

Abstract
Background
Anopheles sinensis is one of the most abundant vectors of malaria and other diseases in Asia. Vector control through the use of insecticides is the front line control method of vector-borne diseases. Pyrethroids are the most commonly used insecticides due to their low toxicity to vertebrates and low repellency. However, the extensive use of insecticides has imposed strong selection pressure on mosquito populations for resistance. High levels of resistance to pyrethroid insecticides and various mutations and haplotypes in the para sodium channel gene that confers knockdown resistance (kdr) have been detected in An. sinensis. Despite the importance of kdr mutations in pyrethroid resistance, the evolutionary origin of the kdr mutations is unknown. This study aims to examine the evolutionary genetics of kdr mutations in relation to spatial population genetic structure of An. sinensis.
Methods
Adults or larvae of Anopheles sinensis were collected from various geographic locations in China. DNA was extracted from individual mosquitoes. PCR amplification and DNA sequencing of the para-type sodium channel gene were conducted to analyse kdr allele frequency distribution, kdr codon upstream and downstream intron polymorphism, population genetic diversity and kdr codon evolution. The mitochondrial cytochrome c oxidase COI and COII genes were amplified and sequenced to examine population variations, genetic differentiation, spatial population structure, population expansion and gene flow patterns.
Results
Three non-synonymous mutations (L1014F, L1014C, and L1014S) were detected at the kdr codon L1014 of para-type sodium channel gene. A patchy distribution of kdr mutation allele frequencies from southern to central China was found. Near fixation of kdr mutation was detected in populations from central China, but no kdr mutations were found in populations from southwestern China. More than eight independent mutation events were detected in the three kdr alleles, and at least one of them evolved multiple times subsequent to their first divergence. Based on sequence analysis of the mitochondrial COI and COII genes, significant and large genetic differentiation was detected between populations from southwestern China and central China. The patchy distribution of kdr mutation frequencies is likely a consequence of geographic isolation in the mosquito populations and the long-term insecticide selection.
Conclusion
Our results indicate multiple origins of the kdr insecticide-resistant alleles in An. sinensis from southern and central China. Local selection related to intense and prolonged use of insecticide for agricultural purposes, as well as frequent migrations among populations are likely the explanations for the patchy distribution of kdr mutations in China. On the contrary, the lack of kdr mutations in Yunnan and Sichuan is likely a consequence of genetic isolation and absence of strong selection pressure. The present study compares the genetic patterns revealed by a functional gene with a neutral marker and demonstrates the combined impact of demographic and selection factors on population structure.
Electronic supplementary material
The online version of this article (doi:10.1186/s13071-016-1513-6) contains supplementary material, which is available to authorized users.
Keywords: Para-type sodium channel gene, Mitochondrial DNA, Evolution, Mutation, Knockdown resistance, Anopheles sinensis
Background
The development of insecticide resistance is regarded as an empirical model of an adaptive trait [1] whose origin and spread depend upon the selection pressure applied to the target population, the fitness cost associated with the resistant allele and the gene flow among populations [2]. Geographical landscape is often considered as an important factor limiting gene flow among populations [3–6]. However, the impact of gene flow barriers imposed by landscape differences and local selection on the spatial distribution of resistance alleles is largely unknown.
Resistance to insecticides in mosquitoes involves multiple mechanisms, including the target-site insensitivity caused by mutations in the para sodium channel gene (knockdown resistance kdr) and the detoxification of insecticides in mosquitoes involving detoxification enzymes (i.e. P450 monooxygenases, glutathione-S-transferases and esterases) that metabolise the insecticide before it reaches its target (metabolic resistance) [7–19]. Mutations in the para sodium channel gene have been shown to be associated with the use of pyrethroids, the most commonly used class of insecticide for malaria control worldwide [13]. The spectrum of mutations at the para sodium channel gene is highly conserved across insect species, indicating convergent evolution [11]. There are two hypotheses that relate to the distribution of kdr mutations: either the mutations arose once and then spread, or have evolved independently in different populations across a broad landscape. For instance, in Anopheles gambiae, the main African malaria vector, non-synonymous mutations at position L1014 of the para sodium channel gene occurred at least 4–5 times independently [1, 20]. Some of the mutation events at the para sodium channel gene were derived from single mutational steps from the common ancestor whereas others were the result of two mutational steps [1, 20]. Multiple origins of kdr mutations were also found in Musca domestica and in the aphid Myzus persicae [21, 22]. An alternative situation was seen in the agricultural pest Bemisia tabaci, where the L925I and T929V mutations appeared to have originated once and then spread through migration and global trade [2, 23].
In An. sinensis, the most abundant and important malaria vector species in southeast Asia [24–32], four non-synonymous mutations at codon L1014 of the para sodium channel gene are known, including the L1014F [31, 33–37], L1014S [17, 35], L1014C [31, 33, 34, 36, 37] and L1014W [35] mutations. There could be various factors influencing the distribution of kdr allele variants in An. sinensis across China and Southeast Asia. For example, the complexity of landscape could impose barriers to gene flow between the mosquito populations. Selection based upon the intensity and duration of pyrethroid use could also determine allelic distribution of kdr. On the other hand, genetic variation based on neutral markers reflects population structure resulting from demographic factors. In this case, the cytochrome c oxidase genes of the mitochondrial genome are used as the neutral reference. Based on the unique characteristics of maternal inheritance, no recombination, high variability relative to nuclear DNA, and low effective population size, the mitochondrial DNA (mtDNA) has been a marker of choice for studies of genetic diversity and population structure.
The central goal of this study was to understand how local selection and migration have shaped the distribution of kdr mutations. The specific objectives were to (i) determine the kdr allele distribution in multiple sites over a large geographic region; (ii) elucidate the evolutionary origin(s) of kdr mutations; and (iii) examine the role of landscape and demographic history of An. sinensis on the evolution of kdr mutations using mitochondrial DNA sequence data.
Methods
Sample collection and preparation
Anopheles sinensis mosquitoes were collected from 15 localities in China, including five sites in central China, seven sites in south China and three sites in southwest China (Fig. 1). For each site, approximately 50 female adult mosquitoes were collected with three to five CDC light-traps. For two sites that had an insufficient number of adult mosquitoes, approximately 50 larval mosquitoes were collected from 20 breeding sites (irrigated rice fields and small ponds with aquatic plants) using the standard 350 ml dippers. The larvae were then reared to adults and identified as An. sinensis based on the published morphological keys [38]. Between 13 and 36 mosquitoes per population were used for DNA analysis, giving a total of 344 mosquitoes (Table 1). Genomic DNA was extracted from each sample following a standard phenol:chloroform method [39]. DNA was re-suspended in TE buffer (10 mM Tris, 0.1 mM EDTA, pH 7.4). Mosquito species identity was further confirmed by amplifications of the ribosomal internal transcribed spacer ITS2 of the nuclear rRNA gene using specific primers for An. sinensis [40, 41].
Fig. 1.
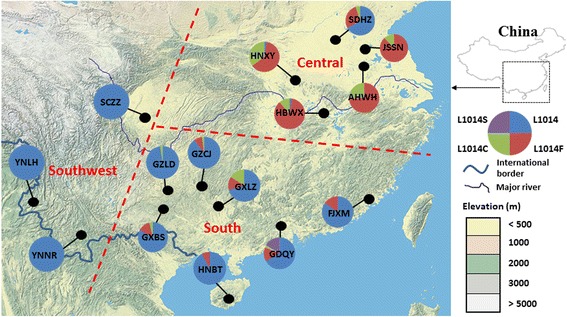
Pie charts showing kdr allele frequency distribution of Anopheles sinensis in China. Site names and abbreviations are as in Table 1
Table 1.
