, inter-individual variation in the numbers of micronucleated PCE’s was estimated using a heterogeneity chi-square calculation. The numbers of micronucleated PCE’s in each treatment group were compared with those of the vehicle control using a 2×2 contingency table to determine chi-square and thus allow statistically significant changes to be identified. A linear trend test was used to evaluate the dose–response relationship observed.
Exposure assessment
Groups of female satellite animals were dosed with vehicle or flubendazole (high dose). Animals were dosed by the same route, dose level and at the same dosing frequency as that described for the micronucleus experiment animals. Plasma was isolated from these animals at 9 and 24h and used to assess systemic exposure to flubendazole and its two significant metabolites. Rat plasma concentrations of flubendazole, reduced flubendazole and hydrolysed flubendazole were determined using a LC-MS-MS method following plasma protein precipitation (see supplementary Method for full details).
Ames tests
Salmonella typhimurium strains TA98, TA100, TA1537, TA1535 and Escherichia coli strain WP2 uvrA (Molecular Toxicology, Inc.) were used in this study. The preincubation method (24) was used. Histidine auxotrophs (strains TA98, TA100, TA1535 and TA1537) of S. typhimurium and the tryptophan auxotroph E. coli WP2 uvrA were thawed from frozen permanent cultures and grown overnight in Oxoid broth No. 2. The test item was diluted in the chosen solvent at concentrations determined in a range-finding test. The bacteria (approximately 1–3×108in 100 μl) were combined with 50 μl of either the test item, the solvent control, or 50 μl of positive control and 500 μl of the phosphate buffer (pH 7.4) and preincubated at 37±1°C for approximately 30min. After the preincubation period, the content of each tube was combined with 2ml of top agar (45°C) containing 50 μM of histidine and tryptophan to give a total volume of 2650 μl. The mixture was then poured onto a 90-mm plate (each well resulting from an independent treatment tube) containing histidine/tryptophan-deficient base agar and was incubated at 37±1°C for 3 days. After incubation the plates were examined for the presence of a precipitate and for signs of toxicity as indicated by a diminished or absent background lawn or reduced colony numbers as compared with the solvent control. The colonies were counted using an automatic colony counter (Sorcerer–Perceptive).
Determination of PoD metrics
The BMD approach was used as a potential improvement on the no observed effect level (NOEL) approach (25,26). BMD has been used as a PoD when carrying out hazard and risk assessment using both cancer and non-cancer endpoints, but has only recently been used for the purpose of genetic toxicology data (26). The International Life Science Institute (ILSI) Genetic Toxicology Technical Committee (GTTC), previously the IVGT, tested this methodology and confirmed its suitability. Another great benefit is the extensive work that the National Institute for Public Health and the Environment (RIVM) did with support from the Food and Drug Administration (FDA). They carried out potency correlations for numerous genotoxic carcinogens using BMD for both genetic toxicology and carcinogenicity endpoints, and found a positive correlation (26–29). NOGEL and Td metrics are not suitable for carrying out these potency ranking correlations due to data limitations as well as limitations in the statistical approaches themselves. Both potency estimates are highly sensitive to experimental design differences, and the NOGEL is also not very accurate and does not provide a measure of uncertainty (29). Therefore, BMD was considered the best choice for carrying out the potency rankings for these benzimidazoles, which then allowed the grouping of these substances, which is the advised approach from the UK COM.(20) Therefore, all in vitro dose responses were assessed using the PROAST software, and the BMD PoD metrics for each compound were defined (29–31).
PROAST version 50.8 (under development) was used to carry out covariate BMD analysis for potency ranking (http://www.proast.nl) (29). The four-parameter ‘full’ exponential model is recommended for the analysis of these continuous micronucleus data, when using the compound as a covariate (25,27,28). During this combined analysis, the shape parameters for maximum response (parameter c) and log-steepness (d) were assumed equal, while background response (a), potency (b) and the additional parameter representing within group variation (var) were covariate dependent (28). Individual fits at the covariate level for all datasets are presented in supplementary Figures 1 and 2.
A BMDL10 refers to the estimate of lower 95% confidence interval of a dose that produces a 10% increase over the back-ground level for continuous endpoints such as frequencies of micronuclei (BMD10), and 10% extra risk for quantal endpoints (25). BMDU10 refers to the upper estimate of this same confidence interval. Ten percent is an arbitrary but common choice (30–33), and changing this to a different benchmark response [BMR, or critical effect size (CES)] has recently been shown to have no effect on the potency rankings (27). In these in vitro potency-ranking plots, the width of the confidence interval between BMDL10–BMDU10 represents the precision to which the BMD10 can be defined. Ranking these by midpoint was chosen as the most suitable way to compare potency across the different chemicals (covariate). The (BMR) of one standard deviation above the spontaneous (control) value (BMD1SD) was used for the in vivo MN dose response data, which is also a recommended approach from the GTTC and International Workgroup on Genotoxicity Testing (30,31). This BMR is recognised to be equivalent to 10% excess risk for individuals below and above the 2nd and 98th percentiles, respectively for micronucleus induction over the negative control frequency in this case (34). Model selection was primarily based upon the P-value for goodness-of-fit to the data and the Akaike’s Information Criterion (AIC). For this study, the best fitting model was selected from among the suite of continuous models, using EPA’s Benchmark Dose Software (i.e. BMDS v2.5). Response data were transformed in order to achieve normally distributed data with homogeneous variance prior to analysis with the BMDS software (32). This same approach to transformation was taken when deriving the flubendazole NOGEL using the one-sided Dunnett’s test (P < 0.05) in the ‘drsmooth’ package of R (32,33,35,36).
Results
In vitro lymphocyte micronucleus tests of flubendazole
The results of the in vitro micronucleus tests of flubendazole are shown in Tables 1 (short exposure with and without rat liver S9) and 2 (long exposure without rat liver S9). These show that flubendazole is a potent aneugen in vitro. After short exposure, statistically significant increases in micronucleated binucleate lymphocytes are seen at doses of flubendazole of 5.0 µg/ml (−S9) and 14.0 µg/ml (+S9). These differences reflect the lower cytotoxicity of flubendazole exposure +S9, which suggests that flubendazole metabolites formed by the rat liver S9 may be less toxic than the parent molecule, as confirmed by the tests on the isolated metabolites shown below. After longer-term exposure, cytotoxicity is increased, as does the aneugenic action of flubendazole. In the first experiment, statistically significant increases in micronuclei were evident at the lowest tested concentration of 0.5 µg/ml. Thus, a second experiment was carried out covering a lower concentration range. In this second experiment the lowest dose where a statistically significant increase in micronuclei was observed was 0.25 µg/ml.
