

Abstract
Heat shock proteins (HSPs) belong to a superfamily of stress proteins that are critical constituents of a complex defense mechanism that enhances cell survival under adverse environmental conditions. Cell protective roles of HSPs are related to their chaperone functions, antiapoptotic and antinecrotic effects. HSPs' antiapoptotic and cytoprotective characteristics, their ability to protect cells from a variety of stressful stimuli, and the possibility of their pharmacological induction in cells under pathological stress make these proteins an attractive therapeutic target for various neurodegenerative diseases; these include Alzheimer's, Parkinson's, Huntington's, prion disease, and others. This review discusses the possible roles of HSPs, particularly HSP70 and small HSPs (alpha A and alpha B crystallins) in enhancing the survival of retinal ganglion cells (RGCs) in optic neuropathies such as glaucoma, which is characterized by progressive loss of vision caused by degeneration of RGCs and their axons in the optic nerve. Studies in animal models of RGC degeneration induced by ocular hypertension, optic nerve crush and axotomy show that upregulation of HSP70 expression by hyperthermia, zinc, geranyl-geranyl acetone, 17-AAG (a HSP90 inhibitor), or through transfection of retinal cells with AAV2-HSP70 effectively supports the survival of injured RGCs. RGCs survival was also stimulated by overexpression of alpha A and alpha B crystallins. These findings provide support for translating the HSP70- and alpha crystallin-based cell survival strategy into therapy to protect and rescue injured RGCs from degeneration associated with glaucomatous and other optic neuropathies.
Keywords: heat shock protein, retina, ganglion cells, optic nerve, glaucoma, neuroprotection
1. Introduction
Heat shock proteins (HSPs) were discovered serendipitously in 1962 by Ritossa in unintentionally overheated drosophila (Ritossa, 1996). HSPs belong to a superfamily of stress proteins, which also include glucose regulating proteins, ubiquitin and the lectin chaperones calnexin and calreticulin. Stress proteins, and HSPs in particular, are critical constituents of a complex defense mechanism that enhances cell survival under adverse environmental conditions. HSPs are highly conserved and are present in all cells in all life forms. They comprise a very heterogeneous group of proteins with significant overlap in functions. These proteins are classified according to their molecular weight into six families: small HSP (12–43 kDa; HSPB), HSP40 (DNAJ), HSP60 (HSPD), HSP70 (HSPA), HSP90 (HSPC), and HSP110 (HSPH; Kampinga et al., 2009). HSPs are expressed and function in cells under normal conditions as molecular chaperones. These proteins play a critical role in protein homeostasis by assisting protein folding, the assembly and disassembly of protein complexes, protein repair or degradation, reduction of protein aggregation, subcellular localization of newly synthesized proteins to target organelles, protein transport across membranes, synaptic transmission, and cytoskeletal organization (Fig. 1; Zimmerman and Minton, 1993; Saibil, 2000; Schröder and Kaufman, 2005; Gottesman, 2003; Ohtsuka and Hata, 2000; Muchowski and Wacker, 2005; Stetler et al., 2010).
Figure 1.

Chaperone-assisted protein folding. In the early phase of newly synthesized protein folding, HSP40 attached to ribosomes (rHSP40) activate and direct cytosolic HSP70 to elongating polypeptides. This co-translational chaperoning may be sufficient for the folding of many proteins. HSP70 also assist post-translational protein folding. Furthermore, HSP70 can direct some proteins to HSP90, for the late phase of folding. Protein misfolding and aggregation caused by stresses can be limited by HSP90, HSP70 and HSP27. The misfolded proteins can be refolded by the refolding activity of HSP70. Proteins that cannot be refolded are directed to the proteasome for degradation. HSP70 is also required for protein translocation across intracellular membranes (endoplasmic reticulum, mitochondria, lysosomes) and import into and export out of the nucleus.