Summary of Anopheles sinensis specimen collection sites in China
| Site ID | Name abbreviation | Province: Locality | Latitude (N) | Longitude (E) | Elevation (m) | Collection date | Life-stages analysed | Specimen genotyped |
|---|---|---|---|---|---|---|---|---|
| 1 | YNLH | Yunnan: Lianghe | 24°51’ | 98°18’ | 1199 | 12-Jun | adult | 26 |
| 2 | YNNE | Yunnan: Ninger | 23°03’ | 101°02’ | 1426 | 7-Jul | adult | 24 |
| 3 | SCNJ | Sichuan: Neijiang | 29°40’ | 104°57’ | 321 | 11-Aug | adult & larva | 22 |
| 4 | GXBS | Guangxi: Baise | 23°23’ | 105°49’ | 1072 | 12-Aug | adult | 27 |
| 5 | GZLD | Guizhou: Luodian | 25°25’ | 106°45’ | 830 | 12-Aug | adult | 21 |
| 6 | GZCJ | Guizhou: Congjiang | 25°44’ | 108°54’ | 566 | 12-Aug | adult | 27 |
| 7 | GXLZ | Guangxi: Liuzhou | 25°45’ | 109°36’ | 352 | 12-Aug | adult | 21 |
| 8 | HNBT | Hainan: Baoting | 18°38’ | 109°46’ | 120 | 12-Jun | adult | 12 |
| 9 | GDQY | Guangdong: Qingyuan | 23°40’ | 113°03’ | 87 | 12-Jun | adult | 20 |
| 10 | FJXM | Fujian: Xiamen | 24°25’ | 118°24’ | 66 | 12-Aug | adult & larva | 22 |
| 11 | HBWX | Hubei: Wuxue | 29°50’ | 115°33’ | 14 | 11-Jul | adult | 23 |
| 12 | HNXY | Henan: Xinyang | 32°06’ | 114°01’ | 117 | 11-Jul | adult | 22 |
| 13 | JSSN | Jiangsu: Suining | 33°52’ | 117°59’ | 24 | 11-Jul | adult | 32 |
| 14 | AHWH | Anhui: Wuhe | 33°09’ | 117°51’ | 17 | 12-Jul | adult | 22 |
| 15 | SDHZ | Shandong: Heze | 34°46’ | 116°06’ | 44 | 12-Aug | adult | 23 |
Amplification and sequencing of the mitochondrial cytochrome c oxidase genes
The COI and COII genes of the mitochondrial genome were amplified, respectively, using degenerate primers 1809 F (5'-CMC TTT CAT CTG GAA TTG CT-3') and 2708R (5'-AAA AAT GTT GAG GGA ARA ATG TTA-3') for COI (~900 bp) and primers COIIF (5'-TCT AAT ATG GCA GAT TAG TGC A-3') and COIIR (5'-ACT TGC TTT CAG TCA TCT AAT G-3') for COII (~800 bp). Reaction mix consisted of 10.5 μl 2X SYBR® Green PCR Master Mix (Thermo Fisher Scientific Inc., Waltham, MA, USA), 1 μl of template DNA (5–20 ng), 0.5 μl of 10 μM each primer, and 8.5 μl water. Conditions for polymerase chain reaction (PCR) were 95 °C for 3 min, 35 cycles of 94 °C for 30 s, 55 °C for 30 s, and 72 °C for 60 s, followed by an additional extension step at 72 °C for 6 min. Products of PCR were visualised on 1 % agarose gel and then purified and sequenced from both ends with the ABI Big Dye Terminator Cycle Sequencing Kit.
Amplification and sequencing of a 1285 bp fragment of the para sodium channel gene
We designed a pair of primers (Forward primer 63 F: 5'-GAC GTT CGT GCT CTG CAT TA-3', Reverse primer 1347R: 5'-GAG CGA TGA TGA TCC GAA AT-3') based on the An. sinensis sodium channel gene sequence (GenBank accession: DQ334052) to amplify a 1285 bp fragment that encompasses the kdr target codon position L1014, an upstream intron (intron-1) and a downstream intron (intron-2). Both primers are located in the coding region. Two internal primers, 566 F (5'-CAC TGC TGT AAA ACC CTG TGT-3') and 855R (5'-CTG TTT GCT AGG CAG TTT GC-3') were used for sequencing. We amplified the 1285 bp fragment from individual specimen and a total of 273 specimens were examined. Reaction mix consisted of 10.5 μl 2X SYBR® Green PCR Master Mix (Thermo Fisher Scientific Inc., Waltham, MA, USA), 1 μl of template DNA (5–20 ng), 0.5 μl of 10 μM each primer, and 8.5 μl water. PCR cycles involved an initial denaturing step at 95 °C for 3 min, 35 cycles of 94 °C for 1 min, 55 °C for 1 min, and 72 °C for 2 min, followed by an additional extension step at 72 °C for 6 min. Amplifications were performed in a Bio-Rad MyCycler Thermal Cycler. Products of PCR were visualised on 1 % agarose gel and then purified and sequenced from both ends with ABI Big Dye Terminator Cycle Sequencing Kit.
Data analysis
This study addressed the central question as to whether the patchy kdr allele distribution in An. sinensis populations from southern China to central China was shaped by geographic isolation and/or by insecticide selection. We asked two questions in particular, i.e., (i) What is the kdr allele frequency distribution pattern and the phylogenetic relationship of kdr haplotypes? and (ii) What is the spatial genetic structure of An. sinensis mosquito populations in China? We used the para sodium channel gene sequence data to address the first question used mtDNA sequence data to address the second.
Phylogenetic relationships of kdr haplotypes
Sequenced 1285 bp fragments of the para sodium channel gene encompassing the kdr codon L1014, intron-1 and intron-2 were aligned in Bioedit version 7.0 [42]. DNA polymorphism including the number of segregating sites, number of haplotypes, haplotype diversity and nucleotide diversity were calculated using DnaSP v5 [43]. The two intron regions were tested for neutrality by Tajima‘s D [44], Fu and Li’s D* and F* [45] and F tests [46]. Partition of the genetic variation among and within populations was examined by AMOVA test implemented in Arlequin v3.5 [47]. A Bayesian approach based on a priori predictions from the coalescent theory implemented in Phase 2.1. [48] was used to reconstruct haplotypes from the population genotypic data in mosquitoes heterozygous at more than one site within the amplified 1285 bp fragment. These haplotypes were confirmed by cloning of the 1285 bp fragment in a subset of 20 individuals. Amplified PCR products from the selected 20 individuals were cloned into pCR2.1TOPO TA vectors (Invitrogen, Carlsbad, CA, USA). Three to five clones were sequenced for each individual to identify haplotypes.
A statistical parsimony network was constructed using the TCS v1.21 software [49] to infer the genealogical relationships among haplotypes based on an analysis of 1285 bp fragments in the samples. The software calculates the frequencies of the haplotypes in the sample and estimates haplotype outgroup probabilities, which correlate with haplotype age [50]. The pairwise comparisons of haplotypes was conducted using the parsimony algorithm described in Templeton et al. [51] to estimate the number of mutational steps between haplotypes using a connection limit at two steps and a connection probability threshold of 0.95.
Spatial genetic structure of An. sinensis
COI and COII gene sequences were aligned using Bioedit version 7.0 [42]. The number of variable sites and haplotypes, as well as haplotype and nucleotide diversity were estimated using Arlequin v3.5 [47] and DnaSP v5 [43]. To determine the genetic structure of the mosquito populations, we used analysis of molecular variance (AMOVA) to partition genetic variation among groups (FCT), among populations within groups (FSC) and among populations among groups (FST). Pairwise FST and ФST among all populations were calculated. Significance values were adjusted for multiple comparisons using the false discovery rate (FDR) approach [52]. Pairwise genetics distances were used to construct unrooted UPGMA tree in Phylip 3.695 [53] to infer population clustering. Isolation-by-distance among populations was explored with Mantel tests [54] using the Isolation by Distance Web Service v3.23 [55]. Euclidian geographical distances among the 15 populations were calculated based on geographical coordinates using great circle calculations to accurately represent distances on a spherical earth. Pairwise genetic distance between populations was calculated as FST/(1- FST) with 10,000 randomizations [56].
To examine the proportion of total genetic variance between groups of populations and identify possible genetic barriers between them, the spatial genetic structure of haplotypes (combined COI and COII sequences) was analysed using the Spatial Analysis of Molecular Variance (SAMOVA 2.0 software) [57]. This software implements an approach, which combines genetic differentiation and geographical distance to define groups of populations that are maximally differentiated from each other, without the constraint for the geographic composition of the groups. The number of initial conditions was set to 100 simulated annealing processes for each configuration of K groups, with K ranging from 2 to 10, and run for 10,000 iterations. For each K, the configuration with the largest FCT values after the 100 independent simulated annealing processes was retained as the best grouping of populations.
Genetic landscape shape (GLS) interpolation analysis
We performed the genetic landscape shape (GLS) interpolation analysis based on the pairwise genetic and geographical distance matrices using the Alleles-In-Space (AIS) software [58]. AIS is a computer program for the joint analysis of inter-individual spatial and genetic information. Detailed patterns of spatial genetic structure across the complete sampled area were visualised using the ‘genetic landscape shape’ interpolation procedure. This procedure produced a 3-dimensional surface plot where X- and Y-axes correspond to geographical locations and surface heights (Z-axes) represent genetic distances. Genetic structure across the landscape was inferred from measured genetic distances using an inverse distance weighted interpolation across a uniform grid laid over the entire sampling area. GLS was made by using a 80 × 80 grid and a raw genetic distance, with a distance weighting parameter α = 1.