Table 1.
In vitro micronucleus test results in human lymphocytes following treatment with flubendazole after short (3h) exposure
| Treatment | Flubendazole (µg/ml) | Cytotoxicity (%) | Mean MNBN cell frequency (%) |
|---|---|---|---|
| 3+21h –S9 | 0.0 (Vehicle) | 0.0 | 0.23 |
| 2.0 | 0.0 | 0.05 | |
| 5.0 | 7.0 | 0.55* | |
| 6.0 | 0.0 | 0.85* | |
| 7.5 | 8.0 | 1.65* | |
| 9.0 | 26.0 | 6.55* | |
| MMC (0.80) | – | 8.70* | |
| 3+21h +S9 | 0.0 (Vehicle) | 0.0 | 0.25 |
| 10.0 | 0.0 | 0.25 | |
| 12.0 | 0.0 | 0.20 | |
| 14.0 | 14.0 | 0.65* | |
| 16.0 | 4.0 | 0.90* | |
| 19.0 | 7.0 | 0.55* | |
| 25.0 | 12.0 | 2.88* | |
| CYC (12.5) | – | 1.40* |
Historical control range for the testing laboratory was 0.1–0.95 and 0–1.1 for –S9 and +S9, respectively. MMC, mitomycin C; CYC, cyclophosphamide.
*Statistically significant.
Table 2.
In vitro micronucleus test results in human lymphocytes following treatment with flubendazole after long (24h +24h) treatment
| Experiment | Flubendazole (µg/ml) | Cytotoxicity (%) | Mean MNBN cell frequency (%) |
|---|---|---|---|
| I | 0.0 (Vehicle) | 0.0 | 0.30 |
| 0.5 | 4.0 | 1.75* | |
| 1.0 | 0.0 | 3.38* | |
| 3.0 | 0.0 | 5.15* | |
| 5.0 | 28.0 | 4.74* | |
| 6.5 | 43.0 | 10.10* | |
| VIN (0.12) | – | 16.99* | |
| II | 0 (Vehicle) | 0.0 | 0.28 |
| 0.050 | 0.0 | 0.10 | |
| 0.075 | 0.0 | 0.30 | |
| 0.100 | 0.0 | 0.55 | |
| 0.250 | 0.0 | 1.15* | |
| 0.500 | 0.0 | 3.15* | |
| 1.00 | 16.0 | 4.25* | |
| VIN (0.08) | – | 4.25* |
VIN, vinblastine sulphate.
*Statistically significant (P > 0.05).
To determine if the micronuclei observed were due to a clastogenic or a aneugenic MOA, a mechanistic study was carried out using centromeric stains (FISH technique) to determine if the micronuclei contained a centromere, where it is assumed that such micronuclei are formed from whole chromosomes as a result of spindle malfunction or lack a centromere and are thus likely to be formed from chromosome fragments. The results are shown in Table 3. As the micronuclei predominantly stain positively for centromeres, flubendazole acts as an aneugen. This was also the case for the micronuclei induced by flubendazole in the presence of S9, indicating that flubendazole metabolism in vitro does not result in the formation of detectably clastogenic metabolites under the conditions used.
Table 3.
Mechanistic study in vitro, to show the percentage of micronuclei with and without centromeres, to determine aneugenicity or clastogenicity, respectively
| Compound | Concentration μg/ml | Fold increases in MN over concurrent vehicle control | % micronuclei with a centromere | % micronuclei without a centromere | Total MN cells scored |
|---|---|---|---|---|---|
| Flubendazole (3h + 21h, −S9) | 7.5 | 7.2 | 80 | 20 | 100 |
| 9.0 | 28.5 | 88 | 12 | 100 | |
| Flubendazole (3h + 21h, +S9) | 16 | 3.6 | 83 | 17 | 100 |
| 25 | 11.5 | 86 | 14 | 100 | |
| Cyclophosphamide | 12.5 | 5.6 | 16 | 84 | 100 |
| Vinblastine | 0.12 | 56.6 | 73 | 11 | 84 |
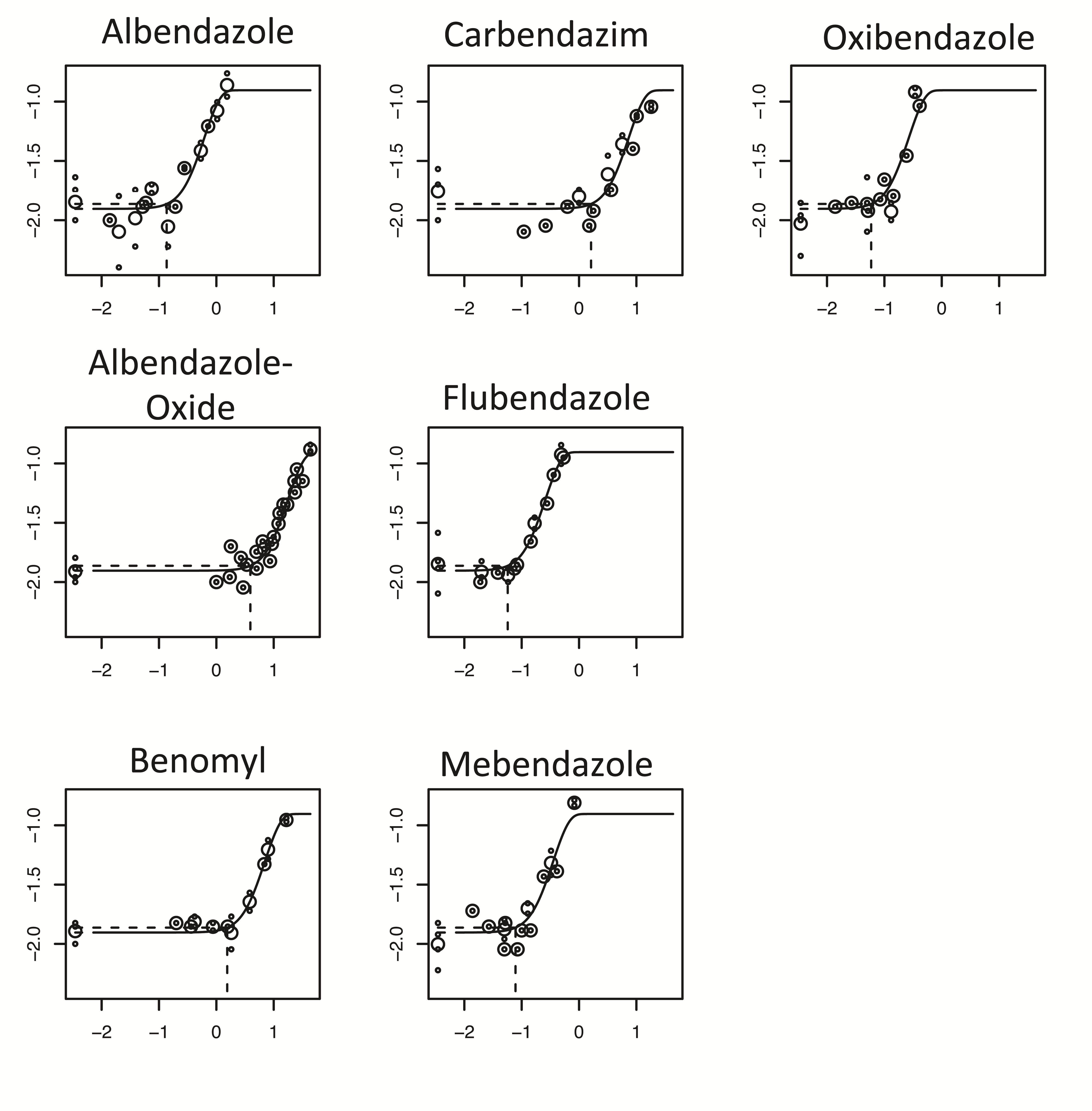
The analysis of thresholds (PoD metrics) for the induction of micronuclei by albendazole, benomyl, carbendazim, flubendazole, mebendazole, oxibendazole and albendazole oxide in CHO-K1 cells (19) are shown in Table 4 and Figure 2. These show that the PoDs for flubendazole, oxibendaolze, mebendazole and albendazole are very similar, and more specifically were equipotent as observed by the overlapping BMDL10–BMDU10 from the covariate BMD analysis. Albendazole oxide was also clearly shown to be less potent than the parent compound due to its lack of overlap in BMD with the other compounds.
