


Abstract
Guessing the boundaries of structural domains has been an important and challenging problem in experimental and computational structural biology. Predictions were based on intuition, biochemical properties, statistics, sequence homology and other aspects of predicted protein structure. Here, we introduced CHOPnet, a de novo method that predicts structural domains in the absence of homology to known domains. Our method was based on neural networks and relied exclusively on information available for all proteins. Evaluating sustained performance through rigorous cross-validation on proteins of known structure, we correctly predicted the number of domains in 69% of all proteins. For 50% of the two-domain proteins the centre of the predicted boundary was closer than 20 residues to the boundary assigned from three-dimensional (3D) structures; this was about eight percentage points better than predictions by ‘equal split’. Our results appeared to compare favourably with those from previously published methods. CHOPnet may be useful to restrict the experimental testing of different fragments for structure determination in the context of structural genomics.
INTRODUCTION
Most proteins contain multiple structural domains. Large-scale sequencing efforts have confirmed that eukaryotes differ from all other kingdoms in the significantly higher proportion of proteins extending over 1500 residues (1–7). These large proteins undoubtedly consist of many structural domains. Structural domains are regions that are either compact, globular modules (‘beads-on-a-string’), or are clearly distinguished from flanking regions such as the membrane regions or long coiled-coil helices separating other domains (8), or tethering proteins (9). Domains can be viewed as semi-independent three-dimensional (3D) units in proteins; they may fold independently (10) and may constitute ‘units of evolution’ (11). Often these domains have particular functions and are recombined in different proteins (12). In fact, we recently proposed (13,42) that almost 60–70% of most non-eukaryotic proteins have multiple domains, and that many multiple domain proteins contain one long (200–400 residues) and many significantly shorter domains (∼100 residues). Many of these short domains may constitute modules enabling higher complexity in regulation (14). Thus, a detailed understanding of the domain organization and domain–domain interactions is essential to advancing our understanding of structure and function. Detailed knowledge of domain boundaries is often particularly relevant for experimental structure determination. Many proteins of known structure constitute fragments of native proteins. More coarse-grained identifications of the approximate map of domain organization benefits sequence analysis, and may also constitute a simple means of increasing the signal-to-noise ratio in yeast-two-hybrid screens by simply running the screen separately with all putative domains, rather than with the full-length protein.
Domains most accurately assigned from 3D structures. Various domain assignment methods and databases have been developed for proteins of known 3D structures. Structural Classification of Proteins (SCOP) is fully based on expert-driven, visual domain assignments (15). All other databases and methods are more or less automated. For instance, the Class Architecture Topology Homology (CATH) database of protein structure relations combines different assignment methods (16), the Dali Domain Dictionary (17) uses the PUU assignment method (11), and MMDB uses VAST (18). Such methods usually define domains as the structurally most compact local region, frequently clipping that stick out from the domain. The only fully automated method that assigns sequence-continuous domains from 3D structures is Protein Informatics System for Modeling [PrISM (19)]. Undoubtedly, automatic domain assignments capture reality much more accurately than any method attempting to define domains without structures. Nevertheless, different methods agree only for ∼81% of all domains (20). While disagreements may indicate errors of the individual methods, they often also reveal that the concept of a structural domain is—albeit powerful—not fully defined.
Predicting domain boundaries from sequence remains an open problem. In the absence of 3D structures, domain assignment becomes much more difficult. Early methods tried to predict domain boundaries without explicitly using alignments, for instance by exploiting interactions between secondary structure units and simulations of protein folding (21), short-range amino acid composition and association preference (22), and predicted inter-residue contact maps (23). Although based on very sound concepts, none of these methods achieved reasonable levels of accuracy. Several later methods explored patterns of conservation in alignments. The most prominent example is a database of putative protein domains [ProDom (24,25,26)] that generates a comprehensive set of protein domain families automatically from the SWISS-PROT and TrEMBL sequence databases (27). DOMO applies successive steps based on similarity in amino acid composition, dipeptide composition, local sequence similarity and multiple sequence alignment similarity to detect domain boundaries (28). DOMAINATION (29) delineates domains through analyzing position-specific iterated database search [PSI-BLAST (30)] alignments. Databases such as Pfam-A (31), SMART (32), TIGRFAMs (33), COG (34), SBASE (35), CDD (36), SUPERFAMILY (37), the CATH-related Gene3D and PFDB (38,39), and other methods (40,41) in some way or other base domain-like assignments on homology. Similarly, CHOP (42) identifies potential domain boundaries through hierarchical searches against databases of more or less well defined domains. Although all these methods provide valuable information about putative domains for proteins with similar sequences, they fail for small families or in the absence of homologous domain assignments. Recently, quite a few novel methods have been developed to predict domain boundaries directly from sequence. The ‘Domain Guess by Size’ (DGS) algorithm (43) ‘guesses’ domain boundaries solely based on the length distribution observed for proteins of known structure. Domain assignment from, sequence through protein folding simulation [SnapDRAGON (44)] performs 100 runs of ab initio protein structure prediction using DRAGON (45), assigns domain boundaries for each model with a fast structure-based domain dissecting method (46), and then predicts the domains through statistical analysis. According to the estimates of the authors, SnapDRAGON is currently the most accurate de novo domain assignment method. However, given the CPU resources needed, it is certainly not a feasible strategy in the context of analysing entire proteomes. More recently, a fast domain prediction method DomSSEA (47) has been proposed. The underlying idea is incredibly simple: align the secondary structure predicted for a query protein against a database of domains assigned from 3D structures (CATH) and simply derive the domain boundaries from the known domain with the most similar secondary structure. Albeit this simplicity, the authors found that DomSSEA correctly identified the number of domains in 72% of all proteins tested, and correctly identified 24% of all domain boundaries within ±20 residues of the boundaries annotated in CATH.
Here, we introduced a novel method that predicts domain boundaries through a neural network using evolutionary information, predicted One-dimensional (1D) structure (secondary structure, solvent accessibility), amino acid flexibility, and amino acid composition. The final predictions of domain boundaries resulted from post-processing the raw network output by removing noisy peaks. We evaluated sustained performance in terms of correctly predicting the number of domains in a protein and in correctly predicting the domain boundary.