Stressful stimuli such as hyperthermia (Morimoto et al., 1990; Ostberg et al., 2002), hypothermia (Cullen and Sarge, 1997), ischemia (Richard et al., 1996), hypoxia (Patel et al., 1995), depletion of ATP (Gabai and Kabakov, 1994), free radicals (Kukreja et al., 1994), desiccation (Hayward et al.,2004), viral infection (Collins and Hightower, 1982; Buccellato et al., 2007), steroid hormones (Norton and Latchman, 1989), and ethanol (Plesset et al, 1982) induce expression of HSPs, which in turn assist in the refolding of denatured proteins and facilitate the synthesis of new proteins to repair damage. Moreover, HSPs can suppress apoptotic pathways by interacting with the proteins associated with signal transduction in active cell death (Fig. 2). Stress-induced HSP expression requires activation of heat shock factors (HSFs) that in turn bind to the heat shock promoter elements and activate HSP transcription (Fig. 3). Plants and vertebrates, unlike yeast and Drosophila, have multiple HSFs. The mammalian HSF family include HSF1, 2, 3, 4, 5, Y and X (Westerheide et al., 2012). Moreover, functionally distinct HSF isoforms generated by alternative splicing allow for an additional regulatory control of HSP gene expression. In vertebrates, HSF1 has been identified as the principal transcription factor (TF) responsible for the regulation of stress-induced HSP expression. HSF1 has demonstrated DNA binding activity, oligomerization, and nuclear localization in response to various environmental stress conditions (Baler et al., 1993; Sarge et al., 1993; Kawazoe et al., 1998). Fibroblasts derived from HSF1-deficient mice had no stress-induced activation of HSP gene transcription, indicating the essential role of HSF1 in the heat shock response (McMillan et al., 1998). Under normal conditions, HSF1 is present in an inert monomeric state, which is supported through transient interactions with chaperones such as HSP70, HSP90 and HSP40 (Fig. 3; Knauf et al., 1996; Kline and Morimoto, 1997). During stress, the emergence of misfolded or aggregated proteins that compete with HSF for association with HSP70 (or HSP90), leads to the dissociation of HSP-HSF complexes and the released HSF1 assembles into trimers able to bind DNA. Acquisition of a transcriptionally inert DNA binding competent state follows by stress-inducible phosphorylation associated with transcriptional activation of HSF1 (Fig. 3). This leads to inducible transcription of HSP genes, synthesis, and accumulation of HSPs, in particular HSP70 and HSP90. Attenuation of the stress-induced response leads to direct binding of HSP70 or HSP90 and HSP40 to HSF1 and suppression of its transcriptional activity. Furthermore, besides the feedback regulation of HSF1 by HSPs, the HSF1 is negatively regulated by HSF binding protein 1, which interacts with both HSF and HSP. Inactivation of HSF1 leads to the dissociation of trimers into HSF1 inert monomers (Ali et al., 1998; Zuo et al., 1995).
Figure 2.
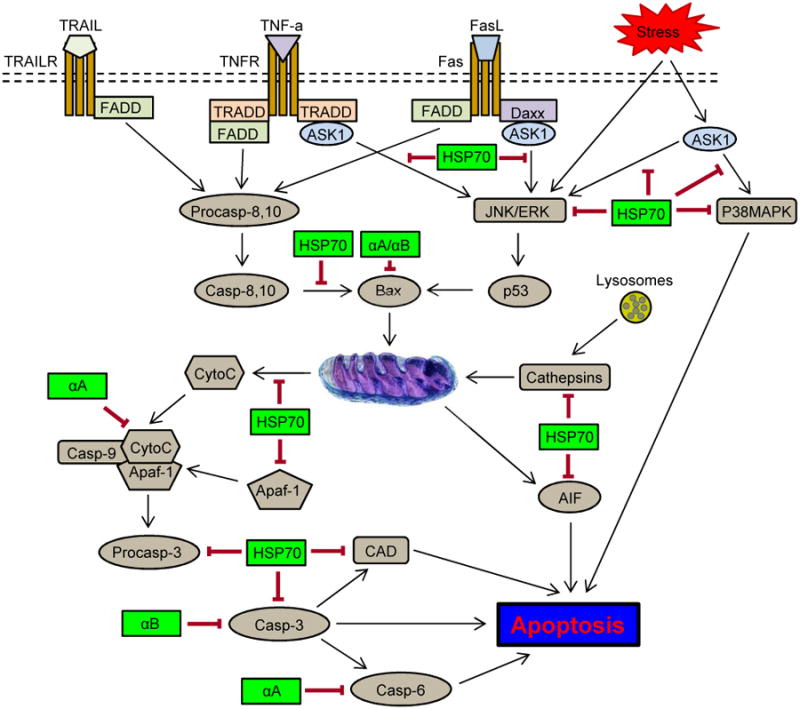
HSPs suppress several intrinsic and extrinsic pathways of apoptotic cell death. The intrinsic mitochondrial pathway of apoptosis can be inhibited by HSP70 at several levels: 1) upstream of the mitochondria, HSP70 modulates the activation of stress-activated kinases such as Akt, JNK or ERK; 2) at the mitochondrial level, HSP70 prevents mitochondrial membrane permeabilization by interaction with BAX and thus, controlling the release of cytochrome c; 3) downstream of mitochondria, HSP70 can interact with apoptosis protease-activating factor-1 (Apaf-1) and consequently, inhibit recruitment of procaspase-9 to the apoptosome; 4) regulation of the enzymatic activity of caspase-activated DNase (CAD). HSP70-mediated inhibition of extrinsic pathway involves: 1) interaction with the death receptors (DR4 and DR5); 2) inhibition of Bid cleavage and consequently, the activation of the mitochondria pathway; 3) binding to apoptosis-inducing factor (AIF) and thus blocking caspase-independent apoptosis; 4) inhibition of cathepsin release by stabilizing lysosome membranes. Cell protective functions of alpha crystallins are associated with interactions of alpha A (αA) and alpha B (αB) with pro-apoptotic factors, such as Bax and Bcl-Xs, as well as interactions between αA and caspase 6 and between αB and caspase 3.
