


Abstract
Chemical synthesis of DNA sequences provides a powerful tool for modifying genes and for studying gene function, structure and expression. Here, we report a simple, high-fidelity and cost-effective PCR-based two-step DNA synthesis (PTDS) method for synthesis of long segments of DNA. The method involves two steps. (i) Synthesis of individual fragments of the DNA of interest: ten to twelve 60mer oligonucleotides with 20 bp overlap are mixed and a PCR reaction is carried out with high-fidelity DNA polymerase Pfu to produce DNA fragments that are ∼500 bp in length. (ii) Synthesis of the entire sequence of the DNA of interest: five to ten PCR products from the first step are combined and used as the template for a second PCR reaction using high-fidelity DNA polymerase pyrobest, with the two outermost oligonucleotides as primers. Compared with the previously published methods, the PTDS method is rapid (5–7 days) and suitable for synthesizing long segments of DNA (5–6 kb) with high G + C contents, repetitive sequences or complex secondary structures. Thus, the PTDS method provides an alternative tool for synthesizing and assembling long genes with complex structures. Using the newly developed PTDS method, we have successfully obtained several genes of interest with sizes ranging from 1.0 to 5.4 kb.
INTRODUCTION
Chemical synthesis of DNA sequences can provide a powerful molecular tool for modifying genes, elucidating gene functions and studying the structure–function relationship of proteins. The recent advances in the area of chemical synthesis of DNA molecules have made it possible to synthesize and assemble genes for improved or novel functions. Also, in many cases, it is highly desirable to use a chemical synthesis method to modify coding sequences to achieve high expression levels. For instance, in recent years, we have used chemical synthesis methods to modify several important genes to improve the stability of their proteins and to enhance their expression levels in desirable expression systems (1–5). Several methods for the synthesis and assembly of DNA sequences based on oligonucleotides have been described previously: a method for enzymatic ligation of oligonucleotides reported in the 1980s (6–11), the FokI method published in 1988 (12), self-priming PCR developed in early 1990s (13–16), the PCR assembly method described in 1995 (17) and further improved in 2002 (19), and the template directed ligation (TDL) method published in 1996 (18). More recently, two methods of synthesis and assembly of long DNA sequences (∼5 kb) have been published. One is called the thermodynamically balanced inside-out (TBIO) PCR-based gene synthesis method, which uses a novel method of primer design to achieve high-fidelity assembly of long gene sequences (20). Another is an improved version of the assembly PCR method described by Smith et al. (21), and is capable of accurately assembling 5–6 kb segments of DNA using synthetic oligonucleotides.
In this paper, we describe a simple and easy to perform, rapid, low error rate and cost-effective PCR-based two-step DNA synthesis (PTDS) method for long genes. Using this newly developed PTDS method, we have successfully synthesized and assembled the vip3a gene named vip3aI (2382 bp in length, GenBank accession no. AF345166) encoding for a Bacillus thuringiensis vegetative insecticidal protein (22), the CrtEBWY gene (5367 bp, GenBank accession no. AY605097) and several other genes. Also, we have compared the PTDS method side by side with several previously published methods with regard to the error rates, costs and DNA product quality. Our data suggest that the PTDS method we have developed is an excellent alternative for the synthesis of long DNA fragments.
MATERIALS AND METHODS
Chemicals, enzymes and strains
High-fidelity Taq DNA polymerase pyrobest, T4 DNA ligase, DL2000 DNA marker, restriction endonucleases, IPTG and X-Gal were purchased from Takara Co., Ltd (Dalian, People's Republic of China). pBluescript II SK vector was purchased from Stratagene (La Jolla, CA). High-fidelity Taq DNA polymerase Pfu was purchased from Promega (Madison, WI). DNA marker (λ-DNA Hind III DNA marker) was obtained from New England Biolabs (Beverly, MA). DNA sequencing kits were purchased from Applied Systems Company (Foster City, CA). Other chemicals and reagents were purchased from Sigma Chemical Co., Ltd (St Louis, MO).
Oligonucleotides and purification
Oligonucleotides were purchased from Shanghai Sangon Biological Engineering Technology and Service Co., Ltd (Shanghai, People's Republic of China). The oligonucleotides were prepared on an Applied Biosystems model 3900 high-flux DNA synthesizer (Foster City, CA) using standard nucleotide cyanoethyl (CE) phosphoramidites. All oligonucleotides were purified by denaturing (7 M urea) PAGE.
Strategies to synthesize the vip3aI gene
To compare the efficiencies, costs and error rates of our PTDS method and the single-step successive PCR method, the two-step successive PCR method (similar to the TBIO method) and the single overlap extension method, we synthesized the vip3aI gene using six groups of oligonucleotides that were chemically synthesized. Four groups of oligonucleotides were designed according to the successive PCR methods. Figures 1 and 1S show the two-step successive PCR-mediated synthesis of the vip3aI gene using 60 and 90mer oligonucleotides, while Figures 2 and 2S show a single-step successive PCR-mediated synthesis of the vip3aI gene also using 60 and 90mer oligonucleotides. (Here and throughout, the ‘S’ indicates a figure in the Supplementary Material.) There was a 20 bp overlap for each of the oligonucleotides used. Two additional groups of oligonucleotides were designed and synthesized for the single-step overlap extension PCR method (17) and the PTDS method. The PTDS and overlap extension PCR methods used to synthesize the vip3aI gene are depicted in Figures 3, 3S, 4 and 4S. Figures 3 and 3S illustrate the two-step overlap extension PCR-mediated synthesis of the vip3aI gene using 60 and 90mer oligonucleotides, while Figures 4 and 4S show the single-step overlap extension PCR-mediated synthesis of the vip3aI gene using these same sets of oligonucleotides.
