


Abstract
The Type IIS restriction endonuclease SapI recognizes the DNA sequence 5′-GCTCTTC-3′ (top strand by convention) and cleaves downstream (N1/N4) indicating top- and bottom-strand spacing, respectively. The asymmetric nature of DNA recognition presented the possibility that one, if not two, nicking variants might be created from SapI. To explore this possibility, two parallel selection procedures were designed to isolate either top-strand nicking or bottom-strand nicking variants from a randomly mutated SapI expression library. These procedures take advantage of a SapI substrate site designed into the expression plasmid, which allows for in vitro selection of plasmid clones possessing a site-specific and strand-specific nick. A procedure designed to isolate bottom-strand nicking enzymes yielded Nb.SapI-1 containing a critical R420I substitution near the end of the protein. The top-strand procedure yielded several SapI variants with a distinct preference for top-strand cleavage. Mutations present within the selected clones were segregated to confirm a top-strand nicking phenotype for single variants Q240R, E250K, G271R or K273R. The nature of the amino acid substitutions found in the selected variants provides evidence that SapI may possess two active sites per monomer. This work presents a framework for establishing the mechanism of SapI DNA cleavage.
INTRODUCTION
Of the greater than 3500 characterized Type II restriction endonucleases, over 200 distinct specificities are commercially available (1). However, only a small number of nicking endonucleases are currently available. Restriction enzymes recognizing an asymmetric DNA sequence are potential subjects for the creation of site-specific and strand-specific nicking endonucleases. SapI recognizes 5′-GCTCTTC-3′ and cleaves one (top strand) and four (bottom strand) bases downstream of this sequence. Enzymes that cut away from an asymmetric sequence are classified as Type IIS (2).
The naturally occurring nicking enzymes, N.BstSEI (3) and N.BstNBI, (4) both nick 4 bp downstream of the 5′-GAGTC-3′ recognition site and are related to the Type IIS restriction enzymes MlyI and PleI which also recognize 5′-GAGTC-3′ and cut downstream either (N5/N5) or (N4/N5), respectively. Analysis of the gene encoding N.BstNBI revealed 32.1 and 33.1% similarity to MlyI and PleI at the amino acid level suggesting that these three enzymes have diverged from a common ancestor (5). Higgins et al. (5) demonstrated that MlyI dimerizes in the presence of its substrate DNA while N.BstNBI remains a monomer. So apparently, the inability of N.BstNBI to perform double-stranded cleavage is due to the loss of the dimerization function. Based on this premise, Besnier and Kong (6) engineered a nicking variant from MlyI by mutating residues within the presumed dimer interface. The double variant (Y491A/K494A) is unable to dimerize and only the top strand of DNA is cleaved (6). Furthermore, the nicking enzyme N.AlwI was created from AlwI by replacement of the putative C-terminal dimerization domain with that of N.BstNBI (7). This domain exchange was successful since the two proteins share 28.7% sequence identity and 41.5% similarity but this method of nicking enzyme engineering may be applied in a very limited number of cases. Most recently, Zhu et al. (8) isolated nicking variants of BsaI and BsmAI by extensive screening of libraries created by random mutagenesis. Although several nicking variants of each enzyme were isolated, the method is not strand specific. For example, only top-strand nicking variants were isolated from the BsmAI mutant library.
Others efforts have resulted in the isolation of nicking endonucleases from the Type II enzymes EcoRV and EcoRI, both of which recognize symmetric sites as homodimers. The Pingoud group (9–11) reported a method for engineering a nicking enzyme from EcoRV by creating two variants with different peptide tags where one variant is catalytically inactive. Upon co-expression, stable homodimers and heterodimers were produced and the heterodimeric proteins were isolated by affinity chromatography. However, such heterodimeric proteins are non-specific with respect to which strand is nicked and therefore the application of such enzymes in DNA manipulation is very limited. Random mutagenesis of EcoRI also yielded some variants that preferentially nick DNA. For example, the EcoRI R200C and R200K variants generate a higher proportion of nicked circular DNA than does the wt EcoRI enzyme (12,13).
In the present study, nicking enzymes were isolated for each strand of the SapI substrate site. The SapI nicking variants were isolated from a random library by a novel selection method, which may be adapted to engineer other Type IIS-derived nicking endonucleases. The key feature of the selection procedure is the presence of a SapI nicking site designed into the expression plasmid directly downstream of the sapIR gene. The method enables selection of enzymes possessing a desired function and does not require prior knowledge of active site residues. In fact, potential active site residues are revealed upon sequencing the selected clones. All four amino acid substitutions resulting in top-strand nicking map to one central region of the SapI polypeptide while the single bottom-strand variant contains a critical substitution very near the C-terminus. Top-strand variants nick after 5′-GCTCTTCN↓-3′ and were named Nt.SapI followed by the number 1, 2, 3 or 4 to distinguish between protein variants. The bottom-strand variant nicks before 5′-↓NNNNGAAGAGC-3′ and was named Nb.SapI-1.
MATERIALS AND METHODS
Materials
All restriction enzymes, DNA modifying enzymes and the substrate pUC19 were supplied by New England Biolabs (NEB). Oligonucleotides were synthesized and PAGE-purified (where indicated) by the NEB Organic Synthesis Division. Bacterial strains were from the NEB strain collection (E. Raleigh and M. Sibley). Plasmid purification was by Qiaprep®spin column.
Construction of expression vector pSAPV6
The expression vector pSAPV6 was derived from pACYCT7 (14). First, four copies of the rrnb transcriptional terminator were inserted preceding the T7 promoter to create pACYCT7-ter. The terminator sequence, T7 promoter and NdeI/BamHI polylinker were isolated from pAII17 (15) by SphI/BamHI excision and inserted into the same sites of pACYCT7. The polylinker of pACYCT7-ter was then modified to contain a strategically located SapI site. Two complementary phosphorylated oligonucleotides were PAGE-purified, annealed and ligated into the BamHI site of pACYCT7-ter. The top strand of the polylinker insert was 5′P-GATCCGCT CTTCGTCGACCCTCAGC (296–332) and the bottom strand was 5′P-GATCGCTGAGGGTCGACGAAGAGCG-3′ (296–333). Proper insertion created one BamHI site followed by the sites for SapI and BbvCI (Figure 2A). The modified polylinker was confirmed by sequencing with the T7 universal primer from NEB (# S1248S). The modified vector was named pSAPV6.
Figure 2.

(A) The polylinker sequence of the T7 expression vector pSAPV6. A randomly mutated sapIR gene library (open box) was ligated into the NdeI/BamHI sites. The SapI and N.BbvCI nicking sites are enabling factors for the selection procedure. (B) The scenario for selection of pSAPV6 expression clones carrying a SapI top-strand nick. The ss adaptor 296–334 is phosphorylated at the 5′ end and was designed to anneal to a 10-base cohesive end created by a SapI top-strand nick followed by nicking the bottom strand with N.BbvCIA. Complementary nucleotides are shown in bold. (C) The scenario for selection of SapI expression clones carrying a SapI bottom-strand nick. The ss adaptor 296–335 was designed to anneal to a 4-base cohesive end created by a SapI bottom-strand nick followed by nicking the top strand with N.BbvCIB. (D) The PCR amplification strategy following ligation of either ss adaptor. Not shown is the Klenow fill-in reaction (to extend from the -ACCC 3′ end) to create a template strand for the PCR step of the Nb.SapI selection procedure.