Figure 3.

HSF1 activation and attenuation. HSF1 in unstressed cells exists in the cytoplasm as an inactive monomer whose activity is repressed by interaction with HSP90, HSP70 and HSP40 as well as phosphorylation on S303 and S307 residues. Upon stress or in the presence of HSP90 inhibitors, HSP90 dissociates from HSF1/HSP70/HSP40 complex, allowing HSF1 trimerization and translocation into the nucleus where it binds to heat shock elements (HSE) in the promoters of stress-induced genes. Post-translational modifications, such as phosphorylation and sumoylation, are involved in regulating the transactivation capacity of HSF1. HSF1 attenuation involves negative feedback from HSPs, which represses the transactivation of DNA-bound HSF1 and the acetylation of K80 in the DNA binding domain (DBD), which inhibits HSF1 DNA binding.
HSF2 and HSF4 appear to be involved in the crosstalk with HSF1, but neither HSF2 nor HSF4 can functionally replace HSF1 or restore the heat shock induced response in HSF1-deficient mice or in cells derived from these animals (McMillan et al., 1998). HSF2 is reported to be involved in the regulation of development- and differentiation-specific gene expression and is not activated in response to stress stimuli. HSF4 has no carboxyl-terminal heptad repeat domain that is essential for the suppression of the trimer formation. Consequently, HSF4 could have constitutive DNA binding activity, which was shown in vitro (Tanabe et al., 1999). However, no HSF4 constitutive binding to the human HSP70 promoter was detected in vivo (Abravaya et al., 1991). HSF4 is required for lens development; mutations of HSF4 lead to cataractogenesis (Fujimoto et al., 2004; Bu et al., 2002). HSF3, 5, X and Y are thus far poorly characterized (Westerheide et al., 2012).
This review focuses on the cell protective roles of HSP70 and alpha crystallins in animal models of retinal ganglion cell (RGC) degeneration and considers the possibility of developing HSP-based therapeutic strategies for chronic degenerative optic neuropathies, such as glaucoma. These strategies are supported by an overwhelming number of studies, which demonstrate the role of HSPs, and particularly HSP70, in enhancing cell survival in models of various neurodegenerative diseases, some of which are described here.
2. The HSP70 superfamily
The HSP70 (HSPA) family is represented in the human genome by 13 members including constitutively expressed HSC70 (HSPA8), as well as the stress-inducible HSP70-1 (HSPA1A, also known as HSP72 and HSPA1) and HSP70-2 (HSPA1B). The HSPA1A and HSPA1B genes are located in tandem along an approximately 15 kb region of human chromosome 6p23.1. Proteins (641 amino acids) encoded by these intronless genes are different by only two amino acids and are believed to be fully interchangeable (Kampinga et al., 2009). In this review, we refer to HSPA1A/HSPA1B as HSP70. This protein is present in the cytosol, nucleus and endoplasmic reticulum (ER) and is synthesized at high levels in response to cellular insults. HSP70 has been recognized as a potential cell-protective protein ever since it was originally described (Livak et al., 1975). A number of studies have shown that HSP70 overexpression protects cells from both apoptotic and necrotic death induced by various insults (Jaattela, 1999; Buzzard et al., 1998). However, it must be acknowledged that even cells with high HSP70 expression may either survive or die, depending on the severity of injury. This indicates that HSP70 expression may be necessary but is not always sufficient to ensure cell survival. Numerous studies have been published on the neuroprotective role of HSP70, but the exact mechanism of neuroprotection by this stress protein remains unknown. The cell protective role of HSP70 could be related to its chaperone functions, or a result of its antiapoptotic and antinecrotic effects (Figs. 1 and 2). HSP70 inhibits c-Jun N-terminal kinase (JNK)-dependent and p38 mitogen-activated protein kinase (MAPK) signaling pathways (Gabai et al., 1997; Park et al., 2001a), blocks the assembly of a functional apoptosome by binding to apoptotic protease activating factor 1 and preventing the recruitment of caspases to the apoptosome complex (Beere et al., 2000), and inhibits caspase-independent cell death by interacting with apoptosis inducing factor (Matsumori et al., 2005).