Demographic history
Demographic history of the 15 populations was elucidated based on the neutrality indices Tajima’s D, Fu’s Fs and Ramos-Onsins and Rozas’s R2 statistic in DnaSP. Significantly negative D, Fs or small R2 values indicated recent population expansion, whereas positive or high values indicated population decline. Population expansion parameter τ (τ = 2 t* μ; where t = time in years, μ = mutation rate per locus) was estimated using the moment method, assuming the infinite sites model [59]. Deviations from a model of population expansion were evaluated by computing the statistical significance of sums of squared deviation (SSD) and Harpending's raggedness index (r) over 1000 simulated samples of pairwise nucleotide differences. All P-values were corrected for multiple testing using the false discovery rate (FDR) approach [52].
Migration rate between populations
Gene flow and migration rates between all pairwise populations were estimated using the Bayesian coalescence-based approach implemented in LAMARC version 2.1.10 [60]. Felsenstein‘84 (F84) substitution model was used with empirical base frequencies [61, 62]. Default prior settings were used for Theta and Bayesian estimation of migration rates. LAMARC analysis consisted of 3 simultaneous searches with automatically adjusted heating temperature using 10 initial chains, with 500 samples and a sampling interval of 20 steps, followed by 2 final chains, with 10,000 samples and sampling interval of 20 steps. One thousand samples were discarded for initial and final burn-in. Migration rate was measured as 4 Nm, a multiplication of LAMARC‘s M and Theta values for the recipient population.
Results
The PCR amplification of para-type sodium channel gene generated a 1285 bp PCR fragment, including a 187 bp exon encompassing the kdr target codon position L1014, its upstream intron 905 bp and exon 74 bp, and its downstream intron 64 bp and exon 55 bp. The PCR amplification of the COI and COII genes of the mitochondrial genome resulted in 900 bp and 774 bp fragments, respectively. In order to elucidate the kdr allele frequency distribution pattern and the phylogenetic relationships of kdr haplotypes, the para sodium channel gene sequence data were analysed for kdr allele frequency distribution, kdr codon upstream and downstream intron polymorphism, population genetic diversity and kdr codon evolution. Because mtDNA is haploid and maternally inherited, isolated populations will drift to different haplotype frequencies faster and achieve approximately twice the level of differentiation comparing to nuclear markers. So, the mtDNA sequence data were analysed for population variations, genetic differentiation, spatial population structure, population expansion and gene flow patterns.
Kdr allele frequency distribution
Three non-synonymous mutations (L1014F, L1014C, and L1014S) were detected at the kdr codon L1014. Overall, the distribution of kdr mutant allele frequencies varied by geographic location. Populations in central China (Anhui, Hubei, Hunan and Jiangsu) reached a near fixation for kdr at codon L1014 mutations, whereas the southern populations (Guangdong, Fujian, Guangxi, Hainan and Guizhou) exhibited a low frequency of kdr mutations (Fig. 1). We did not detect any kdr mutation in the southwestern populations (Yunnan and Sichuan provinces). In central China, L1014F was the predominant mutation and L1014C was also common in three provinces (Henan, Hubei and Anhui) in central China (Fig. 1).
Kdr codon upstream and downstream intron polymorphism
The 1285 bp fragment examined encompasses the kdr target codon position L1014, an upstream intron (intron-1), and a downstream intron (intron-2). Among the 344 total samples, 273 samples were sequenced, and a total of 71 polymorphic sites were observed. Of these sites, 69 were bi-allelic and two (at the second and third nucleotides of the kdr codon L1014) had three variants (Table 2). The two polymorphic sites with three variants were located at nucleotide positions 1161 and 1162 in a template sequence (GenBank KP763726). In intron-1, we observed deletions at nucleotide positions 946 and 949, and one-two base insertions at nucleotide position 230 when compared with a reference sequence (GenBank DQ334052). Among the 71 polymorphic sites, six were located in the exon harboring the kdr codon L1014, 58 were in intron-1, and seven in intron-2. Because of the heterozygous genotypes at the polymorphic sites, haplotypes were predicted by the Phase analysis [63, 64]. The Phase analysis identified a total of 85 haplotypes in 71 polymorphic sites (GenBank KP763726–KP763810). We cloned kdr fragments from 20 individuals with polymorphic sites at the 1285 bp kdr fragment and sequenced a total of 100 clones. Cloned sequences confirmed the haplotypes predicted by the Phase analysis. The 65 polymorphic sites in intron-1 and intron-2 yielded a total of 72 haplotypes.
Table 2.
Polymorphism of kdr intron and neutrality test of 15 Anopheles sinensis populations
| Pop | n | S | K | Pi | Tajima’s D | h | Hd | Fu’s Fs | Fu and Li’s D* | Fu and Li’s F* | τ | SSD | r |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Southwest | |||||||||||||
| YNLH | 42 | 16 | 4.570 | 0.070 | 0.731 | 14 | 0.913 | −1.598 | 0.337 | 0.553 | 5.486 | 0.032* | 0.114* |
| YNNE | 32 | 18 | 4.776 | 0.073 | 0.233 | 13 | 0.917 | −1.637 | 0.909 | 0.815 | 5.496 | 0.025 | 0.095* |
| SCNJ | 44 | 15 | 3.744 | 0.058 | 0.269 | 16 | 0.922 | −4.107* | 0.678 | 0.640 | 5.066 | 0.011 | 0.029 |
| South | |||||||||||||
| GXBS | 22 | 16 | 4.368 | 0.067 | −0.018 | 17 | 0.974 | −9.984** | −0.135 | −0.116 | 4.879 | 0.004 | 0.018 |
| GZLD | 32 | 7 | 3.601 | 0.055 | 3.118 | 4 | 0.575 | 5.319* | 1.269 | 2.138* | 8.051 | 0.260 | 0.541** |
| GZCJ | 40 | 15 | 4.318 | 0.066 | 0.715 | 18 | 0.924 | −5.611* | 0.698 | 0.830 | 5.539 | 0.004 | 0.015 |
| GXLZ | 32 | 11 | 3.216 | 0.049 | 0.563 | 14 | 0.823 | −4.616* | −0.092 | 0.132 | 5.156 | 0.023 | 0.088 |
| HNBT | 30 | 13 | 3.724 | 0.057 | 0.445 | 13 | 0.871 | −3.066* | 0.174 | 0.303 | 5.830 | 0.030 | 0.058 |
| GDQY | 36 | 15 | 5.335 | 0.082 | 1.543 | 11 | 0.883 | 0.529 | 1.137 | 1.493 | 6.641 | 0.021 | 0.071 |
| FJXM | 26 | 17 | 4.028 | 0.062 | −0.338 | 14 | 0.923 | −4.397* | −0.463 | −0.497 | 2.371 | 0.003 | 0.010 |
| Central | |||||||||||||
| HBWX | 42 | 6 | 1.153 | 0.018 | −0.457 | 4 | 0.301 | 1.111 | 0.356 | 0.122 | 3.000 | 0.056* | 0.497 |
| HNXY | 38 | 5 | 0.906 | 0.014 | −0.614 | 4 | 0.368 | 0.377 | 0.204 | −0.052 | 3.000 | 0.038 | 0.310 |
| JSSN | 30 | 6 | 0.759 | 0.012 | −1.424 | 4 | 0.251 | −0.257 | −1.111 | −1.403 | 3.000 | 0.030 | 0.511 |
| AHWH | 62 | 6 | 0.754 | 0.012 | −1.001 | 5 | 0.292 | −0.700 | 1.158 | 0.544 | 3.000 | 0.030 | 0.425 |
| SDHZ | 38 | 29 | 9.757 | 0.150 | 1.431 | 9 | 0.728 | 5.974* | 0.770 | 1.177 | 18.506 | 0.080* | 0.118* |
n number of sequences used, S number of polymorphic (segregating) sites, K average number of pairwise nucleotide differences, Pi nucleotide diversity, h number of Haplotypes, Hd haplotype diversity, τ estimated parameter of population expansion assuming the stepwise growth model (τ = 2 t*MU where t = time in years, MU = mutation rate per locus), SSD the sum of squared deviations; and r Harpending’s raggedness index
*P < 0.05; **P < 0.01 (FDR < 0.02)
The overall haplotype diversity (Hd) and nucleotide diversity (Pi) were 0.82 and 0.07, respectively. Genetic diversity varied significantly among geographical regions (Table 2). In central China, four of the five populations showed low haplotype (0.30 ± 0.05; F(2,11) = 46.3, P < 0.0001) and nucleotide diversity (0.014 ± 0.003, F(2,11) = 47.3, P < 0.0001), whereas in the south and southwest populations, high haplotype (0.92 ± 0.005 and 0.85 ± 0.13) and nucleotide diversity (0.07 ± 0.005 and 0.06 ± 0.03) were detected.