Error-prone PCR mutagenesis of the sapIR gene and creation of a plasmid library
The sapIR gene (1299 bp) was PCR-amplified from the genomic DNA of a Saccharopolyspora species (NEB strain #597) using 0.4 μM oligonucleotide primers containing NdeI and BamHI sites: The forward PCR amplification primer was 5′-AGAGTCTTGCATATGCGGAGGCTTGCTACAC-3′ (298–022, NdeI site is underlined) and the reverse primer was 5′-TGGTTTGGATCCCCTGAAATGGGTTAGGGC-3′ (298–023, BamHI site is underlined). Thermocycling was conducted for 30 cycles (94°C for 30 s, 56°C for 30 s and 72°C for 90 s) in the presence of 1× NEB ThermoPol buffer supplemented with 5 mM MgSO4 and 5 U Taq DNA polymerase. An unequal mixture of dNTPs was added to influence the mutation rate: 0.2 mM dATP, 0.2 mM dGTP, 1.0 mM dCTP and 1.0 mM dTTP. An Applied Biosystems Gene Amp® 2700 instrument was used for thermocycling. The PCR product was purified by Qiagen® spin column, digested with NdeI and BamHI and then subjected to agarose gel purification. The purified gene library was ligated to pSAPV6 prepared by NdeI/BamHI digestion and CIP treatment. After incubation overnight at 16°C, the ligation mix was heated at 65°C for 10 min and drop-dialyzed as preparation for electroporation.
Screening for SapI nicking variants using cell extracts
Variant enzyme screening was accomplished by expression in strain ER2744 [fhuA2 lacZ::T7 gene1 glnV44 e14-rfbD1? relA1? endA1 spoT1? thi-1 Δ(mcrC-mrr)114::IS10] carrying the double methylase clone pBR322-SapIM1M2. Individual transformants from the selection process were inoculated into 10 ml Luria–Bertani (LB) broth containing 30 μg/ml chloramphenicol (Cam) and 100 μg/ml ampicillin (Amp). The cultures were grown at 37°C until late log phase and then induced overnight with 0.1 mM IPTG. The cells were pelleted and the cell extract was produced by partial sonication in 1.0 ml sonication buffer [10 mM Tris–HCl (pH 7.8), 10 mM β-mercaptoethanol and 0.1 mM EDTA]. Nicking activity was assayed by incubating supercoiled pUC19 (one SapI site) with 3 μL cell extract for 45 min at 37°C in the presence of 1× NEB buffer 4 and visualizing a mobility shift by agarose gel electrophoresis.
Determination of strand specificity
The DNA substrate pUC19 was incubated with purified nicking enzyme for 30 min at 37°C in the presence of 1× NEB buffer 4. The nicked DNA product was isolated by electrophoresis in a 1% low-melting (LM) agarose gel (American Bioanalytical). The nicked strand was identified by sequencing the double-stranded template with two primers that converge on the SapI site: primer (266–113) 5′-GGGGGGCGGAGCCTATGGAAAAACGCCAGCAACG-3′ anneals at the pUC origin and was diagnostic for top-strand nicks. M13/pUC sequencing primer (NEB #S1224S) 5′-CGCCAGGGTTTTCCCAGTCACGAC-3′ was diagnostic for bottom-strand nicks. Sequencing was conducted using the AmpliTaq® dideoxy terminator kit (Applied Biosystems) and an ABI 373A sequencing instrument. Note that an additional adenine (A) is added by the terminal transferase activity of AmpliTaq® DNA polymerase (16).
Purification of SapI nicking variants
Clone Nb.33 was transformed into ER2848 [ER2744 with lacIq on F′ (TetR)] (17) carrying pBR322-SapIM1M2. ER2848 [pSAPV6-Nb.33, pBR322-SapIM1M2] was grown at 30°C in a final volume of 3 l. Nb.SapI-1 over-production was induced by the addition of 0.5 mM IPTG during the final 5 h of growth. The cells were pelleted and frozen at −20°C. The cell pellet was resuspended by addition of 60 ml HEPES breakage buffer: 20 mM HEPES (pH 7.7), 200 mM NaCl, 1 mM EDTA, 1 mM DTT, 10% glycerol. The cells were lysed by sonication, which was monitored by Bradford assay measurement of released protein. The clarified lysate was passed through a DEAE–Sepharose column (15 ml resin) pre-equilibrated with HEPES breakage buffer. The flow-through was collected and loaded onto a Heparin–Sepharose FF column pre-equilibrated with HEPES breakage buffer (minus EDTA). Thirty fractions were collected while performing a 0.2–1.0 M NaCl elution gradient. Of these, fractions 3–16 contained significant nicking activity when assayed on pUC19. The active pool (55 ml) was loaded onto a hydroxyapatite Bio-Gel HTP column, prepared by resuspending 3 g resin in 18 ml HEPES breakage buffer minus EDTA. A phosphate gradient was employed to elute protein. Beaker A contained HEPES buffer (minus EDTA) while buffer B contained 0.9 M potassium phosphate (pH 7.7), 200 mM NaCl, 1 mM DTT and 10% glycerol. Twenty-eight fractions were eluted and fractions 9–14 displayed nicking activity. The active pool (20 ml) was subjected to a second DEAE flow-through step to accomplish nucleic acid removal. The second DEAE column (15 ml resin) was equilibrated with 20 mM HEPES (pH 7.7), 200 mM NaCl, 0.3 mM EDTA, 1 mM DTT and 10% glycerol. The final fractionation was by Heparin–Sepharose FF (15 ml resin) using a 0.2–1.0 M NaCl gradient in HEPES breakage buffer (containing 1 mM EDTA). The final pool of 15 ml was dialyzed overnight in SapI storage buffer: 10 mM Tris–HCl (pH 7.5), 300 mM NaCl, 0.1 mM EDTA, 1 mM DTT and 50% glycerol. After dialysis, the final volume was 5.5 ml and the titer of Nb.Sap-1 was 2000 U/ml.
Nt.SapI-1 was purified in a similar manner except the final Heparin–Sepharose column was not necessary. Also, Nt.SapI-1 over-expression from pSAPV6 was conducted in ER2848 carrying pBR322-SapIM1M2 and pSYX33-EarIM1M2. The EarI methylases (5′-CTCTTC-3′) were utilized to provide more complete protection of SapI sites (SYX; K. D. Lunnen and G. Wilson, unpublished results). From a 6 l culture, the final yield was 46 000 U.
