

Abstract
Background & Aims
Severe forms of inflammatory bowel disease (IBD) that develop in very young children can be caused by variants in a single gene. We performed whole-exome sequence (WES) analysis to identify genetic factors that might cause granulomatous colitis and severe perianal disease, with recurrent bacterial and viral infections, in an infant of consanguineous parents.
Methods
We performed targeted WES analysis of DNA collected from the patient and her parents. We validated our findings by a similar analysis of DNA from 150 patients with very early onset IBD not associated with known genetic factors analyzed in Toronto, Oxford, and Munich. We compared gene expression signatures in inflamed vs. non-inflamed intestinal and rectal tissues collected from patients with treatment-resistant Crohn's disease who participated in a trial of ustekinumab. We performed functional studies of identified variants in primary cells from patients and cell culture.
Results
We identified a homozygous variant in the tripartite motif containing 22 gene (TRIM22) of the patient, as well as in 2 patients with a disease similar phenotype. Functional studies showed that the variant disrupted the ability of TRIM22 to regulate nucleotide binding oligomerization domain containing 2 (NOD2)-dependent activation of interferon-beta signaling and NF-κB. Computational studies demonstrated a correlation between the TRIM22–NOD2 network and signaling pathways and genetic factors associated very early onset and adult-onset IBD. The network also associated with antiviral and mycobacterial effectors and markers of inflammation such as fecal calprotectin, c-reactive protein, and Crohn's disease activity index scores.
Conclusion
In WES and targeted exome sequence analyses of an infant with severe IBD, characterized by granulomatous colitis and severe perianal disease, we identified a homozygous variant of TRIM22 that affect the ability of its product to regulate NOD2. Combined computational and functional studies showed that the TRIM22–NOD2 network regulates antiviral and anti-bacterial signaling pathways that contribute to inflammation. Further study of this network could lead to new disease markers and therapeutic targets for patients with very early and adult-onset IBD.
Keywords: VEOIBD, CDAI, genetics, risk, susceptibility
Background
Very early onset inflammatory bowel diseases (VEOIBD) often presents with severe multi-systemic disease that is difficult to treat with conventional therapies. These young patients frequently have novel causal1-3 and risk variants4-8 and common networks are now being established (reviewed in 9). For example, mutations in IL10RA/B genes cause a Mendelian form of VEOIBD with severe colitis and perianal disease10 and mutations in TTC7A cause a severe form of apoptotic enterocolitis1.
Tripartite motif-containing 22 (TRIM22; also known as STAF50) is a RING finger E3 ubiquitin ligase11 that is expressed in the intestine12 and in macrophages13, and has a role in lineage-specific differentiation of lymphocytes14. TRIM22 was originally identified as an interferon inducible protein that possesses antiviral activity15-17 and activates NF-κB signaling13. Here we identify TRIM22 functional variants associated with a distinct VEOIBD phenotype characterized by granulomatous colitis and severe perianal disease and show the TRIM22-NOD2 network as a key antiviral and mycobacterial regulator.
Methods
Abbreviated Methods (see Supplemental Material for Full Description).
Subjects
All experiments were carried out with the approval of the Research Ethics Board (REB) at the Hospital for Sick Children. Informed consent to participate in research was obtained. A copy of the consent is available on the interNational Early Onset Pediatric IBD Cohort Study (NEOPICS) website at http://www.neopics.org/NEOPICS_Documents.html.
Whole exome sequencing
For Patient 1 and her parents (trio), whole exome sequencing (WES) was performed using the Agilent SureSelect Human All Exon 50Mb kit with high-throughput sequencing conducted using the SOLiD 4 System at The Center for Applied Genomics (TCAG) through the Hospital for Sick Children (Toronto, ON). Sanger sequencing was used to verify variant genotypes in the Family 1 and infantile patients from the collaborating institutions were screened for TRIM22 variants.
Validation
In order to validate these findings, we examined WES results from 150 infantile international VEOIBD patients without a genetic diagnosis who were previously sequenced in Toronto (NEOPICS), Oxford, and Munich (Care-For-Rare) and targeted exome sequencing of the TRIM22 gene in ten IL10RA/B, and IL10 negative patients without previous WES.
Computational Analysis
Datasets
Biopsy data were collected at baseline from anti-TNF resistant Crohn's patients enrolled in the ustekinumab trial previously described18. The ustekinumab trial expression data were used for construction of the adult IBD network. RNA-seq from the RISK cohort as previously described19 was used for generation of the pediatric IBD Bayesian network.
Differential expression
To ascertain tissue-specific signatures, we examined inflamed versus non-inflamed tissue from various anatomical regions of the small intestine, colon, and rectum. To identify differential expression signatures, we used an unbiased univariate filter to select top- varying genes and then applied significance analysis of microarrays (SAM)20 to whole-genome expression data collected from biopsy tissue samples and whole blood of individuals in the ustekinumab trial. To control for multiple hypothesis testing, we used the Benjamini & Hochberg adjustment on the raw p-values to control the family-wise error rate and set a false discovery rate threshold of 1% or 5%. All analyses were performed using the R statistical package, version 2.15.24.1
Bayesian Network
We employed Monte Carlo Markov Chain (MCMC)21 simulation to identify potentially thousands of different plausible networks that are then combined to obtain a consensus network. eSNP data were used as priors as follows: genes with cis-eSNP22 are allowed to be parent nodes of genes without cis-eSNPs, but genes without cis-eSNPs are not allowed to be parents of genes with cis-eSNPs.
Key Driver Analysis
Key driver analysis (KDA) takes as input a set of genes (G) and a directed gene network N (e.g. Bayesian network)19,23-26. The objective is to identify the key regulators for the gene sets with respect to the given network. KDA first generates a sub-network NG, defined as the set of nodes in N that are no more than h-layers away from the nodes in G, and then searches the h-layer neighborhood (h=1,..,H) for each gene in NG (HLNg,h) for the optimal h*, such that ESh* = max(ESh,g) ∀g ∈ Ng, h ∈ {1..H} where ESh,g is the computed enrichment statistic for HLNg,h. A node becomes a candidate driver if its HLN is significantly enriched for the nodes in G. Candidate drivers without any parent node (i.e., root nodes in directed networks) are designated as global drivers while the remaining are designated as local drivers.
Pathway Enrichment
eQTL analysis was performed on IBD, UC, and CD GWAS hits from the NHGRI catalog, plus all WTCCC IBD GWAS SNPs with pvalue ≤ 0.001. 10% FDR eGenes were thus extracted, and tested for enrichment of pathways from the Metacore database.
Clinical variable correlation
We used Spearman correlation to examine the relationship between the mRNA expression of TRIM22 in intestine and blood and several key IBD traits. The correlation test was performed with the R function “corr.test”. We applied a simple Bonferroni correction to a 0.01 significance level across all gene-trait correlation tests performed.
Transfection and Luciferase Reporter Assays
NF-κB luciferase reporter plasmids (Promega) and ISRE promoter and IFNPβ promoter luciferase reporter plasmids were obtained from Dr. Hong-bing Shu (Wuhan University). A total of HEK 293 cells (1 × 105) were seeded on 24-well dishes and transfected the following day using Lipofectamine 2000 method. Empty control plasmid was added to ensure that each transfection received the same amount of total DNA. To normalize for transfection efficiency, 0.01 μg of pRL-TK (Renilla luciferase) reporter plasmid was added to each transfection. Approximately 24 hr. after transfection, luciferase assays were performed using a dual-specific luciferase assay kit (Promega). Firefly luciferase activities were normalized on the basis of Renilla luciferase activities. Cells were treated with recombinant TNFα, RSV, or L18-MDP (InvivoGen). Statistical analysis of luciferase assay data consisted of two-way ANOVA followed by Bonferroni post-hoc testing to compare across conditions. Statistical significance was assigned to tests with p<0.05 after Bonferroni correction.
Coimmunoprecipitation and Immunoblot Analysis
For transient transfection and coimmunoprecipitation experiments, HEK 293 cells (1 × 106) were seeded for 24 hr. then transiently transfected for 18‒24 hr. using Lipofectamine 2000 according to the manufacturer's protocols. Transfected cells were lysed in 1 mL of lysis buffer (150mM NaCl, 50mM HEPES, 1% Triton X-100, 10% glycerol, 1.5mM MgCl2, l.0mM EGTA) supplemented with protease and phosphatase inhibitors (aprotinin (1:1000), leupeptin (1:1000), pepsin (1:1000) and, PMSF (1:100). For each immunoprecipitation, a 0.9mL aliquot of the lysate was incubated with 0.5 μg of the indicated antibody and 40 μL of a 1:1 slurry of Protein G Sepharose (Bioshop) for 1 hr. Sepharose beads were washed 3× with 1 mL of high salt lysis buffer containing 0.5 M NaCl. The precipitates were analyzed by standard immunoblot procedures using the following antibodies: mouse monoclonal anti-FLAG (Sigma), anti-HA (Origene), anti-NOD2 (Novus), anti-GFP (Invitrogen), rabbit polyclonal anti-TRIM22 (Abnova) and goat polyclonal anti-NOD2 (Ingenix). For endogenous coimmunoprecipitation experiments, HT29 cells (5 × 107) were stimulated with MDP (10μg/mL) for the indicated times or left untreated. The coimmunoprecipitation and immunoblot experiments were performed as described above.
Results
Case History
Patient 1 was born in Australia to consanguineous parents. She initially presented on day 8 of life with fever, oral ulcers, diarrhea and failure to thrive and later developed severe fistulising perianal disease (Figure 1A) and granulomatous colitis (Figure 1B). She deteriorated despite treatment with corticosteroid and biologic therapy. Symptomatic control was only achieved by faecal steam diversion with combined ileostomy and subtotal colectomy. Patients 2 and 3 had a very similar clinical course with granulomatous colitis and severe perianal disease (Figure 1C-D) requiring ileostomy followed by total proctocolectomy. Both Patients 2 and 3 presented with severe anemia and severe hypoalbuminemia secondary to colitis. Patients 1 and 2 had numerous bacterial and viral infections. The three patients did not have mutations in the IL10RA/B, IL10, XIAP, or NOD2 genes and were all very difficult to treat medically and surgically with their disease non-responsive to anti-TNFα biologic therapy (Table S1A; Supplemental Material for full clinical description).