METHODS
Data
Sequence-unique set of PDB chains. The EVA server [a server automatically evaluating structure prediction methods (48,49)] continuously maintains a set of sequence-unique PDB chains: no pair in that set exceeds a sequence similarity above an HSSP value of 0 (50) (set available at ftp://cubic.bioc.columbia.edu/pub/eva/unique_list.txt). The HSSP curve relates alignment length to pairwise sequence identity or similarity (50,51); for alignments of 100 residues, HSSP = 0 corresponds to 33% pairwise sequence identity, for alignments longer than 250 residues to ∼20%. The version that we used (December 6, 2003) contained 2773 sequence-unique PDB chains.
Structural domains assigned from 3D structures. Structural domains were extracted automatically by the program PrISM (52), and taken from the more or less expert-based assignments from SCOP (15) and CATH (16). Note that we developed separate prediction methods for each assignment scheme, and that we also evaluated performance based on the different assignments. Domain linkers were defined as the regions between two continuous PrISM/CATH/SCOP domains.
Cross-validation of networks. We randomly selected ∼9% of our dataset as internal control sets to optimize free parameters (e.g. number of hidden units, types of input, stop of training), and the rest for training and testing. Note that we refer to the testing set as predictions for proteins pretended to be unknown, i.e. for which we did use 3D data only to evaluate, not to develop. While we used the same protocol to build the training, validation and testing sets for CATH (set_CATH with 1300 proteins) and SCOP (set_SCOP with 2127 proteins) as for PrISM (set_PrISM, 1918 proteins), the actual sets differed slightly since these expert-based databases do not have assignments for all proteins in PDB. For set_PrISM, we excluded NMR and low-resolution X-ray structures (>3.0 Å) from our sequence-unique set of PDB chains. Finally, we compiled the overlap between SCOP and CATH in a set used to establish how well the combination of both methods works (set_SC with 1187 proteins). Since by construction of our dataset, no pair of chains in the set had significant levels of sequence similarity, we could simply split these high-resolution structures at random into training and testing sets. We used 10 splits such that each protein was used for testing exactly once.
Feature extraction and prediction method
Sequence features. As the input to the neural networks we used amino acid composition (averaged over an alignment profile), predicted secondary structure and solvent accessibility (details below). We obtained multiple sequence alignments by searching with PSI-BLAST (30) against all known sequences contained in SWISS-PROT (27), TrEMBL (27) and PDB (53). All hits below a PSI-BLAST E-value of 10−3 were subsequently filtered (50) and included in the sequence profile. The filtered PSI-BLAST alignments were used as input for PROFsec (B. Rost, submitted for publication) for secondary structure prediction, and PROFacc (B. Rost, submitted for publication) for solvent accessibility prediction.
Neural network architecture. We trained a three-layer feed forward artificial neural network using the standard back-propagation algorithm with momentum term (54,55). Since our dataset was rather small, the major problem was to optimally choose input features that were as informative for the prediction task as possible. We accomplished this through the validation set in the following way: first, we trained a network with a group of simple features (evolutionary profiles in window); then we added features grouped by intuition and monitored the increase in performance (on the validation set). Groups of features that yielded no improvement were rejected, all others included and the next group was tested. Note that this strategy by no means guaranteed to find the optimal set of features and that we did not use any information from the test set in order to avoid over-fitting. The final network used 57 input nodes from consecutive sequence segments of 5 residues [shifted through the protein as standard for other prediction methods (56–60)]. For each residue in this local sequence window, three nodes encoded secondary structure (raw output scores from PROFsec representing helix, strand and loop), one node encoded solvent accessibility (PROFacc prediction of relative solvent accessibility) and one node represented the sequence conservation [conservation weight in HSSP files (51,61)]. We also added features for the entire five-residue segment, namely the difference in secondary structure content between the flanking regions of the segment (eight nodes), the difference in solvent accessibility (four nodes), the position of the sequence segment with regards to the N- and C-termini (eight nodes), the flexibility index (62) averaged over the entire five-residue segment and that for the central residues (two nodes), and the amino acid composition in the profile for the entire window (six nodes for residues {P, H, D, Y, V, C}, which are most different between linkers and domains). Finally, we added global nodes representing the length of the protein (four nodes). The hidden layer of the network had three nodes, and the output layer had two nodes: one coding for ‘domain-boundary’ the other for ‘not-domain-boundary’.
Post-processing neural network output. For the combination method (set_SC), we took the more reliable score from the SCOP network and the CATH network as the network output. The raw output from the neural networks had many local peaks. Thus, we had to filter these raw outputs. Towards this end we employed the following five steps. (i) We determined the threshold for the domain boundary network output unit dynamically according to the length (L) of the protein and to the distribution of raw output values for all residues in that protein. Specifically, we compiled the 92nd percentile of the raw output T1 and set the threshold T to
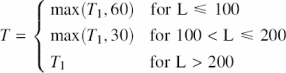
All residues with raw output values above T were considered as domain boundaries. (ii) Next, we smoothed the raw output through averaging over windows of eleven consecutive residues. (iii) For each nine-residue window averages, we assigned the central residue as ‘domain boundary’ if three out of the three residues were predicted as ‘domain-boundary’. (iv) We removed ‘isolated’ predictions, i.e. those for which ‘domain-boundaries’ were not predicted for at least three consecutive residues. (v) Finally, we had to remove conflicts. In particular, we simply predicted a domain boundary exactly in the middle between two adjacent (central residues less than 30 residues apart) boundary predictions. Note that all parameters for these filters were developed using the validation set only.
Measuring performance
All estimates for performance were obtained from the test sets in 10-fold cross-validation. In particular, we never showed any data from the training sets. We evaluated performance in two different ways. First, we monitored the percentage of proteins for which the number of domains was predicted correctly. We separated this number into different ‘classes’ of proteins, namely those with Mobs observed domains and showed the percentages of those that were predicted with Mprd domains (note Mobs = Mprd are those correctly predicted; Mobs < Mprd mark over-predictions and Mobs > Mprd under-predictions). For domain boundary predictions, we measured the distance between predicted and observed domain boundaries as the distance between the central residues of the predicted and observed boundary, and calculated the percentage of correctly predicted domain boundaries.