Purification of wild-type SapI
The IMPACT-CN purification system (NEB) was used to express and purify wild-type SapI. SapI was expressed as a fusion protein from vector pTYB11Δ where the lacI gene had been deleted by AfeI digestion, Klenow fill-in and plasmid reclosure. The sapIR gene was digested by SapI/XbaI and cloned into the SapI/SphI sites of pTYB11Δ to yield a fusion of the intein/chitin tag to the N-terminus of SapI. The sapIR gene was amplified from genomic DNA with a (50:1) mix of Taq and Vent® DNA polymerases (NEB) using forward primer 5′-CCTGGAGCTCTTCTAACATGCGTCGTCTTGCTACCCAACGTCGCGAGGACGCGTAC-3′ (305–273, SapI cloning site is underlined) and reverse primer 5′-GAGCTCTAGAGTTCAGTCCAGTGGTAGTGCTTC-3′ (305–196, XbaI cloning site is underlined). The recombinant SapI clone was transformed into ER2848 carrying pACYC-SapIM1M2 and pSYX-EarIM1M2 for over-expression. A 6 l culture was grown at 29°C until late log phase and then induced with 0.5 mM IPTG overnight at 29°C. The cell pellet was resuspended in chitin column buffer: 20 mM HEPES (pH 7.7), 500 mM NaCl, 1 mM EDTA and 10% glycerol and sonicated. Clarified cell extract was loaded onto 40 ml chitin resin (NEB). After a wash step, the column was flushed quickly with column buffer containing 50 mM DTT. The column was sealed and left at 23°C for 20 h to allow intein-mediated cleavage of the fusion protein. Purified SapI was eluted with 25 ml of column buffer containing 1 mM DTT. After two passes through DEAE–Sepharose at 200 mM NaCl, the pool was dialyzed into SapI storage buffer (minus BSA). The yield of wt SapI was 9.5 ml at 2000 U/ml.
Site-directed mutagenesis of the sapIR gene
Site-directed mutagenesis of the sapIR gene was conducted to identify the individual amino acid substitutions contributing to the nicking phenotype of the selected variants. The overlap extension PCR mutagenesis procedure was used and the mutated sapIR genes were cloned into pSAPV6 for expression. Upstream and downstream primers for this procedure were 298–022 and 298–023 as listed previously. Table 1 displays the results of these mutagenesis studies. Variant E250K was created using forward primer (304–120) 5′-GCACTTATCACACGTAAGCGAAAGATATTCCTG-3′ and reverse primer (304–121) 5′-CAGGAATATCTTTCGCTTACGTGTGATAAGTGC-3′. Variant G271R was created using forward primer (304–170) 5′-GATAGGACCGGCGCGCGTCTTAAAAGTCAAGTG-3′ and reverse primer (304–171) 5′-CACTTGACTTTTAAGACGCGCGCCGGTCCTATC-3′. Variant K273R was created using forward primer (304–122) 5′-GGCGCGGGACTTCGTAGTCAAGTGTGGGAA-3′ and reverse primer (304–123) 5′-TTCCCACACTTGACTACGAAGTCCCGCGCC-3′. Position R420 was randomized using reverse primer (300–045) 5′-GCGGGATCCGTTCAGTCCAGTGGTAGTGCTTCATCGAGAAGTGCGTCTGGNNNTTCCTTCAACTTCTC-3′ (where N is equal mix of A, C, G, T). Primer 300–045 was paired with forward primer 298–022. The BamHI cloning site is underlined. The positions of mutated codons are indicated in bold.
Table 1. Summary of SapI nicking variants.
| Nt.SapI-1 (E250K) | Top strand nicking |
| Nt.SapI-2 (K273R) | Top-strand nicking |
| Nt.SapI-3 (Q240R) | Top-strand nicking |
| Nt.SapI-4 (G271R) | Top-strand nicking |
| Nb.SapI-1 (D34Y/I82V/P168L/R420I) | Bottom-strand nicking |
| R420Na | Nicking phenotype in the context of R340H |
| R420S | Nicking phenotype in the context of T248A |
| R420C | Nicking phenotype |
| R420T | Nicking phenotype in the context of I213V |
| R420L | Nicking phenotype in the context of A71V/T401A |
| R420V | Nicking phenotype |
| R420A | Nicking phenotype in the context of D135G |
| R420G | Nicking phenotype in the context of E243G/L424H |
SapI amino acid substitutions resulting in a DNA nicking phenotype. Single variants E250K, G271R or K273R were created by site-directed mutagenesis using oligonucleotides listed in the Materials and Methods. Variant Q240R was selected directly without any accompanying substitutions. The substitution R420I was studied within the context of the other Nb.SapI-1 substitutions.
aAdditional substitutions at R420 were created by codon randomization (see Materials and Methods).
RESULTS
A method was developed to isolate nicking variants from a library of SapI expression clones possessing random mutations (outlined in Figure 1). The first step of the method is transformation of the library into bacterial cells where the genomic DNA is not protected by DNA methylation of SapI sites. In this step, a stringent selection occurs since RecA+, Lig+ strains are significantly more tolerant of single-strand nicks as compared to double-strand cuts (13). In this genetic selection, survivors are expected to be expressing nicking variants, low activity variants and null variants. The surviving cells are pooled and the plasmid library is isolated after a short outgrowth period. The library plasmid was pSAPV6, which was engineered to possess one SapI substrate site within the polylinker just downstream of the sapIR open reading frame (Figure 2A). Therefore, plasmid clones expressing nicking enzymes may contain a site-specific and strand-specific nick upon isolation from the bacterial cell. This nick is the enabling factor for the in vitro selection procedure.
Figure 1.

An outline of the selection procedures to isolate SapI variants with either top- or bottom-strand nicking activity.
Initial selection procedure for plasmids possessing a site-specific, strand-specific nick
A plasmid library expressing SapI variants was constructed as detailed in the Materials and Methods. The pSAPV6 ligation mix was transformed into ER1992 cells by electroporation and plated on LB agar containing 30 μg/ml chloramphenicol and 80 μg/ml X-gal. ER1992 is a DNA damage indicator strain, which contains the dinD1::lacZ gene fusion (18,19). When cells are plated on media containing X-gal, the extent of blue color development is a measure of the SOS response. A first assumption was made that full-strength nicking variants might induce the SOS response in Escherichia coli while variants possessing double-strand cleavage activity would result in cell lethality. ER1992 does not express the T7 RNA polymerase. Yet, a low level of sapIR gene expression was evident as a mixture of colony phenotypes was observed. Medium-blue colonies reporting DNA damage were pooled in 2 lots (60 colonies each) and inoculated into LB plus Cam. The two 10 ml cultures were grown to saturation at 37°C and plasmid DNA was prepared.