Figure 1.
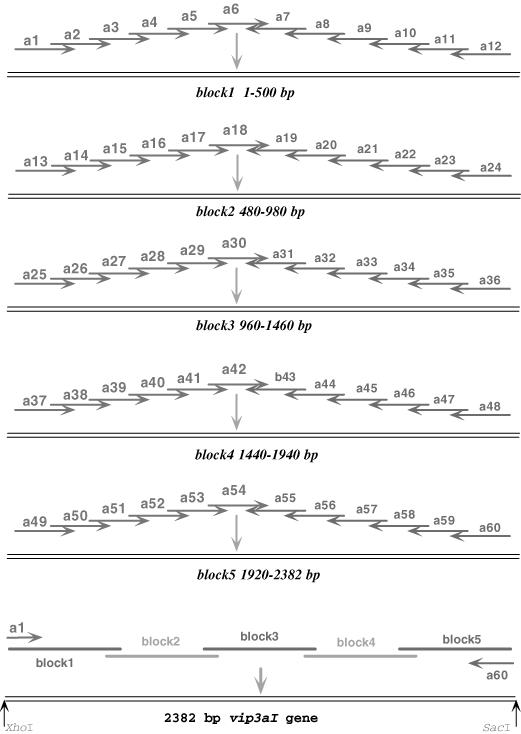
The strategy for the synthesis and assembly of the vip3aI gene using successive PCR. Oligonucleotides of ∼60 bp were assembled by two-step PCR reactions using 1.5 pmol of inner primers and 30 pmol of external primers which contained suitable restriction cleavage sites for cloning.
Figure 2.
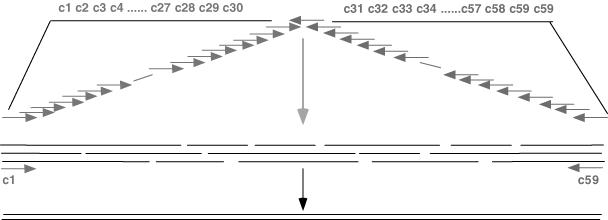
The strategy for the synthesis of the vip3aI gene using the successive PCR method. Oligonucleotides of ∼60 bp were assembled using the single-step PCR method with 1.5 pmol of inner primers and 30 pmol of external primers which contained suitable restriction cleavage sites for cloning.
Figure 3.
The strategy for the synthesis of the vip3aI gene using the overlap extension PCR method. Oligonucleotides of ∼60 bp were assembled using two-step PCR with 1.5 pmol of inner primers and 30 pmol of external primers.
Figure 4.
The strategy for the synthesis of the vip3aI gene using overlap extension PCR. Oligonucleotides of ∼60 bp were assembled by single-step PCR.
PCR reactions and assembly
We synthesized the vip3aI gene using four methods: the single-step successive PCR, two-step successive PCR, single-step overlap extension and PTDS method. For the successive PCR methods, five reactions each containing 12 oligonucleotides were carried out using 1.5 pmol of each inner oligonucleotide and 30 pmol of each outer oligonucleotide, as shown in Figure 1. PCR reactions were done on a PE thermocycler 9600 (Perkin Elmer, Foster City, CA) for 25 cycles with 2.5 U pyrobest Taq polymerase. The PCR conditions were 30 s at 90°C, 45 s at 60°C and 50 s at 72°C for each cycle. The final cycle was followed by an additional 10 min at 72°C to ensure complete extension for all PCR reactions, unless stated otherwise. To obtain the full length of the 2382 bp vip3aI gene, the DNA fragments, four 500 bp segments and one 460 bp segment produced from the five PCR reactions, were mixed and used to assemble the template for the second PCR reaction. The second PCR reaction was performed with the two outermost oligonucleotides as primers. The PCR reaction was carried out in one cycle at 94°C for 1 min (during which 2.5 U pyrobest or Pfu Taq polymerase were added), then 25 cycles of (i) denaturing at 94°C for 30 s, (ii) annealing at 58 or 56°C for 35 s, (iii) extension at 72°C for 3 min. Figure 1S shows the steps involved in the synthesis of the vip3aI gene using 90mer oligonucleotides. The PCR reaction for the gene assembly reaction was essentially the same as for the 60mer oligonucleotides, except that the extension time was 1 min 20 s in the first step.
For the single-step successive PCR procedure, sixty groups of 60mer oligonucleotides were added into a reaction tube, with 1.5 pmol of each of the inner oligonucleotides and 30 pmol of each of the outer oligonucleotides. The PCR reaction was performed on the PE 9600 thermocycler as follows: one cycle at 94°C for 1 min, then 25 cycles of (i) denaturing at 94°C for 30 s, (ii) annealing at 58°C for 40 s, (iii) extension at 72°C for 3 min. The PCR conditions for the gene assembly reaction using thirty-four 90mer oligonucleotides (Figure 2) were essentially the same as for the 60mer oligonucleotides (Figure 2S).