Random control predictions. Given a set of proteins, the number of domains was predicted at random for each protein according to the composition of domain numbers in a particular set. For instance, ∼68% of the proteins were predicted as single-domain proteins, 24% as two-domain proteins and 7% as three-domain. The success rate of random prediction of the number of domains was obtained for the whole dataset, then the test was repeated 100 times, and the average accuracy over all random choices was reported. In order to measure the random background for the domain boundary prediction, we chose Mobs−1 domain boundaries at random in each protein with Mobs domains. Again, we repeated this random cut 100 times and reported the average over all 100 randomizations.
RESULTS
Correctly predicted number of domains for 38% of the multi-domain proteins. The most coarse-grained task of methods detecting domain boundaries (linkers) is to correctly predict the number of domains. We have reliable information about structural domains only for proteins of known structure. Even for these the numbers assigned by different methods and experts differed significantly. For instance, for our cross-validation set, only 93% of the assignments agreed between CATH and SCOP. For the same set, the automatic domain assignment method PrISM agreed for 80 and 73% of all proteins with CATH and SCOP, respectively. Since Protein Data Bank (PDB) is highly biased toward single-domain proteins (depending on the assignment method 60–80%), this number was biased towards more simple cases: only 67–69% of the multi-domain assignments agreed between SCOP and CATH; PrISM agreed with 54 and 44% of the assignments by CATH and SCOP, respectively. While the agreement between automatic and expert-based assignments provided an upper limit for predictions, the lower limit was given by random predictions that correctly identified 8% of all multi-domain proteins. Our prediction method, CHOPnet, correctly predicted the number for ∼38% for all multi-domain proteins. While this was much closer to structure-based assignments than to random predictions (Figure 1A), our method was seemingly outperformed by random for single-domain proteins. When comparing how often CHOPnet over- and under-predicted domains, it appeared that the network leaned toward over-prediction (grey bars in Figure 1B). However, this figure was again biased by the over-representation of single-domain proteins in our dataset: for most multi-domain proteins, the method, in fact, under-predicted domain boundaries (Table 1 and stippled bars in Figure 1B).
Figure 1.

Predicting the number of domains. (A) We compared the success in predicting the number of domains to random predictions. Our method was significantly better than random for all multi-domain proteins. (B) For 69% of all and for 38% of all multi-domain proteins the number of domains predicted by CHOPnet and observed by CATH were identical. Overall, CHOPnet appeared to slightly over-predict the number of domains (solid bars on right higher than those on left). However, the data for multi-domain proteins revealed that it actually under-predicted significantly, in particular the number was under-predicted more often than it was predicted correctly (stippled bar at ‘−1’ higher than that at ‘0’).
Table 1. Domain number prediction accuracy (set_CATH)a.
| Observation (CATH assignment) |
Prediction |
|
|
|---|---|---|---|
| 1 | 2 | 3 | |
| 1 | 73 ± 4 | 25 ± 5 | 2 ± 2 |
| 2 | 54 ± 8 | 41 ± 7 | 5 ± 4 |
| 3 | 50 ± 12 | 29 ± 6 | 21 ± 12 |
aPercentage of proteins observed with N (1–3) domains (rows) and predicted with M domains (column). The diagonal highlights correct predictions in bold. For example, 41% of the two-domain proteins were correctly predicted as such, for 54% the domain boundary was missed incorrectly predicting single domains and 5% were over-predicted to contain three instead of two domains.
Performance overall and for single domains not informative. We found estimates for the performance on single-domain proteins so sensitive to the particular assignment method that they appeared almost meaningless: our basic methods varied between 49 and 63% (Table 2 third column; datasets in second column). Random predictions also varied considerably (63–88%, values in brackets in second column). This problem also over-shadowed estimates for the overall performance, since for all PDB-derived datasets single-domain proteins dominated. We trained different methods on different assignments and observed that the method trained on standard-of-truth X performed best if and only if evaluated on method X (Table 2; note that the networks trained on SCOP to some extent constituted an exception). The method combining SCOP and CATH assignments appeared overall slightly superior for all datasets. Therefore, we chose this one as our final prediction method CHOPnet.
Table 2. Different methods and different gold standardsa.
| Assignment used for training | Data set used for evaluation | Correctly predicted one-domain (random) | Correctly predicted two-domains (random) | Linker position for two-domain proteins (equal split) |
|---|---|---|---|---|
| SCOP | set_SCOP | 49% (83%) | 49% (15%) | 50% (42%) |
| SCOP | set_CATH | 69% (88%) | 44% (11%) | 40% (27%) |
| SCOP | set_Jones | 42% (76%) | 58% (18%) | 53% (49%) |
| CATH | set_SCOP | 58% (88%) | 54% (10%) | 49% (52%) |
| CATH | set_CATH | 52% (88%) | 53% (14%) | 58% (47%) |
| CATH | set_Jones | 42% (76%) | 59% (18%) | 53% (53%) |
| SCOP+CATH | set_SCOP | 73% (88%) | 40% (10%) | 46%(44%) |
| SCOP+CATH | set_CATH | 73% (88%) | 41% (10%) | 47% (39%) |
| SCOP+CATH | set_Jones | 60% (76%) | 50% (18%) | 51% (43%) |
| PrISM | set_PrISM | 63% (63%) | 55% (24%) | 48% (30%) |
| PrISM | set_Jones | 52% (76%) | 52% (18%) | 42% (53%) |
| DomSSEAb | set_Jones | 82% (76%) | 46% (17%) | 49% (49%) |
| DGS-Mb | set_Jones | 100% (76%) | 0% (17%) | 46% (49%) |
aThe leftmost column distinguishes different versions of CHOPnet (trained on SCOP, CATH, SCOP+CATH and PrISM), and previously published methods [DomSSEA (47) and DGS-M (43)]. The second column identifies different test sets [set_Jones taken from (47)], the third and fourth the accuracy in predicting the number of domains (values in brackets give random predictions), and the last column the percentage of linker regions for two-domain proteins predicted within 20 residues of the observation (values in brackets mark performance of equal split).
bAll values taken directly from a previous publication (47).