The in vitro selection process comprises two parallel procedures designed to isolate and amplify the sapIR genes from plasmids carrying a site-specific nick at the SapI cleavage site (Figures 1 and 2). In addition to the SapI site, the expression vector pSAPV6 carries an N.BbvCI nicking site nearby (Figure 2A). Figure 2B displays the expected scenario for isolating the sapIR genes from plasmids containing a SapI top-strand nick. If the library contains any plasmids with a nick after 5′-GCTCTTCG-3′, then digestion with N.BbvCIA will create a linearized plasmid with a pre-defined 10-base cohesive end. Similarly in Figure 2C, plasmids containing a SapI bottom-strand nick (before 5′-CGACGAAGAGC-3′) may be linearized by digestion with N.BbvCIB thus forming a 4-base cohesive end. In both scenarios, a single-stranded (ss) adaptor is ligated and the sapIR genes of interest are amplified by PCR using the strategy displayed in Figure 2D. Single-stranded adaptor 296–334 (Figure 2B) was designed to anneal to the cohesive end produced by SapI top-strand nicking and N.BbvCIA bottom-strand nicking. After ligation of the adaptor, PCR amplification of genes possibly encoding top nicking variants was accomplished using primer 275–224 that anneals to the last 20 nt of adaptor 296–334. The upstream primer in both cases is T7 promoter primer 298–024 with the sequence 5′P-GGGAGATCTCGATCCCGCGAAATTAATACG-3′. Importantly, one additional step is required for the procedure designed to isolate genes encoding bottom-strand variants (Figure 1). After ligation of ss adaptor 296–335 to the 4-base cohesive end (Figure 2C), a Klenow fill-in reaction is required to produce a complementary strand to serve as a template for PCR amplification. Therefore in the Nb.SapI procedure, reverse PCR primer 275–224 anneals to the strand created by the Klenow reaction and not to the ligated adaptor (as in the Nt.SapI procedure).
In the first application of the method, PCR products of the expected size (1493 bp) were observed for both top and bottom selection procedures (data not shown). Templates from mock ligations (where adaptor was not added) did not yield such PCR products. The PCR products were digested with BamHI, isolated by agarose gel electrophoresis and cloned back into pSAPV6 (prepared by SmaI/BamHI digestion and CIP treatment). After the selected gene libraries were ligated to pSAPV6, the ligation reactions were transformed into ER2744 [pBR322-SapIM1M2], a pre-modified expression host. The clone pBR322-SapIM1M2 expresses both SapI methylase genes (14) to modify and protect the SapI recognition sites present within the host genome.
Forty-eight transformants (32 top and 16 bottom) were screened for the expression of SapI nicking variants (see Materials and Methods). Of the 32 ‘top-strand’ clones, none expressed a nicking variant. However, one of the 16 ‘bottom-strand’ clones (variant 33) displayed significant nicking activity as indicated by the conversion of supercoiled pUC19 to relaxed, open circle form (Figure 3A, lane 1). Variant 39 (Figure 3A, lane 7) produced some linearized pUC19 while all other lanes display typical levels of open, circular substrate produced by non-specific nicking activities present in E.coli extract.
Figure 3.
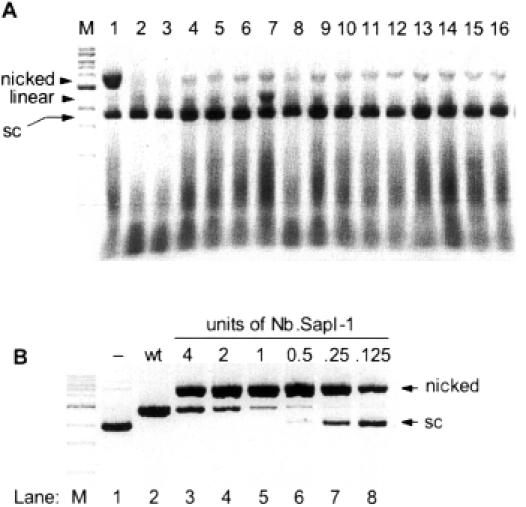
(A) Initial isolation of Nb.SapI-1 (variant 33). Nicking activity is revealed by incubating cell extract with supercoiled pUC19; (sc), supercoiled. Lane M is a 1 kb DNA ladder where the prominent band is a 3 kb band. The nicked form of pUC19 migrates at >3 kb. Lane 1 contains a significant level of nicked pUC19 produced by variant 33 extract. Lanes 2–16 (variants 34–48) contain typical levels of nicked pUC19 produced by the non-specific nicking activity of E.coli extract. Variant 39 (lane 7) displays double-stranded cleavage activity as linearized pUC19 (2686 bp) is observed. (B) DNA nicking activity of purified Nb.SapI-1 (variant 33). Lane 1 is supercoiled pUC19 without enzyme addition (−). Lane 2 is pUC19 linearized by wt SapI. Lanes 3–8 were incubated with 4, 2, 1, 0.5, 0.25 and 0.125 U of Nb.SapI-1 (variant 33) for 60 min at 37°C, respectively.
Characterization of variant 33
Sequencing the sapIR gene of clone 33 revealed mutations resulting in four amino acid substitutions: D34Y/I82V/P168L/R420I. Positions 34, 168 and 420 were individually randomized by site-directed mutagenesis to determine which of these substitutions was responsible for the nicking phenotype. Limited screening of substitutions at position 34 and 168 did not result in a variant possessing nicking activity. However, several nicking variants were produced by randomizing position 420 (Table 1). Variants with pronounced nicking activity were sequenced to discover many allowed substitutions: Asn, Ser, Cys, Thr, Leu, Ile, Val, Ala and Gly. As all these nicking variants possessed some degree of double-strand cleavage activity, none were superior to selected variant 33 (data not shown).
Variant 33 was partially purified by conventional chromatography to allow for more thorough characterization. The final titer of the enzyme was 2000 U/ml, where 1 U is defined as the amount of enzyme required to give complete nicking of 1 μg pUC19 in 60 min at 37°C. Figure 3B displays the nicking/cleavage characteristics of purified variant 33 (also named Nb.SapI-1). As the enzyme level is increased, some double-strand cleavage activity becomes apparent. Various buffer conditions were employed to possibly eliminate this residual double-strand cleavage activity. The following NEB buffers (with distinguishing characteristics in parentheses) were tested at a 1× concentration: N.BstNBI (150 mM KCl), BamHI (150 mM NaCl), BAL-31 nuclease (120 mM NaCl), DpnII (pH 6.0), EcoRI (pH 7.5), λ exonuclease (67 mM glycine–KOH, pH 9.4), MwoI (150 mM NaCl, 50 mM Tris–HCl, pH 7.9), NruI (100 mM KCl), NsiI (pH 8.4), Sau3AI (pH 7.0), ScaI (pH 7.4), TaqI (pH 8.4) and NEB standard buffers 1, 2, 3 and 4. One unit of variant 33 (as defined in NEB buffer 4) was added to each reaction for 60 min at 37°C. None of the conditions allowed complete conversion of supercoiled pUC19 to open, circular form without some linearization (data not shown). Therefore, the reaction conditions recommended for wt SapI (1× NEB buffer 4) appear to be most optimal for nicking variant 33.