In the case of the PCR-based two-step DNA synthesis to synthesize and assemble the vip3aI gene (Figures 3 and 3S), 1.5 pmol of each of the inner oligonucleotides and 30 pmol of each of the two outer oligonucleotides were also used. The PCR reactions were carried out for 25 cycles at 90°C for 30 s, 60°C for 45 s, 72°C for 50 s, with 2.5 U pyrobest or Pfu Taq polymerase. To obtain the full length of the 2382 bp vip3aI gene, four 500 bp and one 460 bp products from the PCR reactions were mixed and used as the template for the second PCR reaction, with the two outermost oligonucleotides as primers. The conditions of the second PCR reaction were one cycle at 94°C for 1 min, and then 25 cycles of (i) 30 s at 94°C, (ii) 35 s at 58°C and (iii) 3 min at 72°C. Figure 3S shows the steps of synthesizing the vip3aI gene using 90mer oligonucleotides. An identical PCR protocol was used for the synthesis and assembly of the vip3aI gene using 60mer oligonucleotides, except that the extension time of the first cycle was 1 min 20 s.
For the single-step overlap extension PCR, similar to Stemmer's method (17), equal amounts of the sixty 60mer (Figure 4) and 90mer (Figure 4S) oligonucleotides were combined and the mixture was subsequently diluted 1000-fold in 50 μl PCR mix containing 2.5 U pyrobest polymerase. The PCR-mediated assembly was performed for 55 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 1 min. For the gene amplification, 1 μl of the assembled mixture was used as the template, with the two outermost oligonucleotides as primers. The PCR reaction was performed at 94°C for 1 min for the first cycle and then 25 cycles of 94°C for 30 s, 58°C for 40 s, 72°C for 3 min.
Analyses of clones and DNA sequences
The molecular cloning of the synthesized DNA fragments was performed according to the standard procedures (23). The PCR amplified products were purified with 1% agarose gel electrophoresis. The full-length vip3aI gene fragments were cloned into a pBluescript II SK vector and then sequenced on a DNA sequencer (ABI377, Perkin Elmer, Applied BioSystems, Foster City, CA). Analyses of this DNA sequence information were performed using the PCGENE program (Intelligenetics, Geneva, Switzerland).
RESULTS
Synthesis and assembly of the vip3aI gene using the successive PCR methods
The entire sequence of the open reading frame encoding for the toxic protein fragment vip3aI is 2370 bp in length (GenBank accession no. AY345166) and the sequence has 70.5% homology with the wild-type vip3a gene (GenBank accession no. L48811). For the synthetic gene used, all codons were optimized for the expression in Pichia pastoris (27,28). In order to improve the efficiency of gene transcription and RNA stability, the G + C content of the vip3aI gene was altered from 30.9 to 46.9%. The sequences for potential hairpin structures and motifs containing consecutive ATs were altered by using degenerating codons (Figure 5S).
We synthesized sixty oligonucleotides that were 60mer in length and were named a1 to a60 (Figures 1 and 6S). Five DNA blocks [named blocks 1, 2, 3 and 4 (500 bp each) and block 5 (460 bp); see Figure 1] were assembled and amplified (Figure 5a, lanes 1 to 5). The PCR was carried out to synthesize the full-length (2382 bp) vip3aI gene using a mixture of the five DNA blocks as the template and the two outermost oligonucleotides, a1 and a60, as the primers. The full-length vip3aI gene fragment obtained from the PCR reaction is shown in Figure 5b (lanes 1 and 2). Almost double the amount of the PCR product was obtained if the annealing temperature was 56°C (Figure 5b, lane 2) rather than 58°C (Figure 5b, lane 1). We also synthesized two 860 bp blocks and one 800 bp block (lanes 6 to 8 of Figure 5a) using thirty-four oligonucleotides that were 90 nt in length and named b1 to b34 (Figures 1S and 7S). The full-length 2382 bp vip3aI gene was assembled and amplified using a mixture of blocks 1 to 3 as the template and the b1 and b34 oligonucleotides as primers. Again, 56°C as the annealing temperature for PCR resulted in more PCR product (lane 4 of Figure 5b) than did 58°C (lane 3 of Figure 5b).
Figure 5.
(A) Four and three blocks were easily amplified using five groups of 60 and 90 nt oligonucleotides separately by two-step successive PCR. M is DNA marker DL2000. Lane 1: 460 bp block; lanes 2 to 5: four 500 bp blocks; lane 6: 750 bp block; lanes 7 and 8: 800 bp blocks. (B) A full-length vip3aI gene was amplified using two-step successive PCR. M is λ-DNA Hind III marker. Lanes 1 and 2: the full-length vip3aI gene amplified from five blocks using 60 nt oligonucleotides (lane 1, annealing temperature 58°C; lane 2, 56°C); lanes 3 and 4: the full-length vip3aI gene amplified from three blocks using 90 nt oligonucleotides (lane 3, annealing temperature 58°C; lane 4 56°C). (C) Synthesis of the vip3aI gene using single-step successive PCR. M is λ-DNA Hind III marker. Lane 1: the full-length vip3aI gene amplified using 60mer oligonucleotides by single-step successive PCR; lane 2: the full-length vip3aI gene amplified using 90mer oligonucleotides by single-step successive PCR; lanes 3 and 4: the amplified vip3aI gene using 30 pmol of the two outermost primers.
We synthesized sixty oligonucleotides that were 60 nt in length, named c1 to c60 (Figures 2 and 8S), and thirty-four oligonucleotides that were 90 nt in length, named d1 to d34 (Figures 2S and 9S). With these two groups of oligonucleotides, a 2382 bp PCR product, the full-length vip3aI gene, was obtained using the single-step PCR method, but the yields of the products were very low (Figure 5c, lanes 1 and 2). After a second PCR-mediated amplification of the single-step PCR products with the outermost oligonucleotides as the primers, we could enrich the DNA products of the 2382 bp vip3aI gene (lanes 3 and 4 of Figure 5c). Our experience is that the single-step PCR method is excellent for synthesizing a DNA fragment that is shorter than 1.0 kb but often produces a very low amount of the target DNA and sometimes smeared DNA bands when the target DNA fragments are >1.0 kb in length.