The Tajima’s D and Fu and Li's D* tests indicated no significant departures from neutrality in all the populations. Similarly, only one test of Fu and Li‘s F* indicated significant departure from neutrality in all the populations. However, significant departures from neutrality were detected by Fu’s Fs in eight populations and seven of them were from the south and southwest (Table 2), which indicated a pattern of haplotype diversity that deviated from the expectation under neutrality. Since the Fu‘s Fs statistic is particularly sensitive to demographic effects, it is difficult to conclude whether positive selection or demographic history (e.g., population expansion) account for the observed pattern. Although the AMOVA indicated that most of the variation (~70 %) occurred among populations among regions (FST = 0.28, P < 0.001), the percentage of variation found among populations within the same region (3.2 %) as well as among the three regions (27.9 %) was also highly significant (FSC = 0.04, P < 0.001; FCT = 0.18, P < 0.001). Both the FST and ФST values (based only on haplotype frequencies) were significant for 61 and 55 out of the 105 pairwise comparisons, respectively, after Bonferroni correction for multiple comparisons (Additional file 1: Table S1). The majority of the differences were detected between the central China populations and the remaining populations.
kdr codon evolution
Among the 65 polymorphic sites detected within intron-1 and intron-2, six (at nucleotide positons: 181, 940, 943, 1197, 1210, 1226 when compared with a template sequence (GenBank KP763726)) showed to be highly polymorphic (>10 %; three sites from intron-1 and three from intron-2), and a total of 18 haplotypes were identified based on these six polymorphic sites (Additional file 1: Table S2). These six polymorphic sites were informative and used with two variable sites of kdr codon L1014 (at nucleotide positons: 1161 and 1162) for constructing a TCS genealogical network. The network revealed at least eight independent mutation events from seven distinct intron haplotypes, giving rise to nine kdr haplotypes (Fig. 2). Haplotypes H1-L1014F and H1-L1014C consist of the majority of the samples from central China and the two largest mutation groups, which are derived from the ancestor H1-L1014 with a single mutational step and two mutational steps, respectively. Haplotypes H07-L1014S and H08-L1014S from south China (Guangdong province) are derived from single mutational steps from two distinct ancestors H07-L1014 and H08-L1014, respectively, but with connections between these kdr haplotypes. Similarly, Haplotypes H04-L1014F and H14-L1014F are results of single mutational steps from two distinct ancestors H04-L1014 and H14-L1014, respectively, but with connection (A-1-G) between them. Haplotypes H04-L1014F (kdr L1014F codon TTT) and H04-L1014F (kdr L1014F codon TTC) are results of single mutational steps from the common ancestor H04-L1014, but with connection (T-5-C) between these kdr haplotypes. The remaining two kdr haplotypes (H05-L1014F and H02-L1014F) are also derived from single mutational steps from the distinct ancestors with a mixed geographical distribution from south to north.
Fig. 2.
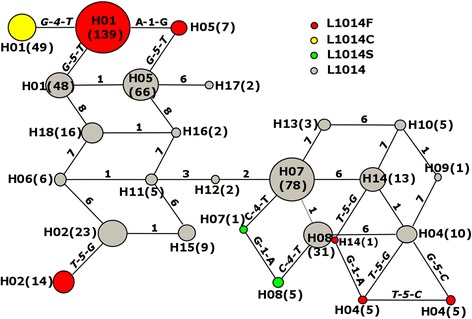
kdr haplotype networks showing the genealogical relationship for Anopheles sinensis. Each haplotype is represented by a circle with size proportional to its frequency in the sample (sample size in parentheses, 2 N = 546). Mutational steps are represented by lines with the indication of the mutation from the immediate ancestral haplotype (kdr mutations in bold). Grey circles: wild-type L1014 allele. Red circles: mutant L1014F allele (G-5-T or G-5-C). Yellow circles: mutant L1014C allele (T-4-G). Green circles: mutant L1014S allele (T-4-C). The numbers from 1 to 8 on the connection lines represent the nucleotide positions at 181, 940, 943, 1161, 1162,1197, 1210 and 1226 when compared with a template sequence (GenBank KP763726)
Mitochondrial DNA sequence variation
A total of 341 sequences for COI and 303 sequences for COII were generated for the 15 populations (Additional file 1: Table S3). COI and COII sequences were aligned giving a total length of 814 and 704 bp, respectively. The COI sequence alignment showed a total of 146 variable sites, of these 78 were parsimony informative. The COII sequence alignment revealed 85 variable sites, including 41 parsimony informative. Nucleotide diversity, number of haplotypes and haplotype diversity were higher in COI (p = 0.88 × 10−2; h = 238; Hd = 0.99; Additional file 1: Table S3) than in COII (p = 0.47 × 10−2; h = 142; Hd = 0.93). Among the 15 populations, An. sinensis from southern China exhibited higher haplotype and nucleotide diversity in COI and COII than those in central China (Additional file 1: Table S3).
Genetic differentiation among populations
AMOVA found a significant population structure in the 15 An. sinensis populations examined, based on the combined COI and COII sequences (FST = 0.027, P < 0.0001). Population differentiation was significant between the southwest and south/central China regions (FST = 0.069, P < 0.001). Both the FST and ФST values (based only on haplotype frequencies) were significant for 30 and 26 out of the 105 pairwise comparisons, respectively, after Bonferroni correction for multiple comparisons (Additional file 1: Table S4). The majority of the differences were detected between the two Yunnan populations (YNLH and YNNE) and the remaining populations.
Spatial genetic structure analysis
SAMOVA analyses with combined COI and COII sequences revealed a distinct decrease in FCT value from K = 2 to 3, and FCT values continue to decrease from K = 4 to 10. Although the FCT value was highest at K = 2, the major decrease on FCT occurred from K = 2 to 3 and from K = 3 to 4, with values only decreasing slightly thereafter. Similar patterns were also evident on FST and FSC values (Additional file 1: Table S5). When K ≥ 5, the FSC value became negative suggesting that there is no genetic barrier between populations within groups (Fig. 3a). The two Yunnan populations (YNLH and YNNE) were assigned to two single population groups when K = 3 with the highest FCT values, suggesting that there is genetic barrier to restrict gene flow from Yunnan to the other populations examined. In general, the patterns found in SAMOVA analyses were consistent with the phylogenetic trees of populations (Fig. 3b). The UPGMA dendogram based on Nei‘s unbiased genetic distances between populations showed two distinctive groups in which the two populations, YNLH and YNNE from Yunnan province constituted a cluster and the remaining populations from south and central China were included in a second cluster (Fig. 3b).
Fig. 3.
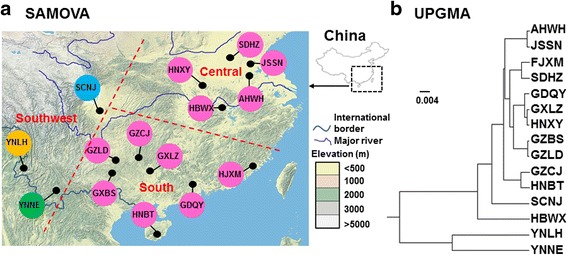
Cluster analysis based on the combined COI and COII sequences in Anopheles sinensis populations. a Colour codes of populations correspond to the five groups defined by SAMOVA; b UPGMA dendogram based on Nei‘s unbiased genetic distance between the 15 populations of An. sinensis
A Mantel test with combined COI and COII sequences revealed a significant correlation between geographical and genetic distances with all populations included (R2 = 0.164, P < 0.01, n = 105), suggesting that the genetic structure observed in the An. sinensis populations could be partially explained by distance isolation (Additional file 2: Figure S1). The genetic landscape shape analysis identified major potential gene flow barriers (Fig. 4). The highest peak of differentiation (P1) was found in southwest China between Yunnan and Guangxi/Sichuan populations, followed by the second highest peak (P2) in southern China between Sichuan and Guangxi/Guizhou/Guangdong. A third peak (P3) was identified between the populations from the south and central China. Low genetic differentiation was detected between populations in the central China region, as shown by the red valleys (Fig. 4).