Variant 33 was expected to be a bottom-strand nicking enzyme as defined by the selection method. The strand specificity was confirmed with sequencing reactions using a nicked pUC19 product as the template (see Materials and Methods). Variant 33 was incubated with 2 μg pUC19 and the nicked product was isolated by agarose gel electrophoresis. The sequencing reaction using primer #S1224S terminated at the expected position 5′…GCTCTTCCGCTA-3′ corresponding to a SapI bottom-strand nick (Figure 4). Note that the final adenine peak is false as it is added by the terminal transferase activity of Taq DNA polymerase. Sequencing the opposite strand of the pUC19 product revealed no evidence for the occurrence of nicking on the top strand. Variant 33 was then renamed Nb.SapI-1. Nb is the proposed nomenclature for Nicking enzyme, bottom strand (2).
Figure 4.
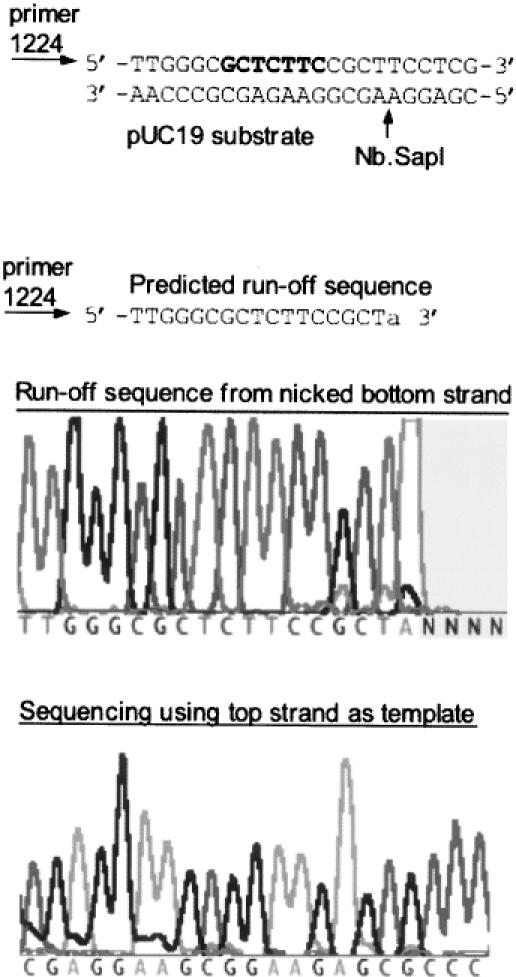
Run-off sequencing to determine the nicking site of Nb.SapI-1 (variant 33). The substrate pUC19 was incubated with purified protein for 30 min at 37°C. The nicked circular DNA product was isolated by agarose gel electrophoresis and sequenced using primers that converge on the SapI site. The sequencing reaction using primer #1224 terminated at 5′-GCTCTTCCGCTA-3′ as expected for a nicked bottom strand. AmpliTaq® DNA polymerase adds an adenine (A) at the end of the extension product (template-independent DNA transferase activity). The SapI recognition site is in bold.
Improved selection procedure for SapI nicking variants
The inability to isolate a top-strand nicking variant in the first procedure was most likely due to the limited size of the initial library (only 120 clones were selected for the first library). Furthermore, it was later determined that genetic selection in ER1992 was not stringent enough to eliminate variants with low double-stranded cleavage activity. For example, a survivor of the selection in ER1992 (variant 39) produced considerable linearization of pUC19 (Figure 3A, lane 7). Ideally, the genetic selection should eliminate all enzymes with measurable double-strand cleavage activity. We chose to repeat the genetic selection step with a strain encoding the T7 RNA polymerase to increase constitutive SapI expression from pSAPV6. In addition, a DNA damage indicator strain was not employed in the improved process since the desired SapI clone would most likely not produce a significant SOS response. This conclusion was made after clone 33 was transformed back into ER1992 to assess blue color development. The blue phenotype produced by variant 33 was very similar to the color of ER1992 transformed with empty vector. Therefore, a pure nicking variant recognizing a 7 bp sequence was expected to escape detection by the SOS indicator system of ER1992.
The same mutagenized sapIR gene library was ligated into pSAPV6 and transformed into ER2848 [ER2744 (lacIq)] by electroporation. Approximately 600 survivors were pooled into 500 ml LB plus Cam. The culture was grown to late log phase at 37°C, harvested by centrifugation and plasmid DNA was prepared by Qiagen® Maxiprep. The in vitro selection process was improved as well, by an agarose gel isolation of the slowly migrating DNA present in the pooled plasmid library. The slowly migrating plasmid DNA (comprising the nicked and the dimer forms) was excised from the gel and purified by adsorption to silica. This step eliminated most of the supercoiled plasmid DNA and enriched the library for nicked expression clones. The adaptor ligation and PCR amplification were carried out as before except for one change in the top-strand procedure. The adaptor was added after heating the N.BbvCIA nicked DNA to 65°C to ensure melting of the 10-base cohesive end (Figure 2A and B). Again, PCR products of the expected size were produced and the sapIR genes of interest were ligated to pSAPV6. The ligation mix was transformed into ER2744 [pBR322-SapM1M2] by electroporation.
Forty-eight ‘top strand’ transformants were analyzed for nicking activity. Of the 48 cell extracts, 5 were positive for significant nicking activity (Figure 3A displays the results of variants 1–19). All five positive clones (2,9,11,15,18) were sequenced to determine the responsible amino acid substitutions. The two most active nicking variants (11 and 15) contained three substitutions in common: K80E, E250K and K273R. Variant 11 carried the additional substitutions Q81R, T109I and L193F and variant 15 carried the additional substitution I414T. Site-directed PCR mutagenesis was conducted to determine which of the common substitutions might be responsible for the nicking phenotype. In this study, both single variants E250K and K273R produced an exclusively top-strand nicked pUC19 product while variant K80E was negative for nicking activity. Mutations present in other selected clones led to the findings that substitutions Q240R and G271R will each independently produce top-strand nicking (see Table 1). The four top-strand nicking variants were named Nt.SapI-1 (E250K), Nt.SapI-2 (K273R), Nt.SapI-3 (Q240R) and Nt.SapI-4 (G271R). Comparative analysis indicated that substitution E250K results in the most active top-strand SapI nicking variant. The improved selection process did not yield additional Nb.SapI variants. However, the fraction of desired clones from the improved top-strand procedure was 5 of 48 (10%) compared to 0 of 48 from the initial procedure.
