

Abstract
Sponges (phylum Porifera) are important members of almost all aquatic ecosystems, and are renowned for hosting often dense and diverse microbial communities. While the specificity of the sponge microbiota seems to be closely related to host phylogeny, the environmental factors that could shape differences within local sponge-specific communities remain less understood. On tropical coral reefs, sponge habitats can span from shallow areas to deeper, mesophotic sites. These habitats differ in terms of environmental factors such as light, temperature, and food availability, as well as anthropogenic impact. In order to study the host specificity and potential influence of varying habitats on the sponge microbiota within a local area, four tropical reef sponges, Rhabdastrella globostellata, Callyspongia sp., Rhaphoxya sp., and Acanthella cavernosa, were collected from exposed shallow reef slopes and a deep reef drop-off. Based on 16S rRNA gene pyrosequencing profiles, beta diversity analyses revealed that each sponge species possessed a specific microbiota that was significantly different to those of the other species and exhibited attributes that are characteristic of high- and/or low-microbial-abundance sponges. These findings emphasize the influence of host identity on the associated microbiota. Dominant sponge- and seawater-associated bacterial phyla were Chloroflexi, Cyanobacteria, and Proteobacteria. Comparison of individual sponge taxa and seawater samples between shallow and deep reef sites revealed no significant variation in alpha diversity estimates, while differences in microbial beta diversity (variation in community composition) were significant for Callyspongia sp. sponges and seawater samples. Overall, the sponge-associated microbiota is significantly shaped by host identity across all samples, while the effect of habitat differentiation seems to be less predominant in tropical reef sponges.
Keywords: 16S rRNA, Microbial diversity, Pyrosequencing, Porifera, Environmental variability, Symbiosis
Introduction
In marine ecosystems, sponges represent common and versatile members of the benthos, with distribution ranges along large environmental gradients and across various habitats including deep sea benthos, seamounts, polar regions, and temperate and tropical coral reefs (Bell, 2008). Many sponges are notable for their diverse and abundant microbial biota, with up to 35% of sponge biomass being made up of microbes (Taylor et al., 2007). Sponge-microbe relationships can include microbial cells as a food source for filter-feeding sponges, carbon- and nitrogen-based nutritional interactions, and the synthesis of secondary metabolites for chemical defence mechanisms (Hentschel et al., 2012; Taylor et al., 2007). Accumulated evidence indicates that much of the sponge microbiota is specific to, or at the very least heavily enriched in, sponge hosts (Lee et al., 2011; Pita et al., 2013; Schmitt, Hentschel & Taylor, 2012; Webster et al., 2010; Simister et al., 2012a). Even studies which detect so-called “sponge-specific” microbes outside the sponge host only find these at very low abundances, with no evidence for these free-living microbes being metabolically active (Taylor et al., 2013; Moitinho-Silva et al., 2014). In addition to the apparent influence of host identity on microbial composition (Easson & Thacker, 2014; Naim et al., 2014; Pita et al., 2013; Reveillaud et al., 2014), marine sponge-associated microbial communities exhibit relatively high temporal and biogeographic stability (e.g., Simister et al., 2013; Hardoim & Costa, 2014; Taylor et al., 2005).
While sponges occupy a range of different depths, knowledge about the influence of depth on the composition of the sponge microbiota still remains rather limited (Olson & Kellogg, 2010; Olson & Gao, 2013; Morrow, Fiore & Lesser, 2016). Spatial dynamics of the host-associated microbiota within coral reef ecosystems, from shallow (0–30 m) to mesophotic (30–150 m) sites, are of great interest because of the potential role of the mesophotic coral ecosystem (MCE) as refugia for both microbial symbionts and their hosts facing threats of environmental change and anthropogenic disturbances (Olson & Kellogg, 2010; Lesser, Slattery & Leichter, 2009; Kahng, Copus & Wagner, 2014). Sponges in particular seem to be very important benthic members of MCEs, with increased growth rates, biomass and coverage compared to their shallow site counterparts (Lesser, Slattery & Leichter, 2009). The habitat of sponges can span from shallow reef ecosystems into these mesophotic zones, which are less influenced by variable abiotic factors such as surface water temperature and salinity, or by direct human impact such as overfishing and pollution (Kahng, Copus & Wagner, 2014; Olson & Kellogg, 2010). Research on thermal stress responses of sponge-associated microbial communities, for example, has already shown the drastic effects of rising water temperatures on the microbial symbionts (Webster, Cobb & Negri, 2008; Simister et al., 2012b). Knowledge of the spatial dynamics of potential microbial refugia could yield new perspectives on the resilience and management of coral ecosystems, which are facing enormous pressures due to increasing global climatic disturbances and anthropogenic influences along highly populated and narrow land-sea transition zones (Olson & Kellogg, 2010; Ainsworth, Thurber & Gates, 2010).
It has been suggested that inter-habitat connectivity of the host-associated microbial biota between the light flooded subsurface and the twilight areas of the MCEs exists because of larval migration, water circulation and the filtering activities of sessile benthic invertebrates that inhabit these zones (Olson & Kellogg, 2010; Slattery et al., 2011; Kahng, Copus & Wagner, 2014; Thacker & Freeman, 2012). The first assessment of in situ sponge-associated communities along an MCE depth gradient suggested host-specific local variations in community structure, which are possibly influenced by prevailing biotic and abiotic factors (Olson & Gao, 2013). A recent study of the Xestospongia muta microbiota with parallel inorganic nutrient and stable isotope analyses demonstrated that changing environmental factors with depth contribute to the microbial 16S rRNA gene-defined microbial compositions in this sponge (Morrow, Fiore & Lesser, 2016).
In this study, we apply high-throughput 16S rRNA gene amplicon pyrosequencing to profile four demosponge species, Rhabdastrella globostellata, Callyspongia sp., Acanthella cavernosa, and Rhaphoxya sp., collected with surrounding seawater to address the following aims: (1) based on all available samples we investigate the degree of host specificity of microbial communities among several tropical sponge species, and (2) based on a habitat-specific subset of samples we estimate the influence of local habitat variation on sponge- and seawater-associated microbial community patterns. We sampled two closely related but environmentally differentiating habitats; a deep drop-off (Guam Blue Hole) for the collection of the mesophotic reef samples and nearby shallow reef slope sites for comparison.
Materials & Methods
Sample processing and sequencing
Samples of four different sponge species R. globostellata (n = 6 specimens), Callyspongia sp. (n = 7), A. cavernosa (n = 3), Rhaphoxya sp. (n = 5) and seawater (n = 5) were collected from closely connected Guam reef sites and depths (shallow exposed reef sites and a deep drop-off) via snorkelling and technical diving (Table 1). The tropical island Guam is known for the presence of a marine karstic limestone sinkhole (i.e., Guam Blue Hole); Taborosi, Jenson & Mylroie (2003). The vertical Blue Hole sinkhole has a vertical shaft of more than 90 m depth at which it merges with a deep drop-off running along the southern part of the Orote peninsula. This site harbours a rich and diverse associated coral reef fauna (Paulay, 2003) with very different environmental conditions from the shallow coral slopes of the surrounding areas. All shallow water samples for R. globostellata and Callyspongia sp. were collected from either Western Shoals or Gab Gab, which are both inside Apra Harbor and separated less than 1 nautical mile. The samples were collected in the same habitat, a shallow fore reef slope, which is dominated by Porites rus corals. Sample collection took place between June 25th 2010 and July 6th 2010, except for the samples C4 & C5 (March 2010) and A2 & A3 (November 2010 and March 2008, respectively) (see Table 1 for the sample accession codes). Sampling was carried out by technical diving, using trimix of helium, nitrogen and oxygen with mixes varying between target depths (oxygen 10–15%, helium 25–45%, nitrogen making up the balance). Decompression stops were carried out with nitrox mixes of 32–40%, which was switched to 75–82% oxygen once a decompression depth of 9 m was reached. Due to the various gas mixes, dive equipment included 5 independent tanks. Given that bottom times were limited to ca. 10–15 min at 90 m including descent time, searching for replicate sponges was very limited, explaining the somewhat limited number of replicates for the deep drop-off sponges. Sea surface temperatures (SST) were obtained from the Coastal Data Information Program (CDIP, http://cdip.ucsd.edu/themes/). SST averaged 29.8 °C in June and 29.7 °C in July 2010. Temperatures at depth (90 m) averaged 25 °C (taken with Dive rite-Intel HE and VR3-trimix dive computers). All samples were frozen, freeze-dried, and then stored at −20 °C prior to further processing. Sponges were identified by Dr Nicole J. de Voogd and vouchers of each species were preserved in 70% ethanol at the Naturalis Biodiversity Center, Leiden, Netherlands. Genomic DNA was extracted from sponge tissue and water filters (1 L each, 0.22 µm filter) by bead-beating in an ammonium acetate buffer, as previously described (Taylor et al., 2004). 16S rRNA gene amplification with primers targeting the V4-V5 region (454MID_533F: GTGCCAGCAGCYGCGGTMA and 454_907RC: CCGTCAATTMMYTTGAGTTT) and purification for pyrosequencing was performed as previously described by Simister et al. (2012b). Pyrosequencing was performed by Macrogen Inc. (Seoul, South Korea) using the Roche GS FLX Titanium system. The obtained raw sequence data can be accessed via the NCBI Sequence Read Archive under accession number SRX838037.
Table 1. Sample data.