Synthesis of the vip3aI gene using the traditional single-step overlap extension PCR method and the PTDS method
We also used both the traditional single-step overlap extension PCR method and PCR-based two-step overlap extension DNA synthesis to assemble the vip3aI gene. To achieve this goal, we synthesized sixty 60mer oligonucleotides (Figure 10S) and thirty-four 90mer oligonucleotides (Figure 11S). The experimental details of the traditional single-step overlap extension PCR are outlined in Figures 4 and 4S. We used 30 pmol of each oligonucleotide for the PCR reactions for the traditional single-step overlap extension PCR method. In this method, PCR reactions contained equal quantities of the oligonucleotides and the assembled and amplified full-length vip3aI gene is shown in Figure 6c (lanes 1 and 2). With the outermost oligonucleotides as primers, we could enrich the DNA products of the 2382 bp vip3aI gene (lanes 3 and 4 of Figure 6c). As shown in Figure 6c, the quantities of the target products from the traditional single-step overlap extension PCR were low and there were some smear and additional bands within the PCR products.
Figure 6.
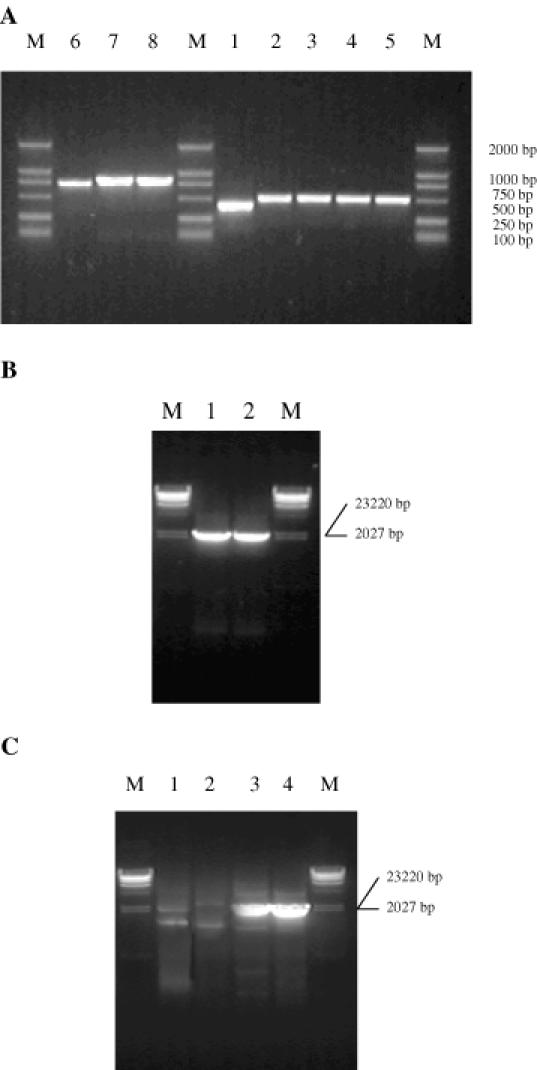
(A) DNA blocks amplified using 60 or 90 nt oligonucleotides separately using the PTDS method. M is DNA marker DL2000. Lane 1: 460 bp block; lanes 2–5: four 500 bp blocks; lane 6: 750 bp block; lanes 7 and 8: two 880 bp blocks. (B) A full-length vip3aI gene amplified using the PTDS method. M is λ-DNA Hind III marker. Lane 1: a full-length vip3aI gene amplified from three blocks that were derived from 90 nt oligonucleotides; lane 2: a full-length vip3aI gene amplified from five blocks that were derived from 60 nt oligonucleotides. (C) A full-length vip3aI gene amplified using single-step overlap extension PCR. M is λ-DNA Hind III marker. Lane 1: a full-length vip3aI gene amplified using 60 nt oligonucleotides by single-step successive PCR; lane 2: a full-length vip3aI gene amplified using 90 nt oligonucleotides by single-step overlap extension PCR; lanes 3 and 4: full-length amplified vip3aI gene fragments using outermost primers.
Figures 3 and 3S outline the details of the two steps of the PTDS method to synthesize the 2382 bp vip3aI gene using the 60 and 90mer oligonucleotides separately. Five groups, each of them having twelve oligonucleotides, were joined together to synthesize five DNA blocks. Figure 6a shows that one 460 bp (lane 1) and four 500 bp (lanes 2 to 5) blocks were obtained. Figure 6b shows that a full-length vip3aI product was derived from the second PCR using the two outermost oligonucleotides as primers and the five DNA block mixture as a template (lane 1). Compared with the traditional single-step overlap extension PCR method, the PTDS method produced a single-band, much higher yield product. Figure 6a also shows that the nearly 880 and 750 bp (lanes 6 to 8) blocks were assembled successfully using 90 nt oligonucleotides. Figure 6b (lane 2) indicates that these three blocks were the assembled full-length vip3aI gene.