Table 4.
Threshold dose (Td) PoD metrics defined for CHO-K1 cells treated with benzimidazoles with CBMN endpoint, with background MNBN% defined along with Td and lower confidence Td–L–CI as calculated in the article by Ermler et al. (19)
| Compound | Td (µg/ml) | Td–L–CI (µg/ml) | BMD10 (µg/ml) | BMDL10 (µg/ml) |
|---|---|---|---|---|
| Albendazole | 0.005 | 0.003 | 0.037 | 0.027 |
| Benomyl | 0.659 | 0.520 | 0.450 | 0.348 |
| Carbendazim | 0.346 | 0.268 | 0.310 | 0.235 |
| Flubendazole | 0.028 | 0.025 | 0.019 | 0.013 |
| Mebendazole | 0.032 | 0.027 | 0.024 | 0.018 |
| Oxibendazole | 0.032 | 0.032 | 0.015 | 0.010 |
| Albendazole oxide | 1.708 | 1.474 | 1.100 | 0.861 |
BMD covariate analysis in PROAST version 50.8, used to define these BMD10 metrics is presented in Figure 2.
Figure 2.

BMD10 analysis of in vitro MN dose responses in Chinese hamster ovary (CHO-K1) cells following exposure to the benzimidazoles, oxibendazole (grey), flubendazole (light blue), mebendazole (pink), albendazole (black), carbendazim (dark blue), benomyl (green) and albendazole oxide (red) (19). The figure was modified from the article (37). The top figure shows that the four parameter (m5-bv) exponential model provided a suitable fit to each dose response using the compound as covariate for BMD analysis. The dotted lines show the BMD10 derivations for each dose response, and these correspond to the mid-point for each line plotted in the bottom figure. These lines in the bottom graph span the BMDL10 to the BMDU10 derived from the top figure using PROAST v50.8. Flubendazole, oxibendazole and mebendazole grouped together along with albendazole. Benomyl and carbendazim also grouped together with albendazole oxide being a lot less potent and forming no groupings. The individual plots are located in supplementary Figure 2.
In vitro micronucleus test of flubendazole metabolites
In vitro micronucleus tests were carried out with the hydrolysed metabolite and the R-reduced and S-reduced metabolites of flubendazole, using flubendazole, albendazole and albendazole sulfoxide as comparators. The most sensitive exposure condition determined for flubendazole of 24h (plus 24h recovery) was used. The results are shown in Tables 5–7 (metabolites) and 8 (comparator compounds). It can be seen that the hydrolysed metabolite is inactive at test concentrations up to 100 µg per ml (54% cytotoxicity). However, there was a clear dose related induction of micronuclei from both the R- and S-reduced metabolites. For the R-reduced metabolite a significant increase in micronuclei was seen at the lowest dose of 1.1 µg per ml, whereas with the S-reduced metabolite, the response with the lowest dose, 1.5 µg per ml, was similar to that of the vehicle control, whereas increases in micronucleated cells were seen at all the higher doses for both enantiomers. Flubendazole, albendazole and albendazole sulfoxide all gave the expected increases in micronuclei with the lowest non-significant doses, 0.1 µg per ml (flubendazole) 0.04 µg per ml (albendazole) and 3.0 µg per ml (albendazole sulfoxide).
Table 5.