With internal sample name, classified taxon, date and site of collection, depth and coordinates for each sample.
| Acc | Sample type | Site | Depth (m) | Date | Coordinates |
|---|---|---|---|---|---|
| A1 | A. cavernosa | Near Blue Hole | 5 | 29-Jun-10 | N13.26.180; E144.37.504 |
| A2 | A. cavernosa | Tanguisson | 77 | 04-Nov-10 | N13.32.620; E144.48.265 |
| A3 | A. cavernosa | Hospital Point | 92 | 17-Mar-08 | N13.30.126; E144.46.092 |
| C1 | Callyspongia sp. | Gab Gab | 4.5 | 06-Jul-10 | N13.26.35; E144.38.36 |
| C2 | Callyspongia sp. | Western Shoals | 4.5 | 06-Jul-10 | N13.27.018; E144.39.120 |
| C3 | Callyspongia sp. | Gab Gab | 4.5 | 06-Jul-10 | N13.26.35; E144.38.36 |
| C4 | Callyspongia sp. | Blue Hole | 80–90 | 23-Jan-08 | N13.26.180; E144.37.504 |
| C5 | Callyspongia sp. | Blue Hole | 80–90 | 23-Jan-08 | N13.26.180; E144.37.504 |
| C6 | Callyspongia sp. | Blue Hole | 88 | 29-Jun-10 | N13.26.180; E144.37.504 |
| C7 | Callyspongia sp. | Blue Hole | 80–90 | 06-Jul-10 | N13.26.180; E144.37.504 |
| RG1 | R. globostellata | Blue Hole | 80–90 | 29-Jun-10 | N13.26.180; E144.37.504 |
| RG2 | R. globostellata | Blue Hole | 80–90 | 29-Jun-10 | N13.26.180; E144.37.504 |
| RG3 | R. globostellata | Blue Hole | 80–90 | 29-Jun-10 | N13.26.180; E144.37.504 |
| RG4 | R. globostellata | Western Shoals | 1.5-3 | 25-Jun-10 | N13.27.018; E144.39.120 |
| RG5 | R. globostellata | Western Shoals | 1.5-3 | 25-Jun-10 | N13.27.018; E144.39.120 |
| RG6 | R. globostellata | Western Shoals | 1.5–3 | 25-Jun-10 | N13.27.018; E144.39.120 |
| RS1 | Rhaphoxya sp. | Blue Hole | 95 | 06-Jul-10 | N13.26.180; E144.37.504 |
| RS2 | Rhaphoxya sp. | Blue Hole | 80–90 | 06-Jul-10 | N13.26.180; E144.37.504 |
| RS3 | Rhaphoxya sp. | Blue Hole | 80–90 | 06-Jul-10 | N13.26.180; E144.37.504 |
| RS4 | Rhaphoxya sp. | Blue Hole | 80-90 | 06-Jul-10 | N13.26.180; E144.37.504 |
| RS5 | Rhaphoxya sp. | Blue Hole | 77 | 29-Jun-10 | N13.26.180; E144.37.504 |
| W1 | Waterfilter | Blue Hole | 88 | 06-Jul-10 | N13.26.180; E144.37.504 |
| W2 | Waterfilter | Blue Hole | 88 | 06-Jul-10 | N13.26.180; E144.37.504 |
| W3 | Waterfilter | Blue Hole | 88 | 06-Jul-10 | N13.26.180; E144.37.504 |
| W4 | Waterfilter | Western Shoals | 3 | 06-Jul-10 | N13.27.018; E144.39.120 |
| W5 | Waterfilter | Western Shoals | 3 | 06-Jul-10 | N13.27.018; E144.39.120 |
Raw sequence processing
Sequences were processed using mothur v.1.33.0 (Schloss, Gevers & Westcott, 2011; Schloss et al., 2009). Pyrosequencing flowgrams were filtered and denoised using the mothur implementation of AmpliconNoise (Quince et al., 2011). Adaptor, MID, and primer sequences were removed from raw sequences. Sequences were removed from the analysis if they were ≤200 bp or contained ambiguous characters, homopolymers longer than 8 bp, more than one MID mismatch, or more than two mismatches to the reverse primer sequence. Unique sequences were aligned against a SILVA alignment (available at http://www.mothur.org/wiki/Silva_reference_alignment). After chimera-checking with UCHIME (Edgar et al., 2011), unique sequences were identified using the Greengenes “gg_13_8_99” reference taxonomy (available at http://www.mothur.org/wiki/Greengenes-formatted_databases). Non-target sequences (e.g. chloroplasts, mitochondria, eukaryotic 18S rRNA) were removed.
Sequence data analyses
After raw data processing, mothur was used to group the obtained high quality sequences into 97% average neighbour sequence-similarity threshold operational taxonomic units (i.e., 97%-OTUs), for calculation of Shannon & inverse Simpson diversity, and rarefaction curves. Reads were evenly subsampled to 2,387 sequences per sample for all alpha diversity calculations (mothur command summary.single & subsample=T; 1,000 iterations). For visualization and interpretation of the microbial community data, we used standardized 97%-OTU abundance information (vegan command decostand & method= hellinger or pa; Bray-Curtis dissimilarities for relative abundance & Jaccard dissimilarities for presence/absence analyses). To estimate the variance of beta diversity, two hypothetical treatments were applied to the dataset: (a) ‘habitat’ (shallow reef slope, deep drop-off) and (b) ‘group’ (Rhabdastrella, Rhaphoxya, Callyspongia, Acanthella, seawater) (Table S1). These treatments were used for analysis of multivariate homogeneity of group dispersions (variances) (Anderson, 2006) with the betadisper (followed by pairwise Tukey’s Honestly Significant Difference tests) and permutest function from the vegan package in R (v. 3.0.2) (Oksanen et al., 2012; R Development Core Team, 2013). We used the adonis function (1000 permutations) from the vegan package to estimate the variances in beta diversity for both treatment groups (Anderson, 2001). Visualization of variations in sponge composition among habitats (i.e., shallow reef slope, deep drop-off) and sponge hosts (R. globostellata, Callyspongia sp., Rhaphoxya sp. and A. cavernosa) were assessed with multivariate non-metric multidimensional scaling (nMDS) using the metaMDS function from the vegan package. Hypothesis-based treatments were added as dispersion ellipses to the ordination plots with the vegan function ordieellipse (0.95 confidence interval). All multivariate analyses were performed with relative abundance and presence/absence data. The contribution of OTUs to average overall pairwise sample dissimilarity in R. globostellata, Callyspongia sp. and seawater specific datasets (‘habitat’ treatment) was assessed using the vegan function simper for similarity percentages (SIMPER). Relative abundance of the 30 most abundant OTUs (each ≥0.5% relative abundance) and relative phylum abundance were visualized with JColorGrid (Joachimiak, Weisman & May, 2006). Representative sequences of the 30 most abundant OTUs were assembled via the get.oturep command in mothur and BLAST searched against the NCBI Nucleotide collection (discontiguous megablast & Models excluded). The best hits and representative sequences for the most abundant OTUs can be accessed via the Figshare online repository (https://dx.doi.org/10.6084/m9.figshare.2366827). Hierarchical clustering based on all OTUs was performed using the vegan package in R via the function vegdist (Bray-Curtis dissimilarity) and hclust (method = average) and subsequently added onto the phylum fingerprint. As described above, all samples were included in the group-based (i.e., sponge and seawater samples) multivariate community (adonis and betadisper) and ordination (nMDS) analysis. In the subsequent habitat-based (shallow reef slope vs. deep drop-off) comparison, A. cavernosaand Rhaphoxya sp. samples were omitted from all analyses, and seawater samples from multivariate community analysis and ordination analyses, due to insufficient numbers of replicates in the dataset for the particular habitats. The 97% OTU abundance table combined with the Greengenes classification for each individual OTU can be accessed via the Figshare online repository (https://dx.doi.org/10.6084/m9.figshare.2063280).
Results
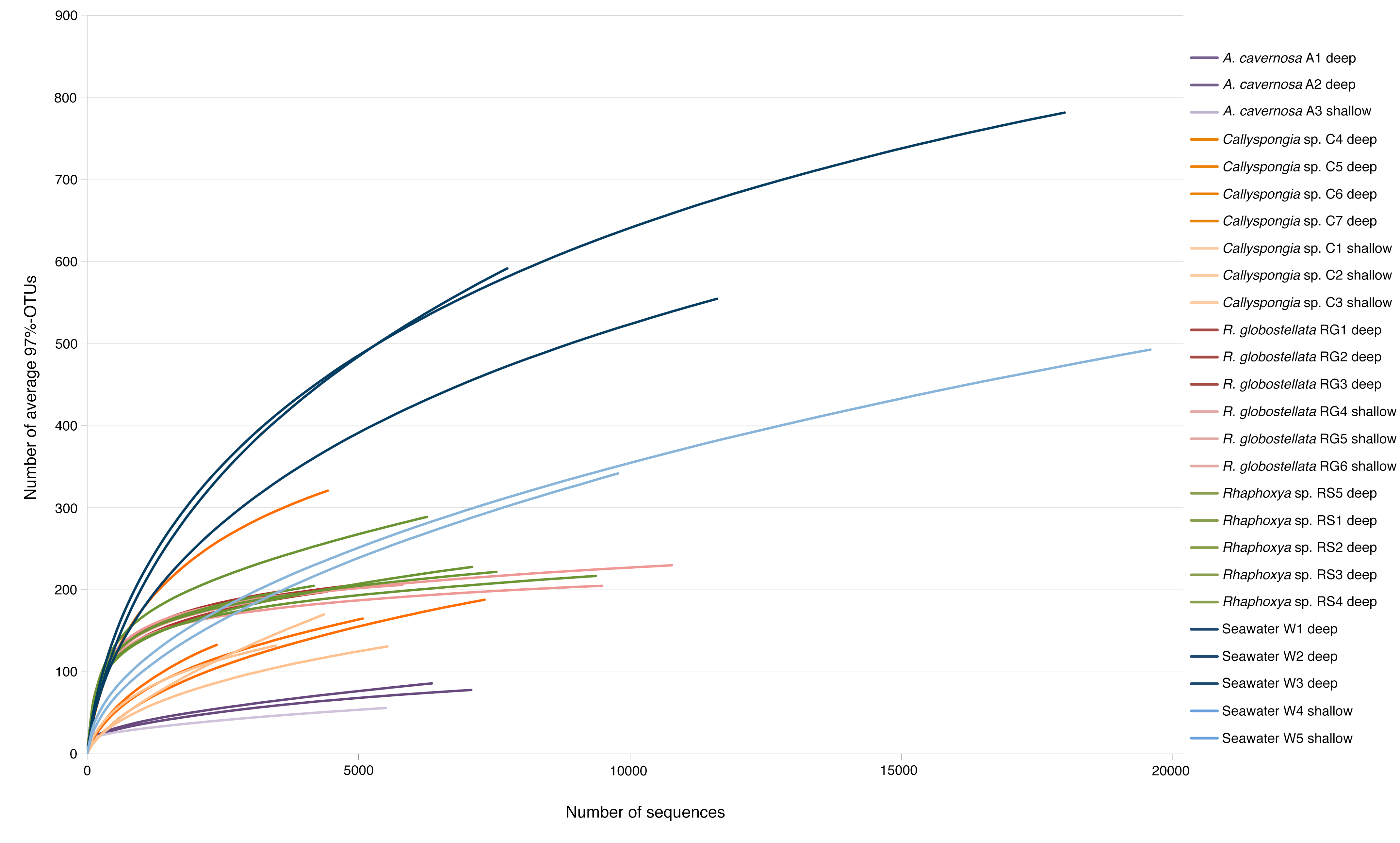
In total, 191,710 sequences were retained after denoising and quality control. Between all sampling groups (four sponge taxa and one seawater group), number of sequences, observed and average 97%-OTUs were higher overall within the seawater group in comparison to the sponge groups (Table 2). Coverage was slightly higher for A. cavernosa and R. globostellata compared to seawater and Callyspongia sp. samples (Table 2). These group-specific observations were also reflected in the rarefaction curves (Fig. S1). Across 26 samples (21 sponges and 5 seawater samples), 2247 OTUs (97% cut-off) were determined. After Greengenes classification, these OTUs were assigned to 33 bacterial and two archaeal phyla (Fig. 1). Pooled seawater samples showed the highest phylum richness, with 30 bacterial and two archaeal phyla identified. The observed phylum-level diversity of R. globostellata and Rhaphoxya sp. was very similar with 18 and 19 bacterial phyla, respectively, and Crenarchaeota as the archaeal phylum present in both sponge species. The main differences in bacterial composition between Rhaphoxya sp. and R. globostellatawere in the relative abundances of Cyanobacteria and Betaproteobacteria (Fig. 1). However, compared to the other three groups, these two sponges were more similar to each other at microbial phylum level. In contrast, Callyspongia sp. exhibited an association with Euryarchaeota and 24 bacterial phyla. A. cavernosa deviated slightly from the so far observed phylum richness, with only 13 bacterial phyla and Crenarchaeota (Fig. 1). Almost half of the occurring phyla were present in all five groups (e.g., Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Cyanobacteria, Nitrospirae, Planctomycetes, Proteobacteria, Spirochaetes, Synergistetes, and Verrucomicrobia), but exhibited group-specific variation (Fig. 1). Hierarchical clustering based on Bray-Curtis dissimilarity distances revealed group-specific clades with high between-group and low within-group dissimilarities (Fig. 1 & Fig. S2A). In addition, individual Bray-Curtis dissimilarity clustering of all five groups separated almost every sample in accordance to the sampled habitat (Figs. S2B–S2E), while subsequent multivariate analyses only confirmed significant differences between habitats for Callyspongia sp. and seawater samples (Table 3).