Figure 1. Clinical Features and Pedigrees of Patients with TRIM22 Variants.
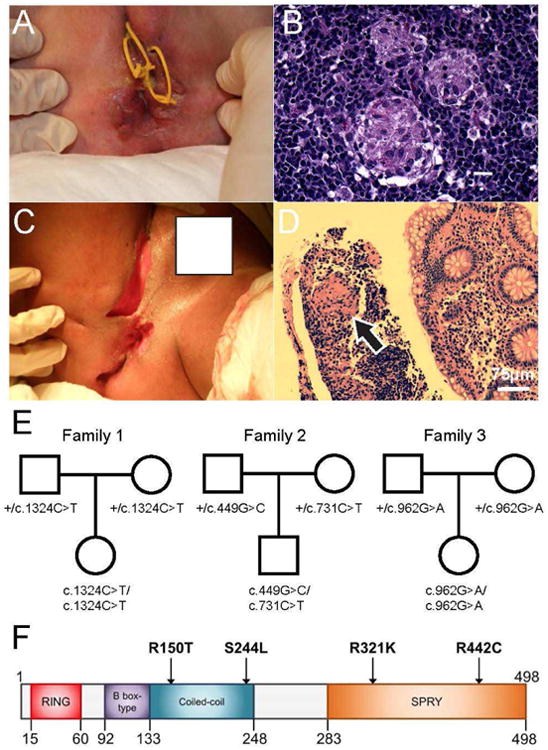
A, B) Clinical presentation and histopathologic findings in colonic biopsy from Patient 1.
Panel A shows severe perianal fistulization with seton placement. Recto-vaginal fistulae were also present. Panel B shows non-caseating granulomas on a background of chronic inflammatory cell infiltrates (H&E stain).
C, D) Clinical presentation and histopathologic findings in colonic biopsy from Patient 2.
Panel C shows multiple, severe cavitating perianal fistulae. Panel D shows a colonic biopsy showing chronic inflammation with non-caseating granuloma (black arrow) (H&E stain, scale bar represents 75 μm).
E) Pedigree analysis of the genetic inheritance pattern for each family trio.
F) Schematic view of the protein domain architecture of TRIM22 with the relative position of each identified variant depicted.
Number indicates amino acid position.
Whole Exome Sequencing
Whole exome sequencing of Patient 1 (and parents) resulted in greater than 100 times mean exome coverage and identified 152,020 variants including 16,097 rare variants (minor allelic frequency MAF < 0.01) predicted to be damaging (Supplemental Figure 1). From this list we identified 12 non-synonymous homozygous variants (Supplemental Table 2) that were inherited in an autosomal recessive manner. Based on known protein function, expression profiles, animal models, mutation conservation, and network analysis (discussed below), this list was narrowed down to a single candidate gene — TRIM22. Patient 1 had a homozygous non-synonymous variant in exon 7 of the TRIM22 gene inherited from both parents (Figure 1E). This missense variant resulted in an arginine to cysteine substitution (c.1324c>T; p.Arg442Cys) predicted to be highly deleterious (Figure 1F and Table S1B). It is highly conserved in a functionally significant region of the exposed beta sheet around the variable loop 3 (V3) in the B30.2 (SPRY) domain thought to be responsible for subcellular localization27, 28 and the 3-dimensional structure suggest it results in an elongated or additional beta strand, which in the wild type, forms a relaxed loop segment.
To validate, we examined WES from 150 VEOIBD patients but did not identify damaging variants in TRIM22. Therefore we performed targeted exome sequencing in ten patients with a similar severe phenotype and identified two additional patients with TRIM22 variants (Patients 2 and 3). Patient 2 had compound heterozygous TRIM22 variants (Figure 1E) including a novel arginine to threonine substitution (c.449G>C, p.Arg150Thr) inherited paternally; and a serine to leucine substitution (c.731C>T; p.Ser244Leu) inherited maternally. Both variants were predicted to be damaging and disrupt the highly conserved coiled-coil region of TRIM22 that facilitates homodimer/heterodimer multimers (Figures 1F, S2, and Table S1B). R150T is in an exposed region and S244L is buried and resides in the spacer-2 region that contains a bipartite nuclear localization sequence and was previously predicted to be a serine phosphorylation site27.
Patient 3 had a homozygous TRIM22 variant inherited from both parents (Figure 1E). This non-synonymous variant resulted in an arginine to lysine substitution (p.Arg321Lys; c.962G>A). The R321K variant was predicted to be deleterious and located in the highly conserved SPRY domain on an exposed residue of a beta sheet that has a functional role in the antiviral activity patch of the SPRY domain27 (Figure 1F; Table S1B).
Integrative Analysis Supports TRIM22 as a Key Regulator in IBD Associated Networks Constructing a VEOIBD-informed network
To gain insight into the role of TRIM22 in the pathogenesis of IBD, we constructed a probabilistic causal network model by integrating 7,568 RNA-sequence derived gene expression traits from ileal biopsies from 322 treatment naïve pediatric Crohn's patients in the RISK cohort19 and expression quantitative trait loci (eQTL). To identify the VEOIBD component of this molecular network, we projected all genes known to be causally associated with VEOIBD9 (Table S3) onto this network, and then identified the most connected subnetwork containing these VEOIBD genes within the full RISK network, resulting in the VEOIBD network comprised of 665 genes, which included TRIM22 (Figure 2A).
Figure 2. TRIM22 - NOD2 Network.
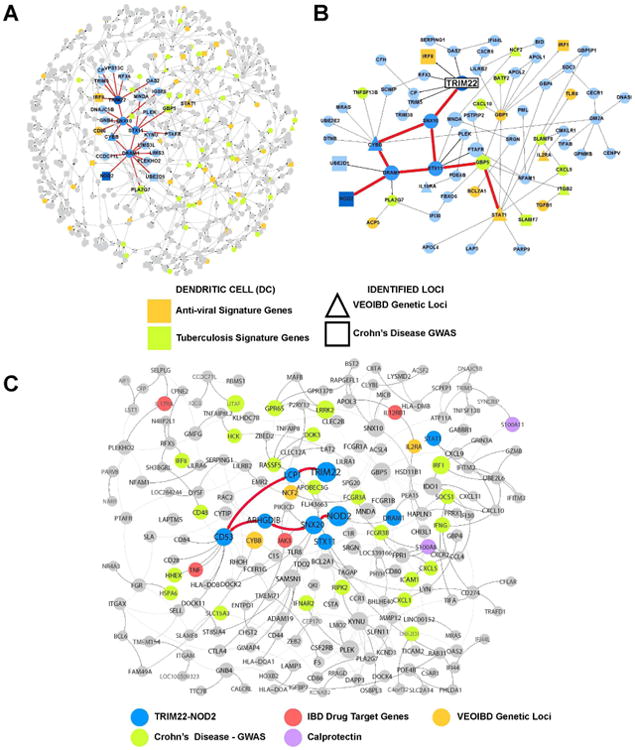
A) VEOIBD loci focused subnetwork within RISK Pediatric Crohn's ileum network.
To define the context of VEOIBD in the ileum network, genes known to be causally associated with VEOIBD (not including TRIM22) were projected onto the RISK network and extended out a path length of two. The resulting VEOIBD focused sub network is comprised of 665 genes and contains TRIM22 and NOD2. The TRIM22-NOD2 paths are highlighted in bold and are connected to the known VEOIBD loci CYBB, and STAT1, as well as the GWAS locus IRF8.
B) Local network structure surrounding the NOD2/TRIM22 paths in the 665-node VEOIBD focused subnetwork.
Representative nodes from the 179-gene TRIM22 network within the 665 node VEOIBD loci focused RISK subnetwork.
C) TRIM22-NOD2 interaction is conserved in the adult IBD intestine.
This network provides the context of the TRIM22/NOD2 interaction in the adult IBD intestine. The 179-gene TRIM22 network was projected and extended out an additional path length on the adult IBD pan intestine Bayesian network. IBD GWAS (green), VEOIBD loci (orange), IBD drug targets approved or in clinical development (red), and calprotectin (S100A8) (purple) are all represented in this conserved subnetwork.
To understand the regulation of this VEOIBD network, we employed a previously established procedure26 to identify key regulator genes that are predicted to modulate the state of the network (Table S4). Interestingly, the most significant key regulators identified in the VEOIBD network included SNX10, STX11, CYBB and DRAM1, which directly connect TRIM22 to the IBD gene NOD2 via two equidistant paths. The first pathway, TRIM22→SNX10→STX11→DRAM1→NOD2, is linked to the known VEOIBD gene STAT1 via STX11 and GBP5, while the second pathway, TRIM22→SNX10→CYBB→DRAM1→NOD2, contains the known VEOIBD gene CYBB (Figure 2B). Both STAT1 and CYBB, in addition to IRF8, which is directly connected to TRIM22, are associated with Mendelian susceptibility to mycobacteria deficiency29. As expected, we did not identify any common DNA variants from the IIBDGC GWAS30 within the TRIM22 locus (Tables S5-6), however, the VEOIBD subnetwork is well represented in genes involved in TRIM22-relevant pathways, including p53, ubiquitin response, and antiviral signaling, which were among the top pathways enriched for genes associated with intestine and blood expression quantitative trait loci (eQTL) that were also associated with adult IBD loci identified in the IIBDGC cohort (Figure S3, Table S7). Further, we found that the VEOIBD network was 2.1 fold enriched (p = 2.3 e-5) for Crohn's genes annotated from adult IBD genetic studies that also give rise to blood eQTLs31 (Table S8).
Adult-onset IBD informed by a TRIM22-centered network
Given preliminary support for adult IBD in the VEOIBD network, we explored whether the TRIM22 component of the VEOIBD network was associated with adult IBD. We constructed an adult IBD causal gene network from gene expression and genotype data generated on 203 intestinal biopsies that included inflamed and non-inflamed tissues from ileum, ascending colon, descending colon, transverse colon, sigmoid and rectum. All of these tissues, in addition to blood, were collected at baseline from 54 anti-TNFα resistant Crohn's patients enrolled in the ustekinumab (IL12/IL23 p40 monoclonal antibody) clinical trial18 (Figure S4). We found that TRIM22 was a significantly up-regulated (1.33, p=1.37e-5), key driver (p = 4.0e-6) gene in inflamed sigmoid and inflamed rectum (1.24, p=1.83e-6), (p = 1.0e-4) compared to the non-inflamed samples of these same tissues.