In vitro micronucleus test of hydrolysed flubendazole
| Compound | Concentration (µg/ml) | Cytotoxicity (%) | Frequency of micronucleated binucleate cells (%) |
|---|---|---|---|
| Vehicle | – | – | 0.65 |
| HF | 5.0 | 0% | 0.5 |
| HF | 20.0 | 1.0% | 0.65 |
| HF | 60.0 | 28.0% | 0.50 |
| HF | 80.0 | 38.0.0% | 0.95 |
| HF | 100.0 | 54.0% | 0.50 |
| Vinblastine | 0.06 | NM | 9.25* |
| Mitomycin C | 0.10 | NM | 27.8* |
NM, not measured; HF, hydrolysed flubendazole.
*Statistically significant P < 0.05.
Table 7.
In vitro micronucleus test of S-reduced flubendazole (combined data from two experiments)
| Concentration (µg/ml) | Cytotoxicity (%) | Frequency of micronucleated binucleate cells (%) |
|---|---|---|
| 0.0a | 0.0 | 0.6a/0.6 |
| 1.5a | 0.0 | 0.65 |
| 4.2a | 3.0 | 1.25* |
| 11.7a | 12.0 | 3.90* |
| 20.0 | 11.0 | 4.00* |
| 32.4a | 0.0 | 5.50* |
| 35.0 | 0.0 | 5.7* |
| 50.0 | 6.0 | 4.15* |
| 55.0 | 27.0 | 6.45* |
| 60.0 | 55.0 | 6.95* |
aData from Experiment 1, all other data is from Experiment 2.
*Statistically significant P < 0.05.
Table 6.
In vitro micronucleus test of R-reduced flubendazole (combined data from two experiments)
| Concentration (µg/ml) | Cytotoxicity (%) | Frequency of microncucleated binucleate cells (%)* |
|---|---|---|
| 0.0 | 0.0 | 0.30a/0.65 |
| 1.1a | 10.0 | 0.85 |
| 3.2a | 18.0 | 2.25 |
| 8.8a | 3.0 | 2.75 |
| 10.0 | 0.0 | 5.05 |
| 20.0 | 0.0 | 4.50 |
| 24.5a | 3.0 | 2.75 |
| 35.0 | 17.0 | 3.95 |
| 40.0 | 22.0 | 3.75 |
| 45.0 | 15.0 | 2.90 |
aData from Experiment 1, all other data is from Experiment 2.
*All treated groups gave increases in micronucleated binucleate cells that were statistically significant relative to the vehicle control P < 0.05.
BMD covariate analysis was used again to rank and group flubendazole, albendazole and their metabolites together based on the potency measure of overlapping BMDL10–BMDU10. Figure 3 shows equipotency and clear grouping of flubendazole, oxibendazole, mebendazole and albendazole, whereas the potency of the S- and R- enantiomers of the reduced flubendazole metabolite along with albendazole sulfoxide formed a separate grouping due to their lower potencies. This is in line with the recent analysis of tests of most of these substances using the MN endpoint in CHO-K1 cells by Ermler et al. (19) (Figure 2).
Figure 3.

BMD10 analysis of in vitro MN dose responses in human lymphocytes albendazole (green) flubendazole (black), R-reduced flubendazole (dark blue), S-reduced flubendazole (light blue) and albendazole sulfoxide (red), in human lymphocytes. The top figure shows that the four parameter (m5-bv) exponential model provided a suitable fit to each dose response using the compound as covariate for BMD analysis. The dotted lines show the BMD10 derivations for each dose response, and these correspond to the mid-point for each line plotted in the bottom figure. These lines in the bottom graph span the BMDL10 to the BMDU10 derived from the top figure using PROAST v50.8. Albendazole and flubendazole were equipotent, and the R- and S-metabolites of flubendazole were less potent and of similar potency to albendazole sulfoxide. Hydrolysed flubendazole showed no dose response using BMD analysis and was omitted from this covariate analysis.
As for flubendazole above, centromeric stains were used to determine if the micronuclei induced by the two reduced metabolite enantiomers contained a centromere and thus to determine if the micronuclei were formed due to aneugenicity and/or clastogenicity. The data are shown in Table 9. Surprisingly, it was found that both compounds showed evidence of both clastogenic and aneugenic activity, in that both centromere positive and centromere negative micronuclei were observed. For the R-enantiomer, approximately equal numbers of centromere positive and centromere negative micronuclei were detected, whereas for the S-enantiomer, approximately 65% of the micronuclei did not contain a centromere. The comparator compounds, including flubendazole, as expected, all showed a predominance of C+ staining micronuclei confirming aneugenicity, whereas the clastogen mitomycin C showed predominantly C− stained micronuclei confirming clastogenicity. Analysis of the micronuclei induced by flubendazole in the presence of S9 using centromeric staining gave almost identical levels of C+ and C− containing micronuclei as tests in the absence of S9, that is predominantly C+ micronuclei indicative of aneugenicity. Thus, if the reduced metabolite is formed by the action of S9 it does not induce sufficient micronuclei due to clastogenicity to make a difference under the conditions of the assay.
Table 9.
Mechanistic study to determine the nature (aneugenic vs. clastogenic) of the micronuclei from the in vitro micronucleus test of the reduced flubendazole enantiomers (R and S)
| Compound | Concentration (µg/ml) | Fold increases in MN over concurrent vehicle control | %C+ micronuclei | %C− micronuclei | Total MN cells scored |
|---|---|---|---|---|---|
| R-reduced flubendazole (RRF) | 3.2 | 7.5 | 40 | 60 | 100 |
| RRF | 8.8 | 9.2 | 57 | 43 | 82 |
| RRF | 24.5 | 14.7 | 49 | 51 | 57 |
| S-reduced flubendazole (SRF) | 4.2 | 2.2 | 39 | 61 | 100 |
| SRF | 11.7 | 6.7 | 33 | 67 | 100 |
| SRF | 32.4 | 9.5 | 37 | 63 | 67 |
| Albendazole | 0.7 | 9.3 | 72 | 28 | 100 |
| Albendazole sulfoxide | 30.0 | 6.2 | 71 | 29 | 100 |
| Flubendazole | 2.0 | 7.1 | 70 | 30 | 100 |
| Mitomycin C | 0.1 | 35 | 7 | 93 | 100 |
Table 8.