Fig. 4.
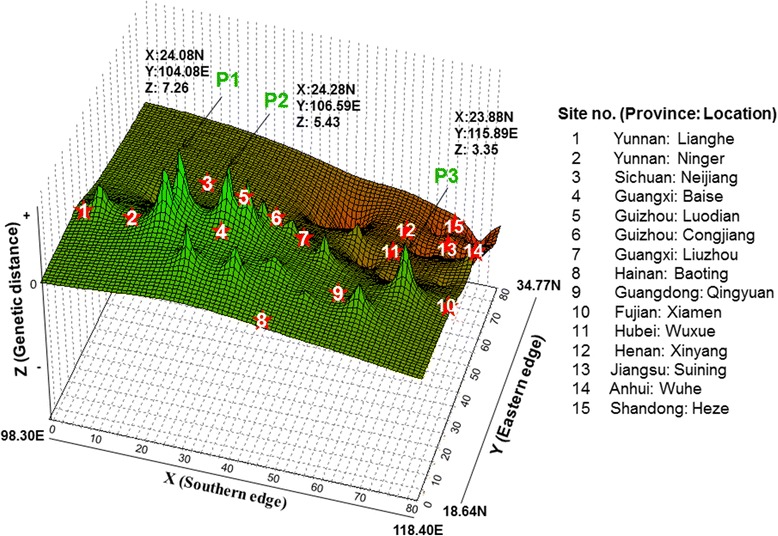
Genetic landscape shape plot showing patterns of spatial genetic distance for 15 populations of Anopheles sinensis. Geographical coordinates are shown in the X (North–south) and Y (East–west) axes. Z axis (height) corresponds to the genetic distance between individuals. Green peaks (P1, P2 and P3) are indicative of areas with high pairwise genetic distances and red valleys are indicative of areas of low pairwise genetic distance
Demographic history and neutrality test based on mtDNA sequences
Tajima’s D and Fu’s Fs tests found that 5 and 13 of the 15 populations were significantly negative, suggesting a recent population expansion or selection (Table 3). R2 statistics found similar results with Fu’s Fs tests. The two populations from Yunnan (YNLH and YNNE) showed nonsignificant D- and F-values, indicating an excess of rare nucleotide site variants compared to what would be expected under a neutral model of evolution. The observed smooth and unimodal mismatch distribution indicates sudden population expansion in 13 out of the 15 populations, and this was consistent with the mismatch distribution derived under the model of sudden expansion (Additional file 2: Figure S2). On the other hand, the multimodal pattern of mismatch distributions observed in the Yunnan populations suggested that no expansion events were detected and population size remained constant over time (Additional file 2: Figure S2).
Table 3.
Analysis of historical demography. Variation of estimated demographic parameters based on mitochondrial COI and COII sequence data of 15 populations of An. sinensis
| Population | N | Tajima’s D | Fu’s Fs | SSD | r | R2 | τ | Time (ky) |
|---|---|---|---|---|---|---|---|---|
| Southwest | ||||||||
| YNLH | 22 | −0.51 | −3.69 | 0.018 | 0.022 | 0.1014 | 5.027 | 144 |
| YNNE | 21 | 0.17 | −1.28 | 0.016 | 0.031 | 0.1325 | 5.178 | 148 |
| SCNJ | 20 | −1.39 | −9.27** | 0.004 | 0.008 | 0.0717** | 5.392 | 154 |
| South | ||||||||
| GXBS | 21 | −1.56* | −9.83** | 0.007 | 0.015 | 0.0719* | 7.85 | 225 |
| GZLD | 20 | −1.1 | −8.24** | 0.005 | 0.009 | 0.0827* | 5.622 | 161 |
| GZCJ | 18 | −1.45 | −9.65** | 0.077 | 0.01 | 0.0683** | 5.877 | 168 |
| GXLZ | 18 | −1.28 | −11.78** | 0.011 | 0.015 | 0.0764* | 6.232 | 178 |
| HNBT | 12 | −1.07 | −4.53* | 0.005 | 0.012 | 0.0872** | 8.249 | 236 |
| GDQY | 19 | −1.17 | −7.90** | 0.007 | 0.01 | 0.0838* | 7.452 | 213 |
| FJXM | 21 | −1.4 | −7.03** | 0.012 | 0.023 | 0.0663** | 6.844 | 196 |
| Central | ||||||||
| HBWX | 20 | −1.67* | −11.93** | 0.007 | 0.019 | 0.0564** | 6.03 | 173 |
| HNXY | 21 | −1.67* | −13.59** | 0.004 | 0.007 | 0.0652** | 5.726 | 164 |
| JSSN | 24 | −1.46 | −18.64** | 0.004 | 0.008 | 0.0613** | 6.477 | 185 |
| AHWH | 17 | −1.62* | −9.60** | 0.037 | 0.015 | 0.0672** | 6.113 | 175 |
| SDHZ | 18 | −1.83* | −11.96** | 0.003 | 0.005 | 0.0621** | 4.841 | 139 |
N number of samples analysed, SSD the sum of squared deviations, R2 Ramos-Onsins & Rozas’s R2, r Harpending’s raggedness index, τ estimated parameter of population expansion assuming the stepwise growth model (τ = 2 t*MU where t = time in years, MU = mutation rate per locus)
*P < 0.05; **P < 0.01 (FDR < 0.02)
Migration and gene flow patterns
LAMARC analysis found historical gene flow rates ranged from 0.01 to 4.85 (Additional file 1: Table S6). The highest migration rates were detected among neighboring populations within each of the five locality groups (Fig. 5). Among the five locality groups, medium levels of migration were observed. The Hainan (HNBT) and Yunnan (YNLH and YNNE) populations were apparently isolated with little gene flow (0.01 < Nem < 0.85). Based on the coalescent analysis, migration was asymmetrical, i.e., higher from the south to the north, and from southwest to the central regions than the opposite directions (Fig. 5).
Fig. 5.
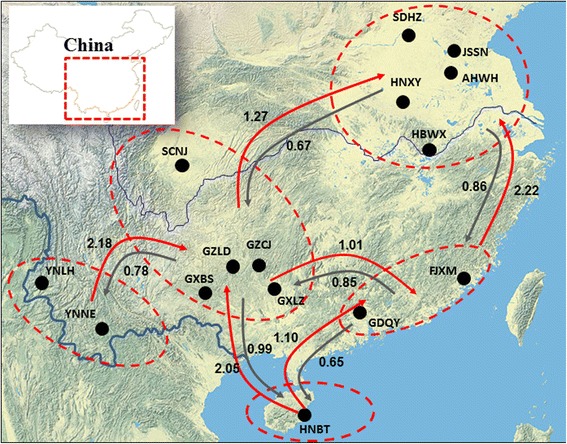
Bayesian estimates of historical asymmetrical migration between populations of Anopheles sinensis. The five locality groups are indicated by dotted circles and arrows indicate the direction of migration rates
Discussion
Near fixation of kdr mutation was detected in populations from central China, but no kdr mutations were found in populations from Yunnan and Sichuan provinces. Absence of kdr mutations in An. sinensis samples from southwestern China and low frequency of kdr mutations in southern China is likely a consequence of geographical isolation in the mosquito populations combined with the absence of strong local selection.
Geographical isolation of An. sinensis populations from southwestern China
Based on mtDNA sequencing, significant genetic differentiation was detected between Yunnan (southwestern China) and populations from south/central China. The AMOVA analysis indicated that the percentage of genetic variation between these two regions (~28 %) is much higher than that among populations within each of the regions (~3 %). Such regional genetic differentiation was also manifested by the observation of more private alleles in each of the regions than within individual populations of a region. Our data show that An. sinensis in Yunnan has a relatively stable population size over time without drastic demographic changes, unlike populations from central and southern China. The strong genetic differentiation and limited gene flow between the Yunnan and central China populations suggest that these two regions are genetically isolated. SAMOVA analysis further confirmed that there is a genetic barrier restricting gene flow between the Yunnan populations and the other populations examined. These patterns were also consistent with the phylogenetic trees of populations. The two Yunnan populations are located in the Yunnan Plateau, a mountainous area in southwest China with high elevations in the northwest and low elevations in the southeast of Yunnan province. Therefore, both physical distance and heterogeneous landscape could be factors inhibiting gene flow between Yunnan and south/central China. However, based on the analysis of DNA sequences surrounding kdr codon L1014, significant genetic differentiation was detected between the central China populations and the remaining populations, suggesting a strong local selection of the kdr resistance allele in the central China populations.