Table 2. Sample sequence statistics.
Sequence and OTU of 97%-OTUs (subsampling size based on the sample with the fewest sequences emphasized in bold = 2,387 reads). Subsequent coverage, richness, and alpha diversity estimates are based on the subsampled dataset.
| Acc | Sample | Reef habitat | Total OTUs | # sequences | Average OTUs | Coverage | Invsimpson | Shannon |
|---|---|---|---|---|---|---|---|---|
| A1 | A. cavernosa | Shallow slope | 56 | 5,500 | 40 | 0.99 | 6.04 ± 0.69 | 2.32 ± 0.1 |
| A2 | A. cavernosa | Deep drop-off | 78 | 7,076 | 50 | 0.99 | 4.04 ± 0.54 | 2.12 ± 0.12 |
| A3 | A. cavernosa | Deep drop-off | 86 | 6,351 | 55 | 0.99 | 5.61 ± 0.81 | 2.4 ± 0.11 |
| C1 | Callyspongia sp. | Shallow slope | 131 | 5,530 | 87 | 0.98 | 1.56 ± 0.12 | 1.13 ± 0.16 |
| C2 | Callyspongia sp. | Shallow slope | 132 | 3,474 | 114 | 0.98 | 3.12 ± 0.29 | 1.94 ± 0.16 |
| C3 | Callyspongia sp. | Shallow slope | 170 | 4,361 | 114 | 0.97 | 1.63 ± 0.13 | 1.14 ± 0.16 |
| C4 | Callyspongia sp. | Deep drop-off | 165 | 5,077 | 117 | 0.98 | 2.02 ± 0.2 | 1.58 ± 0.17 |
| C5 | Callyspongia sp. | Deep drop-off | 188 | 7,322 | 105 | 0.98 | 1.56 ± 0.12 | 1.1 ± 0.16 |
| C6 | Callyspongia sp. | Deep drop-off | 133 | 2,387 | 133 | 0.97 | 2.36 ± 0.24 | 1.74 ± 0.17 |
| C7 | Callyspongia sp. | Deep drop-off | 321 | 4,433 | 258 | 0.96 | 5.45 ± 0.84 | 3.2 ± 0.19 |
| RG1 | R. globostellata | Deep drop-off | 206 | 4,917 | 181 | 0.99 | 62.86 ± 8.51 | 4.53 ± 0.08 |
| RG2 | R. globostellata | Deep drop-off | 198 | 4,423 | 173 | 0.98 | 63.06 ± 7.14 | 4.47 ± 0.08 |
| RG3 | R. globostellata | Deep drop-off | 199 | 3,635 | 183 | 0.98 | 66.03 ± 8.09 | 4.54 ± 0.08 |
| RG4 | R. globostellata | Shallow slope | 205 | 9,488 | 167 | 0.99 | 37 ± 7.31 | 4.31 ± 0.1 |
| RG5 | R. globostellata | Shallow slope | 206 | 5,803 | 181 | 0.99 | 64.79 ± 8.73 | 4.55 ± 0.08 |
| RG6 | R. globostellata | Shallow slope | 230 | 10,773 | 177 | 0.99 | 57.45 ± 8.95 | 4.49 ± 0.09 |
| RS1 | Rhaphoxya sp. | Deep drop-off | 289 | 6,262 | 214 | 0.97 | 43.3 ± 8.24 | 4.47 ± 0.11 |
| RS2 | Rhaphoxya sp. | Deep drop-off | 228 | 7,092 | 172 | 0.98 | 51.53 ± 7.47 | 4.39 ± 0.09 |
| RS3 | Rhaphoxya sp. | Deep drop-off | 205 | 4,175 | 181 | 0.98 | 60.78 ± 9.04 | 4.52 ± 0.08 |
| RS4 | Rhaphoxya sp. | Deep drop-off | 222 | 7,539 | 177 | 0.99 | 62.81 ± 8.27 | 4.52 ± 0.08 |
| RS5 | Rhaphoxya sp. | Deep drop-off | 217 | 9,372 | 169 | 0.99 | 56.58 ± 7.89 | 4.44 ± 0.08 |
| W1 | Waterfilter | Deep drop-off | 555 | 11,606 | 276 | 0.94 | 5.09 ± 0.86 | 3.27 ± 0.2 |
| W2 | Waterfilter | Deep drop-off | 592 | 7,740 | 334 | 0.92 | 4.18 ± 0.66 | 3.18 ± 0.21 |
| W3 | Waterfilter | Deep drop-off | 782 | 18,007 | 345 | 0.93 | 6.94 ± 1.35 | 3.74 ± 0.19 |
| W4 | Waterfilter | Shallow slope | 493 | 19,586 | 174 | 0.96 | 4.52 ± 0.72 | 2.83 ± 0.17 |
| W5 | Waterfilter | Shallow slope | 342 | 9,781 | 160 | 0.96 | 2.53 ± 0.3 | 2.15 ± 0.19 |
Figure 1. Phylum abundance and distribution.

Heatmap of the relative abundance of 16S rRNA gene amplicon sequences taxonomically classified to phylum level. The dendrogram is based on Bray-Curtis dissimilarities (relative abundances of 97%-OTUs).
Table 3. Multivariate analyses.
Analysis of Bray-Curtis (relative abundance) and Jaccard (presence-absence) dissimilarities among all samples (group based) and the R. globostellata, Callyspongia sp., and seawater specific subsets (habitat based). Results represent the three groups and ordination ellipses from Figs. 3A–3C and seawater with the number of 97%-OTUs available in each individual dataset for the betadisper/permutest (Dispersion) and adonis (PERMANOVA) analyses (1,000 permutations each). Attached are the nMDS stress values of the multivariate ordination from Figs. 3A–3C and seawater (seawater ordination not shown). Significant differences in bold.
| Data matrix | Sample groups | Dispersion | PERMANOVA | ||||||
|---|---|---|---|---|---|---|---|---|---|
| OTUs | df | F value | p value | F value | R2 | p value | nMDS stress | ||
| Relative abundance | Sponges & water (groups) | 2,247 | 21 | 1.75 | >0.99 | 22.09 | 0.81 | <0.001 | 0.08 |
| R. globostellata (habitat) | 341 | 4 | 0.01 | >0.99 | 1.80 | 0.31 | 0.1 | 0.01 | |
| Callyspongia sp. (habitat) | 673 | 5 | 0.29 | >0.99 | 2.85 | 0.36 | <0.001 | <0.01 | |
| Seawater (habitat) | 1,537 | 3 | 5.23 | 0.02 | 6.28 | 0.68 | 0.008 | <0.01 | |
| Presence-absence | Sponges & water (groups) | 2,247 | 21 | 31.38 | 0.02 | 6.15 | 0.54 | <0.001 | 0.09 |
| R. globostellata (habitat) | 341 | 4 | 0.50 | >0.99 | 1.64 | 0.290 | 0.1 | <0.01 | |
| Callyspongia sp. (habitat) | 673 | 5 | 0.26 | >0.99 | 1.85 | 0.270 | <0.001 | <0.01 | |
| Seawater (habitat) | 1537 | 3 | 0.62 | >0.99 | 2.4 | 0.45 | 0.1 | <0.01 | |
The most abundant OTUs (n = 30) included members of the Proteobacteria (Alpha-, Beta-, Delta-, and Gamma-), Cyanobacteria, Acidobacteria, Chloroflexi, “Poribacteria”, Actinobacteria, Nitrospirae and PAUC34f (Fig. 2). The distribution of the three most abundant OTUs (OTU0001: Betaproteobacteria, uncultured order EC94; OTU0002: Cyanobacteria, Synechococcus; OTU0003: Cyanobacteria, Prochlorococcus) was mostly limited to Callyspongia sp. and seawater samples. Among the abundant R. globostellata OTUs were representatives of Chloroflexi class SAR202, the candidate phylum “Poribacteria,” various Acidobacteria and Proteobacteria, and the uncultured sponge symbiont PAUC34f. Interestingly, while most abundant OTUs in R. globostellata and Rhaphoxya sp. were largely shared, and evenly distributed overall, the abundant OTU0016 (Synechococcus) in R. globostellata was not detected in Rhaphoxya sp. On the contrary, the latter sponge species featured an Acidobacteria OTU (OTU0033) that was predominantly shared with A. cavernosa instead of R. globostellata (Fig. 2). Additionally, three OTUs were highly abundant only in A. cavernosa: OTU0004 (Acidobacteria, PAUC26f), OTU0007 (Gammaproteobacteria, HTCC2089), and OTU0027 (Alphaproteobacteria, Rhodobacteraceae) (Fig. 2).