We identified the homologous TRIM22 component of the above adult IBD network by first identifying all genes within a path length of 5 to TRIM22 in the VEOIBD network, which included NOD2, leaving a network comprised of 179 genes (referred to here as the TRIM22 network; Table S9). This TRIM22 network contained many macrophage/monocyte expressed genes associated with mycobacterial and anti-viral responses (including genes involved in autophagy, NF-κB pathway and apoptosis; Figure S5; Table S10). Strikingly, of the 480 key driver genes we identified in the adult IBD network, 62 of these genes, including STX11, CYBB, and DRAM1 were represented in the pediatric TRIM22 network, 7.2-fold enrichment over what is expected by chance (p = 6.5 e-38) (Table S11). Further, the TRIM22 containing co-expression module from the blood of ustekinumab patients, which was 12.7-fold enriched for response to virus (p = 6.3e-14), was 9-fold enriched in the pediatric TRIM22 network (p < 1e-16). Further supporting the pediatric TRIM22 network's association to viral response was a 3.2-fold enrichment (p = 2.5e-5) of eQTLs associated with dendritic cell response to stimulation with flu or interferon beta32 and an 8.3-fold enrichment (p = 1.1e-16) of genes in a tuberculosis infection signature in dendritic cells30.
The homologous TRIM22 network in the adult IBD network was then identified by projecting nodes from the TRIM22 network onto the adult IBD network, identifying those nodes that were either in the TRIM22 network or directly connected to such nodes (Figure 2C). In this TRIM22 pediatric oriented adult IBD network, TRIM22 is in close proximity to several drug targets that are either approved or in clinical development for IBD, including TNFα, the target of adalimumab (humira), certolizumab (cimzia), infliximab (remicade), and colimumab (simponi); IL12Rβ, a chain shared between the receptors that bind IL12/23, the molecular target of ustekinumab; JAK3, the target of tofacitinib; and IL17RA, the target of brodalumab.
Since S100A8 (calprotectin), a stool biomarker for inflammation, is also co-localized with TRIM22 in this network33-35 (Figure 2C), we decided to further investigate the role of TRIM22 in treatment refractory patients, exploring associations between TRIM22 and IBD-associated clinical traits in the ustekinumab clinical trial (Tables S12). We found that expression levels of TRIM22 in the colon were positively correlated with fecal calprotectin regardless of biopsy inflammation status. Overall, both inflamed and non-inflamed baseline intestine levels of TRIM22 revealed a correlation to blood c-reactive protein (CRP) of which consistently high levels is associated with subsequent development of perianal fistulae in Crohn's patients36 and response to anti-TNFα37. TRIM22 expression levels in blood were positively correlated with Crohn's disease activity index (CDAI) (Tables S13).
Taken together, the intestinal network models provide causal support for the TRIM22 regulation of NOD2 and the correlation of the TRIM22-NOD2 pathway with predicted disease regulators, functionally relevant VEO and adult onset IBD genetic loci, effectors of anti-viral and mycobacterial function, correlation with inflammatory clinical variables and validated IBD drug targets.
Identification of TRIM22 as a NOD2 Interacting Protein
As NOD2 signaling plays a critical role in Crohn's disease susceptibility38, 39; we further investigated this in silico TRIM22-NOD2 interaction. Immunofluorescence showed that NOD2 and TRIM22 colocalized in colonic biopsies and in the cytoplasm of transient transfected HEK293 cells (Figure S6A-C). This NOD2 immunostaining was blocked by a NOD2-specific peptide and TRIM22 did not colocalize with NOD1 (Figure S6D-F) another member of the NOD family confirming the specificity of this TRIM22-NOD2 interaction. Immunostaining of colonic biopsy samples from Patients 1-3 with TRIM22 variants showed altered expression and localization of NOD2 and TRIM22 (Figure S6A-B).
TRIM22 Influences NOD2 Signaling Pathways
NOD2 senses muramyl dipeptide (MDP), a conserved structure in peptidoglycan present in most Gram-negative and Gram-positive bacteria. We found that TRIM22 was weakly associated with endogenous NOD2 in co-immunoprecipitation experiments in unstimulated HT29 cells, a human intestinal epithelial cell line, and this association increased after stimulation with MDP (Figure 3A). Furthermore, transiently transfected TRIM22 and NOD2 were found to reciprocally co-immunoprecipitate and these interactions were reduced with the TRIM22 variants (Figure 3B-C and S7A).
Figure 3. TRIM22 Interacts with and Activates NOD2 Signaling.
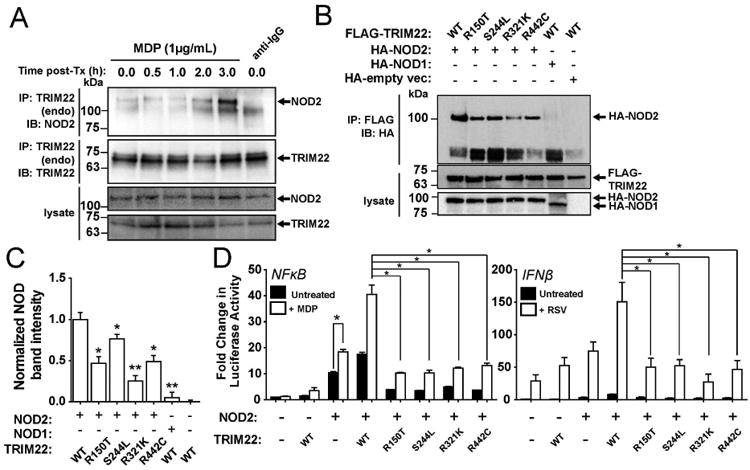
A) Endogenous NOD2 and TRIM22 co-immunoprecipitate following MDP stimulation.
HT29 cells were stimulated with MDP (1 μg/mL) for various time periods. Following treatment,cells were lysed and endogenous TRIM22 was immunoprecipitated using anti-TRIM22 antibody conjugated to Protein G-Sepharose. Bound levels of endogenous NOD2 were evaluated using Western blotting. As stimulation time with MDP increased, the binding of endogenous NOD2 toTRIM22 increased. Immunoprecipitation with IgG bound to Protein G-Sepharose was used as anegative IP control.
B) TRIM22 interacts with NOD2.
HEK293T cells were transiently co-transfected with HA-NOD2 and either wildtype (WT) orvariant (R150T, S244L, R321K, and R442C) FLAG-epitope tagged TRIM22. Twenty-four hours post-transfection, cells were lysed followed by immunoprecipitation of the FLAG-TRIM22 byanti-FLAG-agarose. Reduced binding of NOD2 to TRIM22 was observed in varying degrees for each variant compared to the wildtype protein. HA-NOD1 and an empty vector construct were used as negative controls.
C) Quantification of NOD2 co-immunoprecipitation with wild-type or mutant
TRIM22. Densitometry analysis was performed on Western blots performed in 3B. Each TRIM22 point mutation significantly reduced NOD2 co-immunoprecipitation (Student's t-test,*p<0.05, **p<0.01, n=3).
D) TRIM22 mutants fail to activate NF-κB and IFNβ downstream of NOD2. HCT116 cells were transiently co-transfected with (or without) NOD2, TRIM22-WT, or TRIM22 point mutants (R150T, S244L, R321K, R442C). Twenty-four hours post transfection, cells were stimulated with either MDP (10 μg/mL) or respiratory syncytial virus (RSV) (0.1 μg) for 18 hr before NF-κB or IFNPβ promoter activation was evaluated by luciferase assay. Cells transfected with TRIM22 mutants showed significantly reduced NF-κB and IFNPβ promoter activation following stimulation compared to wild type TRIM22 transfected controls (*p < 0.001 after Bonferronipost-hoc testing, n=3).
Since TRIM22 co-localized and co-immunoprecipitated with NOD2, we next determined if TRIM22 influenced MDP-induced NF-κB signaling via NOD2. In reporter assays, co-expression of TRIM22 and NOD2 enhanced MDP-induced activation of the NF-κB (Figure 3D) in HEK293 cells. As TRIM22 regulates viral activity15-17 and NOD2 functions as cytoplasmic viral pathogen recognition receptor by triggering interferon-β (IFNβ) activation in response to Respiratory Syncytial Virus (RSV)40, 41, we next determined whether TRIM22 also regulated NOD2-dependent RSV-induced IFNβ signaling. As shown in Figure 3D, overexpression of TRIM22 and NOD2 both enhanced RSV-induced activation of the IFNβ promoter and the Interferon Stimulated Response Element (ISRE). Neither wildtype (WT)-TRIM22 nor the TRIM22-R442C variant enhanced TNFα gene expression in response to TNFα (Figure S7B). As TNFα activates NF-κB target genes independent of NOD2 signaling42, this suggested that TRIM22 regulation of MDP-induced NF-κB activation is NOD2-dependent.
TRIM22 Variants Influence NOD2 Signaling
We next examined the role of TRIM22 to regulate NOD2-dependent NF-κB and IFNβ signaling. Luciferase assays using HEK293 cells and real-Time PCR experiments using HCT116 showed that expression of wild type TRIM22 potentiated NOD2 signaling in response to both MDP-induced NF-κB and RSV-induced IFNβ activation (Figures 3D and S7C-F). However, the TRIM22 variants (Figures 3D and S7C-E) and TRIM22 knockdown using short hairpin RNA (shRNA; Figure S8A) showed impaired NOD2-dependent NF-κB and IFNβ signaling (Figure S8B). MDP-mediated NOD2-dependent signaling was also significantly reduced in only Patient 2 with the heterozygous TRIM22 variants in the coiled-coil domain (Figure S9 and Supp. Material). Together this data suggests that TRIM22 is a positive regulator of antiviral and antibacterial NOD2-dependent signaling pathways.
TRIM22 mediates K63-linked Polyubiquitination of NOD2
As TRIM22 is a RING finger E3 ubiquitin ligase11 and NOD2 has high confidence predicted ubiquitination sites at K436 and K445 (CKSAAP_UbSITE43), we next determined if TRIM22 mediated ubiquitination of NOD2. Immunoprecipitation experiments using a HEK293 cell line co-expressing HA-NOD2 and Flag-TRIM22 showed that overexpression of TRIM22 enhanced NOD2 polyubiquitination (Figure 4A). The TRIM22 variants (Figure 4A and S10A) were unable to mediate this NOD2 polyubiquitination. TRIM22 did not increase polyubiquitination of other components (MAVS or RIPK2) of the NOD2 signaling pathways known to be ubiquitinated44, 45 (Figure S10B). Using the TRIM22 shRNA HT29 stable cell line, we found that only wildtype TRIM22 rescued NOD2 polyubiquitination and not the TRIM22-R442C variant (Figure 4B). Taken together, this suggested that TRIM22 specifically mediates NOD2 polyubiquitination.