Effects of insecticide-associated selection
For the two Yunnan populations, neutrality tests (Fu’s Fs statistic) did not provide evidence of selection at the kdr introns. This finding concurs with a relatively low rate of insecticide use in agriculture and indoor spraying in Yunnan as compared to central China. Apart from limited migrations, the absence of strong selective pressure may in part explain the lack of insecticide-resistance alleles in Yunnan. Lower genetic variation was observed in the introns of the kdr locus in populations from central China when compared to the other populations. These populations showed a high frequency of kdr mutation (L1014F), suggesting that low variation at the adjacent introns could be a consequence of a recent selective sweep associated with intense insecticide use following the selection and/or fixation of the resistance genotypes. A selective sweep occurs when an allele rapidly increases its frequency due to positive or directional selection. Through genetic hitchhiking, the frequency of linked alleles in the flanking regions of the locus under selection can also increase, thus reducing genetic variation [65, 66].
It must be noted that among the different neutrality tests, Fu’ FS statistic shows the greatest power in detecting departures from neutrality under a genetic hitchhiking model [46]. The high Fu’s FS estimates for central China could reflect the impact of strong selection by the increased use of insecticide not only on the kdr locus, but also on the other gene regions. In the past half-century, insecticides are known to have been extensively used for agricultural and vector control purposes in central China [33]. Rice is the major agricultural crop in these study sites, with 1–2 harvests per year. Due to severe insect pest damage, insecticide use for pest control has been very intensive, with several rounds of sprays administered during each growing season. Since the mid-1980s, pyrethroids have been the dominant insecticides with pyrethroids-treated areas constituting more than one third of the total insecticide-treated areas in central China [37, 67, 68]. In addition to their agricultural use, pyrethroids have had various public health applications, e.g., as indoor sprays or incense, impregnated in bed nets, or as tools in public sanitation [25, 37, 67, 68]. The large scale agricultural use and indoor residual spraying of pyrethroid insecticides have probably generated selective pressure at the sodium channel gene in local An. sinensis populations. Anopheles sinensis from central China were found with extremely high resistant to pyrethroids, organochlorine, organophosphates and carbamates [31, 33]. Thus, it is not surprising for selection to have operated beyond the upstream and downstream introns of the kdr locus and towards parts of the mosquito’s genome in central China.
Multiple origins of kdr mutations
Information on whether resistance-associated mutations evolved independently vs single emergency and then spatially spread is valuable to elucidate the relative importance of mutation and migration in the spread of insecticide resistance. We detected three non-synonymous mutations at codon L1014 of the sodium channel gene. The majority of samples from central China harbored the kdr mutation L1014F, whereas the L1014S mutation was only found in Guangdong Province, southern China. The L1014C mutation was detected in both southern and central China. This result is consistent with previous study [37] of kdr genotyping in An. sinensis which found that the L1014F mutation was largely distributed in central China. Analysis of an upstream intron (intron-1) and a downstream intron (intron-2) of the kdr codon suggests at least eight independent origins of kdr alleles in An. sinensis populations from south to central China. Multiple origins of kdr alleles were also reported in the Afrotropical mosquito vector An. gambiae [1]. The kdr haplotypes H01-1014 F and H01-L1014C from the same ancestor were the most widespread in An. sinensis populations from central China and evolved multiple times subsequent to their first divergence. This phenomenon could be explained by the selection pressure of insecticides experienced in these populations, which favors kdr resistant alleles. Therefore, the frequency of haplotype (H01-L1014) carrying the wild-type allele is much lower than that of haplotype (H01-L1014F) harbouring kdr mutations. However, the kdr haplotype L1014S from two distinct ancestors was detected only in the Guangdong population, suggesting a recent origin of this mutation that is unique to the populations in southern China.
Conclusions
Our results indicate multiple origins of the kdr insecticide-resistant alleles in An. sinensis from southern and central China. Local selection related to intense and prolonged use of insecticide for agricultural purposes, as well as frequent migrations among populations are likely the explanations for the patchy distributions of kdr mutations in China. On the contrary, the lack of kdr mutations in Yunnan and Sichuan is likely a consequence of genetic isolation and absence of strong selection pressure. The present study compares the genetic patterns revealed by a functional gene with a neutral marker and demonstrates the combined impact of demographic and selection factors on population structure.
Acknowledgements
We thank Ying Wang, Jianhua Duan, Zhengbo He, Jiangyan Li, Yiji Li, Lin Zhou, Tielong Xu and Fengyang Fu for assistance with mosquito collection, two anonymous reviewers for critical review and valuable suggestions, Amruta Dixit and Elizabeth Hemming for their editorial assistance. This work is supported by grants from the U.S. National Institutes of Health (U19 AI089672, R03TW008237 and D43TW009527) and from the Key Projects of Science Research in Education Institutes of Anhui Colleges and Universities (KJ2015A146), the Science Research Innovation Team Projects of Anhui Colleges and Universities (2016–40).
Additional files
kdr L1014 codon region DNA based population differentiation for population pairs (estimates of F ST below diagonal and Ф ST above diagonal). Table S2. Distribution of Intron-1 and intron-2 haplotypes of kdr allele in the 15 Anopheles sinensis populations. Table S3. Haplotype and nucleotide diversity of the mitochondrial cytochrome c oxidase gene. Table S4. Mitochondrial DNA based population differentiation for population pairs (estimates of F ST below diagonal and Ф ST above diagonal). Table S5. List of grouping results from the spatial analysis of molecular variance (SAMOVA) showing values for variation among groups (F CT), among populations among groups (F ST) and among populations within groups (F SC). Table S6. Estimates of gene flow (migration rate) among the 15 Anopheles sinensis populations in China. (DOCX 44 kb)
Isolation by distance, relationship between geographical and genetic distances based on mtDNA sequences in the Anopheles sinensis populations. a, All populations; b, All populations without Yunnan samples. Figure S2. Observed (Obs) and expected (Exp) mismatch distributions showing the frequencies of pairwise differences. The observed distributions (red dot lines) are compared for their goodness-of-fit to a Poisson distribution under a model of sudden expansion illustrated by the overlaid curve (green solid lines). a, All populations; b, All populations without Yunnan; and c, Two populations from Yunnan province. (PDF 336 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors have contributed significantly to this study. Conceived and designed the experiments: GY, DZ and QF. Performed the experiments: XChan, DZ, QQ, and GZhu. Analysed the data: DZ, EL, MB, XW, ML, GZhou, and GY. Wrote and revised the manuscript: XC, DZ, GY, XChen, and LC. All authors read and revised the manuscript and approved the final version for submission.
Contributor Information
Xuelian Chang, Email: xuelianchang@126.com.
Daibin Zhong, Email: dzhong@uci.edu.
Eugenia Lo, Email: eugenia.lo@uci.edu.
Qiang Fang, Email: fq333@sohu.com.
Mariangela Bonizzoni, Email: mbonizzo@uci.edu.
Xiaoming Wang, Email: xiaomiw1@uci.edu.
Ming-Chieh Lee, Email: mingchil@uci.edu.
Guofa Zhou, Email: zhoug@uci.edu.
Guoding Zhu, Email: jipdzhu@hotmail.com.
Qian Qin, Email: qinqian@wzmc.edu.cn.
Xiaoguang Chen, Email: xgchen2001@hotmail.com.
Liwang Cui, Email: luc2@psu.edu.
Guiyun Yan, Email: guiyuny@uci.edu.