Figure 2. Most abundant 97%-OTUs.

Fingerprint of the 30 most abundant 97%-OTUs (>0.5%) based on relative abundance for each individual OTU. Greengenes classifications from phylum to species level are provided on the left. On the right are relative abundances of the individual 97%-OTUs in relation to all detected OTUs.
Non-metric multidimensional scaling plots created with all samples showed a high degree of sample-specific pooling (Fig. 3A). The variation among sample-specific groups was significant for the relative abundance and presence-absence datasets (Table 3). Pairwise comparisons of mean group dispersions revealed the significant contributions of seawater and Callyspongia sp. samples to the differences between the groups (Tables S2 and S3).The habitat-based multivariate analysis of variance was significant for Callyspongia sp. and seawater samples (Table 3). The nMDS ordination showed apparent habitat-related community clusters for both (Callyspongia sp. and R. globostellata) sponge taxa (Figs. 3B and 3C). The observation of habitat-specific clusters in the ordination is also present in the individual hierarchical clustering approaches (Figs. S2B–S2D). In addition, for seawater (habitat-based PERMANOVA) the relative abundance, and the sponge & seawater (group-based permanova) presence-absence groups the significant multivariate spread (Dispersion) might contribute to the observed significant variance effects (Table 3).
Figure 3. 97%-OTU community structure.

nMDS ordinations based on Bray-Curtis (relative abundance) dissimilarities (97%-OTUs). (A) all samples and ordination ellipse for each sample group, (B) only Callyspongia sp. with ordination ellipse for each habitat, and (C) R. globostellata with ordination ellipse for each habitat.
The graphical summary of the OTUs (collapsed to high taxonomic ranks) with the highest average abundance in SIMPER in each of the sample groups presents three distinct microbial communities, with only slight variations between the two habitats (Fig. 4). For example, R. globostellata harbours an abundant Chloroflexi community and is the only sample group containing the candidate phylum “Poribacteria.” In contrast, Callyspongia sp. was dominated by members of Betaproteobacteria and seawater samples by Cyanobacteria and Gammaproteobacteria. In all three sample types, Cyanobacteria were slightly more abundant in samples from the shallow reef slope habitat. In contrast to the overall balanced taxonomic contribution patterns between habitats at higher taxonomic ranks, the analysis of 97%-OTUs showed individual habitat contributions and that several OTUs with a high abundance among all samples were also main contributors to the overall dissimilarity among the shallow and very deep sample groups (Table 4). The most prominent feature among all three analyzed sample groups was the dominance of Synechococcus (phylum Cyanobacteria) in the shallow reef slope habitats. In the deep drop-off Callyspongia sp. and seawater samples a second cyanobacterium, genus Prochlorococcus (OTU0003), was most dominant. The dominant Synechococcus OTUs were also separated in R. globostellata (OTU0016) and Callyspongia sp. and seawater (OTU0002, OTU0037, OTU0041). Additionally, compared to Callyspongia sp. and seawater, which exhibit dominant OTUs either in shallow reef slopes or deep habitats, the main contributing OTUs in R. globostellata dominate completely the specimens from shallow habitat (Table 4). The contrasting and habitat-dependent patterns were also prominent in the average species richness and the two alpha diversity indices among the three groups (Figs. 5A–5C); R. globostellata represents the sponge with the highest evenness, richness and OTU dominance compared to Callyspongia sp. and seawater samples. Additionally, all three sample groups exhibit higher richness and diversity values in the deep drop-off habitats and individual variations among the shallow reef slopes and deep drop-off habitats.
Figure 4. SIMPER contributions to Bray-Curtis dissimilarities between habitats.

Mean sequence abundances of the 97%-OTUs which contribute the most to overall Bray-Curtis dissimilarities, as calculated by SIMPER among open and deep R. globostellata, Callyspongia sp., and seawater samples. Individual 97%-OTUs were collapsed at class level and only considered with an overall abundance of >1%.
Table 4. Most abundant SIMPER OTUs.
Average abundance and cumulative contribution of the top 15 dominant 97%-OTUs contributing at least to 70% of the Bray-Curtis dissimilarities (SIMPER). Calculated among shallow reef slopes and deep reef drop-off R. globostellata, Callyspongia sp. and seawater samples. Microbial taxonomy is based on the Greengenes 97%-OTU classification from phylum to species level (if applicable). Habitat-specific OTU prevalence emphasized in bold; cusum: ordered cumulative contribution.
| Average abundance | |||||
|---|---|---|---|---|---|
| OTUs | Deep | Shallow | Cusum | Microbial taxonomy | |
| R. globostellata | Otu0016 | 45.67 | 534.67 | 0.09 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus |
| Otu0006 | 109.33 | 529.33 | 0.17 | Actinobacteria, Acidimicrobiia, Acidimicrobiales, wb1_P06 | |
| Otu0017 | 72.67 | 216.00 | 0.19 | Nitrospirae, Nitrospira, Nitrospirales, Nitrospiraceae | |
| Otu0072 | 1.33 | 146.33 | 0.22 | Gemmatimonadetes, Gemm-2 | |
| Otu0034 | 42.33 | 184.33 | 0.24 | Proteobacteria, Gammaproteobacteria, Thiotrichales, Piscirickettsiaceae | |
| Otu0054 | 0.67 | 107.00 | 0.26 | Poribacteria | |
| Otu0042 | 21.67 | 125.33 | 0.28 | Proteobacteria, Gammaproteobacteria, Thiotrichales, Piscirickettsiaceae | |
| Otu0020 | 106.00 | 195.33 | 0.30 | Chloroflexi, SAR202 | |
| Otu0089 | 6.00 | 105.33 | 0.32 | Bacteroidetes, Rhodothermi, Rhodothermales, Rhodothermaceae, Salisaeta | |
| Otu0013 | 91.00 | 182.00 | 0.34 | Proteobacteria, Gammaproteobacteria, Chromatiales, Ectothiorhodospiraceae | |
| Otu0046 | 38.00 | 126.00 | 0.35 | Gemmatimonadetes, Gemm-2 | |
| Otu0021 | 66.33 | 151.33 | 0.37 | PAUC34f | |
| Otu0008 | 199.67 | 242.33 | 0.38 | Poribacteria | |
| Otu0093 | 11.00 | 95.00 | 0.40 | Gemmatimonadetes, Gemm-2 | |
| Otu0012 | 53.00 | 127.33 | 0.41 | Proteobacteria, Gammaproteobacteria | |
| Callyspongia sp. | Otu0001 | 3147.25 | 3153.67 | 0.41 | Proteobacteria, Betaproteobacteria, EC94 |
| Otu0002 | 52.75 | 628.67 | 0.57 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus | |
| Otu0003 | 585.00 | 21.33 | 0.71 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Prochlorococcus | |
| Otu0005 | 117.00 | 68.67 | 0.72 | Proteobacteria, Alphaproteobacteria, Rickettsiales, Pelagibacteraceae | |
| Otu0073 | 37.25 | 38.00 | 0.73 | Proteobacteria | |
| Otu0152 | 35.75 | 10.33 | 0.74 | Proteobacteria, Gammaproteobacteria, Oceanospirillales, Endozoicimonaceae | |
| Otu0037 | 1.00 | 24.00 | 0.75 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus | |
| Otu0011 | 31.25 | 49.00 | 0.75 | Actinobacteria, Acidimicrobiia, Acidimicrobiales, OCS155 | |
| Otu0041 | 0.75 | 19.33 | 0.76 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus | |
| Otu0025 | 7.00 | 24.00 | 0.76 | Proteobacteria, Alphaproteobacteria, Rhodobacterales, Rhodobacteraceae | |
| Otu0232 | 7.25 | 13.67 | 0.77 | Proteobacteria, Gammaproteobacteria, Oceanospirillales, Endozoicimonaceae | |
| Otu0044 | 8.00 | 21.33 | 0.77 | Proteobacteria, Alphaproteobacteria, Rhodobacterales, Rhodobacteraceae | |
| Otu0284 | 12.50 | 1.33 | 0.77 | Proteobacteria, Gammaproteobacteria, Alteromonadales, Shewanellaceae, Shewanella | |
| Otu0059 | 10.50 | 20.33 | 0.78 | Bacteroidetes, Flavobacteriia, Flavobacteriales, Flavobacteriaceae | |
| Otu0157 | 13.75 | 2.33 | 0.78 | Planctomycetes, Planctomycetia, Pirellulales, Pirellulaceae | |
| Seawater | Otu0002 | 314.00 | 7548.00 | 0.34 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus |
| Otu0003 | 5147.67 | 60.00 | 0.58 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Prochlorococcus | |
| Otu0005 | 711.33 | 630.50 | 0.60 | Proteobacteria, Alphaproteobacteria, Rickettsiales, Pelagibacteraceae | |
| Otu0037 | 3.00 | 432.50 | 0.62 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus | |
| Otu0011 | 398.33 | 628.50 | 0.64 | Actinobacteria, Acidimicrobiia, Acidimicrobiales, OCS155 | |
| Otu0041 | 5.00 | 393.00 | 0.66 | Cyanobacteria, Synechococcophycideae, Synechococcales, Synechococcaceae, Synechococcus | |
| Otu0025 | 96.67 | 462.00 | 0.67 | Proteobacteria, Alphaproteobacteria, Rhodobacterales, Rhodobacteraceae | |
| Otu0058 | 4.33 | 328.00 | 0.69 | Firmicutes, Bacilli, Bacillales, Bacillaceae, Bacillus | |
| Otu0071 | 32.67 | 234.50 | 0.70 | Cyanobacteria, Chloroplast, Chlorophyta, Mamiellaceae | |
| Otu0061 | 198.33 | 18.50 | 0.70 | Proteobacteria, Deltaproteobacteria, Sva0853, SAR324 | |
| Otu0032 | 257.33 | 111.00 | 0.71 | Proteobacteria, Alphaproteobacteria, Rickettsiales, Pelagibacteraceae | |
| Otu0044 | 71.67 | 244.00 | 0.72 | Proteobacteria, Alphaproteobacteria, Rhodobacterales, Rhodobacteraceae | |
| Otu0076 | 177.67 | 11.50 | 0.73 | Proteobacteria, Alphaproteobacteria, Sphingomonadales, Erythrobacteraceae, Erythrobacter | |
| Otu0080 | 131.67 | 35.00 | 0.73 | Proteobacteria, Alphaproteobacteria, Rhodobacterales, Rhodobacteraceae | |
| Otu0148 | 4.67 | 102.00 | 0.74 | Planctomycetes, OM190, CL500-15 | |
Figure 5. Alpha diversity comparisons between habitats.