Figure 4. TRIM22 Mediates K63-linked NOD2 Polyubiquitination.
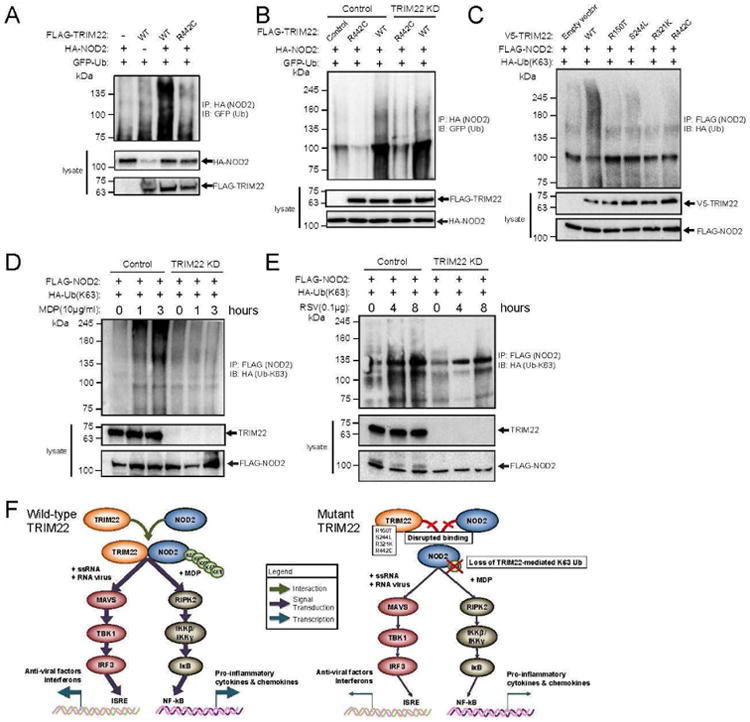
A) TRIM22 enhances NOD2 polyubiquitination in HEK293 cells.
HEK293T cells were transiently co-transfected with HA-NOD2, GFP-ubiquitin, and either wildtype (WT) or variant (R442C) FLAG-TRIM22. Twenty-four hours post-transfection, cells were lysed followed by immunoprecipitation of HA-NOD2. Ubiquitination of NOD2 was evaluated by immunoblotting for GFP (ubiquitin). Co-transfection with wildtype TRIM22 enhanced ubiquitination of NOD2, which was abrogated with the R442C variant TRIM22. All experiments were carried out in triplicate, independent experiments.
B) Transgenic TRIM22 rescues NOD2 polyubiquitination in TRIM22-shRNA-HT29 cells.
HT29 cells stably transduced with control (scramble) or TRIM22 shRNA were transiently co-transfected with HA-NOD2 and either wildtype (WT) or variant (R442C) FLAG-TRIM22. Twenty-four hours post-transfection, cells were lysed followed by immunoprecipitation of the HA-NOD2. Evaluation of NOD2 poly-ubiquitination by GFP immunoblotting demonstrated rescued ubiquitination following transfection of WT-TRIM22 in the TRIM22 shRNA cell line that was not observed following transfection of the variant R442C. All experiments were carried out in triplicate, independent experiments.
C) Variants in TRIM22 abrogates K63-linked NOD2 polyubiquitination.
HEK293T cells were transiently co-transfected with FLAG-NOD2, HA-(K63-specific) ubiquitin, and either wildtype (WT) or variant (R150T, S244L, R321K, or R442C) V5-TRIM22. Twenty-four hours post-transfection, cells were lysed followed by immunoprecipitation of the FLAG-NOD2. Evaluation of NOD2 K63 specific-ubiquitination by HA immunoblotting demonstrated significantly reduced ubiquitination in cells co-transfected with TRIM22 variants compared to wildtype. All experiments were carried out in triplicate, independent experiments.
D and E) TRIM22-shRNA reduces MDP- and RSV-induced K63-linked NOD2 polyubiquitination.
HEK293T cells stably transduced with control (scramble) or TRIM22 shRNA were transfected with HA-tagged K63-specific ubiquitin and FLAG-NOD2. Forty-eight hours after transfection, cell lysates were stimulated by MDP (D) or RSV (E) for indicated the time points followed by lysis and immunoprecipitation using anti-FLAG-agarose. K63-poly-ubiquination of NOD2 was evaluated by immunoblotting using anti-HA antibody. Following stimulation by either MDP or RSV, NOD2 K63-specific ubiquitination increased over treatment time. However, in TRIM22 knockdown cells, this ubiquitination was significantly reduced or absent. All experiments were carried out in triplicate independent experiments and blots are representative.
F) Schematic overview of the proposed impact of TRIM22 and its identified variants on the NOD2 signaling axis.
Wildtype TRIM22 binds and mediates the K63-specific polyubiquitination of NOD2, potentiating downstream signaling through the MAVS and RIPK2 signaling axes depending on antigen. TRIM22 variants disrupt binding to NOD2, abrogating this polyubiquination thus attenuating downstream pro-inflammatory signaling
K63-linked polyubiquitination regulates the localization and signaling activity whereas K48-linked polyubiquitination often mediates degradation46. NOD2 has been previously shown to be K48-linked ubiquitinated by TRIM27 and results in NOD2 degradation47. Using ubiquitin mutants, we showed that overexpression of TRIM22 mediated K63-linked but not K48-linked polyubiquitination of NOD2 while the TRIM22 variants were unable to mediate K63- or K48-linked NOD2 polyubiquitination (Figure 4C and S11A). We next determined that TRIM22 is required for MDP- and RSV-induced K63-linked NOD2 polyubiquitination and knockdown of TRIM22 reduced this MDP- and RSV–induced K63-linked (Figure 4D-E), but not K48-linked NOD2 polyubiquitination (Figures S11B-C).
Discussion
TRIM proteins are involved in a number of biological processes, including cell proliferation, apoptosis, innate immunity, autoimmunity, and inflammatory response48, 49. Variants in TRIM genes result in monogenic diseases including familial Mediterranean fever (FMF) and Opitz syndrome type 1, and are implicated in a number of cancers and autoimmune diseases including multiple sclerosis, rheumatoid arthritis, and systemic lupus erythematosus48, 49. TRIM22 belongs to a subfamily of TRIM proteins that contain a SPRY domain and are thought to have evolved to limit innate immune responses to viruses20. Mutations in the SPRY domain of TRIM20 (pyrin) result in autoimmune diseases including FMF50. Overexpression of TRIM22 is known to activate NF-κB in a dose-dependent manner and both the N-terminal RING domain and C-terminal SPRY domain are crucial for TRIM22-mediated NF-κB activation13 and antiviral activity15-17, and are implicated in the pathogenesis of autoimmune diseases51. Patients 1 and 3 both had homozygous variants in the SPRY domain and Patient 2 had compound heterozygous variants both predicted to disrupt the coiled-coil domain and had loss of NOD2-dependent MDP signaling. The characterized TRIM22 variants resulted in impaired NOD2 binding and K63-linked NOD2 polyubiquitination. The predicted NOD2 polyubiquitination sites described earlier are in the NACHT domain, a conserved domain required for NOD2 conformational change in response to MDP sensing through the LRR domain or interaction with binding adaptor proteins for NOD252. Therefore, as all identified TRIM22 variants disrupt both NOD2-dependent NF-κB and IFNβ signaling, this suggests that K63-linked polyubiquitination of NOD2 meditated by TRIM22 is critical for NOD2 function (Figure 4F).
NOD2 has long been recognized as a critical player in Crohn's disease pathogenesis38, 39 where it is proposed to regulate innate immunity through NF-κB-induced pro-inflammatory responses triggered by PGN (reviewed in53). Numerous studies have shown that the loss of function variants in NOD2 associated with Crohn's disease result in the loss of NF-κB-induced pro-inflammatory cytokine response to MDP (reviewed in54, 55) and therefore mirror the defect we observe in Patient 2 with TRIM22 mutations in the coiled-coil domain. Similarly, mutations in XIAP (also an E3 ubiquitin ligase) are associated with IBD with granulomatous colitis and perianal disease56, 57 and are associated with loss of NOD2-dependent mediated NF-κB signalling58-60 and XIAP is also regulated by TRIM proteins61. XIAP deficiency usually presents in early childhood but there are cases of patients presenting with IBD at over 40 years of age9 with and without hemophagocytic lymphohistiocytosis (HLH) (reviewed in 62). Furthermore, XIAP not only regulates NOD2-dependent innate immunity responses but also is proposed to regulate inflammasome and adaptive immunity62. Loss of function NOD2 variants associated with IBD have low penetrance and do not cause the severe early onset disease observed in this group with either TRIM22- or XIAP-deficiency. Therefore it is most likely that in Patients 1 and 3, with TRIM22 SPRY domain variants, defects in other pathways regulated by TRIM22 result in the early onset and severity of disease. Alternatively hypomorphic TRIM22 variants act in an oligogenic manner with mutations in other genes similar to what has been observed with hypomorphic XIAP mutations63 and as observed in Patient 3 who is also a carrier of a XIAP mutation. Together, this suggests that loss of NOD2-dependent mediated signaling in response to MDP contributes to a severe form of IBD and that other yet defined pathways must also be involved in disease pathogenesis. With regards to TRIM22 specifically, we anticipate that other pathways not identified in these studies play a role in disease pathogenesis.