3. HSP70 in neurodegenerative diseases
3.1. HSP70 and neurodegenerations associated with toxicity from misfolded/aggregated proteins
Several human neurodegenerative diseases are caused by protein misfolding and aggregation. These include: Alzheimer disease (AD), Parkinson disease (PD), amyotrophic lateral sclerosis, prion diseases [Creutzfeld-Jacob Disease, Gerstmann-Straussler-Scheinker syndrome, fatal familial insomnia, Kuru, Alpers syndrome], polyglutamine (polyQ) diseases [Huntington disease (HD), spinocerebellar ataxias (SCA), also known as Machado-Joseph disease, spinobulbar muscular atrophy (SBMA or Kennedy disease), and dentatorubropallidoluysian atrophy].
Overexpression of HSP70, in combination with HSP40, has been shown to reduce an accumulation of abnormal polyQ protein and to increase cell survival in a variety of cellular models of polyQ diseases (Cummings et al., 1998; Kobayashi et al., 2000; Wyttenbach et al., 2001). According to the “chaperone hypothesis of polyQ disease”, the normal endogenous levels of HSP70/HSP40 may be sufficient to control the damaging effects of polyQ-expanded proteins for decades. However, the balance between cellular chaperone capacity and production of polyQ-expanded proteins may be shifted during aging, leading to toxic aggregation pathways and subsequently triggering the onset of disease (Muchowski et al., 2000). The beneficial effects of HSP70 have been verified in studies with mouse models of polyQ diseases (Cummings et al., 2001; Li et al., 1998a), suggesting that this chaperone may have a broad protective effect against the toxicity associated with protein misfolding, oligomerization, and aggregation. In a mouse model of SCA, characterized by loss of motor coordination caused by the degeneration of Purkinje cells and brain stem neurons, overexpression of HSP70 suppresses neuronal degeneration and improves motor function. Cerebella of crossbred SCA1 mice with over-expression of inducible HSP70 demonstrated numerous Purkinje cells with thicker and more arborized dendritic branches than SCA1 neurons (Cummings et al., 2001). The effect of increasing HSP70 expression was studied in a mouse model for SBMA, an inherited motor neuron disease caused by the expansion of the polyQ. In SBMA, nuclear inclusions containing mutant androgen receptor protein were found mostly in the brainstem motor nuclei and spinal motor neurons (Li et al., 1998a; Li et al., 1998b). Nuclear inclusions, which are common pathological features in polyQ diseases, are co-localized with many components of ubiquitin-proteasome and molecular chaperones and indicate that misfolding, aggregation and altered degradation of the mutant protein are associated with the pathogenesis of polyQ diseases (Stenoien et al., 1999; Waelter et al., 2001). SBMA transgenic mice that were cross-bred with HSP70 overexpressing mice showed considerable amelioration of motor function. The level of mutant androgen receptor protein was significantly reduced, suggesting efficient degradation of mutant androgen receptor protein (Adachi et al., 2003).
HSP70 is protective against polyQ-induced toxicity in HD models (Carmichael et al., 2000; Sittler et al., 2001; Dedeoglu et al., 2002). Striatal lesion sizes were evaluated in homozygous (HSP70+/+), heterozygous (HSP70+/−) and wild-type controls neurotoxicity induced by malonate and 3-nitropropionic acid (3-NP) in animal models of HD which received 3-NP or malonate (Dedeoglu et al., 2002). Compared to homozygous and heterozygous HSP70 overexpressing mice, controls showed significantly larger striatal lesions after 3-NP or malonate injections. In contrast, ubiquitous overexpression of HSP70 in the R6/2 mouse model of HD has no effect on the solubility of aggregates and does not alter the course of the neurological phenotype (Hay et al., 2004).