Analysis of error rates of the synthesized vip3aI gene using different methods
We cloned the chemically synthesized vip3aI gene using 60 and 90mer oligonucleotides using the two-step successive PCR, single-step successive PCR, traditional single-step overlap extension PCR and PTDS methods. In order to compare the error rates of these methods, eight kinds of synthesized vip3aI DNA sequences produced from these methods were cloned and sequenced. The results are shown in Table 1. Overall, the numbers of errors using 90mer oligonucleotides were significantly higher than those using 60mer oligonucleotides for all the methods tested. The reason for this may be that the automatic DNA synthesizer produces more errors for longer oligonucleotides than for shorter oligonucleotides during chemical synthesis. As shown in Table 1, the error rate for the PTDS method is several times lower (1.26% per 1000 bp) than those of all the other methods with which it was compared, including the single-step overlap extension method. For the CrtEBIWY gene (5397 bp in length, GenBank accession no. AY605097), which we have synthesized and assembled using the PTDS method, the error rate is as low as 2.2 bp out of 1000 bp, which further supports the high fidelity of the PTDS method. We have used the PTDS method to synthesize many other genes recently, and all the ones we have sequenced have low rates of errors. Table 2 summarizes the genes and DNA fragments that have been successfully synthesized and assembled in our lab using the PTDS method.
Table 1. Comparison of error rates of different methods using different sized oligonucleotides.
| Clones | Length of oligos (nt) | Two-step successive PCR method | Single-step successive PCR method | PTDS method | Single-step overlap extension PCR method | ||||
|---|---|---|---|---|---|---|---|---|---|
| Number of errors | Error rate (/1000) | Number of errors | Error rate (/1000) | Number of errors | Error rate (/1000) | Number of errors | Error rate (/1000) | ||
| 1 | 60 | 10 | 4.20 | 19 | 7.98 | 3 | 1.26 | 9 | 3.78 |
| 90 | 24 | 10.08 | 28 | 11.75 | 11 | 4.62 | 19 | 7.98 | |
| 2 | 60 | 11 | 4.62 | 19 | 7.98 | 3 | 1.26 | 10 | 4.20 |
| 90 | 26 | 10.92 | 28 | 11.75 | 14 | 5.88 | 17 | 7.14 | |
| 3 | 60 | 11 | 4.62 | 17 | 7.14 | 4 | 1.68 | 8 | 3.36 |
| 90 | 26 | 10.92 | 27 | 11.34 | 14 | 5.88 | 20 | 8.40 | |
| 4 | 60 | 12 | 5.04 | 20 | 8.40 | 4 | 1.68 | 8 | 3.36 |
| 90 | 28 | 11.75 | 30 | 12.59 | 16 | 6.72 | 18 | 7.56 | |
| 5 | 60 | 10 | 4.20 | 20 | 8.40 | 2 | 0.84 | 13 | 5.46 |
| 90 | 26 | 10.92 | 28 | 11.75 | 16 | 6.72 | 22 | 9.24 | |
| Average | 60 | 11 | 4.62 | 19 | 7.98 | 3 | 1.26 | 9 | 3.78 |
| 90 | 26 | 10.92 | 28 | 11.75 | 14 | 5.88 | 19 | 7.98 | |
Table 2. Comparison of the error rates of the PTDS and successive PCR methods.
| GenBank number | Length (bp) | Error rate | Characteristics of synthesized DNA fragment |
|---|---|---|---|
| PTDS method | |||
| AY345166 | 2370 | 1.26 | Synthesized vip3aI gene |
| AY605097 | 5397 | 2.20 | Synthesized fusion CrtEBIWY gene of CrtE, crtB, crtI, crtW and crtZ |
| AF274585 | 1017 | 2.10 | Synthesized ACC deaminase gene |
| AY136629 | 1104 | 1.50 | Synthesized DNA OF heat-sensitive lambda citS857 repressor and VP16 fusion protein |
| AY136630 | 1941 | 1.40 | Synthesized DNA OF carboxypeptidase Y and myrosinase fusion protein |
| AY136633 | 1236 | 1.30 | Synthesized DNA of HSF3 DNA-binding domain and VP16 activation domain fusion protein |
| AY136634 | 1473 | 1.50 | Synthesized DNA of lac repressor VP16 activation domain fusion protein |
| AY136635 | 1515 | 1.20 | Synthesized DNA of NahR and VP16 activation domain fusion protein |
| AY137197 | 1116 | 1.60 | Synthesized DNA of virG DNA-binding domain and VP16 activation domain fusion protein |
| AY183360 | 726 | 1.80 | Synthesized virGI gene |
| AY183361 | 735 | 1.30 | Synthesized DNA of duplicated CaMV35 and TMV omega-prime leader sequence fusion promoter |
| AY345165 | 1002 | 0.90 | Synthesized DNA of CrtW/CrtY fusion protein |
| Successive PCR method | |||
| AY345166 | 2370 | 7.98 | Synthesized vip3aI gene |
| AF274974 | 1003 | 5.40 | Synthesized kanamycin resistance gene |
| AF537267 | 1848 | 4.55 | Synthesized insect toxin CryIA(c) gene |
| AF542235 | 1347 | 3.60 | Synthesized phytase (phyA-sh) mRNA |
| AF547224 | 1347 | 5.10 | Synthesized phytase (phyA) mRNA |
| AY136631 | 1398 | 4.85 | Synthesized thermostabile phytase |
| AY136632 | 837 | 3.10 | Synthesized DNA of Gal4 DNA-binding domain and VP16 activation domain fusion protein |
| AY136636 | 1347 | 6.50 | Synthesized phytase mRNA |
| AY137196 | 1347 | 5.60 | Synthesized phytase (fphy-sh) mRNA |
| AY137198 | 1237 | 4.20 | Synthesized phytase mRNA |
| AY137201 | 723 | 3.20 | Synthesized DNA of CBF1 DNA-binding domain and VP16 activation domain fusion protein |
| AY182953 | 1434 | 4.95 | Synthesized phytase (phyI) mRNA |
| AY182954 | 1434 | 5.55 | Synthesized phytase (phy2I) mRNA |
| AY182955 | 1332 | 4.90 | Synthesized phytase (FphyI3) mRNA, |
| AY192160 | 827 | 3.60 | Synthesized multi-copy enhancer promoter element |
We used both PTDS and successive PCR methods to synthesize a large number of genes and the error rates are listed. It is clear that the PTDS method produces much lower error rates than the successive PCR method.