Domain boundaries predicted better than random. For all proteins that were assigned two domains by CHOPnet, we analysed how close the predicted and observed linker regions were (Figure 2). Using CATH as predictions for SCOP observations (Figure 2A) and SCOP as predictions for CATH observations (Figure 2B), both found almost all linkers within ±20 residues. In contrast, the automatic PrISM assignments agreed only for ∼80% of the linkers with SCOP (Figure 2A) and CATH (Figure 2B). In practice, this level constitutes an upper limit for methods predicting domain boundaries in the absence of structure. Conversely, the lower limit is provided by a random prediction. Following the work of David Jones' group (2), we tested two different random predictions, namely a ‘random split’ of each two-domain protein into two fragments, and an ‘equal split’ introducing a domain boundary in the middle. We then monitored how often a domain boundary was predicted closer than ±D residues from the observed boundary (Figure 2). Note that the absolute value of the performance of ‘random split’ and ‘equal split’ depends on the number of correct predictions and the particular assignment method (therefore the values differ slightly for the different data in Figure 2). At ±20 residues, ‘random split’ correctly predicted ∼16% and ‘equal split’ ∼39-44% of the boundaries. CHOPnet correctly assigned 46–51% of the boundaries at ±20 residues (Figure 2). Thus, our method consistently outperformed equal split.
Figure 2.

Accuracy of predicting the precise domain boundary. For all two-domain proteins for which the domain number was predicted correctly, the distance between predicted and observed domain boundaries was measured, and the accuracy of the prediction was calculated for thresholds ranging from 0–20 residues and compared with that of two trivial predictions: ‘random split’ and ‘equal split’. The left panel (A) gives the results for using SCOP assignments as standard-of-truth. The lines for CATH and PrISM mark the agreement between SCOP and these two methods that are based on known 3D structures. The two panels on the right (B and C) are based on using CATH assignments as standard-of-truth. Panels (A and B) were based on the cross-validation section common between CATH and SCOP (set_SC), while (C) was based on the test set used to evaluate DomSSEA (47).
Correct number and location of boundaries for 19% of two-domain proteins. If assessed on all proteins in our dataset, our method correctly predicted both the number of domains and the boundary within ±20 residues for 19% of all two-domain proteins (Table 3). On average, the distributions of domain lengths from CHOPnet and from CATH (Figure 3), SCOP, and PrISM (data not shown) were surprisingly similar. The major difference was that CHOPnet slightly over-predicted short domains.
Table 3. Overall prediction of domain number and boundary (±20 residues).
| Number of domains observed | Percentage of proteins with correctly predicted number of domains | Percentage of proteins with correctly predicted number and location of domains (±20 residues) |
|---|---|---|
| 1 | 73 | 73 |
| 2 | 41 | 19 |
| 3 | 21 | 0 |
Figure 3.

Length distribution of predicted and observed domains. Overall, the length of structural domains predicted by CHOPnet (black line with open circles) was rather similar to that assigned from 3D structures by CATH (light grey with plus signs). We compiled the distribution with 10 residue bins. The obvious noise in the distributions revealed the limited size of our datasets (1187 PDB chains).
DISCUSSION
Domain assignment is more accurate for proteins with fewer domains. Our method predicted the number of protein domains much better for single-domain than for multi-domain proteins (Figure 1, Table 1). Nevertheless, two-domain proteins were identified much more accurately than could have been done through simple ‘guesses by length’ or random predictions. Furthermore, for about 50% of all boundaries in two-domain proteins, our predictions fall within ±20 residues of the boundaries assigned from the known 3D structures (Figure 2). The combination of both errors (wrong number and wrong location) dropped the percentage of three-domain proteins for which both the number and location were correctly predicted to 0% (Table 3). Since there are supposedly two to five times more single-domain proteins in PDB than in nature (42), the estimates for multi-domain proteins are more likely to reflect the sustained performance of CHOPnet. Thus, in context of chopping entire proteomes, we need methods that reduce the number of unknown linkers by pre-processing the sequences. Candidates for such pre-processing are methods based on homology to known structure domains or reliably annotated sequence domains. We previously developed CHOP to dissect proteins into structural domain-like fragments according to sequence homology known domains (42,63). CHOP was able to process 69% of the proteins in 62 entirely sequenced proteomes. Over two-thirds of these proteins have more than one domain. Single-domain proteins are clearly over-represented in the 31% of the proteins for which we could not chop due to missing homology information. Although CHOP may have identified a considerable fraction of the structural domains in these proteomes, it undoubtedly missed many domain boundaries. We hope that CHOPnet will allow the reliable dissection of these remaining multi-domain fragments. CHOPnet becomes increasingly unreliable for proteins with more domains. However, many of the fragments with domain boundaries undetected by CHOP are likely to have fewer domains, i.e. instead of having to break down the entire protein, CHOPnet may only have to identify one boundary. We have also begun to experiment with a combination of CHOPnet and CHOP hoping that the combination of both will increase the reliability of chopping.
Major problems: limited and contradictory data. In our final network, we encoded various sequence and structure features into 57 input nodes. Presumably, there are other ways of encoding the input information that better represent the necessary information. For example, the information from multiple sequence alignments was encoded only in 11 nodes over the sequence window of five residues (five for sequence weight, six for amino acid composition of the sequence profile). A 20-dimensional vector for the full information contained in substitution profiles of the alignment might perform more accurately. However, we failed to successfully explore more input information due to the rather limited size of the dataset. Therefore, we assume that our method will become more accurate when high-resolution structures for more multi-domain proteins will become available. Another major limitation for our method was the inconsistency of domain assignment methods. If expert-based annotations agree for only two-thirds of the multi-domain proteins, then the implicit assumption for developing a prediction method, namely that there is a bio–physical reason for domain formation is partially flawed. This strongly impacts the ability of methods such as neural networks in extracting such rules. Our differently trained networks all performed best on the assignment scheme used as standard-of-truth. Thus, the networks extracted some of the rules underlying the different assignment programs; there is no objective means of telling which one is better. While all PrISM domains are consecutive in sequence, 5% of the domains in SCOP and 15% of those in CATH are not consecutive in sequence. Our neural networks largely failed on most of these non-consecutive domains, no matter on which assignment schema we trained.