Our computational studies further support our functional studies demonstrating that TRIM22 influences both NOD2-dependent IFNβ and NF-κB signaling pathways. While the loss of function TRIM22 variants in our VEOIBD patients may manifest as an immunodeficiency, the inflammation and tissue damage that ensues from a dysregulated response to microbial burden, may resemble the profile of adult-onset IBD. The functional outcome of pro-inflammatory NF-κB activation may differ depending on the cell population in which TRIM22 is expressed, with circulating plasmacytoid dendritic cells (DCs) and monocytes predominating in the blood and resident intestinal DCs, macrophages and enterocytes in the intestine. We see from the intestine network that the regulation of causal TRIM22 results in modulation of a mycobacterial and viral response subnetwork that may also result in feedback to TRIM22 expression. Thus, either due to immunodeficiency arising from the TRIM22 variants or excessive inflammation in non-TRIM22 variant carried by anti-TNFα refractory patients, TRIM22 driven NOD2 activation of NF-κB in response to MDP or viral stimuli, contributes to dysregulation of NF-KB and IFNβ driven pathways as reflected in the networks. Together, this demonstrates that the TRIM22-NOD2 network plays a critical role in the development of IBD.
Investigation of VEO and adult onset IBD in treatment refractory disease will elucidate new treatment opportunities by revealing complexity of disease network dysregulation. Causal and correlative based network analysis allows for consideration of the synergistic effects between genes in disease, which can be a greater determinant than evaluation of single target genes, to inform disease drivers and opportunities for combinatorial therapy. Thus network analysis can be focused by novel variants, as seen with TRIM22 identified through WES in VEOIBD patients to generate hypothesis and potentially identify biomarkers and guide drug discovery and repositioning in both VEO and adult-onset IBD patient subsets.
Supplementary Material
Table S1. Clinical (A) and Genetic Features (B) of Patients with TRIM22.
MAF minor allelic frequency based on 1000 Genomes and 6503 healthy individuals genotyped in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu). The variant prediction are based Polyphen218, SIFT (http://sift.jcvi.org), GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/), and PhyloP (http://ccg.vital-it.ch/mga/hg19/phylop/phylop.html).
Table S2. Autosomal Recessive Variants Identified by WES in Patient 1.
Table S3: VEO Genes
See excel sheet
Table S4. VEOIBD Key Drivers
See excel sheet
Table S5. IIBDGC GWAS Enrichment in TRIM22 Locus
See excel sheet.
Table S6. IIBDGC GWAS Enrichment in TRIM22 eQTL Enrichment
See excel sheet
Table S7. eQTL Pathway Enrichment
See excel sheet
Table S8. Crohn's GWAS Enriched Blood eQTL Overlap with the VEO Network
Table S9. Annotation of 179 VEO Network
See excel sheet
Table S10. Functional Categories of Genes in the TRIM22 Sub network
Table S11. Inflamed Sigmoid and Rectum Key Drivers
Table S12. Correlations of the Expression of TRIM22 to the Clinical Response Characteristics Stratified by Distinct Intestine Anatomy Sites (Includes Both Inflamed and Non Inflamed Samples).
Table S13. Correlation of the Expression of TRIM22 in blood to CDAI
Figure S1: Variant Identification Pipeline for WES of Patient 1.
Whole exome sequencing identified 152,020 exonic variants in Patient 1. Eliminating low-quality and synonymous variants further refined the WES variant set. To isolated potential causative variants, only rare and predicated damaging (by dbNSFP) variants were included in inheritance analysis. Variants identified according to each inheritance model are shown.
Figure S2: Heterozygous TRIM22 Variants R150T and S244L Occur in the Coiled-coil Domain and May Affect Domain Oligomerization.
A) Protein sequence for TRIM22 indicating the position of the two coiled-coil regions termed region 1 (green) and region 2 (purple) including the position of R150 and S244.
B, C) PrOCoil analysis of coiled coil region 1 (B) and region 2 (C) for TRIM22 with the corresponding variants reveals potential alterations in domain oligomerization patterns. PrOCoil evaluates the relative weighted contribution of each amino acid to the oligomeric character of the coiled-coil domain. The introduction of the R150T and S244L variants alter the weighted effect of positions near the site of variant. These alterations have potential to impact the ability of the coiled-coil domain to fold correctly.
Figure S3: Blood and Intestine Derived eQTLs: Gene Set Enrichment Analysis of Genes Controlled by IBD SNPs (Metacore).
See Table S7 for genes in pathway.
Top enriched gene sets among genes controlled by IBD SNPs: NHGRI catalog entries for IBD, UC and CD, plus all WTCCC IBD GWAS SNPs with p-value ≤ 0.001. 362 genes showed IBD SNP regulation in the blood, 98 in the intestine (56 genes were shared between the two tissues). Log10 p-value of enrichment on the horizontal axis: intestine (blue bar) and blood (orange bar).
Different Gene Set database categories are reported in each panel: TRIM22 Pathways: See below
1. Transcription P53 signaling (TRIM22 is a P53 target gene)
4. Immune response: Antiviral actions of interferons
9. Ubiquitin Pathway
Figure S4: Computational Design.
A) Inflamed and non inflamed tissue biopsies were collected from anti TNFα refractory Crohn's patients enrolled in clinical trial for ustekinumab (anti P40- IL12/IL23).
B) Bayesian pan intestine network was constructed from microarray expression profiling on biopsies collected from various regions of the intestine.
C) Differential expression was performed on inflamed vs. non-inflamed for each region of the intestine collected.
D) Differential expression signatures of genes up regulated in inflamed sigmoid and inflamed rectum were projected on Bayesian ileum network for key driver analysis.
Figure S5: First Neighbors of TRIM22 and NOD2 in VEOIBD Intestine Network.
Genes within one path of TRIM22 in the VEOIBD RISK network have putative functional roles in autophagy.
Figure S6: TRIM22 and NOD2 Co-localize in Healthy Controls and Disrupted in Patient Tissue Sections.
A, B) Immunofluorescence analysis of TRIM22 and NOD2 in colonic biopsy samples from Healthy Controls and Patients.
TRIM22 (red) and NOD2 (green) localization was analyzed in the villus (A) and crypts (B) of both healthy controls and Patients 1-3. TRIM22 and NOD2 colocalization was observed in the both the villus and crypts of the healthy control biopsy samples. Both TRIM22 and NOD2 expression and colocalization were reduced in the patient biopsy samples Scale bars represent10 μm.
C) Exogenous TRIM22 co-localize with Exogenous NOD2.
HEK293T cells seeded on coverslips were co-transfected with FLAG-TRIM22 and HA-NOD2. TRIM22 (red) and NOD2 (green) immunostaining show strong cytoplasmic colocalization. Scale bars represent 10μm.
D) TRIM22 does not co-localize with NOD1.
To evaluate the specificity of the observed TRIM22 and NOD2 co-localization, immunofluorescence microscopy was performed on HEK293T cells transiently expressing FLAG-TRIM22 and HA-NOD1, a NOD family member that does not interact with TRIM22. TRIM22 (red) and NOD1 (green) staining did not show significant overlap. Scale bars represent 10μm.
E, F) Peptide inhibition of NOD2 in colonic biopsy samples of Healthy Controls and Patients.
To demonstrate NOD2 antibody specificity, endogenous NOD2 immunostaining was performed on health and disease control biopsy samples as well as Patients 1-3 in the presence or absence of a NOD2 antibody peptide inhibitor. Without peptide incubation, NOD2 showed a cytoplasmic expression in healthy controls and disease controls, and is reduced in the patients. The specific NOD2 peptide is blocking the antigen-antibody binding and therefore no staining is detected when the peptide is added concurrently.
Figure S7: TRIM22 Variants and shRNA Abrogate NOD2 Signaling.
A) NOD2 co-immunoprecipitates TRIM22 from in vitro cell lysates after transient co-expression.
HEK293T cells were transfected with HA-epitope tagged NOD2 and either wild type (WT) or mutant (R150T, S244L, R321K, R442C) FLAG-epitope tagged TRIM22 constructs. Twenty-four hours post-transfection, cells were lysed and NOD2 was immunoprecipitated with anti-HA antibody. Western blotting identified bound proteins. WT-TRIM22 interacted with and pulled-down NOD2; however, mutant TRIM22 showed reduced or absent NOD2 binding. NOD1, which does not interact with TRIM22, and an empty vector construct were included as negative controls. All lucerifase assay and Western blotting experiments were carried out in triplicate independent experiments and blots are representative.
B) TRIM22 overexpression has no effect on TNFα mRNA levels following stimulation by TNFα.
HCT116 cells were transiently transfected with control (empty vector), TRIM22-WT, or TRIM22-R442C plasmids. Twenty-four hours post-transfection, cells were stimulated for 18 hr with TNFα (20 ng/mL). mRNA of TNFα were assessed by Q-PCR. No significant increase in TNFα mRNA was observed following TNFα stimulation of cells overexpressing TRIM22.
C, D, E) TRIM22 mutants reduce mRNA level of genes downstream of the NOD2 signalling.
HCT116 cells were transiently co-transfected with NOD2 and TRIM22-WT or TRIM22 mutants (R150T, S244L, R321K, R442C). Twenty-four hours post-transfection, cells were stimulated with MDP (10 μg/mL) (C) or RSV (0.1 μg) (D, E) for 18 hr and then qPCR were performed. Cells transfected with TRIM22 mutants showed significantly reduced expression of TNFα (C), IL-6 (D), and ISG15 (E) (*p < 0.001 after Bonferroni post-hoc testing).
F) Overexpression of TRIM22 increases ISRE activation. HEK293T cells were transiently transfected with control (empty vector), TRIM22, or NOD2. ISRE reporter activation was evaluated by luciferase assay following stimulation with respiratory syncytial virus (RSV) (0.1 μg). TRIM22 overexpression significantly increased ISRE reporter activation compared to empty vector control (*p<0.001 after Bonferroni post-hoc testing). All experiments were carried out in triplicate, independent experiments.
Figure S8: TRIM22 shRNA Knockdown.
A) To demonstrate the degree of knockdown present in cells expressing TRIM22 shRNA,HEK293T cells were transiently transfected with control (scramble) or various TRIM22 shRNA clones for 48 hours. Western blotting analysis was performed using anti-TRIM22 antibodies with β-actin staining as a loading control.
B) Knockdown of TRIM22 reduces mRNA level of genes downstream of the NOD2. mRNA ofTNFα, IL-6, and ISG15were assessed by qPCR in HT29 cells stably transduced with control (scramble) or TRIM22 shRNA following stimulation by MDP (10 μg/mL) or respiratory syncytial virus (RSV) (0.1 μg). (*p<0.001 after Bonferroni post-hoc testing).
Figure S9: TRIM22 Variants Abrogates NOD2 Signaling in Patient 2 with Coiled-coil Variants.