References
- 1.Pinto J, Lynd A, Vicente JL, Santolamazza F, Randle NP, Gentile G, Moreno M, Simard F, Charlwood JD, do Rosário VE, et al. Multiple origins of knockdown resistance mutations in the afrotropical mosquito vector Anopheles gambiae. PLoS ONE. 2007;2(11) doi: 10.1371/journal.pone.0001243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gauthier N, Clouet C, Perrakis A, Kapantaidaki D, Peterschmitt M, Tsagkarakou A. Genetic structure of Bemisia tabaci Med populations from home-range countries, inferred by nuclear and cytoplasmic markers: impact on the distribution of the insecticide resistance genes. Pest Manag Sci. 2014;70(10):1477–91. doi: 10.1002/ps.3733. [DOI] [PubMed] [Google Scholar]
- 3.Coster SS, Babbitt KJ, Cooper A, Kovach AI. Limited influence of local and landscape factors on finescale gene flow in two pond-breeding amphibians. Mol Ecol. 2015;24(4):742–58. doi: 10.1111/mec.13062. [DOI] [PubMed] [Google Scholar]
- 4.Cushman S, Shirk A, Landguth E. Landscape genetics and limiting factors. Conserv Genet. 2013;14(2):263–74. doi: 10.1007/s10592-012-0396-0. [DOI] [Google Scholar]
- 5.Igawa T, Oumi S, Katsuren S, Sumida M. Population structure and landscape genetics of two endangered frog species of genus Odorrana: different scenarios on two islands. Heredity. 2013;110(1):46–56. doi: 10.1038/hdy.2012.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP. Putting the ‘landscape’ in landscape genetics. Heredity. 2006;98(3):128–42. doi: 10.1038/sj.hdy.6800917. [DOI] [PubMed] [Google Scholar]
- 7.WHO . Global plan for insecticide resistance management in malaria vectors. Geneva: World Health Organization; 2012. [Google Scholar]
- 8.Pasteur N, Raymond M. Insecticide resistance genes in mosquitoes: their mutations, migration, and selection in field populations. J Hered. 1996;87(6):444–9. doi: 10.1093/oxfordjournals.jhered.a023035. [DOI] [PubMed] [Google Scholar]
- 9.Wondji CS, Priyanka De Silva WA, Hemingway J, Ranson H, Parakrama Karunaratne SH. Characterization of knockdown resistance in DDT- and pyrethroid-resistant Culex quinquefasciatus populations from Sri Lanka. Trop Med Int Health. 2008;13(4):548–55. doi: 10.1111/j.1365-3156.2008.02033.x. [DOI] [PubMed] [Google Scholar]
- 10.Brengues C, Hawkes NJ, Chandre F, McCarroll L, Duchon S, Guillet P, Manguin S, Morgan JC, Hemingway J. Pyrethroid and DDT cross-resistance in Aedes aegypti is correlated with novel mutations in the voltage-gated sodium channel gene. Med Vet Entomol. 2003;17(1):87–94. doi: 10.1046/j.1365-2915.2003.00412.x. [DOI] [PubMed] [Google Scholar]
- 11.Dong K, Du Y, Rinkevich F, Nomura Y, Xu P, Wang L, Silver K, Zhorov BS. Molecular biology of insect sodium channels and pyrethroid resistance. Insect Biochem Mol Biol. 2014;50:1–17. doi: 10.1016/j.ibmb.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Donnelly M, Corbel V, Weetman D, Wilding C, Williamson M, Black W. Does kdr genotype predict insecticide-resistance phenotype in mosquitoes. Trends Parasitol. 2009;25(5):213–9. doi: 10.1016/j.pt.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 13.Martinez-Torres D, Chandre F, Williamson MS, Darriet F, Berge JB, Devonshire AL, Guillet P, Pasteur N, Pauron D. Molecular characterization of pyrethroid knockdown resistance (kdr) in the major malaria vector Anopheles gambiae s.s. Insect Mol Bio. 1998;7(2):179–84. doi: 10.1046/j.1365-2583.1998.72062.x. [DOI] [PubMed] [Google Scholar]
- 14.Nwane P, Etang J, Chouaibou M, Toto J, Koffi A, Mimpfoundi R, Simard F. Multiple insecticide resistance mechanisms in Anopheles gambiae s.l. populations from Cameroon, Central Africa. Parasit Vectors. 2013;6(1):41. doi: 10.1186/1756-3305-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nwane P, Etang J, Chouaibou M, Toto J, Mimpfoundi R, Simard F. Kdr-based insecticide resistance in Anopheles gambiae s.s populations in Cameroon: spread of the L1014F and L1014S mutations. BMC Res Notes. 2011;4:463. doi: 10.1186/1756-0500-4-463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranson H, Jensen B, Vulule JM, Wang X, Hemingway J, Collins FH. Identification of a point mutation in the voltage-gated sodium channel gene of Kenyan Anopheles gambiae associated with resistance to DDT and pyrethroids. Insect Mol Bio. 2000;9(5):491–7. doi: 10.1046/j.1365-2583.2000.00209.x. [DOI] [PubMed] [Google Scholar]
- 17.Verhaeghen K, Van Bortel W, Trung HD, Sochantha T, Keokenchanh K, Coosemans M. Knockdown resistance in Anopheles vagus, An. sinensis, An. paraliae and An. peditaeniatus populations of the Mekong region. Parasit Vectors. 2010;3(1):59. doi: 10.1186/1756-3305-3-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kasai S, Scott J. Over expression of cytochrome P450 CYP6D1 is associated with monooxygenases-mediated pyrethroid resistance in house flies from Georgia. Pestic Biochem Physiol. 2000;68:34–41. doi: 10.1006/pest.2000.2492. [DOI] [Google Scholar]
- 19.Liu N. Insecticide resistance in mosquitoes: impact, mechanisms, and research directions. Annu Rev Entomol. 2015;60(1):537–59. doi: 10.1146/annurev-ento-010814-020828. [DOI] [PubMed] [Google Scholar]
- 20.Etang J, Vicente JL, Nwane P, Chouaibou M, Morlais I, Do Rosario VE, Simard F, Awono-Ambene P, Toto JC, Pinto J. Polymorphism of intron-1 in the voltage-gated sodium channel gene of Anopheles gambiae s.s. populations from Cameroon with emphasis on insecticide knockdown resistance mutations. Mol Ecol. 2009;18(14):3076–86. doi: 10.1111/j.1365-294X.2009.04256.x. [DOI] [PubMed] [Google Scholar]
- 21.Anstead JA, Williamson MS, Denholm I. Evidence for multiple origins of identical insecticide resistance mutations in the aphid Myzus persicae. Insect Biochem Mol Biol. 2005;35(3):249–56. doi: 10.1016/j.ibmb.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Rinkevich FD, Hedtke SM, Leichter CA, Harris SA, Su C, Brady SG, Taskin V, Qiu X, Scott JG. Multiple origins of kdr-type resistance in the house fly, Musca domestica. PLoS ONE. 2012;7(12) doi: 10.1371/journal.pone.0052761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alon M, Benting J, Lueke B, Ponge T, Alon F, Morin S. Multiple origins of pyrethroid resistance in sympatric biotypes of Bemisia tabaci (Hemiptera: Aleyrodidae) Insect Biochem Mol Biol. 2006;36(1):71–9. doi: 10.1016/j.ibmb.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 24.Chareonviriyaphap T, Bangs MJ, Ratanatham S. Status of malaria in Thailand. Southeast Asian J Trop Med Public Health. 2000;31(2):225–37. [PubMed] [Google Scholar]
- 25.Cui F, Raymond M, Qiao CL. Insecticide resistance in vector mosquitoes in China. Pest Manag Sci. 2006;62(11):1013–22. doi: 10.1002/ps.1288. [DOI] [PubMed] [Google Scholar]
- 26.Jung J, Jung Y, Min GS, Kim W. Analysis of the population genetic structure of the malaria vector Anopheles sinensis in South Korea based on mitochondrial sequences. Am J Trop Med Hyg. 2007;77(2):310–5. [PubMed] [Google Scholar]
- 27.Jung J, Lee E, Kim W. Isolation and characterization of polymorphic microsatellite markers of Anopheles sinensis, a malaria vector mosquito in the East Asia region. Mol Ecol Notes. 2006;6(4):1272–4. doi: 10.1111/j.1471-8286.2006.01512.x. [DOI] [Google Scholar]