CHOPnet appeared to perform favourably in comparison to other methods. Although we evaluated the performance of our method through measures previously established, it was not straightforward to meaningfully compare our estimates to those published by others. Previous results were based on different datasets, benchmark standards, cross-validation and evaluation procedures. For example, a previous study using neural networks to predict domain boundaries reported a coverage of 36% at 58% accuracy for all predicted domain boundaries (64). The dataset came from 99 domain linkers defined by SCOP in 74 multi-domain proteins, and the sequence redundancy was removed at BLAST E-values of 10−70 for training and 10−20 for evaluation. At such high levels of sequence similarity between the training and the testing set, we can predict boundaries through sequence homology at higher levels of accuracy/coverage. Since the neural network used only sequence information as input it may well be that it simply found all boundaries with homology to training proteins and missed all others. However, the paper did not address this possibility. The ‘Domain Guess by Size’ [DGS (43)] was reported to reach an accuracy of 50% for proteins shorter than 400 residues, given the gold standard from MMDB (65). This was surprisingly accurate given that the only input to the program was the protein length. However, for this value, predictions were considered as correct, if one of the top 10 predictions was within ±20 residues of the observed domain boundary. Accuracy was much lower if only the top hit was assessed: when we applied DGS to our dataset, all proteins in that set were predicted as single-domain proteins by the top-ranking prediction [data not shown; note: a similar result was previously observed (47)]. SnapDRAGON (44) was evaluated with similar criteria for separating training and testing set as applied for this work. Since the dataset differed, comparing the numerical values has rather limited validity. Nevertheless, the numerical results were the following: SnapDRAGON correctly identified 47%, CHOPnet ∼70% of the single-domain proteins. Comparing the numbers for the success in correctly identifying the boundary region was even more difficult as was illustrated by the differences for the ‘equal split’ scenario. ‘Equal split’ accuracy was 30% for SnapDRAGON's data set (all multi-domain proteins in continuous set), 50% for DomSSEA's set (two-domain proteins), and 27–53% for our different sets (two-domain proteins). One possible base for comparison could then be the difference between the accuracy of the actual prediction method and the ‘equal split’ background. SnapDRAGON was nine percentage points more accurate than ‘equal split’ for all multi-domain proteins (39 versus 30%). Since the accuracy of ‘equal split’ is more likely to drop for proteins with more than two domains, it is not unlikely that the difference between prediction and random would be lower than this for two-domain proteins. Depending on the dataset, CHOPnet was four to eight percentage points better than equal split for two-domain proteins. The only more or less straightforward comparisons were those to DomSSEA as we tested on the same dataset (in cross-validation mode, Methods) and to DGS (43) as that was tested carefully on the same dataset by the Jones group. Both DomSSEA and DGS clearly outperformed CHOPnet in correctly identifying single-domain proteins as such (Table 2). DomSSEA was slightly less accurate for multi-domain proteins. In terms of correctly identifying the linker region, DGS was outperformed by ‘equal split’; DomSSEA performed at par with ‘equal split’, and CHOPnet about eight percentage points better (Table 2). While we could read these data to mean that all methods fail to consistently capture important information, in reality the vast majority of domains in entirely sequenced proteomes appear to extend over 70–130 residues (13,42). In other words, most domains have similar lengths. Thus, ‘equal split’ is not really that uneducated a guess as it might seem. In fact, CHOPnet is slightly closer to the ‘best we can do automatically’ (PrISM assignments in Figure 2A and B) than to ‘random splits’ (open triangles in Figure 2). Note that the Jones group received their values for DomSSEA and DGS under the explicit assumption that the proteins contained two domains (exclusive alignment against two-domain proteins). In contrast, we applied CHOPnet pretending not to know the number and evaluated then. It is unclear how this difference would affect the performance of DomSSEA and DGS; however, using the explicit knowledge of the number of domains did not improve CHOPnet markedly.
Target selection in structural genomics needs predictions of structural domains. The largest funding for structural genomics worldwide originates from the Science & Technology Agency in Japan and is concentrated at the RIKEN Structural Genomics Initiative (RSGI) at the Institute of Physical and Chemical Research (66). The second largest funding originates from the National Institute of General Medical Sciences (NIGMS) at the National Institute of Health (NIH) in the USA. The NIGMS protein structure initiative (PSI) formulates as one of the goals of structural genomics ‘to determine representative structures from all protein families’ (67). The seemingly simple task for computational biology then is to cluster all proteins into sequence–structure families, such that one experimental structure per family optimally covers protein space. The basic concepts to solve this task were laid out before structural genomics started (68–74). Meanwhile structural genomics has proven that we have to revise many of the initial assumptions. For example, we cannot adequately realize the concept of one representative structure per sequence–structure family without dissecting proteins into structural domains before we cluster into such families (42,75,76). Furthermore, while about 3000 sequence–structure families allow the prediction of 3D structure through comparative modelling for about one-third of all residues in 62 entirely sequenced proteomes (13,42,77), we need to experimentally determine representative structures for almost 10 times more families to double this coverage (J. Liu and B. Rost, unpublished data; C.A. Orengo, unpublished data). Another surprising result is that consortia have began running out of high-hanging fruits, in particular, we have to begin chopping proteins into domain-like fragments to further large-scale experimental efforts. Homology-based methods alone will not suffice. Thus, we need de novo prediction methods. The good news is that structural genomics efforts will provide large-scale experimental means of refining such methods by trial and error.
CONCLUSIONS
We introduced CHOPnet, a novel method for predicting structural domain boundaries in the absence of homology to structurally known domains. The good news is that CHOPnet appeared superior to previously published methods and it was significantly better than random by all measures that we investigated. For example, for ∼50% of the two-domain proteins that were correctly predicted as having two domains by CHOPnet, the boundary predictions were within ±20 residues of the boundaries assigned by PrISM from the 3D structures (Figure 2). The bad news is that CHOPnet correctly identified only 41% of the two-domain proteins as such (Table 1), and for only 21% of the two-domain proteins were both the number and the location of the boundaries correct (Table 3). Undoubtedly, these numbers were rather low. However, no alternative method appears available that performs significantly better. In particular, the expert-curated solution to grouping proteins into fragment-based families, Pfam-A bases its fragmentation on functional rather than structural criteria. Previously, we found that Pfam-A agrees in the number of domains assigned with PrISM for ∼41% of all proteins (48). This number is similar to that for CHOPnet (Table 1). And even if Pfam-A were more accurate in identifying structural domains, a considerable fraction of putative targets for structural genomics would remain untouched by Pfam-based fragment parsing. The crucial question to be answered experimentally is to which extent predictions from methods such as CHOPnet will help advancing large-scale experimental efforts toward structure determination. Structural genomics consortia will tell.