Number of (A) average 97%-OTUs and alpha diversity estimates, (B) inverse Simpson’s and (C) Shannon index for shallow reef slopes and deep reef drop-off R. globostellata, Callyspongia sp. and seawater samples. Top, middle, and bottom lines of the boxes represent the 25th, 50th (median), and 75th percentiles, respectively. The end of the whiskers represent the 5th and 95th percentiles, respectively. Blue and red habitat colors correspond with the color code in Fig. 3. Measurements of observed average OTUs, inverse Simpson’s index and Shannon index were analyzed using a one-way analysis of variance (ANOVA, p < 0.05) using habitat as a fixed factor. Significant results are marked by an asterisk.
Discussion
Host specificity of the microbiota of four MCE sponges
In the present study divergent patterns between two different habitats were visible in the tropical sponge and seawater microbiota. Callyspongia sp. and seawater samples appear to have an intrinsic microbial community composition, which is variable enough to significantly separate the intra-species communities by their shallow reef slope or deep drop-off habitats. On the other hand R. globostellata exhibits an observable difference in community variation that is not significant. In addition, we could also observe significant microbial specificity across all analyzed sponge taxa independent of habitat. Since the shallow collection sites for Callyspongia sp. and R. globostellata inside Apra Harbor represent the same shallow water fore reef slope habitat dominated by Porites rus corals, it is unlikely that the observed differences in beta diversity between shallow reef slope or deep drop-off habitats are due to collection of Callyspongia sp. from the two shallow Apra Harbor sites.
Little is known about the R. globostellata and Rhaphoxya sp. microbial communities. Culture-dependent approaches reported only Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria (Alpha & Gamma) from R. globostellata (Lafi, Garson & Fuerst, 2005; Steinert et al., 2014). In addition, Lafi et al. (2009) found the candidate phylum “Poribacteria” in this demosponge. While the first high-throughput-sequencing amplicon screening detected 16 bacterial phyla associate with R. globostellata (Schmitt et al., 2012), we can now increase the number to a total of 23 microbial phyla. For Rhaphoxya sp., virtually nothing has been known about the associated microbiota. However, this sponge taxon is already the focus of natural products research, which hints at a chemically active symbiotic microbiota (Wright et al., 2012). Here we present phylum and OTU composition patterns in R. globostellata and Rhaphoxya sp. that are surprisingly similar. While designation of the two sponge taxa as either high microbial abundance (HMA) or low microbial abundance (LMA) sponges is lacking in the literature, the associated microbial phylotypes are congruent with the microbial phyla commonly found in association with HMA sponges (i.e., Proteobacteria, Chloroflexi, Acidobacteria, Actinobacteria, and the candidate phylum “Poribacteria”) (Schmitt et al., 2011; Gloeckner et al., 2014).
Recent transmission electron microscopy, DAPI cell-counting and 16S rRNA gene amplicon sequencing results assigned different members of the demosponges A. cavernosa and Callyspongia sp. to the LMA group, with abundant Proteobacteria (Alpha, Beta, & Gamma) and Cyanobacteria (Synechococcus) microbial community members (Gloeckner et al., 2014; Giles et al., 2013; Jeong, Kim & Park, 2013). We also found that our Callyspongia sp. specimens exhibited an abundant occurrence of Synechococcus (Cyanobacteria), Actinobacteria, Bacteroidetes and Proteobacteria (Beta & Gamma). Within the Betaproteobacteria, the order EC94 exhibited the highest overall abundance in our Callyspongia sp. samples and among all OTUs. Reports on this order are rare, but recent community analyses on different demosponges found this microbial taxon to be dominant in the deep sea sponge Inflatella pellicula and several shallow water sponges from Korea (Jeong, Kim & Park, 2015; Jackson et al., 2013; Jeong, Kim & Park, 2013).
Overall, sponge and seawater community structures correlated significantly with host identity. This underlines the common view that host identity is an important factor for the composition of sponge-associated microbial communities (e.g., Cárdenas et al., 2014; Cleary et al., 2013; Easson & Thacker, 2014; Pita et al., 2013). Recent studies on several sponge species showed that the microbiota of LMA sponges, in particular, exhibits a low degree of similarity among the investigated sponge species (Giles et al., 2013; Blanquer, Uriz & Galand, 2013). The sponge genera Acanthella and Callyspongia were previously found to include LMA sponges (Gloeckner et al., 2014), and the bacterial community ordination, phylum composition and OTU structures of the present study separates these two sponge taxa very distinctly from the other two investigated sponges (Rhabdastrella, Rhaphoxya). In addition, the observed microbial community patterns in Callyspongia sp. appear to be more closely related to the seawater samples (see OTU and phylum composition). This high similarity with seawater communities is a well-known feature of LMA sponges (e.g., Taylor et al., 2007; Schmitt et al., 2011; Blanquer, Uriz & Galand, 2013). In contrast, HMA members are commonly more closely related to each other, especially after the removal of potential environmental sources of variation (Blanquer, Uriz & Galand, 2013). Finally, four sponge samples (two A. cavernosa and two Callyspongia sp.) have been sampled at different points in time. However, a temporal effect on the community structure is not evident, which is in accordance with recent research showing the low seasonal variability of the sponge-microbiota (Erwin et al., 2012; Erwin et al., 2015), although other similar studies have indicated the temporal variability of sponge-associated bacteria (White et al., 2012; Anderson, Northcote & Page, 2010).
Depth-dependent microbial community patterns in MCE sponges
Knowledge about variability of the sponge microbiota along environmental gradients on local spatial scales is still scarce (Olson & Kellogg, 2010). A T-RFLP and clone library study on three MCE sponges identified a trend in community composition along a depth gradient, but could not identify the bacteria which caused these variations (Olson & Gao, 2013). Correspondingly, a recent 16S rRNA gene amplicon analysis found significant shifts in the X. muta-associated microbial community along a depth gradient from 10 to 90 m and demonstrated that environmental factors may influence the sponge microbiota (Morrow, Fiore & Lesser, 2016). A sponge transplantation experiment showed little overall effect between different habitats (Cárdenas et al., 2014), and comparisons between sponges obtained from different habitats (marine lake vs. coastal system & intertidal vs. subtidal) showed the importance of host relatedness and habitat as determinants of microbial community structure (Weigel & Erwin, 2015; Cleary et al., 2013).
In the present study, Callyspongia sp. and seawater microbial communities were significantly different when comparing shallow reef slope and deep drop-off habitats. In contrast, based on the multivariate statistical tests, R. globostellata microbial communities were not significantly affected by the different habitats (shallow versus deep), which leads to the conclusion that in this particular case the observed habitat-specific nMDS and hierarchical clustering patterns are random variations of the microbial community composition. In order to sample in two environmentally very distinct but closely related habitats, we focused sampling on “shallow” and “very deep” depths as categorized by Brazeau, Lesser & Slattery (2013). Irradiance, but also nutrient availability and water temperature, are dependent on both depth and changing environmental factors along spatial gradients from shallow to mesophotic coral reefs (see Lesser, Slattery & Leichter, 2009; Olson & Gao, 2013). The observed temperature difference between shallow reef slope and deep drop-off sites averaged 4 °C, similar to that reported by Lesser et al. (2010); difference of 4 °C between 3 m and 91 m depth). While temperature is one indication for environmental differences between the two habitats, it seems unlikely that the observed difference of 4 °C will affect sponge-microbe communities between the two habitats to the extent reported in this study. Studies examining the effect of elevated temperatures found no change (at sub-lethal temperatures) in sponge bacterial communities during short-term experiments (Webster, Cobb & Negri, 2008; Simister et al., 2012b). It is likely that other local environmental factors such as light have a considerable effect on the small-scale patterns observed here, especially as similar studies on mesophotic reefs by Lesser, Slattery & Leichter (2009) and Lesser et al. (2010) did observe pronounced differences in light along similar depth gradients.