In vitro tests of comparator benzimidazoles
| Compound | Concentration (µg/ml) | Cytotoxicity (%) | Frequency of micronucleated binucleate cells (%) |
|---|---|---|---|
| Albendazole | 0.00 | 0.0 | 0.45 |
| 0.04 | 11.0 | 0.45 | |
| 0.16 | 18.0 | 1.20* | |
| 0.70 | 32.0 | 4.20* | |
| 3.00 | 12.0 | 4.00* | |
| Albendazole sulfoxide | 0.0 | 0.0 | 0.60 |
| 3.0 | 1.0 | 0.30 | |
| 10.0 | 3.0 | 1.85* | |
| 30.0 | 23.0 | 3.70* | |
| 40.0 | 20.0 | 5.50* | |
| Flubendazole | 0.000 | 0.0 | 0.40 |
| 0.025 | 4.0 | 0.20 | |
| 0.100 | 2.0 | 0.40 | |
| 0.400 | 6.0 | 4.75* | |
| 2.00 | 24.0 | 5.30* |
*Statistically significant P < 0.05.
Ames tests of the flubendazole metabolites
The results of the Ames tests of hydrolysed flubendazole and reduced flubendazole (racemate) can be accessed online (supplementary Tables 1a and 1b). Neither compound showed any dose related increase in revertants in any test strain used.
In vivo bone marrow micronucleus test
A previous toxicity test of the new formulation of flubendazole provided data on plasma exposures of the parent compound (Table 10). This showed that exposure to flubendazole is sub-linear with small increases in exposure between 65 and 400mg/kg. The results of the rat bone marrow micronucleus test using similar doses are shown in Table 11. It can be seen that contrary to previously published negative bone marrow micronucleus tests of flubendazole with alternative formulations (8), the new formulation of flubendazole induces micronuclei at all tested doses. In addition, there was a decrease in the PCE/NCE ratio with increasing dose, indicative of bone marrow toxicity as might be expected from exposure to an aneugen and the resulting cell cycle delay. The BMD analysis shown in Figure 4, using the BMDS software, estimates the likely PoD from these data. Thus for the administered dose, the BMD1SD is calculated as 18.97mg/kg and the BMDL1SD as 14.65mg/kg.
Table 10.
Data on flubendazole exposure from a previous single dose rat study
| Dose (mg/kg) | C max (µg/ml) | AUC 0–24 (µg·h/ml) |
|---|---|---|
| 65.0 | 0.91 | 15.10 |
| 130.0 | 0.90 | 16.48 |
| 402.0 | 1.33 | 24.92 |
Table 11.
In vivo rat micronucleus test
| Dose (mg/kg) | % Micronucleated PCE | % PCE |
|---|---|---|
| 0 | 0.21 | 45.83 |
| 65.0 | 1.35* | 31.74 |
| 130.0 | 2.31* | 33.00 |
| 400.0 | 1.94* |
*Statistically significant.
Figure 4.

In vivo MN dose responses from the rat study outlined in Table 4. BMDS was used to defined BMD1SD metrics, and log transformed response data provided a normal distribution and homogeneous variance as measured through the Shapiro–Wilks and Bartlett’s test, respectively, so were used for the analysis (35). The polynomial model was the most suitable for these data. A BMD1SD, 18.9699 and BMDL1SD, 14.6483 were derived from this model. A reference dose (RfD) calculated using this value would be calculated a scaling factor of 0.16 for rat to human, 60kg person, and an uncertainty factor of 10 for unknown differences; [(14.6483×0.16×60)/10] × 1000 = 14.06mg/person/day. Note that if the uncertainty is taken into account by use of the lower bound of the BMD1SD (BMDL1SD) compared with the BMD1SD or the NOGEL, then this factor of 10 is removed and you obtain a reference dose = 140.62mg/person/day.
Table 12 shows exposure to flubendazole and its two major metabolites 9h after administration of the highest dose of 400mg/kg. This shows that animals have detectable exposure to all three compounds with the non-genotoxic hydrolysed metabolite showing by far the greatest exposure compared with the other two molecules.
Table 12.
Exposure data including metabolites from the in vivo micronucleus test
| Compound | Mean plasma concentration, ng/ml (9ha after dose of 400mg/kg) |
|---|---|
| Flubendazole | 610 |
| Reduced flubendazole | 105 |
| Hydrolysed flubendazole | 20400 |
aNine hours represents the approximate Tmax from a previous pharmacokinetic experiment.
Discussion
This article shows that the benzimidazole flubendazole is a potent aneugen in vitro in human lymphocytes. Flubendazole is extensively metabolised in vivo to a hydrolysed form and small amounts of a reduced form (R- and S-enantiomers). In vitro micronucleus tests of these metabolites show that the hydrolysed form (by far the most prevalent metabolite) is not genotoxic, but both the S- and R-form of the reduced metabolite, when tested in isolation, showed both aneugenic and clastogenic properties. However, in vitro micronucleus tests of flubendazole showed the same high level of aneugenicity in the presence and absence of rat liver S9, suggesting that any clastogenicity from the reduced metabolite was not detectable under these conditions. Both metabolites were, like flubendazole, inactive in the Ames test.
In vivo tests of a new oral ASD formulation of flubendazole, which provides higher systemic bioavailability than currently marketed formulations, show that this compound is also a potent aneugen in vivo, as shown by the results of a bone marrow micronucleus test in the rat. The previous bone marrow micronucleus test of flubendazole, using the marketed oral GI formulation that results in little systemic availability, was negative. The results in this article show the dramatic difference changes in formulation can make to a compound’s toxicity profile, if it provides for greater systemic exposure. This has previously been demonstrated with flubendazole in developmental toxicity tests (38).
Flubendazole has been in use for many years as an anti-helmintic treatment for GI parasitic infections, both for humans and various animal species (6). The usual dosage of flubendazole in humans for this indication is 100mg once or twice a day for 3 days with systemic exposures resulting in plasma Cmax ‘s of <1ng/ml, although higher exposures have been noted if the compound is given with food. Thus, using the original commercial oral GI formulation, this compound is poorly bioavailable. Although toxic to GI-located parasites, there is little reported local GI or systemic toxicity in patients or in the animal species used in toxicology testing. The original genotoxicity tests of flubendazole provided for pharmaceutical registration of this indication showed a lack of mutagenicity in the Ames test and also a lack of clastogenicty or aneugenicity in vivo in the mouse bone marrow test.