Acknowledgments
ACKNOWLEDGEMENTS
Thanks to Dariusz Przybylski, Rajesh Nair and Yanay Ofran (all from Columbia University) for helpful comments. Thanks to Russell Marsden and David Jones (both from University College London) for providing their original dataset for cross-reference. We are grateful to both anonymous reviewers for their constructive comments that helped to improve the manuscript considerably! Thanks also to our experimental colleagues at the Northeast Structural Genomics Consortium (NESG) for their advice and strong support of our project. In particular, thanks to Guy Montelione (Rutgers) for his invaluable optimism in leading the NESG team and to the team around Tom Acton (Rutgers) who will test our domain predictions. Thanks also to all those who deposit their experimental data in public databases, and to those who maintain these databases. The work of J.L. and B.R. was supported by the grants 1-P50-GM62413-01, RO1-GM63029-01, R01-GM64633-01 and R01-LM07329-01 from the National Institutes of Health (NIH).
REFERENCES
- 1.The genome international sequencing consortium. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 2.Venter J.C., Adams,M.D., Myers,E.W., Li,P.W., Mural,R.J., Sutton,G.G., Smith,H.O., Yandell,M., Evans,C.A., Holt,R.A. et al. (2001) The human genome. Science, 291, 1304–1351. [DOI] [PubMed] [Google Scholar]
- 3.The C. elegans Sequencing Consortium (1998) Genome sequence of the nematode C. elegans: a platform for investigating biology. Science, 282, 2012–2018. [DOI] [PubMed] [Google Scholar]
- 4.Adams M.D., Celniker,S.E., Holt,R.A., Evans,C.A., Gocayne,J.D., Amanatides,P.G., Scherer,S.E., Li,P.W., Hoskins,R.A., Galle,R.F. et al. (2000) The genome sequence of Drosophila melanogaster. Science, 287, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 5.Dujon B. (1996) The yeast genome project: what did we learn? TIGS, 12, 263–270. [DOI] [PubMed] [Google Scholar]
- 6.Goffeau A., Barrell,B.G., Bussey,H., Davis,R.W., Dujon,B., Feldmann,H., Galibert,F., Hoheisel,J.D., Jacq,C., Johnston,M. et al. (1996) Life with 6000 genes. Science, 274, 546–567. [DOI] [PubMed] [Google Scholar]
- 7.Rost B. (2002) Did evolution leap to create the protein universe? Curr. Opin. Struct. Biol., 12, 409–416. [DOI] [PubMed] [Google Scholar]
- 8.Abrahams J.P., Leslie,A.G., Lutter,R. and Walker,J.E. (1994) Structure at 2.8 Å resolution of F1-ATPase from bovine heart mitochondria. Nature, 370, 621–628. [DOI] [PubMed] [Google Scholar]
- 9.Brunger A.T. (2001) Structure of proteins involved in synaptic vesicle fusion in neurons. Annu. Rev. Biophys. Biomol. Struct., 30, 157–171. [DOI] [PubMed] [Google Scholar]
- 10.Jaennicke R. (1987) Folding and association of proteins. Prog. Biophys. Mol. Biol., 49, 117–237. [DOI] [PubMed] [Google Scholar]
- 11.Holm L. and Sander,C. (1994) Parser for protein folding units. Proteins, 19, 256–268. [DOI] [PubMed] [Google Scholar]
- 12.Teichmann S.A., Rison,S.C., Thornton,J.M., Riley,M., Gough,J. and Chothia,C. (2001) Small-molecule metabolism: an enzyme mosaic. TIBTECH, 19, 482–486. [DOI] [PubMed] [Google Scholar]
- 13.Liu J., Acton,T., Goldsmith,S., Honig,B., Montelione,G.T. and Rost,B. (2004) Automatic target selection for structural genomics on eukaryotes. Proteins, 56, 188–200. [DOI] [PubMed] [Google Scholar]
- 14.Dueber J.E., Yeh,B.J., Chak,K. and Lim,W.A. (2003) Reprogramming control of an allosteric signaling switch through modular recombination. Science, 301, 1904–1908. [DOI] [PubMed] [Google Scholar]
- 15.Lo Conte L., Brenner,S.E., Hubbard,T.J., Chothia,C. and Murzin,A.G. (2002) SCOP database in 2002: refinements accommodate structural genomics. Nucleic Acids Res., 30, 264–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orengo C.A., Bray,J.E., Buchan,D.W., Harrison,A., Lee,D., Pearl,F.M., Sillitoe,I., Todd,A.E. and Thornton,J.M. (2002) The CATH protein family database: a resource for structural and functional annotation of genomes. Proteomics, 2, 11–21. [PubMed] [Google Scholar]
- 17.Holm L. and Sander,C. (1998) Dictionary of recurrent domains in protein structures. Proteins, 33, 88–96. [DOI] [PubMed] [Google Scholar]
- 18.Marchler-Bauer A., Addess,K.J., Chappey,C., Geer,L., Madej,T., Matsuo,Y., Wang,Y. and Bryant,S.H. (1999) MMDB: Entrez's 3D structure database. Nucleic Acids Res., 27, 240–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang A.S. and Honig,B. (2000) An integrated approach to the analysis and modeling of protein sequences and structures. III. A comparative study of sequence conservation in protein structural families using multiple structural alignments. J. Mol. Biol., 301, 691–711. [DOI] [PubMed] [Google Scholar]
- 20.Jones S., Stewart,M., Michie,A.D., Swindells,M.B., Orengo,C.A. and Thornton,J.M. (1998) Domain assignment for protein structures using a consensus approach: characterization and analysis. Protein Sci., 7, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Busetta B. and Barrans,Y. (1984) The prediction of protein domains. Biochim. Biophys. Acta, 790, 117–124. [DOI] [PubMed] [Google Scholar]
- 22.Vonderviszt F. and Simon,I. (1986) A possible way for prediction of domain boundaries in globular proteins from amino acid sequence. Biochem. Biophys. Res. Commun., 139, 11–17. [DOI] [PubMed] [Google Scholar]