Within Callyspongia sp. and seawater samples, either Cyanobacteria or Proteobacteria OTUs were mainly responsible for the differences observed by SIMPER. In particular, cyanobacterial Synechococcus OTUs were among the main contributors to the shallow microbiota, whereas only one Prochlorococcus OTU was a dominant cyanobacterium contributing to the deep group from mesophotic depths. The predominance of photoautotrophs in tropical filter-feeding sponges is intuitive given the widespread prevalence of cyanobacteria in the ocean. LMA sponges in particular are known to harbour cyanobacteria in high abundance (Bayer, Kamke & Hentschel, 2014), and cyanobacteria comprise one of the most abundant sponge-associated phyla, with well-established sponge-specific symbionts (e.g., Synechococcus spongarium) (Hentschel, Usher & Taylor, 2006; Taylor et al., 2007; Simister et al., 2012a). Moreover, LMA sponges exhibit higher water filtering capabilities, presumably due to a less dense mesohyl and less complex aquiferous system compared to HMA sponges (Weisz, Lindquist & Martens, 2008). Therefore, the congruent microbial community patterns between Callyspongia sp. and seawater samples observed here, in contrast to those of R. globostellata, could be correlated with physiological differences between HMA and LMA sponges (see Weisz, Lindquist & Martens, 2008; Thacker & Freeman, 2012). Besides the potential phototrophic activities in the sponge pinacoderm (outer tissue) and mesohyl (inner sponge matrix), nitrogen-fixing cyanobacteria may inhabit a niche within the complex nitrogen cycle in sponges, with a mutual benefit due to nutrient supply by the sponge and secondary metabolite production by the cyanobacteria (Taylor et al., 2007; Wilkinson & Fay, 1979; Arillo et al., 1993). Given the dominance of Cyanobacteria in our Callyspongia sp. specimens and seawater samples, with Synechococcus dominant in the shallow and Prochlorococcus in the deep sponges, the spatial pattern could be shaped predominantly by cyanobacterial lineages. This distribution pattern of microbes in Callyspongia sp. could also indicate a seasonal vertical distribution pattern, in which one genus dominates the shallow high-light water column, while the other genus is temporarily mainly present in the low-light area below. While temporal shifts of Cyanobacteria in sponges have been observed to varying degrees previously, the combined effect of time and depth on these and other chlorophototrophs in sponges remains uncertain (White et al., 2012; Erwin et al., 2012; Hardoim & Costa, 2014; Taylor et al., 2004). A recent analysis on stable isotopes indicated that, with increasing depth, the inorganic nutrients dependency in sponges shifts from photoautotrophy to heterotrophy, in accordance with a significant shift in the associated microbiota (Morrow, Fiore & Lesser, 2016). Conversely, in the same sponge species at a different location an observed stable isotopic enrichment correlated with a larger microbial community similarity across different sampling depths. In addition, other factors should be considered to explain the observed differences. Since the same sponge species show differences in growth rates and species richness at different depths (Lesser, Slattery & Leichter, 2009), different biotic and abiotic niches may be available for symbionts depending on their habitat. Such ecologically-based niche differentiation for symbionts and hosts is known for the tropical corals Seriatopora hystrix (Bongaerts et al., 2010) and Montastraea cavernosa (Lesser et al., 2010; Brazeau, Lesser & Slattery, 2013). Recent oligotyping of Nitrospira symbionts associated with sponges collected along large horizontal and vertical gradients provided further evidence for such patterns of differential enrichment of closely related microbial variants (Reveillaud et al., 2014).
In R. globostellata, one cyanobacterial OTU (OTU0016) is predominant within the deep sheltered communities, but also present at high abundance in the shallow specimens of this species (Table 4). Moreover, it was also largely absent from all other sponge and seawater samples. The BLAST search against the NCBI nucleotide collection revealed that this particular OTU is highly similar to Candidatus Synechococcus spongiarum, a symbiotic cyanobacterium found in many sponges (Hentschel, Usher & Taylor, 2006). Compared to this, the main cyanobacterial OTUs found in Callyspongia sp. and seawater (OTU0002 & OTU0003) yielded different BLAST results (uncultured Synechococcus sp. clone & Prochlorococcus sp., respectively). This corresponds to the theory that LMA sponges generally acquire their microbial symbionts via horizontal transmission from the surrounding environment, while HMA sponges possess a more individual microbial community that does not mirror the surrounding seawater microbiome as closely as their low abundance counterparts (Gloeckner et al., 2014; Hentschel, Usher & Taylor, 2006). The detection of microbes within different reproductive stages of seven sponge species led to the hypothesis that HMA sponges can maintain parts of their symbiotic microbiota via vertical transmission (Schmitt et al., 2008). Interestingly, the major microbial drivers contributing to the observed differences in beta diversity are all located in the deep sponges. Given that the abundant microbial phyla in R. globostellata (i.e., Acidobacteria, Chloroflexi, Cyanobacteria, Gemmatimonadetes, and Alphaproteobacteria) are known to contain (bacterio)chlorophyll-based phototrophic lineages (Zeng et al., 2014), it is possible that photoheterotrophic bacteria also play a considerable role in this host-specific microbiota. However, while the function of Chloroflexi in sponges is yet unclear, the distribution of members of this phylum within their hosts from different depths suggests that they may not be phototrophically active within the sponges (Olson & Gao, 2013).
Conclusion
The present study suggests that sponge-specific communities in tropical coral ecosystems are predominantly influenced by host identity. Moreover, the variance between Callyspongia-associated microbial communities from two different habitats (i.e., shallow reef slopes and deep drop-off reefs) is large enough to observe significant differences. However, the actual environmental factors contributing to the observed habitat-dependent variances remain uncertain, although we speculate that temperature may be less likely to have caused the variations in the sponge microbiota between shallow and deep specimens. While sponge-microbe communities show an overall stability along large geographic and temporal gradients, local environmental factors may have an effect on the small-scale patterns observed here. To further test the hypothesis of differential sponge-associated microbial communities along local depth gradients, functional and temporal aspects should be considered in future in situ studies. Moreover, since temporal turnover of phytoplankton is faster in the tropics (Soininen, 2010), tropical sponges with dominant phototrophic microbial communities most likely provide ideal conditions to design spatio-temporal studies on phototrophic host-symbiont dynamics.
Supplemental Information
Rarefied 97%-OTU 16S rRNA gene amplicon data for each sample.
{kind=link}
Dendrograms showing the Bray-Curtis dissimilarity of microbial communities of (A) the complete sample dataset, (B) Callyspongia sp., (C) R. globostellata, (D) seawater, (E) A. cavernosa, (F) Rhaphoxya sp. sponge specimens based on 97%-OTU amplicon subsets.
{kind=link}
Habitat file used for grouping of samples in multivariate analyses (nMDS & adonis).
Tukey multiple comparisons of means for the group based betadisper analysis. 95% family-wise confidence level—diff giving the difference in the observed means, lwr giving the lower end point of the interval, upr giving the upper end point and p adj giving the p-value after adjustment for the multiple comparisons.
Pairwise comparisons of the group based permutation test for homogeneity of multivariate dispersions. Observed p-value below diagonal, permuted p-value above diagonal.
Acknowledgments
We thank the UOG Marine Laboratory boat crews and Mr. Jim Pinson for the collection of sponges from the twilight zone. We also thank two anonymous reviewers for valuable comments and suggestions.
Funding Statement
GS acknowledges funding for microbiological analyses at the University of Auckland in the authors’ laboratory by the German Academic Exchange Service (DAAD) short term fellowship ‘Microbial Symbiosis and Diversity in Marine Sponges’ from 02/2013 to 06/2013. PJS acknowledges funding by the National Cancer Institute (grant 5U56CA096278) and NIH MBRS SCORE (grant S06-GM-44796). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional Information and Declarations
Competing Interests
The authors declare there are no competing interests.
Author Contributions
Georg Steinert conceived and designed the experiments, performed the experiments, analyzed the data, wrote the paper, prepared figures and/or tables.
Michael W. Taylor and Peter J. Schupp conceived and designed the experiments, contributed reagents/materials/analysis tools, reviewed drafts of the paper.
Peter Deines, Rachel L. Simister and Michael Hoggard performed the experiments, reviewed drafts of the paper.
Nicole J. de Voogd reviewed drafts of the paper, taxonomic classification of the sponges.
DNA Deposition
The following information was supplied regarding the deposition of DNA sequences:
The obtained raw sequence data can be accessed via the NCBI Sequence Read Archive under accession number SRX838037.
Data Availability
The following information was supplied regarding data availability:
The 97% OTU abundance table combined with the Greengenes classification for each individual OTU can be accessed via the Figshare online repository— 10.6084/m9.figshare.2063280.
The best hits and representative sequences for the most abundant OTUs can be accessed via the Figshare online repository (10.6084/m9.figshare.2366827).