Representative FACS blots and quantification of TNFα production in monocytes following stimulation with NOD2 and TLR4 ligand. Monocytes obtained from Patient 2 did not generate TNFα production in monocytes following stimulation with NOD2 and TLR4 ligand. Monocytes obtained from patient in response to L18-MDP stimulation but demonstrate normal reactivity to LPS. The patient's father and healthy donors are shown as controls. Monocytes are gated for single, live, CD14+, HLA-DR+ cells.ΔTNF (%) is determined by subtracting frequency of TNF-positive monocytes in stimulated from unstimulated conditions. Overnight travel of blood sample from Patient 2 and father of Patient 2 caused high baseline TNF without affecting results calculated asΔTNF (%). Grey background indicates normal range. Similar results with patient 2 were obtained in two independent experiments performed ten months apart. Open symbols L18-MDP stimulation, closed symbols LPS stimulation. Similar results were not obtained from Patient 1 and Patient 3 was not tested as she did not consent to further testing.
Figure S10: TRIM22 Directly Mediates the Polyubiquitination of NOD2 but not MAVS or RIPK2.
A) Wildtype TRIM22 enhances polyubiquitination of NOD2 in HEK293 cells.
HEK293 cells were transiently co-transfected with HA-NOD2, GFP-ubiquitin, and either wildtype (WT) or variant (R150T, S244L, R321L or R442C) FLAG-TRIM22. Following cell lysis, NOD2 was immunoprecipitated using anti-HA antibody bound to Protein G-Sepharose and NOD2 ubiquitination was observed by GFP immunoblotting. Co-transfection with WT-TRIM22 increased NOD2 ubiquitination that was lost in cells transfected with the TRIM22 variants.
B) TRIM22 does not mediate the ubiquitination of MAVS or RIPK2.
To determine if TRIM22 also regulates downstream NOD2 signaling modulators, HEK23T cells were co-transfected with FLAG-TRIM22, GFP-ubiquitin, and either HA-MAVS or HA-RIPK2. Twenty-four hours post-transfection, cells were lysed followed by immunoprecipitation of MAVS or RIPK2 using anti-HA-agarose. Immunoblotting for GFP-ubiquitin demonstrated no significant increase compared to empty TRIM22 vector controls. All experiments were carried out in triplicate independent experiments and blots are representative.
Figure S11: TRIM22 Does Not Mediate K48-linked Polyubiquitination of NOD2.
A) TRIM22 does not mediate K48-specific polyubiquitination of NOD2.
HEK293T cells were transiently co-transfected with FLAG-NOD2, HA-(K48-specific) ubiquitin, and either wildtype (WT) or variant (R150T, S244L, R321K, or R442C) V5-TRIM22. Twenty-four hours post-transfection, cells were lysed and NOD2 was immunoprecipitation with anti-FLAG-agarose. Immunoblotting for HA-tagged ubiquitin demonstrated no significant increase in cells transfected with WT-TRIM22 compared to empty vectors controls. Likewise, TRIM22 variants did not show significant K48-ubiquitin staining.
B, C) TRIM22-shRNA does not reduce MDP and RSV induced K48-linked polyubiquitination of NOD2.
HEK293T cells stably transduced with control (scramble) or TRIM22 shRNA were transfected with a HA-tagged K48-specific ubiquitin construct and FLAG-NOD2. Forty-eight hours post transfection, cell lysates were stimulated with MDP (B) or RSV (C) for the indicated time points followed by cell lysis and immunoprecipitation of NOD2 by anti-FLAG-agarose. In both control and TRIM22 knockdown cells, stimulation did not result in increased NOD2 K48-specific poly-ubiquitination. Western blotting experiments were carried out in triplicate, independent experiments.
Acknowledgments
Grant Support: The authors thank the patients and their families described here from Canada and Australia. The authors thank Karoline Fielder and Dr. Amanda Charlton for assistance with patient related materials. Thanks to Neil Warner for critical reading of the manuscript. CK/DK were supported by the Deutsche Forschungsgemeinschaft (SFB1054) and BioSysNe. HHU is supported by the Crohn's & Colitis Foundation of America (CCFA). TS is supported by the Deutsche Forschungsgemeinschaft (SCHW1730/1-1). QL and AE are supported by a Crohn's and Colitis Canada (CCC), Canadian Association of Gastroenterology (CAG), and Canadian Institute of Health Research (CIHR) Fellowship. AMM is funded by a CIHR – Operating Grant (MOP119457) and AMM, CK, SBS, ES, and HHU are funded by the Leona M. and Harry B. Helmsley Charitable Trust to study VEOIBD.
Footnotes
QL, CHL, LAP, LAM, CT, AE, TS, DRS, JZ, BZ, YZ, KH, AD, GH, BK, RM, ZAA, CG, DK carried out all the experiments under supervision of MC, RD, CB, SBS, CK, MH, RN, JB, BSN, MW, KJG, HHU, EES, and AMM. EC, TDW, FLD, EH CMR, CD, AMG, KJG provided clinical care or clinical testing. All authors provided critical insight and interpretation of the experimental data. The manuscript was written by LAP, QL, CHL, and EES and AMM with contributions from all authors.
Conflict of Interest: none
Author names in bold designate shared co-first authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Avitzur Y, Guo C, Mastropaolo LA, et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology. 2014;146:1028–39. doi: 10.1053/j.gastro.2014.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elkadri A, Thoeni C, Deharvengt SJ, et al. Mutations in Plasmalemma Vesicle Associated Protein Result in Sieving Protein-Losing Enteropathy Characterized by Hypoproteinemia, Hypoalbuminemia, and Hypertriglyceridemia. Cell Mol Gastroenterol Hepatol. 2015;1:381–394 e7. doi: 10.1016/j.jcmgh.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Janecke AR, Heinz-Erian P, Yin J, et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum Mol Genet. 2015;24:6614–23. doi: 10.1093/hmg/ddv367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dhillon SS, Fattouh R, Elkadri A, et al. Variants in nicotinamide adenine dinucleotide phosphate oxidase complex components determine susceptibility to very early onset inflammatory bowel disease. Gastroenterology. 2014;147:680–689 e2. doi: 10.1053/j.gastro.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Dhillon SS, Mastropaolo LA, Murchie R, et al. Higher activity of the inducible nitric oxide synthase contributes to very early onset inflammatory bowel disease. Clin Transl Gastroenterol. 2014;5:e46. doi: 10.1038/ctg.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Moran CJ, Walters TD, Guo CH, et al. IL-10R polymorphisms are associated with very-early-onset ulcerative colitis. Inflamm Bowel Dis. 2013;19:115–23. doi: 10.1002/ibd.22974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muise AM, Walters T, Xu W, et al. Single nucleotide polymorphisms that increase expression of the guanosine triphosphatase RAC1 are associated with ulcerative colitis. Gastroenterology. 2011;141:633–41. doi: 10.1053/j.gastro.2011.04.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Muise AM, Xu W, Guo CH, et al. NADPH oxidase complex and IBD candidate gene studies: identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut. 2012;61:1028–35. doi: 10.1136/gutjnl-2011-300078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Uhlig HH, Schwerd T, Koletzko S, et al. The Diagnostic Approach to Monogenic Very Early Onset Inflammatory Bowel Disease. Gastroenterology. 2014 doi: 10.1053/j.gastro.2014.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Glocker EO, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–45. doi: 10.1056/NEJMoa0907206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan Z, Gao B, Xu W, et al. Identification of TRIM22 as a RING finger E3 ubiquitin ligase. Biochem Biophys Res Commun. 2008;374:502–6. doi: 10.1016/j.bbrc.2008.07.070. [DOI] [PubMed] [Google Scholar]
- 12.Sawyer SL, Emerman M, Malik HS. Discordant evolution of the adjacent antiretroviral genes TRIM22 and TRIM5 in mammals. PLoS Pathog. 2007;3:e197. doi: 10.1371/journal.ppat.0030197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu S, Gao B, Duan Z, et al. Identification of tripartite motif-containing 22 (TRIM22) as a novel NF-kappaB activator. Biochem Biophys Res Commun. 2011;410:247–51. doi: 10.1016/j.bbrc.2011.05.124. [DOI] [PubMed] [Google Scholar]
- 14.Obad S, Olofsson T, Mechti N, et al. Expression of the IFN-inducible p53-target gene TRIM22 is down-regulated during erythroid differentiation of human bone marrow. Leuk Res. 2007;31:995–1001. doi: 10.1016/j.leukres.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Barr SD, Smiley JR, Bushman FD. The interferon response inhibits HIV particle production by induction of TRIM22. PLoS Pathog. 2008;4:e1000007. doi: 10.1371/journal.ppat.1000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eldin P, Papon L, Oteiza A, et al. TRIM22 E3 ubiquitin ligase activity is required to mediate antiviral activity against encephalomyocarditis virus. J Gen Virol. 2009;90:536–45. doi: 10.1099/vir.0.006288-0. [DOI] [PubMed] [Google Scholar]
- 17.Di Pietro A, Kajaste-Rudnitski A, Oteiza A, et al. TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J Virol. 2013;87:4523–33. doi: 10.1128/JVI.02548-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandborn WJ, Gasink C, Gao LL, et al. Ustekinumab induction and maintenance therapy in refractory Crohn's disease. N Engl J Med. 2012;367:1519–28. doi: 10.1056/NEJMoa1203572. [DOI] [PubMed] [Google Scholar]
- 19.Haberman Y, Tickle TL, Dexheimer PJ, et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J Clin Invest. 2014;124:3617–33. doi: 10.1172/JCI75436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madigan Da, Y J. Bayesian graphical models for discrete data. International Statistical Review. 1995;63:215–232. [Google Scholar]