- 28.Liu XB, Liu QY, Guo YH, Jiang JY, Ren DS, Zhou GC, Zheng CJ, Zhang Y, Liu JL, Li ZF, et al. The abundance and host-seeking behavior of culicine species (Diptera: Culicidae) and Anopheles sinensis in Yongcheng city, People‘s Republic of China. Parasit Vectors. 2011;4(1):221. doi: 10.1186/1756-3305-4-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y, Yang M, Fan Y, Wu J, Ma Y, Xu J. Population structure of the malaria vector Anopheles sinensis (Diptera: Culicidae) in China: two gene pools inferred by microsatellites. PLoS ONE. 2011;6(7) doi: 10.1371/journal.pone.0022219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinka M, Bangs M, Manguin S, Chareonviriyaphap T, Patil A, Temperley W, Gething P, Elyazar I, Kabaria C, Harbach R, et al. The dominant Anopheles vectors of human malaria in the Asia-Pacific region: occurrence data, distribution maps and bionomic precis. Parasit Vectors. 2011;4(1):89. doi: 10.1186/1756-3305-4-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhong D, Chang X, Zhou G, He Z, Fu F, Yan Z, Zhu G, Xu T, Bonizzoni M, Wang MH, et al. Relationship between knockdown resistance, metabolic detoxification and organismal resistance to pyrethroids in Anopheles sinensis. PLoS ONE. 2013;8(2) doi: 10.1371/journal.pone.0055475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu G, Xia H, Zhou H, Li J, Lu F, Liu Y, Cao J, Gao Q, Sattabongkot J. Susceptibility of Anopheles sinensis to Plasmodium vivax in malarial outbreak areas of central China. Parasit Vectors. 2013;6(1):176. doi: 10.1186/1756-3305-6-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang X, Zhong D, Fang Q, Hartsel J, Zhou G, Shi L, Fang F, Zhu C, Yan G. Multiple resistances and complex mechanisms of Anopheles sinensis mosquito: a major obstacle to mosquito-borne diseases control and elimination in China. PLoS Negl Trop Dis. 2014;8(5) doi: 10.1371/journal.pntd.0002889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang S, Jung J, Lee S, Hwang H, Kim W. The polymorphism and the geographical distribution of the knockdown resistance (kdr) of Anopheles sinensis in the Republic of Korea. Malaria J. 2012;11(1):151. doi: 10.1186/1475-2875-11-151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tan WL, Li CX, Wang ZM, Liu MD, Dong YD, Feng XY, Wu ZM, Guo XX, Xing D, Zhang YM, et al. First detection of multiple knockdown resistance (kdr)-like mutations in voltage-gated sodium channel using three new genotyping methods in Anopheles sinensis from Guangxi Province, China. J Med Entomol. 2012;49(5):1012–20. doi: 10.1603/ME11266. [DOI] [PubMed] [Google Scholar]
- 36.Tan WL, Wang ZM, Li CX, Chu HL, Xu Y, Dong YD, Wang ZC, Chen DY, Liu H, Liu DP, et al. First report on co-occurrence knockdown resistance mutations and susceptibility to beta-cypermethrin in Anopheles sinensis from Jiangsu province, China. PLoS ONE. 2012;7(1) doi: 10.1371/journal.pone.0029242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y, Yu W, Shi H, Yang Z, Xu J, Ma Y. Historical survey of the kdr mutations in the populations of Anopheles sinensis in China in 1996–2014. Malaria J. 2015;14(1):120. doi: 10.1186/s12936-015-0644-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong X. The mosquito fauna of Yunnan (Volume one) Kunming: Yunnan Publishing Group Corporation, Yunnan Science & Technology Press; 2010. [Google Scholar]
- 39.Severson DW. RFLP analysis of insect genomes. In: Crampton JM, Beard CB, Louis C, editors. The molecular biology of insect disease vectors. London: Chapman & Hall; 1997. pp. 309–20. [Google Scholar]
- 40.Gao Q, Beebe NW, Cooper RD. Molecular identification of the malaria vectors Anopheles anthropophagus and Anopheles sinensis (Diptera: Culicidae) in central China using polymerase chain reaction and appraisal of their position within the Hyrcanus group. J Med Entomol. 2004;41(1):5–11. doi: 10.1603/0022-2585-41.1.5. [DOI] [PubMed] [Google Scholar]
- 41.Joshi D, Park MH, Saeung A, Choochote W, Min GS. Multiplex assay to identify Korean vectors of malaria. Mol Ecol Resour. 2010;10(4):748–50. doi: 10.1111/j.1755-0998.2010.02835.x. [DOI] [PubMed] [Google Scholar]
- 42.Hall TA. A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8. [Google Scholar]
- 43.Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003;19(18):2496–7. doi: 10.1093/bioinformatics/btg359. [DOI] [PubMed] [Google Scholar]
- 44.Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu YX, Li WH. Statistical tests of neutrality of mutations. Genetics. 1993;133(3):693–709. doi: 10.1093/genetics/133.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25. doi: 10.1093/genetics/147.2.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Excoffier L, Lischer HE. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour. 2010;10(3):564–7. doi: 10.1111/j.1755-0998.2010.02847.x. [DOI] [PubMed] [Google Scholar]
- 48.Stephens M, Smith NJ, Donnelly P. A new statistical method for haplotype reconstruction from population data. Am J Hum Genet. 2001;68(4):978–89. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Clement M, Posada D, Crandall KA. TCS: a computer program to estimate gene genealogies. Mol Ecol. 2000;9(10):1657–9. doi: 10.1046/j.1365-294x.2000.01020.x. [DOI] [PubMed] [Google Scholar]
- 50.Donnelly P, Tavaré S. The ages of alleles and a coalescent. Adv Appl Probab. 1986;18:1–19. doi: 10.2307/1427237. [DOI] [Google Scholar]
- 51.Templeton AR, Crandall KA, Sing CF. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics. 1992;132(2):619–33. doi: 10.1093/genetics/132.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 53.Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.7a. Seattle: Department of Genome Sciences, University of Washington; 2009. [Google Scholar]
- 54.Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27(2 Part 1):209–20. [PubMed] [Google Scholar]
- 55.Jensen JL, Bohonak AJ, Kelley ST. Isolation by distance, web service. BMC Genet. 2005;6:13. doi: 10.1186/1471-2156-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rousset F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics. 1997;145(4):1219–28. doi: 10.1093/genetics/145.4.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dupanloup I, Schneider S, Excoffier L. A simulated annealing approach to define the genetic structure of populations. Mol Ecol. 2002;11(12):2571–81. doi: 10.1046/j.1365-294X.2002.01650.x. [DOI] [PubMed] [Google Scholar]
- 58.Miller MP. Alleles in space (AIS): computer software for the joint analysis of interindividual spatial and genetic information. J Hered. 2005;96(6):722–4. doi: 10.1093/jhered/esi119. [DOI] [PubMed] [Google Scholar]
- 59.Rogers AR. Genetic evidence for a pleistocene population explosion. Evolution. 1995;49(4):608–15. doi: 10.2307/2410314. [DOI] [PubMed] [Google Scholar]
- 60.Kuhner MK. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics. 2006;22(6):768–70. doi: 10.1093/bioinformatics/btk051. [DOI] [PubMed] [Google Scholar]
- 61.Kishino H, Hasegawa M. Evaluation of the maximum likelihood estimate of the evolutionary tree topologies from DNA sequence data, and the branching order in hominoidea. J Mol Evol. 1989;29(2):170–9. doi: 10.1007/BF02100115. [DOI] [PubMed] [Google Scholar]
- 62.Felsenstein J. PHYLIP (Phylogeny Inference Package) Seattle: Department of Genetics, University of Washington; 1993. [Google Scholar]
- 63.Stephens M, Donnelly P. A comparison of Bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet. 2003;73(5):1162–9. doi: 10.1086/379378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. doi: 10.1093/bioinformatics/btp187. [DOI] [PubMed] [Google Scholar]
- 65.Gentile G, Santolamazza F, Fanello C, Petrarca V, Caccone A, della Torre A. Variation in an intron sequence of the voltage-gated sodium channel gene correlates with genetic differentiation between Anopheles gambiae s.s. molecular forms. Insect Mol Biol. 2004;13(4):371–7. doi: 10.1111/j.0962-1075.2004.00494.x. [DOI] [PubMed] [Google Scholar]
- 66.Weill M, Chandre F, Brengues C, Manguin S, Akogbeto M, Pasteur N, Guillet P, Raymond M. The kdr mutation occurs in the Mopti form of Anopheles gambiae s.s. through introgression. Insect Mol Biol. 2000;9(5):451–5. doi: 10.1046/j.1365-2583.2000.00206.x. [DOI] [PubMed] [Google Scholar]
- 67.Huang J, Qiao F, Zhang L, Rozelle S. Farm pesticides, rice production, and human health in China. In: EEPSEA, editor. Research report no2001-RR3. Singapore: Economy and Environment Program for Southeast Asia; 2001. pp. 1–58. [Google Scholar]
- 68.Wang DQ, Xia ZG, Zhou SS, Zhou XN, Wang RB, Zhang QF. A potential threat to malaria elimination: extensive deltamethrin and DDT resistance to Anopheles sinensis from the malaria-endemic areas in China. Malaria J. 2013;12(1):164. [DOI] [PMC free article] [PubMed]