References
- Ainsworth, Thurber & Gates (2010).Ainsworth TD, Thurber RV, Gates RD. The future of coral reefs: a microbial perspective. Trends in Ecology & Evolution. 2010;25:233–240. doi: 10.1016/j.tree.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Anderson (2001).Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecology. 2001;26:32–46. doi: 10.1111/j.1442-9993.2001.01070.pp.x. [DOI] [Google Scholar]
- Anderson (2006).Anderson MJ. Distance-based tests for homogeneity of multivariate dispersions. Biometrics. 2006;62:245–253. doi: 10.1111/j.1541-0420.2005.00440.x. [DOI] [PubMed] [Google Scholar]
- Anderson, Northcote & Page (2010).Anderson SA, Northcote PT, Page MJ. Spatial and temporal variability of the bacterial community in different chemotypes of the New Zealand marine sponge Mycale hentscheli. FEMS Microbiology Ecology. 2010;72:328–342. doi: 10.1111/j.1574-6941.2010.00869.x. [DOI] [PubMed] [Google Scholar]
- Arillo et al. (1993).Arillo A, Bavestrello G, Burlando B, Sarà M. Metabolic integration between symbiotic cyanobacteria and sponges: a possible mechanism. Marine Biology. 1993;117:159–162. doi: 10.1007/BF00346438. [DOI] [Google Scholar]
- Bayer, Kamke & Hentschel (2014).Bayer K, Kamke J, Hentschel U. Quantification of bacterial and archaeal symbionts in high and low microbial abundance sponges using real-time PCR. FEMS Microbiology Ecology. 2014;89:679–690. doi: 10.1111/1574-6941.12369. [DOI] [PubMed] [Google Scholar]
- Bell (2008).Bell JJ. The functional roles of marine sponges. Estuarine, Coastal and Shelf Science. 2008;79:341–353. doi: 10.1016/j.ecss.2008.05.002. [DOI] [Google Scholar]
- Blanquer, Uriz & Galand (2013).Blanquer A, Uriz MJ, Galand PE. Removing environmental sources of variation to gain insight on symbionts vs. transient microbes in high and low microbial abundance sponges. Environmental Microbiology. 2013;15:3008–3019. doi: 10.1111/1462-2920.12261. [DOI] [PubMed] [Google Scholar]
- Bongaerts et al. (2010).Bongaerts P, Riginos C, Ridgway T, Sampayo EM, Van Oppen MJH, Englebert N, Vermeulen F, Hoegh-Guldberg O. Genetic divergence across habitats in the widespread coral Seriatopora hystrix and its associated Symbiodinium. PLoS ONE. 2010;5:e1936. doi: 10.1371/journal.pone.0010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazeau, Lesser & Slattery (2013).Brazeau DA, Lesser MP, Slattery M. Genetic structure in the coral, Montastraea cavernosa: assessing genetic differentiation among and within mesophotic reefs. PLoS ONE. 2013;8:e1936. doi: 10.1371/journal.pone.0065845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas et al. (2014).Cárdenas CA, Bell JJ, Davy SK, Hoggard M, Taylor MW. Influence of environmental variation on symbiotic bacterial communities of two temperate sponges. FEMS Microbiology Ecology. 2014;88:516–527. doi: 10.1111/1574-6941.12317. [DOI] [PubMed] [Google Scholar]
- Cleary et al. (2013).Cleary DFR, Becking LE, De Voogd NJ, Pires ACC, Polónia ARM, Egas C, Gomes NCM. Habitat- and host-related variation in sponge bacterial symbiont communities in Indonesian waters. FEMS Microbiology Ecology. 2013;85:465–482. doi: 10.1111/1574-6941.12135. [DOI] [PubMed] [Google Scholar]
- Easson & Thacker (2014).Easson CG, Thacker RW. Phylogenetic signal in the community structure of host-specific microbiomes of tropical marine sponges. Frontiers in Microbiology. 2014;5:1–11. doi: 10.3389/fmicb.2014.00532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar et al. (2011).Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erwin et al. (2015).Erwin PM, Coma R, López-Serdino P, Serrano E, Ribes M. Stable symbionts across the HMA-LMA dichotomy: low seasonal and interannual variation in sponge-associated bacteria from taxonomically diverse hosts. FEMS Microbiology Ecology. 2015;91(10) doi: 10.1093/femsec/fiv115. fiv115. [DOI] [PubMed] [Google Scholar]
- Erwin et al. (2012).Erwin PM, Pita L, López-Legentil S, Turon X. Stability of sponge-associated bacteria over large seasonal shifts in temperature and irradiance. Applied and Environmental Microbiology. 2012;78:7358–7368. doi: 10.1128/AEM.02035-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles et al. (2013).Giles EC, Kamke J, Moitinho-Silva L, Taylor MW, Hentschel U, Ravasi T, Schmitt S. Bacterial community profiles in low microbial abundance sponges. FEMS Microbiology Ecology. 2013;83:232–241. doi: 10.1111/j.1574-6941.2012.01467.x. [DOI] [PubMed] [Google Scholar]
- Gloeckner et al. (2014).Gloeckner V, Wehrl M, Moitinho-Silva L, Gernert C, Schupp PJ, Pawlik JR, Lindquist NL, Erpenbeck D, Wörheide G, Hentschel U. The HMA-LMA dichotomy revisited: an electron microscopical survey of 56 sponge species. Biological Bulletin. 2014;227:78–88. doi: 10.1086/BBLv227n1p78. [DOI] [PubMed] [Google Scholar]
- Hardoim & Costa (2014).Hardoim CCP, Costa R. Temporal dynamics of prokaryotic communities in the marine sponge Sarcotragus spinosulus. Molecular Ecology. 2014;23:3097–3112. doi: 10.1111/mec.12789. [DOI] [PubMed] [Google Scholar]
- Hentschel et al. (2012).Hentschel U, Piel J, Degnan SM, Taylor MW. Genomic insights into the marine sponge microbiome. Nature Reviews Microbiology. 2012;10:641–654. doi: 10.1038/nrmicro2839. [DOI] [PubMed] [Google Scholar]
- Hentschel, Usher & Taylor (2006).Hentschel U, Usher KM, Taylor MW. Marine sponges as microbial fermenters. FEMS Microbiology Ecology. 2006;55:167–177. doi: 10.1111/j.1574-6941.2005.00046.x. [DOI] [PubMed] [Google Scholar]
- Jackson et al. (2013).Jackson SA, Flemer B, McCann A, Kennedy J, Morrissey JP, O’Gara F, Dobson ADW. Archaea appear to dominate the microbiome of Inflatella pellicula deep sea sponges. PLoS ONE. 2013;8:e1936. doi: 10.1371/journal.pone.0084438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, Kim & Park (2013).Jeong I-H, Kim K-H, Park J-S. Analysis of bacterial diversity in sponges collected off Chujado, an island in Korea, using barcoded 454 pyrosequencing: analysis of a distinctive sponge group containing Chloroflexi. Journal of Microbiology. 2013;51:570–577. doi: 10.1007/s12275-013-3426-9. [DOI] [PubMed] [Google Scholar]
- Jeong, Kim & Park (2015).Jeong J-B, Kim K-H, Park J-S. Sponge-specific unknown bacterial groups detected in marine sponges collected from Korea through barcoded pyrosequencing. Journal of Microbiology and Biotechnology. 2015;25:1–10. doi: 10.4014/jmb.1406.06041. [DOI] [PubMed] [Google Scholar]
- Joachimiak, Weisman & May (2006).Joachimiak MP, Weisman JL, May BCH. JColorGrid: software for the visualization of biological measurements. BMC Bioinformatics. 2006;7:225. doi: 10.1186/1471-2105-7-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahng, Copus & Wagner (2014).Kahng SE, Copus JM, Wagner D. Recent advances in the ecology of mesophotic coral ecosystems (MCEs) Current Opinion in Environmental Sustainability. 2014;7:72–81. doi: 10.1016/j.cosust.2013.11.019. [DOI] [Google Scholar]
- Lafi et al. (2009).Lafi FF, Fuerst JA, Fieseler L, Engels C, Goh WWL, Hentschel U. Widespread distribution of poribacteria in demospongiae. Applied and Environmental Microbiology. 2009;75:5695–5699. doi: 10.1128/AEM.00035-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafi, Garson & Fuerst (2005).Lafi FF, Garson MJ, Fuerst JA. Culturable bacterial symbionts isolated from two distinct sponge species (Pseudoceratina clavata and Rhabdastrella globostellata) from the Great Barrier Reef display similar phylogenetic diversity. Microbial Ecology. 2005;50:213–220. doi: 10.1007/s00248-004-0202-8. [DOI] [PubMed] [Google Scholar]
- Lee et al. (2011).Lee OO, Wang Y, Yang J, Lafi FF, Al-Suwailem A, Qian P-Y. Pyrosequencing reveals highly diverse and species-specific microbial communities in sponges from the Red Sea. The ISME Journal: Multidisciplinary Journal of Microbial Ecology. 2011;5:650–664. doi: 10.1038/ismej.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesser, Slattery & Leichter (2009).Lesser MP, Slattery M, Leichter JJ. Ecology of mesophotic coral reefs. Journal of Experimental Marine Biology and Ecology. 2009;375:1–8. doi: 10.1016/j.jembe.2009.05.009. [DOI] [Google Scholar]
- Lesser et al. (2010).Lesser MP, Slattery M, Stat M, Ojimi M, Gates RD, Grottoli A. Photoacclimatization by the coral Montastraea cavernosa in the mesophotic zone: light, food, and genetics. Ecology. 2010;91:990–1003. doi: 10.1890/09-0313.1. [DOI] [PubMed] [Google Scholar]
- Moitinho-Silva et al. (2014).Moitinho-Silva L, Bayer K, Cannistraci CV, Giles EC, Ryu T, Seridi L, Ravasi T, Hentschel U. Specificity and transcriptional activity of microbiota associated with low and high microbial abundance sponges from the Red Sea. Molecular Ecology. 2014;23:1348–1363. doi: 10.1111/mec.12365. [DOI] [PubMed] [Google Scholar]
- Morrow, Fiore & Lesser (2016).Morrow KM, Fiore CL, Lesser MP. Environmental drivers of microbial community shifts in the giant barrel sponge, Xestospongia muta, over a shallow to mesophotic depth gradient. Environmental Microbiology. 2016 doi: 10.1111/1462-2920.13226. Epub ahead of print Feb 15 2016. [DOI] [PubMed] [Google Scholar]
- Naim et al. (2014).Naim MA, Morillo JA, Sørensen SJ, Waleed AAS, Smidt H, Sipkema D. Host-specific microbial communities in three sympatric North Sea sponges. FEMS Microbiology Ecology. 2014;90:390–403. doi: 10.1111/1574-6941.12400. [DOI] [PubMed] [Google Scholar]
- Oksanen et al. (2012).Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR. vegan: community ecology package. (R Packag. version 2.0-2) 2012 Available at http://cran.r-project.org/package=vegan .