Cost analysis of syntheses of the vip3aI gene using different methods
In Shanghai, People's Republic of China, the cost of oligonucleotide synthesis is currently USD 0.2 (RMB yuan 1.5) for oligonucleotides of length <60 nt, and USD 0.7 (RMB yuan 6.0) per base for lengths >90 nt. The latter is four times the former. For the vip3aI gene, the total number of nucleotides to be synthesized was ∼3500 using 60mer oligonucleotides, and ∼3000 using 90mer oligonucleotides. The total cost was nearly USD 710 for the 60mer route but USD 2130 when taking the 90mer route. Thus, using the 60mer oligonucleotide strategy to synthesize the vip3aI gene is only one-third of the cost of the 90mer oligonucleotide strategy (Table 3). If the error factor and the cost to correct the errors are taken into consideration, the relative cost for the 60mer strategy will reduce further because almost 5-fold fewer errors were produced using the 60mer strategy (Table 1).
Table 3. Cost comparison of four different methods of synthesizing the vip3aI gene using 60 or 90 nt oligonucleotides.
| Length of oligos | 60 nt | 90 nt |
|---|---|---|
| Two-step successive PCR method | ||
| Number of oligos | 60 (oligoA ) | 34 (oligoB) |
| Number of overlaps (bases) | 1180 | 660 |
| Total chemical syntheses (bases) | 3562 | 3032 |
| Unit price (USD) | 0.20 | 0.70 |
| Total price (USD) | 712.4 | 2122.4 |
| Single-step successive PCR method | ||
| Number of oligos | 59 (oligoC ) | 34 (oligoD) |
| Number of overlaps (bases) | 1160 | 660 |
| Total chemical syntheses (bases) | 3542 | 3042 |
| Unit price (USD) | 0.20 | 0.70 |
| Total price (USD) | 708.4 | 2129.4 |
| PTDS method | ||
| Number of oligos | 60 (oligoE ) | 34 (oligoF ) |
| Number of overlaps (bases) | 1180 | 640 |
| Total chemical syntheses (bases) | 3562 | 3022 |
| Unit price (USD) | 0.20 | 0.70 |
| Total prices (USD) | 712.4 | 2115.4 |
| Single-step overlap extension PCR method | ||
| Number of oligos | 60 (oligoE ) | 34 (oligoF ) |
| Number of overlaps (bases) | 1180 | 640 |
| Total chemical syntheses (bases) | 3562 | 3022 |
| Unit price (USD) | 0.20 | 0.70 |
| Total prices (USD) | 712.4 | 2115.4 |
DISCUSSION
Based on the data presented in this paper, we believe that the PTDS method offers a simple, rapid, high-fidelity and low-cost alternative method of synthesizing and assembling long DNA fragments. To perform the PTDS method successfully, we recommend 60mer oligonucleotides with 20 nt sequence overlap for each and a Tm of ∼56°C for the first step of the PTDS method. We suggest that 12 oligonucleotides be grouped to synthesize a DNA block that is ∼500 bp in length. For the second PCR reaction of the PTDS method, the synthesized DNA blocks are mixed and assembled to produce the template DNA to synthesize a full-length gene using the two outermost oligonucleotides as primers. The concentrations recommended are 1.5 pmol for the inner oligonucleotides and 30 pmol for the outer oligonucleotides for the first PCR reaction. The concentrations recommended for the two outermost oligonucleotides for assembling and amplifying a full-length gene (the second step of the PTDS method) are 30 pmol. For the first step of the PTDS method, PCR can be conducted for 25 cycles with 2.5 U Pfu Taq polymerase (each cycle consisting of 30 s at 90°C, 45 s at 60°C, 50 s at 72°C). For the second PCR reaction, we recommend one cycle at 94°C for 5 min and (2.5 U pyrobest Taq polymerase should be added in this step). We recommend 25 cycles for the PCR reaction, each cycle at 94°C for 30 s, 56°C for 35 s, 72°C for 2–6 min (variable according to the length of the gene to be synthesized).
Our experiments have demonstrated that the PTDS procedures are simple and easy to perform to obtain DNA blocks that are ∼500 bp and to assemble the resulting DNA blocks to produce a full-length gene using PCR with the two outermost oligonucleotides. We have demonstrated that the PTDS method is highly suitable for synthesizing DNA fragments that are 0.3–5.0 kb in length. It may also work well for longer DNA fragments, but that possibility needs to be investigated.
We used 25 PCR amplification cycles in the PTDS method, whereas the number of amplification cycles in traditional overlap extension PCR was over 50 using Stemmer's method (17). In the successive PCR and TBIO methods, the number of amplification cycles was more than 25. The reduced number of PCR amplification cycles for the PTDS method should reduce the error rate, thereby producing DNA with higher fidelity. If errors are revealed through DNA sequencing analysis, we can correct them using the overlap extension method (17). For both the successive PCR method and the TBIO method, we can use an oligonucleotide that is one of the oligonucleotides already used, but we need to chemically synthesize another, new oligonucleotide. For the PTDS method, both the sense- and antisense oligonucleotides to be used for correction have already been synthesized, which reduces the cost of, and time for, synthesis of new oligonucleotides considerably. The cost-effectiveness of the PTDS method is more obvious when long DNA fragments are synthesized.