- 22.Schadt EE, Molony C, Chudin E, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:e107. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tran LM, Zhang B, Zhang Z, et al. Inferring causal genomic alterations in breast cancer using gene expression data. BMC Syst Biol. 2011;5:121. doi: 10.1186/1752-0509-5-121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang IM, Zhang B, Yang X, et al. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol Syst Biol. 2012;8:594. doi: 10.1038/msb.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang X, Zhang B, Molony C, et al. Systematic genetic and genomic analysis of cytochrome P450 enzyme activities in human liver. Genome Res. 2010;20:1020–36. doi: 10.1101/gr.103341.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer's disease. Cell. 2013;153:707–20. doi: 10.1016/j.cell.2013.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kelly JN, Barr SD. In Silico Analysis of Functional Single Nucleotide Polymorphisms in the Human TRIM22 Gene. PLoS One. 2014;9:e101436. doi: 10.1371/journal.pone.0101436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Herr AM, Dressel R, Walter L. Different subcellular localisations of TRIM22 suggest species-specific function. Immunogenetics. 2009;61:271–80. doi: 10.1007/s00251-009-0357-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol. 2014;26:454–70. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–24. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Westra HJ, Peters MJ, Esko T, et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat Genet. 2013;45:1238–43. doi: 10.1038/ng.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee MN, Ye C, Villani AC, et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science. 2014;343:1246980. doi: 10.1126/science.1246980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sandborn WJ, Ghosh S, Panes J, et al. Tofacitinib, an oral Janus kinase inhibitor, in active ulcerative colitis. N Engl J Med. 2012;367:616–24. doi: 10.1056/NEJMoa1112168. [DOI] [PubMed] [Google Scholar]
- 34.Ho GT, Lee HM, Brydon G, et al. Fecal calprotectin predicts the clinical course of acute severe ulcerative colitis. Am J Gastroenterol. 2009;104:673–8. doi: 10.1038/ajg.2008.119. [DOI] [PubMed] [Google Scholar]
- 35.Leach ST, Yang Z, Messina I, et al. Serum and mucosal S100 proteins, calprotectin (S100A8/S100A9) and S100A12, are elevated at diagnosis in children with inflammatory bowel disease. Scand J Gastroenterol. 2007;42:1321–31. doi: 10.1080/00365520701416709. [DOI] [PubMed] [Google Scholar]
- 36.P226. Consistently high C reactive protein is associated with subsequent development of perianal fistulae in patients with Crohn's disease. J Crohns Colitis. 2015;9(Suppl 1):S188. [Google Scholar]
- 37.Murdoch T, O'Donnell S, Silverberg MS, et al. Biomarkers as potential treatment targets in inflammatory bowel disease: A systematic review. Can J Gastroenterol Hepatol. 2015;29:203–8. doi: 10.1155/2015/389548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hugot JP, Chamaillard M, Zouali H, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature. 2001;411:599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 39.Ogura Y, Bonen DK, Inohara N, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature. 2001;411:603–6. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 40.Kim YG, Park JH, Reimer T, et al. Viral infection augments Nod1/2 signaling to potentiate lethality associated with secondary bacterial infections. Cell Host Microbe. 2011;9:496–507. doi: 10.1016/j.chom.2011.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabbah A, Chang TH, Harnack R, et al. Activation of innate immune antiviral responses by NOD2. Nat Immunol. 2009;10:1073–80. doi: 10.1038/ni.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Q, Verma IM. NF-kappaB regulation in the immune system. Nat Rev Immunol. 2002;2:725–34. doi: 10.1038/nri910. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Chen YZ, Wang XF, et al. Prediction of ubiquitination sites by using the composition of k-spaced amino acid pairs. PLoS One. 2011;6:e22930. doi: 10.1371/journal.pone.0022930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castanier C, Zemirli N, Portier A, et al. MAVS ubiquitination by the E3 ligase TRIM25 and degradation by the proteasome is involved in type I interferon production after activation of the antiviral RIG-I-like receptors. BMC Biol. 2012;10:44. doi: 10.1186/1741-7007-10-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang S, Wang B, Humphries F, et al. Pellino3 ubiquitinates RIP2 and mediates NOD2-induced signaling and protective effects in colitis. Nat Immunol. 2013;14:927–36. doi: 10.1038/ni.2669. [DOI] [PubMed] [Google Scholar]
- 46.Walczak H, Iwai K, Dikic I. Generation and physiological roles of linear ubiquitin chains. BMC Biol. 2012;10:23. doi: 10.1186/1741-7007-10-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zurek B, Schoultz I, Neerincx A, et al. TRIM27 negatively regulates NOD2 by ubiquitination and proteasomal degradation. PLoS One. 2012;7:e41255. doi: 10.1371/journal.pone.0041255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jefferies C, Wynne C, Higgs R. Antiviral TRIMs: friend or foe in autoimmune and autoinflammatory disease? Nat Rev Immunol. 2011;11:617–25. doi: 10.1038/nri3043. [DOI] [PubMed] [Google Scholar]
- 49.Kawai T, Akira S. Regulation of innate immune signalling pathways by the tripartite motif (TRIM) family proteins. EMBO Mol Med. 2011;3:513–27. doi: 10.1002/emmm.201100160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chae JJ, Wood G, Masters SL, et al. The B30.2 domain of pyrin, the familial Mediterranean fever protein, interacts directly with caspase-1 to modulate IL-1beta production. Proc Natl Acad Sci U S A. 2006;103:9982–7. doi: 10.1073/pnas.0602081103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hattlmann CJ, Kelly JN, Barr SD. TRIM22: A Diverse and Dynamic Antiviral Protein. Mol Biol Int. 2012;2012:153415. doi: 10.1155/2012/153415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zurek B, Proell M, Wagner RN, et al. Mutational analysis of human NOD1 and NOD2 NACHT domains reveals different modes of activation. Innate Immun. 2012;18:100–11. doi: 10.1177/1753425910394002. [DOI] [PubMed] [Google Scholar]
- 53.Shaw MH, Kamada N, Warner N, et al. The ever-expanding function of NOD2: autophagy, viral recognition, and T cell activation. Trends Immunol. 2011;32:73–9. doi: 10.1016/j.it.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strober W, Watanabe T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn's disease. Mucosal Immunol. 2011;4:484–95. doi: 10.1038/mi.2011.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Philpott DJ, Sorbara MT, Robertson SJ, et al. NOD proteins: regulators of inflammation in health and disease. Nat Rev Immunol. 2014;14:9–23. doi: 10.1038/nri3565. [DOI] [PubMed] [Google Scholar]
- 56.Marsh RA, Rao K, Satwani P, et al. Allogeneic hematopoietic cell transplantation for XIAP deficiency: an international survey reveals poor outcomes. Blood. 2013;121:877–83. doi: 10.1182/blood-2012-06-432500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeissig Y, Petersen BS, Milutinovic S, et al. XIAP variants in male Crohn's disease. Gut. 2014 doi: 10.1136/gutjnl-2013-306520. [DOI] [PubMed] [Google Scholar]
- 58.Abbott DW, Yang Y, Hutti JE, et al. Coordinated regulation of Toll-like receptor and NOD2 signaling by K63-linked polyubiquitin chains. Mol Cell Biol. 2007;27:6012–25. doi: 10.1128/MCB.00270-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Damgaard RB, Fiil BK, Speckmann C, et al. Disease-causing mutations in the XIAP BIR2 domain impair NOD2-dependent immune signalling. EMBO Mol Med. 2013;5:1278–95. doi: 10.1002/emmm.201303090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krieg A, Correa RG, Garrison JB, et al. XIAP mediates NOD signaling via interaction with RIP2. Proc Natl Acad Sci U S A. 2009;106:14524–9. doi: 10.1073/pnas.0907131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ryu YS, Lee Y, Lee KW, et al. TRIM32 protein sensitizes cells to tumor necrosis factor (TNFalpha)-induced apoptosis via its RING domain-dependent E3 ligase activity against X-linked inhibitor of apoptosis (XIAP) J Biol Chem. 2011;286:25729–38. doi: 10.1074/jbc.M111.241893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Latour S, Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol. 2015;39:115–123. doi: 10.1016/j.semcdb.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 63.Rigaud S, Lopez-Granados E, Siberil S, et al. Human X-linked variable immunodeficiency caused by a hypomorphic mutation in XIAP in association with a rare polymorphism in CD40LG. Blood. 2011;118:252–61. doi: 10.1182/blood-2011-01-328849. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Clinical (A) and Genetic Features (B) of Patients with TRIM22.
MAF minor allelic frequency based on 1000 Genomes and 6503 healthy individuals genotyped in the NHLBI Exome Sequencing Project (http://evs.gs.washington.edu). The variant prediction are based Polyphen218, SIFT (http://sift.jcvi.org), GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/), and PhyloP (http://ccg.vital-it.ch/mga/hg19/phylop/phylop.html).
Table S2. Autosomal Recessive Variants Identified by WES in Patient 1.
Table S3: VEO Genes
See excel sheet
Table S4. VEOIBD Key Drivers
See excel sheet
Table S5. IIBDGC GWAS Enrichment in TRIM22 Locus
See excel sheet.
Table S6. IIBDGC GWAS Enrichment in TRIM22 eQTL Enrichment
See excel sheet
Table S7. eQTL Pathway Enrichment
See excel sheet
Table S8. Crohn's GWAS Enriched Blood eQTL Overlap with the VEO Network
Table S9. Annotation of 179 VEO Network
See excel sheet
Table S10. Functional Categories of Genes in the TRIM22 Sub network
Table S11. Inflamed Sigmoid and Rectum Key Drivers
Table S12. Correlations of the Expression of TRIM22 to the Clinical Response Characteristics Stratified by Distinct Intestine Anatomy Sites (Includes Both Inflamed and Non Inflamed Samples).
Table S13. Correlation of the Expression of TRIM22 in blood to CDAI
Figure S1: Variant Identification Pipeline for WES of Patient 1.
Whole exome sequencing identified 152,020 exonic variants in Patient 1. Eliminating low-quality and synonymous variants further refined the WES variant set. To isolated potential causative variants, only rare and predicated damaging (by dbNSFP) variants were included in inheritance analysis. Variants identified according to each inheritance model are shown.
Figure S2: Heterozygous TRIM22 Variants R150T and S244L Occur in the Coiled-coil Domain and May Affect Domain Oligomerization.
A) Protein sequence for TRIM22 indicating the position of the two coiled-coil regions termed region 1 (green) and region 2 (purple) including the position of R150 and S244.
B, C) PrOCoil analysis of coiled coil region 1 (B) and region 2 (C) for TRIM22 with the corresponding variants reveals potential alterations in domain oligomerization patterns. PrOCoil evaluates the relative weighted contribution of each amino acid to the oligomeric character of the coiled-coil domain. The introduction of the R150T and S244L variants alter the weighted effect of positions near the site of variant. These alterations have potential to impact the ability of the coiled-coil domain to fold correctly.