- Olson & Gao (2013).Olson JB, Gao X. Characterizing the bacterial associates of three Caribbean sponges along a gradient from shallow to mesophotic depths. FEMS Microbiology Ecology. 2013;85:74–84. doi: 10.1111/1574-6941.12099. [DOI] [PubMed] [Google Scholar]
- Olson & Kellogg (2010).Olson JB, Kellogg CA. Microbial ecology of corals, sponges, and algae in mesophotic coral environments. FEMS Microbiology Ecology. 2010;73:17–30. doi: 10.1111/j.1574-6941.2010.00862.x. [DOI] [PubMed] [Google Scholar]
- Paulay (2003).Paulay G. Marine biodiversity of Guam and the Marianas: overview. Micronesica. 2003;35–36:653–660. [Google Scholar]
- Pita et al. (2013).Pita L, Turon X, López-Legentil S, Erwin PM. Host rules: spatial stability of bacterial communities associated with marine sponges (Ircinia spp.) in the Western Mediterranean Sea. FEMS Microbiology Ecology. 2013;86:268–276. doi: 10.1111/1574-6941.12159. [DOI] [PubMed] [Google Scholar]
- Quince et al. (2011).Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ. Removing noise from pyrosequenced amplicons. BMC Bioinformatics. 2011;12:38. doi: 10.1186/1471-2105-12-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2013).R Development Core Team . R: a language and environment for statistical computing. Vienna: R Foundation of Statistical Computing; 2013. Available at http://www.r-project.org/ [Google Scholar]
- Reveillaud et al. (2014).Reveillaud J, Maignien L, Eren MA, Huber JA, Apprill A, Sogin ML, Vanreusel A. Host-specificity among abundant and rare taxa in the sponge microbiome. The ISME Journal. 2014;8:1198–1209. doi: 10.1038/ismej.2013.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss, Gevers & Westcott (2011).Schloss PD, Gevers D, Westcott SL. Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PLoS ONE. 2011;6:e1936. doi: 10.1371/journal.pone.0027310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss et al. (2009).Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt et al. (2008).Schmitt S, Angermeier H, Schiller R, Lindquist N, Hentschel U. Molecular microbial diversity survey of sponge reproductive stages and mechanistic insights into vertical transmission of microbial symbionts. Applied and Environmental Microbiology. 2008;74:7694–7708. doi: 10.1128/AEM.00878-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt et al. (2011).Schmitt S, Deines P, Behnam F, Wagner M, Taylor MW. Chloroflexi bacteria are more diverse, abundant, and similar in high than in low microbial abundance sponges. FEMS Microbiology Ecology. 2011;78:497–510. doi: 10.1111/j.1574-6941.2011.01179.x. [DOI] [PubMed] [Google Scholar]
- Schmitt, Hentschel & Taylor (2012).Schmitt S, Hentschel U, Taylor MW. Deep sequencing reveals diversity and community structure of complex microbiota in five Mediterranean sponges. Hydrobiologia. 2012;687:341–351. doi: 10.1007/s10750-011-0799-9. [DOI] [Google Scholar]
- Schmitt et al. (2012).Schmitt S, Tsai P, Bell J, Fromont J, Ilan M, Lindquist N, Perez T, Rodrigo A, Schupp PJ, Vacelet J, Webster N, Hentschel U, Taylor MW. Assessing the complex sponge microbiota: core, variable and species-specific bacterial communities in marine sponges. The ISME Journal. 2012;6:564–576. doi: 10.1038/ismej.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simister et al. (2012a).Simister RL, Deines P, Botté ES, Webster NS, Taylor MW. Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environmental Microbiology. 2012a;14:517–524. doi: 10.1111/j.1462-2920.2011.02664.x. [DOI] [PubMed] [Google Scholar]
- Simister et al. (2013).Simister RL, Taylor MW, Rogers KM, Schupp PJ, Deines P. Temporal molecular and isotopic analysis of active bacterial communities in two New Zealand sponges. FEMS Microbiology Ecology. 2013;85:195–205. doi: 10.1111/1574-6941.12109. [DOI] [PubMed] [Google Scholar]
- Simister et al. (2012b).Simister RL, Taylor MW, Tsai P, Fan L, Bruxner TJ, Crowe ML, Webster N. Thermal stress responses in the bacterial biosphere of the Great Barrier Reef sponge, Rhopaloeides odorabile. Environmental Microbiology. 2012b;14:3232–3246. doi: 10.1111/1462-2920.12010. [DOI] [PubMed] [Google Scholar]
- Slattery et al. (2011).Slattery M, Lesser MP, Brazeau D, Stokes MD, Leichter JJ. Connectivity and stability of mesophotic coral reefs. Journal of Experimental Marine Biology and Ecology. 2011;408:32–41. doi: 10.1016/j.jembe.2011.07.024. [DOI] [Google Scholar]
- Soininen (2010).Soininen J. Species turnover along abiotic and biotic gradients: patterns in space equal patterns in time? Bioscience. 2010;60:433–439. doi: 10.1525/bio.2010.60.6.7. [DOI] [Google Scholar]
- Steinert et al. (2014).Steinert G, Whitfield S, Taylor MW, Thoms C, Schupp PJ. Application of diffusion growth chambers for the cultivation of marine sponge-associated bacteria. Marine Biotechnology. 2014;16:594–603. doi: 10.1007/s10126-014-9575-y. [DOI] [PubMed] [Google Scholar]
- Taborosi, Jenson & Mylroie (2003).Taborosi D, Jenson JW, Mylroie JE. Zones of enhanced dissolution and associated cave morphology in an uplifted carbonate island karst aquifer, northern Guam, Mariana Islands. Speleogenesis and Evolution of Karst Aquifers. 2003;1:1–16. [Google Scholar]
- Taylor et al. (2007).Taylor MW, Radax R, Steger D, Wagner M. Sponge-associated microorganisms: evolution, ecology, and biotechnological potential. Microbiology and Molecular Biology Reviews. 2007;71:295–347. doi: 10.1128/MMBR.00040-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor et al. (2004).Taylor MW, Schupp PJ, Dahllöf I, Kjelleberg S, Steinberg PD. Host specificity in marine sponge-associated bacteria, and potential implications for marine microbial diversity. Environmental Microbiology. 2004;6:121–130. doi: 10.1046/j.1462-2920.2003.00545.x. [DOI] [PubMed] [Google Scholar]
- Taylor et al. (2005).Taylor MW, Schupp PJ, De Nys R, Kjelleberg S, Steinberg PD. Biogeography of bacteria associated with the marine sponge Cymbastela concentrica. Environmental Microbiology. 2005;7:419–433. doi: 10.1111/j.1462-2920.2004.00711.x. [DOI] [PubMed] [Google Scholar]
- Taylor et al. (2013).Taylor MW, Tsai P, Simister RL, Deines P, Botte E, Ericson G, Schmitt S, Webster NS. “Sponge-specific” bacteria are widespread (but rare) in diverse marine environments. The ISME Journal: Multidisciplinary Journal of Microbial Ecology. 2013;7:438–443. doi: 10.1038/ismej.2012.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker & Freeman (2012).Thacker RW, Freeman CJ. Sponge-microbe symbioses: recent advances and new directions. Advances in Marine Biology. 2012;62:57–111. doi: 10.1016/B978-0-12-394283-8.00002-3. [DOI] [PubMed] [Google Scholar]
- Webster, Cobb & Negri (2008).Webster NS, Cobb RE, Negri AP. Temperature thresholds for bacterial symbiosis with a sponge. The ISME Journal. 2008;2:830–842. doi: 10.1038/ismej.2008.42. [DOI] [PubMed] [Google Scholar]
- Webster et al. (2010).Webster NS, Taylor MW, Behnam F, Lücker S, Rattei T, Whalan S, Horn M, Wagner M. Deep sequencing reveals exceptional diversity and modes of transmission for bacterial sponge symbionts. Environmental Microbiology. 2010;12:2070–2082. doi: 10.1111/j.1462-2920.2009.02065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel & Erwin (2015).Weigel BL, Erwin PM. Intraspecific variation in microbial symbiont communities of the sun sponge, Hymeniacidon heliophila, from intertidal and subtidal habitats. Applied and Environmental Microbiology. 2015;82:650–658. doi: 10.1128/AEM.02980-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz, Lindquist & Martens (2008).Weisz JB, Lindquist N, Martens CS. Do associated microbial abundances impact marine demosponge pumping rates and tissue densities? Oecologia. 2008;155:367–376. doi: 10.1007/s00442-007-0910-0. [DOI] [PubMed] [Google Scholar]
- White et al. (2012).White JR, Patel J, Ottesen A, Arce G, Blackwelder P, Lopez JV. Pyrosequencing of bacterial symbionts within Axinella corrugata sponges: diversity and seasonal variability. PLoS ONE. 2012;7:e1936. doi: 10.1371/journal.pone.0038204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson & Fay (1979).Wilkinson CR, Fay P. Nitrogen fixation in coral reef sponges with symbiotic cyanobacteria. Nature. 1979;279:527–529. doi: 10.1038/279527a0. [DOI] [Google Scholar]
- Wright et al. (2012).Wright AD, Schupp PJ, Schrör J-P, Engemann A, Rohde S, Kelman D, De Voogd N, Carroll A, Motti CA. Twilight zone sponges from Guam yield theonellin isocyanate and psammaplysins I and J. Journal of Natural Products. 2012;75:502–506. doi: 10.1021/np200939d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng et al. (2014).Zeng Y, Feng F, Medová H, Dean J, Koblížek M. Functional type 2 photosynthetic reaction centers found in the rare bacterial phylum Gemmatimonadetes. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:7795–7800. doi: 10.1073/pnas.1400295111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Rarefied 97%-OTU 16S rRNA gene amplicon data for each sample.
Dendrograms showing the Bray-Curtis dissimilarity of microbial communities of (A) the complete sample dataset, (B) Callyspongia sp., (C) R. globostellata, (D) seawater, (E) A. cavernosa, (F) Rhaphoxya sp. sponge specimens based on 97%-OTU amplicon subsets.
Habitat file used for grouping of samples in multivariate analyses (nMDS & adonis).
Tukey multiple comparisons of means for the group based betadisper analysis. 95% family-wise confidence level—diff giving the difference in the observed means, lwr giving the lower end point of the interval, upr giving the upper end point and p adj giving the p-value after adjustment for the multiple comparisons.
Pairwise comparisons of the group based permutation test for homogeneity of multivariate dispersions. Observed p-value below diagonal, permuted p-value above diagonal.
Data Availability Statement
The following information was supplied regarding data availability:
The 97% OTU abundance table combined with the Greengenes classification for each individual OTU can be accessed via the Figshare online repository— 10.6084/m9.figshare.2063280.
The best hits and representative sequences for the most abundant OTUs can be accessed via the Figshare online repository (10.6084/m9.figshare.2366827).