- 23.Kikuchi T., Nemethy,G. and Scheraga,H.A. (1988) Prediction of the location of structural domains in globular proteins. J. Protein Chem., 7, 427–471. [DOI] [PubMed] [Google Scholar]
- 24.Corpet F., Servant,F., Gouzy,J. and Kahn,D. (2000) ProDom and ProDom-CG: tools for protein domain analysis and whole genome comparisons. Nucleic Acids Res., 28, 267–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Servant F., Bru,C., Carrere,S., Courcelle,E., Gouzy,J., Peyruc,D. and Kahn,D. (2002) ProDom: automated clustering of homologous domains. Brief Bioinform., 3, 246–251. [DOI] [PubMed] [Google Scholar]
- 26.Corpet F., Gouzy,F. and Kahn,D. (1998) The ProDom database of protein domain families. Nucleic Acids Res., 26, 323–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bairoch A. and Apweiler,R. (2000) The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Res., 28, 45–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gracy J. and Argos,P. (1998) DOMO: a new database of aligned protein domains. TIBS, 23, 495–497. [DOI] [PubMed] [Google Scholar]
- 29.George R.A. and Heringa,J. (2002) Protein domain identification and improved sequence similarity searching using PSI-BLAST. Proteins, 48, 672–681. [DOI] [PubMed] [Google Scholar]
- 30.Altschul S.F., Madden,T.L., Schaffer,A.A., Zhang,J., Zhang,Z., Miller,W. and Lipman,D.J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res., 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bateman A., Birney,E., Cerruti,L., Durbin,R., Etwiller,L., Eddy,S.R., Griffiths-Jones,S., Howe,K.L., Marshall,M. and Sonnhammer,E.L. (2002) The Pfam protein families database. Nucleic Acids Res., 30, 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Letunic I., Goodstadt,L., Dickens,N.J., Doerks,T., Schultz,J., Mott,R., Ciccarelli,F., Copley,R.R., Ponting,C.P. and Bork,P. (2002) Recent improvements to the SMART domain-based sequence annotation resource. Nucleic Acids Res., 30, 242–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haft D.H., Selengut,J.D. and White,O. (2003) The TIGRFAMs database of protein families. Nucleic Acids Res., 31, 371–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tatusov R.L., Natale,D.A., Garkavtsev,I.V., Tatusova,T.A., Shankavaram,U.T., Rao,B.S., Kiryutin,B., Galperin,M.Y., Fedorova,N.D. and Koonin,E.V. (2001) The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res., 29, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vlahovicek K., Murvai,J., Barta,E. and Pongor,S. (2002) The SBASE protein domain library, release 9.0: an online resource for protein domain identification. Nucleic Acids Res., 30, 273–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchler-Bauer A., Anderson,J.B., DeWeese-Scott,C., Fedorova,N.D., Geer,L.Y., He,S., Hurwitz,D.I., Jackson,J.D., Jacobs,A.R., Lanczycki,C.J. et al. (2003) CDD: a curated Entrez database of conserved domain alignments. Nucleic Acids Res., 31, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gough J., Karplus,K., Hughey,R. and Chothia,C. (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J. Mol. Biol., 313, 903–919. [DOI] [PubMed] [Google Scholar]
- 38.Shepherd A.J., Martin,N.J., Johnson,R.G., Kellam,P. and Orengo,C.A. (2002) PFDB: a generic protein family database integrating the CATH domain structure database with sequence based protein family resources. Bioinformatics, 18, 1666–1672. [DOI] [PubMed] [Google Scholar]
- 39.Buchan D.W., Rison,S.C., Bray,J.E., Lee,D., Pearl,F., Thornton,J.M. and Orengo,C.A. (2003) Gene3D: structural assignments for the biologist and bioinformaticist alike. Nucleic Acids Res., 31, 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulikowski C.A., Muchnik,I., Yun,H.J., Dayanik,A.A., Zhang,D., Song,Y. and Montelione,G.T. (2001) Protein structural domain parsing by consensus reasoning over multiple knowledge sources and methods. Medinfo, 10, 965–969. [PubMed] [Google Scholar]
- 41.Sauder J.M., Arthur,J.W. and Dunbrack,R.L.,Jr (2000) Large-scale comparison of protein sequence alignment algorithms with structure alignments. Proteins, 40, 6–22. [DOI] [PubMed] [Google Scholar]
- 42.Liu J. and Rost,B. (2004) CHOP proteins into structural domain-like fragments. Proteins, 55, 678–688. [DOI] [PubMed] [Google Scholar]
- 43.Wheelan S.J., Marchler-Bauer,A. and Bryant,S.H. (2000) Domain size distributions can predict domain boundaries. Bioinformatics, 16, 613–618. [DOI] [PubMed] [Google Scholar]
- 44.George R.A. and Heringa,J. (2002) SnapDRAGON: a method to delineate protein structural domains from sequence data. J. Mol. Biol., 316, 839–851. [DOI] [PubMed] [Google Scholar]
- 45.Aszodi A., Gradwell,M.J. and Taylor,W.R. (1995) Global fold determination from a small number of distance restraints. J. Mol. Biol., 251, 308–326. [DOI] [PubMed] [Google Scholar]
- 46.Taylor W.R. (1999) Protein structural domain identification. Protein Eng., 12, 203–216. [DOI] [PubMed] [Google Scholar]
- 47.Marsden R.L., McGuffin,L.J. and Jones,D.T. (2002) Rapid protein domain assignment from amino acid sequence using predicted secondary structure. Protein Sci., 11, 2814–2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eyrich V., Martí-Renom,M.A., Przybylski,D., Fiser,A., Pazos,F., Valencia,A., Sali,A. and Rost,B. (2001) EVA: continuous automatic evaluation of protein structure prediction servers. Bioinformatics, 17, 1242–1243. [DOI] [PubMed] [Google Scholar]