It is known that flubendazole can bind to tubulin at the colchicine binding site, interfering with tubulin polymerisation, disrupting microtubule formation and interfering with normal mitosis. Due to the close chemical similarity with other aneugenic benzimidazoles, such as albendazole and mebendazole (flubendazole is a fluoro analogue of mebendazole), the aneugenic activity of flubendazole is not a surprise. The UK Committee on Mutagens has considered that the available data for these compounds can be regarded as belonging to a common mechanistic group with a similar MOA (20). As the target for all three compounds is a protein, rather than DNA, it is accepted that such compounds have a threshold below which no aneugenicity is induced (13,14). Using various analytical functions to determine PoDs in vitro and in vivo and making comparisons with related compounds from literature sources, it has been shown in this article that flubendazole, albendazole and mebendazole share similar PoD values for aneugenicity derived from in vitro micronucleus tests. This correlation for all of these antihelmintics was also clearly shown in the recent study in Chinese hamster CHO-K1 cells, where oxibendazole also showed a closely linked PoD for MNBN, but where thiabendazole was inactive, as shown in Table 4 [derived from reference (19)].
With regard to the metabolites of flubendazole, the hydrolysed form was inactive in in vitro micronucleus tests. The two enantiomers of the reduced metabolite (R, S) were equipotent with albendazole sulfoxide, and all were significantly less potent that their respective parent compounds.
In terms of PoD metrics for micronucleus induction in vivo after oral dosing with the new ASD formulation of flubendazole, the BMDL1SD metric from the rat in vivo study was used to derive a reference dose (RfD) (32,34) of 14.06mg/subject/day (Figure 4). However, it could be argued that using this metric in place of the NOGEL or BMD1SD could reduce the need for an uncertainty factor of 10 (as included in the original calculation), resulting in a revised RfD of 140.62mg/subject/day (33).
Analysis of plasma samples from rats dosed orally with flubendazole using the new ASD formulation from the in vivo micronucleus test have shown that the principal metabolite is the hydrolysed form (Table 4). The reduced metabolite is only found at 100ng/ml after dosing with 400mg/kg of flubendazole. Thus, if patients show a similar profile of metabolite formation to the rat, it is likely that plasma exposure to the reduced metabolite will be very low, although it would be helpful for risk assessment purposes to determine if it is likely that this metabolite could accumulate in specific tissues.
Flubendazole has been tested for carcinogenicity in both rats and mice using formulated diet at doses equivalent to a maximum of 20mg/kg in rats and 30mg/kg in mice. Neither showed evidence of carcinogenicity in any tissue including the GI tract (8). Although systemic exposure data were not reported in these tests, separate studies in the Wistar rat, after an oral dose of 40mg/kg, using a microsuspension of flubendazole, showed that after 4h, plasma levels of flubendazole reached 81ng/ml. In a separate study in the same strain given 10mg/kg of a microcrystalline suspension of 14C-labelled flubendazole, plasma levels reached 0.27µg/ml. The amount of total radioactivity present in the liver, lung, kidney, muscle and fat did not exceed 3.1 µg/g of tissue (39). It should be noted that the level observed in plasma after dosing with 10mg/kg of flubendazole is similar to the POD metrics for aneuploidy calculated for this compound in in vitro tests.
Analysis of faeces after oral dosing with flubendazole shows that the vast majority of the administered dose (>85%) is retained in the GI tract and appears in the faeces as the unchanged parent molecule (40). Thus, these studies do not provided definitive evidence of a lack of carcinogenicity in organs exposed via the systemic circulation at higher exposures of flubendazole and its metabolites such as those obtained after oral doses of flubendazole using formulations such as that described in this article.
Albendazole and mebendazole have been tested for carcinogenicity in lifetime studies in the rat and mouse (41–43). Both compounds were deemed negative for carcinogenicity in both test species. In the case of mebendazole carcinogenicity studies, there have been some criticisms of the study design due to poor survival and less than optimal histology. However, these data have been regarded as sufficient to define non-carcinogenicity by regulatory authorities. For the carcinogenicity studies of albendazole, an assessment by a joint FAO/WHO Expert committee on Food Additives noted that questions had been raised regarding the statistical analysis of the data from the rat and mouse carcinogenicity studies and the use of historical controls in the examination of the incidence of tumours. Thus, these studies were reviewed and were deemed to be satisfactory (41). Thus, these studies have passed scrutiny by many regulatory authorities around the world, including those in the USA, sufficient to allow their unrestricted use, including mass administration in the case of albendazole. Thus, although both compounds are aneugenic, this does not predict their lack of carcinogenicity under the conditions of the rodent tests provided. Both compounds are in widespread human use as anti-parasitic therapies. Albendazole is of particular interest as it is used in MDA programmes to control filariasis, involving almost a billion patients to date, who have been dosed with the compound in a small number of doses annually, but repeated over many years of treatment.
The proposed therapeutic use of flubendazole will be for the administration of a low number of doses to affected patients. A formulation that can achieve higher systemic exposure, such as used in this article, will have the potential to kill macrofilaria resulting also in loss of microfilaria, without the need for repeated dosing with similar compounds (such as albendazole) over more than a decade, as is currently the case. In addition it has the potential to avoid the encephalopathy and brain dysfunction seen in patients co-infected with Onchocerca and L. loa and treated using ivermectin alone or in combination with albendazole. Whether the chosen dose will produce an exposure of flubendazole that exceed the threshold for aneugenicity, is unknown. In addition, there will be exposure in patients to low levels of the R-reduced metabolites, which have shown both clastogenic and aneugenic potential when tested in isolation in vitro. However, taking into account the minimal duration of the intended dosing period and the lack of carcinogenicity of closely related compounds with the same aneugenic MOA this therapy is unlikely to pose a carcinogenic risk to patients. Mebendazole is also used in single doses (e.g. Vermox® at 500mg) for particular parasitic infections also resulting in exposures that most likely exceed the aneugenic threshold (human PK studies have shown that at high therapeutic doses, oral bioavailability is around 2%) (44).