Characterization of Nt.SapI-1 (E250K)
The strand specificity of variant E250K was confirmed by incubating pUC19 with purified protein followed by sequencing both strands of the nicked product (see Materials and Methods). The sequencing results are shown in Figure 6. The sequencing reaction using primer 266–113 terminated at 5′…GGAAGCA-3′. This truncated sequence is the product of using a nicked top strand as template. Top-strand nicking occurs between the first and second nucleotide downstream of the SapI recognition sequence. Note that the final adenine peak is false as it is added by the terminal transferase activity of Taq DNA polymerase. The activity of purified Nt.SapI-1 (E250K) was assayed to investigate the characteristics of double-stranded cleavage. When 8 U of enzyme were incubated with 1 μg of pUC19 for 60 min at 37°C, there was no detectable linearization of pUC19 (Figure 5B). The specific activity of variant E250K is estimated to be higher than that of wt SapI. This feature has been documented previously in the creation of the nicking enzyme N.AlwI where the specific activity is 20-fold higher than wt AlwI (7). Despite the high specific activity of variant E250K, the DNA damage indicator strain ER1992 did not report the induction of SOS when this clone was introduced.
Figure 6.

Run-off sequencing to determine the nicking site of Nt.SapI-1 (E250K). The substrate pUC19 was incubated with purified protein for 30 min at 37°C. The nicked circular DNA product was isolated by agarose gel electrophoresis and sequenced using primers that converge on the SapI site. AmpliTaq® DNA polymerase adds an adenine (A) at the end of the extension product (template-independent DNA transferase activity). The SapI recognition site is in bold.
Figure 5.
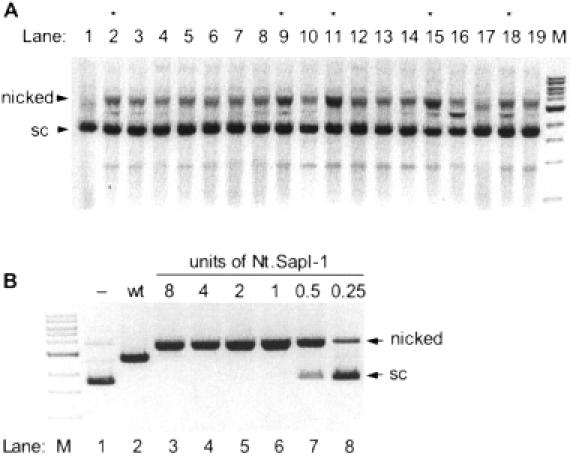
(A) Isolation of Nt.SapI variants containing the substitution E250K. Nicking activity is revealed by incubating cell extract with supercoiled pUC19. Variants #11 and 15 (lanes 11 and 15) carry the common substitutions K80E, E250K and K273R. All variants highlighted by an asterisk were sequenced to identify amino acid substitutions contributing to the nicking phenotype. Lane M is a 1 kb DNA ladder where the prominent band is 3 kb. (B) DNA nicking activity of purified Nt.SapI-1. Lane 1 supercoiled pUC19 without enzyme addition (−). Lane 2 is pUC19 linearized by wt SapI. Lanes 3–8 were incubated with 8, 4, 2, 1, 0.5 0.25 U of Nt.SapI-1 for 60 min at 37°C, respectively.
Nicking characteristics of wt SapI
Wild-type SapI was purified using the IMPACT-CN purification system (see Materials and Methods). The final enzyme concentration was adjusted to 2000 U/ml, which corresponds to 0.04 mg/ml or 800 nM monomer. A previous study by Bath et al. (20) evaluated the cleavage characteristics of SapI on substrates containing one or two sites. The conclusion was that SapI cleaves each site within each substrate at similar rates. This suggests that SapI does not need to interact with two recognition sites to accomplish DNA cleavage, in contrast to FokI, e.g. (20–22). Furthermore, Bath et al. (20) concluded that SapI did not generate appreciable amounts of nicked product when 4.8 nM (12 U/ml) enzyme was incubated with 5 nM plasmid substrate. The nicking characteristics of wt SapI were re-evaluated in this study using a lower ratio of enzyme:substrate. In this study, 1 nM SapI was incubated with 8 nM supercoiled pUC19 at 37°C in 1× NEB buffer 4 plus 0.1 mg/ml BSA and aliquots were taken every 5 min throughout a 40 min reaction. The results of this time course reaction are displayed in Figure 7A. The first lane is an aliquot taken before enzyme addition to show the inherent amount of nicked substrate. As the reaction proceeds, linear product and nicked product both increase and then appear to become constant. This phenomenon was observed for enzyme concentrations from 0.1 to 1.0 nM (data not shown). Complete conversion to linear form within 40 min was only observed when the enzyme:substrate ratio was ≥1:2 (data not shown).
Figure 7.
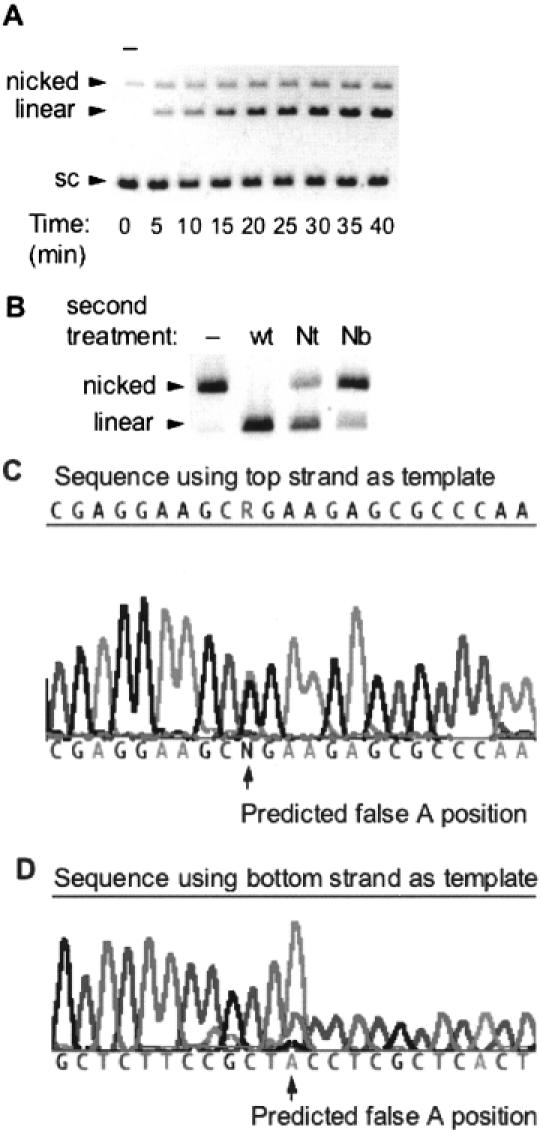
(A) DNA nicking/cleavage characteristics of wt SapI. SapI at a concentration of 1 nM was incubated with 8 nM pUC19 in a 200 μl reaction incubated at 37°C for 40 min. Aliquots of 20 μl were withdrawn every 5 min and added to 5 μl stop solution. Lane 1 is an aliquot taken before enzyme addition (−). (B) Characterization of the nicks created by incubating limiting amounts of wt SapI with pUC19 [as displayed in (A)]. Pre-nicked pUC19 was gel-purified and then subsequently incubated with Nt.SapI-1 (lane 3) or Nb.SapI-1 (lane 4) to determine the strand specificity of the pre-existing nick. Lane 2 is a control digest with excess wt SapI. (C) Sequence of pUC19 nicked by wt SapI. The R indicates a mixed peak of G from circular templates and A from linearized (nicked) top strands. (D) Sequence results using the bottom strand as template. Note the large false A peak (from nicked templates) at the same position as a small T peak produced from circular templates.