Figure S3: Blood and Intestine Derived eQTLs: Gene Set Enrichment Analysis of Genes Controlled by IBD SNPs (Metacore).
See Table S7 for genes in pathway.
Top enriched gene sets among genes controlled by IBD SNPs: NHGRI catalog entries for IBD, UC and CD, plus all WTCCC IBD GWAS SNPs with p-value ≤ 0.001. 362 genes showed IBD SNP regulation in the blood, 98 in the intestine (56 genes were shared between the two tissues). Log10 p-value of enrichment on the horizontal axis: intestine (blue bar) and blood (orange bar).
Different Gene Set database categories are reported in each panel: TRIM22 Pathways: See below
1. Transcription P53 signaling (TRIM22 is a P53 target gene)
4. Immune response: Antiviral actions of interferons
9. Ubiquitin Pathway
Figure S4: Computational Design.
A) Inflamed and non inflamed tissue biopsies were collected from anti TNFα refractory Crohn's patients enrolled in clinical trial for ustekinumab (anti P40- IL12/IL23).
B) Bayesian pan intestine network was constructed from microarray expression profiling on biopsies collected from various regions of the intestine.
C) Differential expression was performed on inflamed vs. non-inflamed for each region of the intestine collected.
D) Differential expression signatures of genes up regulated in inflamed sigmoid and inflamed rectum were projected on Bayesian ileum network for key driver analysis.
Figure S5: First Neighbors of TRIM22 and NOD2 in VEOIBD Intestine Network.
Genes within one path of TRIM22 in the VEOIBD RISK network have putative functional roles in autophagy.
Figure S6: TRIM22 and NOD2 Co-localize in Healthy Controls and Disrupted in Patient Tissue Sections.
A, B) Immunofluorescence analysis of TRIM22 and NOD2 in colonic biopsy samples from Healthy Controls and Patients.
TRIM22 (red) and NOD2 (green) localization was analyzed in the villus (A) and crypts (B) of both healthy controls and Patients 1-3. TRIM22 and NOD2 colocalization was observed in the both the villus and crypts of the healthy control biopsy samples. Both TRIM22 and NOD2 expression and colocalization were reduced in the patient biopsy samples Scale bars represent10 μm.
C) Exogenous TRIM22 co-localize with Exogenous NOD2.
HEK293T cells seeded on coverslips were co-transfected with FLAG-TRIM22 and HA-NOD2. TRIM22 (red) and NOD2 (green) immunostaining show strong cytoplasmic colocalization. Scale bars represent 10μm.
D) TRIM22 does not co-localize with NOD1.
To evaluate the specificity of the observed TRIM22 and NOD2 co-localization, immunofluorescence microscopy was performed on HEK293T cells transiently expressing FLAG-TRIM22 and HA-NOD1, a NOD family member that does not interact with TRIM22. TRIM22 (red) and NOD1 (green) staining did not show significant overlap. Scale bars represent 10μm.
E, F) Peptide inhibition of NOD2 in colonic biopsy samples of Healthy Controls and Patients.
To demonstrate NOD2 antibody specificity, endogenous NOD2 immunostaining was performed on health and disease control biopsy samples as well as Patients 1-3 in the presence or absence of a NOD2 antibody peptide inhibitor. Without peptide incubation, NOD2 showed a cytoplasmic expression in healthy controls and disease controls, and is reduced in the patients. The specific NOD2 peptide is blocking the antigen-antibody binding and therefore no staining is detected when the peptide is added concurrently.
Figure S7: TRIM22 Variants and shRNA Abrogate NOD2 Signaling.
A) NOD2 co-immunoprecipitates TRIM22 from in vitro cell lysates after transient co-expression.
HEK293T cells were transfected with HA-epitope tagged NOD2 and either wild type (WT) or mutant (R150T, S244L, R321K, R442C) FLAG-epitope tagged TRIM22 constructs. Twenty-four hours post-transfection, cells were lysed and NOD2 was immunoprecipitated with anti-HA antibody. Western blotting identified bound proteins. WT-TRIM22 interacted with and pulled-down NOD2; however, mutant TRIM22 showed reduced or absent NOD2 binding. NOD1, which does not interact with TRIM22, and an empty vector construct were included as negative controls. All lucerifase assay and Western blotting experiments were carried out in triplicate independent experiments and blots are representative.
B) TRIM22 overexpression has no effect on TNFα mRNA levels following stimulation by TNFα.
HCT116 cells were transiently transfected with control (empty vector), TRIM22-WT, or TRIM22-R442C plasmids. Twenty-four hours post-transfection, cells were stimulated for 18 hr with TNFα (20 ng/mL). mRNA of TNFα were assessed by Q-PCR. No significant increase in TNFα mRNA was observed following TNFα stimulation of cells overexpressing TRIM22.
C, D, E) TRIM22 mutants reduce mRNA level of genes downstream of the NOD2 signalling.
HCT116 cells were transiently co-transfected with NOD2 and TRIM22-WT or TRIM22 mutants (R150T, S244L, R321K, R442C). Twenty-four hours post-transfection, cells were stimulated with MDP (10 μg/mL) (C) or RSV (0.1 μg) (D, E) for 18 hr and then qPCR were performed. Cells transfected with TRIM22 mutants showed significantly reduced expression of TNFα (C), IL-6 (D), and ISG15 (E) (*p < 0.001 after Bonferroni post-hoc testing).
F) Overexpression of TRIM22 increases ISRE activation. HEK293T cells were transiently transfected with control (empty vector), TRIM22, or NOD2. ISRE reporter activation was evaluated by luciferase assay following stimulation with respiratory syncytial virus (RSV) (0.1 μg). TRIM22 overexpression significantly increased ISRE reporter activation compared to empty vector control (*p<0.001 after Bonferroni post-hoc testing). All experiments were carried out in triplicate, independent experiments.
Figure S8: TRIM22 shRNA Knockdown.
A) To demonstrate the degree of knockdown present in cells expressing TRIM22 shRNA,HEK293T cells were transiently transfected with control (scramble) or various TRIM22 shRNA clones for 48 hours. Western blotting analysis was performed using anti-TRIM22 antibodies with β-actin staining as a loading control.
B) Knockdown of TRIM22 reduces mRNA level of genes downstream of the NOD2. mRNA ofTNFα, IL-6, and ISG15were assessed by qPCR in HT29 cells stably transduced with control (scramble) or TRIM22 shRNA following stimulation by MDP (10 μg/mL) or respiratory syncytial virus (RSV) (0.1 μg). (*p<0.001 after Bonferroni post-hoc testing).
Figure S9: TRIM22 Variants Abrogates NOD2 Signaling in Patient 2 with Coiled-coil Variants.
Representative FACS blots and quantification of TNFα production in monocytes following stimulation with NOD2 and TLR4 ligand. Monocytes obtained from Patient 2 did not generate TNFα production in monocytes following stimulation with NOD2 and TLR4 ligand. Monocytes obtained from patient in response to L18-MDP stimulation but demonstrate normal reactivity to LPS. The patient's father and healthy donors are shown as controls. Monocytes are gated for single, live, CD14+, HLA-DR+ cells.ΔTNF (%) is determined by subtracting frequency of TNF-positive monocytes in stimulated from unstimulated conditions. Overnight travel of blood sample from Patient 2 and father of Patient 2 caused high baseline TNF without affecting results calculated asΔTNF (%). Grey background indicates normal range. Similar results with patient 2 were obtained in two independent experiments performed ten months apart. Open symbols L18-MDP stimulation, closed symbols LPS stimulation. Similar results were not obtained from Patient 1 and Patient 3 was not tested as she did not consent to further testing.
Figure S10: TRIM22 Directly Mediates the Polyubiquitination of NOD2 but not MAVS or RIPK2.
A) Wildtype TRIM22 enhances polyubiquitination of NOD2 in HEK293 cells.
HEK293 cells were transiently co-transfected with HA-NOD2, GFP-ubiquitin, and either wildtype (WT) or variant (R150T, S244L, R321L or R442C) FLAG-TRIM22. Following cell lysis, NOD2 was immunoprecipitated using anti-HA antibody bound to Protein G-Sepharose and NOD2 ubiquitination was observed by GFP immunoblotting. Co-transfection with WT-TRIM22 increased NOD2 ubiquitination that was lost in cells transfected with the TRIM22 variants.
B) TRIM22 does not mediate the ubiquitination of MAVS or RIPK2.
To determine if TRIM22 also regulates downstream NOD2 signaling modulators, HEK23T cells were co-transfected with FLAG-TRIM22, GFP-ubiquitin, and either HA-MAVS or HA-RIPK2. Twenty-four hours post-transfection, cells were lysed followed by immunoprecipitation of MAVS or RIPK2 using anti-HA-agarose. Immunoblotting for GFP-ubiquitin demonstrated no significant increase compared to empty TRIM22 vector controls. All experiments were carried out in triplicate independent experiments and blots are representative.
Figure S11: TRIM22 Does Not Mediate K48-linked Polyubiquitination of NOD2.
A) TRIM22 does not mediate K48-specific polyubiquitination of NOD2.
HEK293T cells were transiently co-transfected with FLAG-NOD2, HA-(K48-specific) ubiquitin, and either wildtype (WT) or variant (R150T, S244L, R321K, or R442C) V5-TRIM22. Twenty-four hours post-transfection, cells were lysed and NOD2 was immunoprecipitation with anti-FLAG-agarose. Immunoblotting for HA-tagged ubiquitin demonstrated no significant increase in cells transfected with WT-TRIM22 compared to empty vectors controls. Likewise, TRIM22 variants did not show significant K48-ubiquitin staining.
B, C) TRIM22-shRNA does not reduce MDP and RSV induced K48-linked polyubiquitination of NOD2.
HEK293T cells stably transduced with control (scramble) or TRIM22 shRNA were transfected with a HA-tagged K48-specific ubiquitin construct and FLAG-NOD2. Forty-eight hours post transfection, cell lysates were stimulated with MDP (B) or RSV (C) for the indicated time points followed by cell lysis and immunoprecipitation of NOD2 by anti-FLAG-agarose. In both control and TRIM22 knockdown cells, stimulation did not result in increased NOD2 K48-specific poly-ubiquitination. Western blotting experiments were carried out in triplicate, independent experiments.