In summary, this article shows that flubendazole is a potent aneugen, in vitro and in vivo. The latter contrasts with previously negative findings in vivo where the formulation did not provided significant systemic exposure. New approaches have been used to establish PoD metrics for flubendazole and its metabolites, which will be useful when therapeutic doses of flubendazole are established for treating serious cases of co-infection with specific filarial parasites.
Supplementary data
Supplementary Method, Tables 1a and 1b and Figures 1 and 2 are available at Mutagenesis Online.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgements
The contribution of Covance Ltd staff in carrying out the cytogenetic studies reported in this article, is acknowledged, as is Accelera Pty Ltd for carrying out the Ames tests. Robert Don (DNDi) is thanked for his support of this work. We are grateful to Janssen Pharmaceutica N.V., Beerse, Belgium (a Johnson and Johnson Company) for supply of the flubendazole metabolites. The Drugs for Neglected Diseases initiative acknowledges financial support from the Bill & Melinda Gates Foundation (USA) for this work. The authors would like to acknowledge Dr John Wills (Health Canada post-doctoral fellow) who provided support in generating Figure 2.
References
- 1. WHO (2015) Fact Sheet No 2012. http://www.who.int/mediacentre/factsheets/fs102/en/ (accessed September 22, 2015). [Google Scholar]
- 2. Onchocerciasis—River Blindness, Fact Sheet No 95. http://www.who.int/mediacentre/factsheets/fs095/en/ [Google Scholar]
- 3. WHO (2007) Onchocerciasis meeting of the International Task Force for Disease Education, Weekly Epidemiological Record, 1st June 2007, Nos 22/23, 82, pp. 197–208. [Google Scholar]
- 4. Zouré H. G. M. Wanji S. Noma M. Amazigo U. V. Diggle P. J. Tekle A. H. and Remme J. H (2011) The geographic distribution of Loa loa in Africa: results of large-scale implementation of the Rapid Assessment Procedure for Loiasis (RAPLOA). PLoS Negl. Trop. Dis., 5, e1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Simonsen P. E. Pedersen E. M. Rwegoshora R. T. Malecela M. N. Derua Y. A. and Magesa S. M (2010) Lymphatic filariasis control in Tanzania: effect of repeated mass drug administration with ivermectin and albendazole on infection and transmission. PLoS Negl. Trop. Dis., 4, e696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feldmeier H. Bienzle U. Dohring E. and Dietrich M (1982) Flubendazole versus mebendazole in intestinal helminthic infections. Acta Trop., 39, 185–189. [PubMed] [Google Scholar]
- 7. Mackenzie C. D. and Geary T. G (2011) Flubendazole: a candidate macrofilaricide for lymphatic filariasis and onchocerciasis field programs. Expert Rev. Anti. Infect. Ther., 9, 497–501. [DOI] [PubMed] [Google Scholar]
- 8. EMEA (2006) Flubendazole (Extrapolation to Poultry), summary report (4). Committee for Medicinal products for Veterinary Use. EMEA/CVMP/33128/2006- FINAL, http://www.ema.europa.eu/docs/en_GB/document_library/Maximum_Residue_Limits_-_Report/2009/11/WC500014292.pdf. [Google Scholar]
- 9. Spagnuolo P.A., Hu J., Hurren R., et al. (2010) The antihelmintic flubendazole inhibits microtubule function through a mechanism distinct from Vinca alkaloids and displays preclinical activity in leukemia and myeloma. Blood, 115, 4824–4833. [DOI] [PubMed] [Google Scholar]
- 10. Nianjun H. Cerepnalkoski L. Nwankwo J. O. Dews M. and Landolph J. R (1994) Induction of chromosomal aberrations, cytotoxicity, and morphological transformation in mammalian cells by the antiparasitic drug flubendazole and the antineoplastic drug harringtonine. Toxicol Sci., 22, 304–313. [PubMed] [Google Scholar]
- 11. COM (2010) Guidance Statement: Thresholds for In Vivo Mutagens. Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment. https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/315698/assessment_of_threshold_for_in_vivo_mutagens.pdf [Google Scholar]
- 12. COM (2000) Statement on Thresholds for Aneugens. Extrapolation of Data from Somatic Cells to Germ Cells. Committee on Mutagenicity of Chemicals in Food, Consumer Products and the Environment. Annual Report 2000. Published HMSO, London, pp 88–90. [Google Scholar]
- 13. Elhajouji A. Lukamowicz M. Cammerer Z. and Kirsch-Volders M (2011) Potential thresholds for genotoxic effects by micronucleus scoring. Mutagenesis, 26, 199–204. [DOI] [PubMed] [Google Scholar]
- 14. Elhajouji A. Tibaldi F. and Kirsch-Volders M (1997) Indication for thresholds of chromosome non-disjunction versus chromosome lagging induced by spindle inhibitors in vitro in human lymphocytes. Mutagenesis, 12, 133–140. [DOI] [PubMed] [Google Scholar]
- 15. Elhajouji A. Van Hummelen P. and Kirsch-Volders M (1995) Indications for a threshold of chemically-induced aneuploidy in vitro in human lymphocytes. Environ. Mol. Mutagen., 26, 292–304. [DOI] [PubMed] [Google Scholar]
- 16. Bolt H. M. and Degen G. H (2004) Human carcinogenic risk evaluation, part II: contributions of the EUROTOX specialty section for carcinogenesis. Toxicol. Sci., 81, 3–6. [DOI] [PubMed] [Google Scholar]
- 17. Parry E. M., Parry J. M., Corso C., et al. (2002) Detection and characterization of mechanisms of action of aneugenic chemicals. Mutagenesis, 17, 509–521. [DOI] [PubMed] [Google Scholar]
- 18. COM (1994) Thresholds for Aneuploidy Inducing Chemicals, in, The 1993 Annual Report of the Committees on Toxicity, Mutagenicity, Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Published HMSO , London, pp 36–38. [Google Scholar]