The nicked product observed in Figure 7A was isolated from the gel in order to analyze the nicks generated at the SapI cleavage site. The strand specificity was assessed by a second incubation with a 16-fold unit excess of wt SapI, Nt.SapI-1 or Nb.SapI-1 (Figure 7B). The results indicate that the nicks generated by wt SapI are not strand specific. Incubation of the ‘pre-nicked’ substrate with Nt.SapI or Nb.SapI allows the following conclusion, which is based on the amount of nicked substrate that is resistant to linearization. Approximately 70% of the nicks are on the bottom strand and 30% are on the top strand. This finding is corroborated by the results obtained from run-off sequencing reactions. Figure 7C displays the sequencing results of using the top strand as template and Figure 7D displays the results of using the bottom strand as template. In both sequence traces, the presence of a mixed peak indicates a mixture of templates where a fraction is nicked. Although the trace data is not quantitative, the more severe drop off of peak height in Figure 7D indicates that bottom-strand nicking is more prevalent than top-strand nicking. Finally, incubating the pre-nicked substrate with an excess of wt SapI (Figure 7B, lane 2) verified that the SapI recognition/cleavage site had not been damaged during the first digestion.
DISCUSSION
Strand-specific selection methods were designed and carried out resulting in rapid isolation of SapI nicking variants without prior knowledge of the enzyme architecture or even putative active site residues. These methods are based on directly selecting for a desired function rather than rational design of an enzyme of interest. The genetic selection step exploited the extreme difference in host DNA damage caused by the Type IIS restriction endonuclease SapI relative to its nicking variants. The in vitro selection steps took advantage of the site- and strand-specific nick carried by plasmid clones expressing a SapI nicking variant. This method may be universally applied to isolate site-specific nicking enzymes from other Type IIS restriction enzymes by simple modification of the library expression vector.
The method of Zhu et al. (8) depends critically on a genetic selection step to eliminate variants with double-strand cleavage activity before embarking on a screening procedure where the strand specificity of the nicking variant is left to chance. In the current work, a genetic selection step is also employed, but further in vitro enrichment steps are enabled by the presence of a nicking site within the library expression vector. After enrichment of the library via agarose gel electrophoresis, individual nicking variants are isolated by strand-specific procedures. The current method is also more practical than the method used to engineer N.AlwI (7) where similarity to a naturally occurring nicking enzyme is required for domain swapping.
The type and position of the amino acid substitutions from this work (Table 1) give some indication of the active site or sites within SapI. The four SapI variants with top-strand nicking activity contain amino acid substitutions within or near a sequence which conforms to the consensus PD(X)8–20D/EXK for Type II catalytic centers. Thus, the amino acids E250, E263 and K265 may form an active site for bottom-strand cleavage. The acidic residues within this consensus sequence most likely form a divalent-cation binding site, with Mg2+ being the essential cofactor in nearly every Type II enzyme studied (23). If E250 is in fact a metal chelating residue, it is easily envisioned that an E250K substitution would destroy the catalytic function of this center.
The critical substitution (R420I) identified in Nb.SapI-1 resides near the end of the protein and does not fall within or near a recognizable consensus sequence. As active sites of Type II restriction enzymes are often unrecognizable by sequence alone (23), the C-terminal domain of SapI may contain a second active site where R420 plays a critical role in DNA cleavage. Alternatively, R420 may be part of a salt bridge for bringing the N- and C-terminal domains together to form an active site for top-strand cleavage. However, we cannot rule out the possibility that R420 may play a role in transient dimer formation. Previous data revealed that SapI cleaves a two-site substrate at the same rate as a one-site substrate (20) suggesting that dimerization is not a requirement for double-strand cleavage. Also, our preliminary gel filtration data indicate that SapI remains a monomer in the presence or absence of substrate DNA. Further studies are planned to determine whether SapI cleaves both DNA strands as a monomeric protein. This phenomenon has never been verified for a Type IIS restriction enzyme but there is precedence in the LAGLIDADG family of homing endonucleases. PI-SceI acts as a monomer, using two catalytic centers to cleave a loosely defined asymmetric sequence (24). The nicking characteristics of wt SapI are not consistent with the cleavage mechanism proposed for the Type IIS enzyme MlyI (5,6) where bottom-strand cleavage occurs upon subunit dimerization. In the case of MlyI, a high concentration of nicked intermediate is formed (5) and this intermediate is nicked exclusively on the top strand (JCS, data not shown). In the case of wt SapI, a low concentration of nicked intermediate is formed (Figure 7A) and either strand may be nicked (Figure 7B, C and D).
The methods described in this work may be easily applied for the selection of other nicking variants derived from Type IIS restriction endonucleases. The availability of more site-specific nicking enzymes will expand the molecular biology toolbox and allow for the development of new technologies. Already, many technologies are making use of the nicking enzymes currently available. Multiple isothermal DNA amplification methods have been reported which rely on the production of site-specific nicks (25–27). Another application is in the preparation of nicked duplex DNA or gapped DNA for studying DNA mismatch repair (28,29). Furthermore, assays for DNA or RNA detection may be designed to exploit the property of single-stranded cleavage of either DNA–DNA hybrids or DNA–RNA hybrids (30). And finally, new methods of in vitro DNA recombination are possible. For example, long cohesive ends can be prepared to increase the efficiency of DNA fragment assembly into cloning vectors. Such a procedure replaces a restriction enzyme digestion and does not require a ligation step prior to cell transformation.
Acknowledgments
ACKNOWLEDGEMENTS
We thank Richard Roberts and Elisabeth Raleigh for critical reading of the manuscript, Stephen Picone for advice on purification of the SapI nicking variants, Laurie Mazzola and Jennifer Ware for DNA sequencing and Roger Knott for oligonucleotide synthesis. We are especially grateful for the environment and support provided by Don Comb. This work was supported in part by NIH grant R41GM070212-01 to S.Y.X.