Because Pfu DNA polymerase exhibits the lowest error rate among all thermostable DNA polymerases (24,25), the enzyme has been the choice for the DNA synthesis and assembly in many of the methods previously described. However, while we have found Pfu DNA polymerase to be excellent for amplification of DNA fragments that are <1.0 kb, it becomes difficult to synthesize DNA fragments that are >2.0 kb in length. Another high-fidelity DNA polymerase, pyrobest, is excellent for synthesizing DNA fragments that are >2.0 kb. Thus, for the PTDS method we recommend the Pfu enzyme for synthesizing 500 or 800 bp DNA blocks in the first step of the method because it has the lowest error rate, while the pyrobest enzyme is recommended for assembling full-length genes that are >2.0 kb in length.
There are some major differences between the PTDS method and the Smith method (21). First, the Smith method involves at least four major steps: (i) phosphorylation of oligonucleotides; (ii) Taq ligase-mediated ligation of the oligonucleotides; (iii) polymerase cycline assembly of ligation products into a full-length gene (35 to 70 cycles) and (iv) PCR amplification of the assembled full-length gene. For the PTDS method, only two major steps are needed: (i) PCR-mediated synthesis of ∼500 bp DNA fragments and (ii) PCR-mediated synthesis of the full-length gene. Because fewer steps are involved and because only two PCR enzymes are used for the entire procedure, the PTDS method is much simpler and easier to perform, more rapid (5–7 days for the PTDS method but 14 days for the Smith method) and also less expensive than the Smith method. Second, because the Smith method uses 42mer oligonucleotides while the PTDS method uses 60mer oligonucleotides, the cost associated with the PTDS method can be further reduced. Third, the Smith method uses functional screening to select the clones and in so doing will underestimate the error rates. Also, although functional screening is ideal for selecting clones with the correct DNA sequence, in many cases functional screening is impossible to perform. For instance, if one wants to synthesize a flowering gene that will be expressed in plants, functional screening to select genes with the correct DNA sequence at the gene assembly and synthesis stages is not practical. Thus, the application of the Smith method would be limited to the synthesis of genes whose functions can be easily assayed if the error rate based on non-discriminable clones for the Smith method were high (i.e. higher than for the previously published methods). On the other hand, the low error rate for the PTDS method is based on non-discriminable clones, and the method should therefore be applicable to the synthesis of any gene or DNA fragment regardless of whether its function can be easily assayed or not.
In general, chemical-synthesis-mediated gene assembly should eliminate/reduce secondary structures, contrary repeats and repetitive sequences, and adjust G + C composition. However, in some cases (26), such as DNA shuffling, these structures, repeats or high G + C composition should be preserved. When using some of the previously published gene synthesis methods, such as TBIO, successive PCR and TDL, it is difficult to synthesize long genes with these types of structures or sequences directly in a single step. On the other hand, based on our experience, the PTDS method, which synthesizes shorter DNA fragments (∼500 bp in length) as a first step and then assembles these DNA blocks, can make it easier to synthesize and assemble long genes with complex structures.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to Shanghai YongYe Bio-engineering Co., Ltd for providing some instruments and technical help. This research was supported by the Key Project Fund of the Science and Technology Commission Foundation of Shanghai City, People's Republic of China (No. 993913002) and the Key Project Fund of the Shanghai Municipal Committee of Agriculture (No. 99-1-8).
DDBJ/EMBL/GenBank accession no. AF345166
REFERENCES
- 1.Peng R.H., Xiong,A.S, Li,X., Fan,H.Q., Yao,Q.H., Guo,M.J. and Zhang,S.L. (2002) High expression of a heat-stable phytase in Pichia pastoris. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao, 34, 725–730. [PubMed] [Google Scholar]
- 2.Peng R.H., Xiong,A.S., Li,X., Fan,H.Q., Huang,X.M. and Yao,Q.H. (2001) PCR-aided synthesis and stable expression in E.coli of the cryIA (c)Bt gene. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao, 33, 219–224. [PubMed] [Google Scholar]
- 3.Peng R., Xiong,A., Li,X., Fuan,H. and Yao,Q. (2003) A delta-endotoxin encoded in Pseudomonas fluorescens displays a high degree of insecticidal activity. Appl. Microbiol Biotechnol., 63, 300–306. [DOI] [PubMed] [Google Scholar]
- 4.Xiong A.S, Peng,R.H, Li, X, Fan,H.Q, Yao,Q.H, Guo,M.J. and Zhang,S.L. (2003) The study of signal peptide sequence affecting expression of heterologous protein in Pichia pastoris. Sheng Wu Hua Xue Yu Sheng Wu Wu Li Xue Bao. 35, 154–160. [PubMed] [Google Scholar]
- 5.Zha D., Eipper,A. and Reetz,M.T. (2003) Assembly of designed oligonucleotides as an efficient method for gene recombination: a new tool in directed evolution. Chembiochem, 4, 34–39. [DOI] [PubMed] [Google Scholar]