- 19. Ermler S. Scholze M. and Kortenkamp A (2013) Seven benzimidazole pesticides combined at sub-threshold levels induce micronuclei in vitro. Mutagenesis, 28, 417–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. COM (2008) Staement: Benzimidazoles: An Approach to Defining a Common Aneugenic Grouping, In: The 2007 Annual Report of the Committees on Toxicity, Mutagenicity, Carcinogenicity of Chemicals in Food, Consumer Products and the Environment. Published by the Food Standards Agency/Department of Health, pp, 146–153 [Google Scholar]
- 21. Bentley K. S. Kirkland D. Murphy M. and Marshall R (2000) Evaluation of thresholds for benomyl- and carbendazim-induced aneuploidy in cultured human lymphocytes using fluorescence in situ hybridization. Mutat. Res., 464, 41–51. [DOI] [PubMed] [Google Scholar]
- 22. Gollapudi B. B., Johnson G. E., Hernandez L. G., et al. (2013) Quantitative approaches for assessing dose–response relationships in genetic toxicology studies. Environ. Mol. Mutagen., 54, 8–18. [DOI] [PubMed] [Google Scholar]
- 23. OECD (2010) Test No. 487: In Vitro Mammalian Cell Micronucleus Test, OECD Guidelines for the Testing of Chemicals, Section 4, OECD Publishing, Paris. doi: http://dx.doi.org/10.1787/9789264091016-en [Google Scholar]
- 24. Gatehouse D., Haworth S., Cebula T., et al. (1994) Recommendations for the performance of bacterial mutation assays. Mutat. Res., 312, 217–233. [DOI] [PubMed] [Google Scholar]
- 25. Slob W. and Setzer R. W (2014) Shape and steepness of toxicological dose–response relationships of continuous endpoints. Crit. Rev. Toxicol., 44, 270–297. [DOI] [PubMed] [Google Scholar]
- 26. Hernández L. G. Slob W. van Steeg H. and van Benthem J (2011) Can carcinogenic potency be predicted from in vivo genotoxicity data? A meta-analysis of historical data. Environ. Mol. Mutagen., 52, 518–528. [DOI] [PubMed] [Google Scholar]
- 27. Bemis J. Wills J. Bryce S. Torous D. Dertinger D. and Slob W (2015) Comparison of in vitro and in vivo clastogenic potency based on benchmark dose analysis of flow cytometric micronucleus data in its current form. Mutagenesis, 10.1093/mutage/gev041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hernández L. G. Van Benthem J. and Johnson G. E (2013) A mode-of-action approach for the identification of genotoxic carcinogens. PLoS One, 8, e64532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Slob W. (2002) PROAST: Software for Dose–Response Modeling and Benchmark Dose Analysis. RIVM, http://www.rivm.nl/en/Library/Scientific/Models/PROAST (accessed September 22, 2015). [Google Scholar]
- 30. MacGregor J. T., Frötschl R., White P. A., et al. (2015) IWGT Report on quantitative approaches to genotoxicity risk assessment I. Methods and metrics for defining exposure–response relationships and points of departure (PoDs). Mutat. Res. Gen. Toxicol., 783, 55–65. [DOI] [PubMed] [Google Scholar]
- 31. MacGregor J. T., Frötschl R., White P. A., et al. (2015) IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutat. Res. Gen. Toxicol, 783, 66–78. [DOI] [PubMed] [Google Scholar]
- 32. Johnson G. E., Soeteman-Hernandez L. G., Gollapudi B. B., et al. (2014) Derivation of point of departure (PoD) estimates in genetic toxicology studies and their potential applications in risk assessment. Environ. Mol. Mutagen., 55, 609–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Johnson G. E., Slob W., Doak S. H., et al. (2015) New approaches to advance the use of genetic toxicology analyses for human health risk assessment. Toxicol. Res., 4, 667–676. [Google Scholar]
- 34. Crump K. S. (1995) Calculation of benchmark doses from continuous data. Risk Anal., 15, 79–89. [Google Scholar]
- 35. Hixon G. and Bichteler A (2013) drsmooth: Dose–Response Modeling with Smoothing Splines. http://CRAN.R-project.org/package=drsmooth (accessed September 22, 2015). [Google Scholar]
- 36. Avancini D. Menzies G. Morgan C. Wills J. Johnson G. E. White P. A. and Lewis P. D (2016) MutAIT—a online genetic toxicology data portal and analysis tools. Mutagenesis, 31, 323–328. [DOI] [PubMed] [Google Scholar]
- 37. Johnson G. E. Wills J. Doak S. H. Soeteman-Hernandez L. G. and White P. A (2015) Empirical analysis of BMD metrics in genetic toxicology part I: in vitro BMD comparisons for potency ranking and to support MOA evaluation. Mutagenesis, in press. [DOI] [PubMed] [Google Scholar]
- 38. Yoshimura H. (2003) Effect of oral dosing vehicles on the developmental toxicity of flubendazole in rats. Reprod. Toxicol., 17, 377–385. [DOI] [PubMed] [Google Scholar]
- 39. Bolt H. M. Foth H. Hengstler J. G. and Degen G. H (2004) Carcinogenicity categorization of chemicals-new aspects to be considered in a European perspective. Toxicol. Lett., 151, 29–41. [DOI] [PubMed] [Google Scholar]
- 40. IPCS Inchem (1987) 770 Flubendazole (WHO Food Additive Series 31. http://www.inchem.org/documents/jecfa/jecmono/v31je02.htm (accessed September 22, 2015). [Google Scholar]
- 41. IPCS Inchem (1990) Joint FAO/WHO Expert Committee on Food Additives, WHO Food additive Series 25, 666 Albendazole. http://www.inchem.org/documents/jecfa/jecmono/v25je02.htm (accessed September 22, 2015). [Google Scholar]
- 42. EMEA Vetrinary Medicines Evaluation Unit (1999) Mebendazole, Summary Report (1), EMEA/MRL/625/99-FINAL. http://www.ema.europa.eu/docs/en_GB/document_library/Maximum_Residue_Limits_-_Report/2009/11/WC500014872.pdf [Google Scholar]
- 43. EMEA/CVMP (2001) Mebendazole Summary Report (2) EMEA/MRL/781/01-FINAL March 2001. http://www.ema.europa.eu/docs/en_GB/document_library/Maximum_Residue_Limits_-_Report/2009/11/WC500014873.pdf [Google Scholar]
- 44. FSCJ (2010) Food Safety Commission of Japan: Risk Assessment Report Mebendazole (veterinary medicines). http://www.fsc.go.jp/english/evaluationreports/vetmedicine/mebendazole_fs156_2010.pdf (accessed September 22, 2015). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.