REFERENCES
- 1.Roberts R.J., Vincze,T., Posfai,J. and Macelis,D. (2003) REBASE: restriction enzymes and methyltransferases. Nucleic Acids Res., 31, 418–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roberts R.J., Belfort,M., Bestor,T., Bhagwat,A.S., Bickle,T.A., Bitinaite,J., Blumenthal,R.M., Degtyarev,S.Kh., Dryden,D.T.F., Dybvig,K. et al. (2003) A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res., 31, 1805–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abdurashitov M.A., Belichenko,O.A., Shevchenko,A.V. and Degtyarev,S.K. (1996) N.BstSE–a site-specific nickase from Bacillus stearothermophilus SE-589. Mol. Biol. (Mosk), 30, 1261–1267. [PubMed] [Google Scholar]
- 4.Morgan R.D., Calvet,C., Demeter,M., Agra,R. and Kong,H. (2000) Characterization of the specific DNA nicking activity of restriction endonuclease N.BstNBI. Biol. Chem., 381, 1123–1125. [DOI] [PubMed] [Google Scholar]
- 5.Higgins L.S., Besnier,C. and Kong,H. (2001) The nicking endonuclease N.BstNBI is closely related to Type IIS restriction endonucleases MlyI and PleI. Nucleic Acids Res., 29, 2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Besnier C.E. and Kong,H. (2001) Converting MlyI endonuclease into a nicking enzyme by changing its oligomerization state. EMBO Rep., 9, 782–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu Y., Lunnen,K.D. and Kong,H. (2001) Engineering a nicking endonuclease N.AlwI by domain swapping. Proc. Natl Acad. Sci. USA, 98, 12990–12995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Z., Samuelson,J.C., Zhou,J., Dore,A. and Xu,S.-Y. (2004) Engineering strand-specific DNA nicking enzymes from the Type IIS restriction endonucleases BsaI, BsmBI and BsmAI. J. Mol. Biol., 337, 1888–1896. [DOI] [PubMed] [Google Scholar]
- 9.Stahl F., Wende,W., Jeltsch,A. and Pingoud,A. (1996) Introduction of asymmetry in the naturally symmetric restriction endonuclease EcoRV to investigate intersubunit communication in the homodimeric protein. Proc. Natl Acad. Sci. USA, 93, 6175–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wende W., Stahl,F. and Pingoud,A. (1996) The production and characterization of artificial heterodimers of the restriction endonuclease EcoRV. Biol. Chem., 377, 625–632. [DOI] [PubMed] [Google Scholar]
- 11.Stahl F., Wende,W., Wenz,C., Jeltsch,A. and Pingoud,A. (1998) Intra- vs intersubunit communication in the homodimeric restriction enzyme EcoRV: Thr 37 and Lys 38 involved in indirect readout are only important for the catalytic activity of their own subunit. Biochemistry, 37, 5682–5688. [DOI] [PubMed] [Google Scholar]
- 12.Heitman J. and Model,P. (1990) Substrate recognition by the EcoRI endonuclease. Proteins: Struct. Func. Genet., 7, 185–197. [DOI] [PubMed] [Google Scholar]
- 13.Heitman J., Ivanenko,T. and Kiss,A. (1999) DNA nicks inflicted by restriction endonucleases are repaired by a RecA- and RecB-dependent pathway in Escherichia coli. Mol. Microbiol., 33, 1141–1151. [DOI] [PubMed] [Google Scholar]
- 14.Xu S.-Y., Xiao,J.-P., Ettwiller,L., Holden,M., Aliotta,J., Poh,C.L., Dalton,M., Robinson,D.P., Petronzio,T.R., Moran,L. et al. (1998) Cloning and expression of the ApaLI, NspI, NspHI, SacI, ScaI, and SapI restriction-modification systems in Escherichia coli. Mol. Gen. Genet., 260, 226–231. [DOI] [PubMed] [Google Scholar]
- 15.Kong H., Kucera,R.B. and Jack,W.E. (1993) Characterization of a DNA polymerase from the hyperthermophile archaea Thermococcus litoralis. Vent DNA polymerase, steady state kinetics, thermal stability, processivity, strand displacement, and exonuclease activities. J. Biol. Chem., 268, 1965–1975. [PubMed] [Google Scholar]
- 16.Magnuson V.L., Ally,D.S., Nylund,S.J., Karanjawala,Z.E., Rayman,J.B., Knapp,J.I., Lowe,A.L., Ghosh,S. and Collins,F.S. (1996) Substrate nucleotide-determined non-templated addition of adenine by Taq DNA polymerase: implications for PCR-based genotyping and cloning. Biotechniques, 21, 700–709. [DOI] [PubMed] [Google Scholar]
- 17.Raleigh E.A. (2003) U.S. Patent No. 6,569,669.
- 18.Heitman J. and Model,P. (1991) SOS induction as an in vivo assay of enzyme-DNA interactions. Gene, 103, 1–9. [DOI] [PubMed] [Google Scholar]
- 19.Fomenkov A., Xiao,J.-P., Dila,D., Raleigh,E. and Xu,S.-Y. (1994) The ‘endo-blue method’ for direct cloning of restriction endonuclease genes in E. coli. Nucleic Acids Res., 22, 2399–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bath A.J., Milson,S.E., Gormley,N.A. and Halford,S.E. (2002) Many Type IIS restriction endonucleases interact with two recognition sites before cleaving DNA. J. Biol. Chem., 277, 4024–4033. [DOI] [PubMed] [Google Scholar]
- 21.Bitinaite J., Wah,D.A., Aggarwal,A.K. and Schildkraut,I. (1998) FokI dimerization is required for DNA cleavage. Proc. Natl Acad. Sci. USA, 95, 10570–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanamee E.S., Santagata,S. and Aggarwal,A.K. (2001) FokI requires two specific DNA sites for cleavage. J. Mol. Biol., 309, 69–78. [DOI] [PubMed] [Google Scholar]
- 23.Pingoud A. and Jeltsch,A. (2001) Structure and function of Type II restriction endonucleases. Nucleic Acids Res., 29, 3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Christ F., Schoettler,S., Wende,W., Steuer,S., Pingoud,A. and Pingoud,V. (1999) The monomeric homing endonuclease PI-SceI has two catalytic centres for cleavage of the two strands of its DNA substrate. EMBO J., 18, 6908–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Ness J., Van Ness,L.K. and Galas,D.J. (2003) Isothermal reactions for the amplification of oligonucleotides. Proc. Natl Acad. Sci. USA, 100, 4504–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Walker G.T. and Linn,C.P. (1996) Detection of Mycobacterium tuberculosis DNA with thermophilic strand displacement amplification and fluorescence polarization. Clin. Chem., 42, 1604–1608. [PubMed] [Google Scholar]
- 27.Walker G.T., Little,M.C., Nadeau,J.G. and Shank,D.D (1992) Isothermal in vitro amplification of DNA by a restriction enzyme/DNA polymerase system. Proc. Natl Acad. Sci. USA, 89, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang H. and Hays,J.B. (2000) Preparation of DNA substrates for in vitro mismatch repair. Mol. Biotechnol., 15, 97–104. [DOI] [PubMed] [Google Scholar]
- 29.Wang H. and Hays,J.B. (2001) Simple and rapid preparation of gapped plasmid DNA for incorporation of oligomers containing specific DNA lesions. Mol. Biotechnol., 19, 133–140. [DOI] [PubMed] [Google Scholar]
- 30.Zheleznaya L.A., Perevyazova,T.A., Zheleznyakova,E.N. and Matvienko,N.I. (2002) Some properties of site-specific nickase BspD6I and the possibility of its use in hybridization analysis of DNA. Biochemistry (Mosc.), 67, 498–502. [DOI] [PubMed] [Google Scholar]