- 6.Smith J., Cook,E., Fotheringham,I., Pheby,S., Derbyshire,R., Eaton,M.A., Doel,M., Lilley,D.M., Pardon,J.F., Patel,T., Lewis,H. and Bell,L.D. (1982) Chemical synthesis and cloning of a gene for human beta-urogastrone. Nucleic Acids Res., 10, 4467–4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Edge M.D., Greene,A.R., Heathcliffe,G.R., Moore,V.E., Faulkner,N.J., Camble,R., Petter,N.N., Trueman,P., Schuch,W. and Hennam,J. (1983) Chemical synthesis of a human interferon-alpha 2 gene and its expression in Escherichia coli. Nucleic Acids Res., 11, 6419–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jay E., MacKnight,D., Lutze-Wallace,C., Harrison,D., Wishart,P., Liu,W.Y., Asundi,V., Pomeroy-Cloney,L., Rommens,J. and Eglington,L. (1984) Chemical synthesis of a biologically active gene for human immune interferon-gamma. Prospect for site-specific mutagenesis and structure-function studies. J. Biol. Chem., 259, 6311–6317. [PubMed] [Google Scholar]
- 9.Sproat B.S. and Gait,M.J. (1985) Chemical synthesis of a gene for somatomedin C. Nucleic Acids Res., 13, 2959–2977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ecker D.J., Khan,M.I., Marsh,J., Butt,T.R. and Crooke,S.T. (1987) Chemical synthesis and expression of a cassette adapted ubiquitin gene. J. Biol. Chem., 262, 3524–3527. [PubMed] [Google Scholar]
- 11.Ashman K., Matthews,N. and Frank,R.W. (1989) Chemical synthesis, expression and product assessment of a gene coding for biologically active human tumour necrosis factor alpha. Protein Eng., 2, 387–391. [DOI] [PubMed] [Google Scholar]
- 12.Mandecki W. and Bolling,T.J. (1988) FokI method of gene synthesis. Gene, 68, 101–107. [DOI] [PubMed] [Google Scholar]
- 13.Dillon P.J. and Rosen,C.A. (1990) A rapid method for the construction of synthetic genes using the polymerase chain reaction. Biotechniques, 9, 298–300. [PubMed] [Google Scholar]
- 14.Prodromou C. and Pearl,L.H. (1992) Recursive PCR: a novel technique for total gene synthesis. Protein Eng., 5, 827–829. [DOI] [PubMed] [Google Scholar]
- 15.Ciccarelli R.B., Gunyuzlu,P., Huang,J., Scott,C. and Oakes,F.T. (1991) Construction of synthetic genes using PCR after automated DNA synthesis of their entire top and bottom strands. Nucleic Acids Res., 19, 6007–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayashi N., Welschof,M., Zewe,M., Braunagel,M., Dubel,S., Breitling,F. and Little,M. (1994) Simultaneous mutagenesis of antibody CDR regions by overlap extension and PCR. Biotechniques, 17, 310–315. [PubMed] [Google Scholar]
- 17.Stemmer W.P., Crameri,A., Ha,K.D., Brennan,T.M. and Heyneker,H.L. (1995) Single-step assembly of a gene and entire plasmid from large numbers of oligonucleotides. Gene, 164, 49–53. [DOI] [PubMed] [Google Scholar]
- 18.Strizhov N., Keller,M., Mathur,J., Koncz-Kalman,Z., Bosch,D., Prudovsky,E., Schell,J., Sneh,B., Koncz,C. and Zilberstein,A. (1996) A synthetic cryIC gene, encoding a Bacillus thuringiensis delta-endotoxin, confers Spodoptera resistance in alfalfa and tobacco. Proc. Natl Acad. Sci. USA, 93, 15012–15017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David M.H. and Jacek,L. (2002) DNAWorks: an automated method for designing oligonucleotides for PCR-based gene synthesis. Nucleic Acids Res., 30, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao X., Yo,P., Keith,A., Ragan,T.J. and Harris,T.K. (2003) Thermodynamically balanced inside-out (TBIO) PCR-based gene synthesis: a novel method of primer design for high-fidelity assembly of longer gene sequences. Nucleic Acids Res., 31, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith H.O., Hutchison,C.A., III, Pfannkoch,C. and Venter,J.C. (2003) Generating a synthetic genome by whole genome assembly: phiX174 bacteriophage from synthetic oligonucleotides. Proc. Natl Acad. Sci. USA, 100, 15440–15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Estruch J.J., Warren,G.W., Mullins,M.A., Nye,G.J. and Craig,J.A. (1996) Vip3A, a novel Bacillus thuringiensis vegetative insecticidal protein with a wide spectrum of activities against lepidopteran insects. Proc. Natl Acad. Sci. USA, 93, 5389–5394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sambrook J., Fritsch,E.F. and Maniatis,T. (1989) Molecular Cloning: A Laboratory Manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York. [Google Scholar]
- 24.Cline J., Braman,J.C. and Hogrefe,H.H. (1996) PCR fidelity of pfu DNA polymerase and other thermostable DNA polymerases. Nucleic Acids Res., 24, 3546–3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andre P., Kim,A., Khrapko,K. and Thilly,W.G. (1997) Fidelity and mutational spectrum of Pfu DNA polymerase on a human mitochondrial DNA sequence. Genome Res., 7, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sreekrishna K., Brankamp,R.G., Kropp,K.E., Blankenship,D.T., Tsay,J.T., Smith,P.L., Wierschke,J.D., Subramaniam,A. and Birkenberger,L.A. (1997) Strategies for optimal synthesis and secretion of heterologous proteins in the methylotrophic yeast Pichia pastoris. Gene, 190, 55–62. [DOI] [PubMed] [Google Scholar]
- 27.Zhao X., Huo,K.K. and Li,Y.Y. (2000) Synonymous codon usage in Pichia pastoris. Chin. J. Biotechnol., 16, 308–311. [PubMed] [Google Scholar]
- 28.Sharp P.M, Tuohy,T.M.F. and Mosurski,K.R. (1986) Condon usage in yeast: cluster analysis clearly differentiates highly and lowly expression genes. Nucleic. Acids Res., 14, 5125–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.