- 49.Koh I.Y.Y., Eyrich,V.A., Marti-Renom,M.A., Przybylski,D., Madhusudhan,M.S., Narayanan,E., Graña,O., Valencia,A., Sali,A. and Rost,B. (2003) EVA: evaluation of protein structure prediction servers. Nucleic Acids Res., 31, 3311–3315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rost B. (1999) Twilight zone of protein sequence alignments. Protein Eng., 12, 85–94. [DOI] [PubMed] [Google Scholar]
- 51.Sander C. and Schneider,R. (1991) Database of homology-derived structures and the structural meaning of sequence alignment. Proteins, 9, 56–68. [DOI] [PubMed] [Google Scholar]
- 52.Yang A.S. and Honig,B. (2000) An integrated approach to the analysis and modeling of protein sequences and structures. II. On the relationship between sequence and structural similarity for proteins that are not obviously related in sequence. J. Mol. Biol., 301, 679–689. [DOI] [PubMed] [Google Scholar]
- 53.Berman H.M., Westbrook,J., Feng,Z., Gillliland,G., Bhat,T.N., Weissig,H., Shindyalov,I.N. and Bourne,P.E. (2000) The Protein Data Bank. Nucleic Acids Res., 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rumelhart D.E., Hinton,G.E. and Williams,R.J. (1986) In Rumelhart,D.E. and McClelland,J.L. (eds), Parallel Distributed Processing. MIT Press, Cambridge, UK, Vol. 1, pp. 318–362. [Google Scholar]
- 55.Rost B. and Sander,C. (1993) Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol., 232, 584–599. [DOI] [PubMed] [Google Scholar]
- 56.Rost B. and Sander,C. (1994) Combining evolutionary information and neural networks to predict protein secondary structure. Proteins, 19, 55–72. [DOI] [PubMed] [Google Scholar]
- 57.Rost B. and Sander,C. (1994) Conservation and prediction of solvent accessibility in protein families. Proteins, 20, 216–226. [DOI] [PubMed] [Google Scholar]
- 58.Rost B. (1996) PHD: predicting one-dimensional protein structure by profile based neural networks. Methods Enzymol., 266, 525–539. [DOI] [PubMed] [Google Scholar]
- 59.Rost B., Casadio,R. and Fariselli,P. (1996) Topology prediction for helical transmembrane proteins at 86% accuracy. Protein Sci., 5, 1704–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ofran Y. and Rost,B. (2003) Predict protein-protein interaction sites from local sequence information. FEBS Lett., 544, 236–239. [DOI] [PubMed] [Google Scholar]
- 61.Rost B., Sander,C. and Schneider,R. (1994) PHD–an automatic server for protein secondary structure prediction. CABIOS, 10, 53–60. [DOI] [PubMed] [Google Scholar]
- 62.Vihinen M., Torkkila,E. and Riikonen,P. (1994) Accuracy of protein flexibility predictions. Proteins, 19, 141–149. [DOI] [PubMed] [Google Scholar]
- 63.Liu J. and Rost,B. (2004) CHOP: parsing proteins into structural domains. Nucleic Acids Res., 32, W569–W571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Miyazaki S., Kuroda,Y. and Yokoyama,S. (2002) Characterization and prediction of linker sequences of multi-domain proteins by a neural network. J. Struct. Funct. Gen., 2, 37–51. [DOI] [PubMed] [Google Scholar]
- 65.Wheeler D.L., Church,D.M., Edgar,R., Federhen,S., Helmberg,W., Madden,T.L., Pontius,J.U., Schuler,G.D., Schriml,L.M., Sequeira,E. et al. (2004) Database resources of the National Center for Biotechnology Information: update. Nucleic Acids Res., 32, D35–D40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yokoyama S., Hirota,H., Kigawa,T., Yabuki,T., Shirouzu,M., Terada,T., Ito,Y., Matsuo,Y., Kuroda,Y., Nishimura,Y. et al. (2000) Structural genomics projects in Japan. Nature Struct. Biol., (Suppl 7), 943–945. [DOI] [PubMed] [Google Scholar]
- 67.Norvell J.C. and Machalek,A.Z. (2000) Structural genomics programs at the US National Institute of General Medical Sciences. Nature Struct. Biol., (Suppl 7), 931. [DOI] [PubMed] [Google Scholar]
- 68.Gaasterland T. (1998) Structural genomics taking shape. TIGS, 14, 135. [DOI] [PubMed] [Google Scholar]
- 69.Sali A. (1998) 100 000 protein structures for the biologist. Nature Struct. Biol., 5, 1029–1032. [DOI] [PubMed] [Google Scholar]
- 70.Rost B. (1998) Marrying structure and genomics. Structure, 6, 259–263. [DOI] [PubMed] [Google Scholar]
- 71.Brenner S.E., Barken,D. and Levitt,M. (1999) The PRESAGE database for structural genomics. Nucleic Acids Res., 27, 251–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Teichmann S.A., Chothia,C. and Gerstein,M. (1999) Advances in structural genomics. Curr. Opin. Struct. Biol., 9, 390–399. [DOI] [PubMed] [Google Scholar]
- 73.Vitkup D., Melamud,E., Moult,J. and Sander,C. (2001) Completeness in structural genomics. Nature. Struct. Biol., 8, 559–566. [DOI] [PubMed] [Google Scholar]
- 74.Linial M. and Yona,G. (2000) Methodologies for target selection in structural genomics. Prog. Biophys. Mol. Biol., 73, 297–320. [DOI] [PubMed] [Google Scholar]
- 75.Liu J. and Rost,B. (2002) Target space for structural genomics revisited. Bioinformatics, 18, 922–933. [DOI] [PubMed] [Google Scholar]
- 76.Hurley J.H., Anderson,D.E., Beach,B., Canagarajah,B., Ho,Y.S., Jones,E., Miller,G., Misra,S., Pearson,M., Saidi,L. et al. (2002) Structural genomics and signaling domains. TIBS, 27, 48–53. [DOI] [PubMed] [Google Scholar]
- 77.Pieper U., Eswar,N., Braberg,H., Madhusudhan,M.S., Davis,F.P., Stuart,A.C., Mirkovic,N., Rossi,A., Marti-Renom,M.A., Fiser,A. et al. (2004) MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res., 32, D217–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]