Protein misfolding, aggregation, and inability to degrade specific neuronal proteins in addition to mitochondrial dysfunction and oxidative stress, were implicated in the pathogenesis of PD, a movement disorder characterized by degeneration of dopaminergic neurons in the substantia nigra pars compacta. The potential beneficial effects of HSP70 gene therapy for the treatment of PD was shown in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of idiopathic PD (Dong et al., 2005). Histological, biochemical, and behavioral data showed that recombinant adeno-associated virus (AAV) mediated delivery of HSP70 to dopamine neurons protects the dopaminergic system against MPTP-induced neuron loss. Directed expression of HSP70 prevented dopaminergic neuronal loss associated with α-synuclein (a protein implicated in the pathogenesis of PD) toxicity in Drosophila model for PD. In contrast, when chaperone function was compromised the α-synuclein-induced dopaminergic neuronal loss was enhanced (Auluck et al., 2002). By crossing α-synuclein transgenic mice with HSP70-overexpressing mice, a significant reduction in high molecular weight and insoluble α-synuclein species was observed. This indicates that HSP70 can alter α-synuclein misfolding and protect against the development of abnormal α-synuclein aggregation (Klucken et al., 2004).
3.2. HSP70 and neurodegeneration induced by ischemia
Although the endogenous level of inducible HSP70 in the brain is increased in response to stress such as ischemia (Nowak, 1990; Sharp et al., 1991; Planas et al., 1997), boosting the levels of HSP70 reduces brain injury in rat stroke models of both global and focal ischemia (Kelly et al., 2002; Davis and Antonawich, 1997; Yenari et al., 1998; Martin et al., 2000; Kelly et al., 2001; Zhang et al., 2001). Twenty-four hours after occlusion of the middle cerebral artery in transgenic mice constitutively expressing the human inducible HSP70, hippocampal pyramidal neurons had normal morphology with no evidence of pyknosis, whereas in the wild-type control mice, pyramidal neurons of the ipsilateral hippocampus were found to be pyknotic (Plumier et al., 1997). In a similar study, the extent of cerebral infarction was analyzed in heterozygous transgenic mice overexpressing the rat HSP70 and their wild type littermates were subjected to permanent focal cerebral ischemia by intraluminal blockade of the middle cerebral artery. The HSP70 transgenic mice were protected against cerebral infarction at 6 and 24 hours after the procedure, suggesting that HSP70 can protect the brain against ischemic damage (Rajdev et al., 2000). HSP70 gene transfer into the striatum with adenovirus vectors was also reported to reduce ischemic neuronal damage resulting from global cerebral ischemia in mice (Kelly et al., 2001). Furthermore, pharmacological stimulation of HSP70 and HSP25 expression by geldanamycin protected neurons in the brain from focal ischemia and improved post-ischemic behavioral outcomes (Lu et al., 2002).
4. HSP70 and RGC survival
4.1. HSPs and challenges in developing clinically relevant neuroprotection for glaucoma
The degeneration of RGCs and their axons in the optic nerve is the cause of visual deficits in optic neuropathies; this includes glaucoma, which affects millions of people and if left untreated can lead to blindness. Chronic forms of the disease usually progress over years or decades. Currently, reduction of intraocular pressure (IOP) remains the main strategy to slow progression of the disease. However, glaucomatous neuropathy often continues to progress even after IOP has been reduced, especially in advanced cases of the disease. It is clear that new strategies are required to supplement or even replace IOP reduction in some patients to decrease rate of RGCs loss and subsequent visual disability. Since the exact molecular pathways of RGC death are not yet understood, several directions of RGC neuroprotection are being investigated, including supplying neurotrophins (Mansour-Robaey et al., 1994; Cheng et al., 2002; Martin et al., 2003), blocking glutamate excitotoxicity (Hare et al., 2001; Schori et al., 2001), overexpressing proteins regulating the cellular redox state (Munemasa et al., 2009a) stabilizing Ca2+ homeostasis (Zhang et al., 2003; Wood et al., 2003; Hains et al., 2005), inhibiting nitric oxide production (Le and Lipton, 2001; Neufeld et al., 2002), preventing apoptosis (McKinnon et al., 2002; Huang et al., 2005), and modulating immunologic status (Anderson et al., 2005; Bakalash et al., 2003). Although many of these strategies protect RGCs in the laboratory setting, they were primarily designed as proof of principle studies and targeted a specific protein or pathway associated with glaucomatous neurodegeneration in animal models. One of these strategies aimed at reducing excitotoxicity with memantine (an N-methyl-D-aspartate receptor antagonist) has been evaluated in clinical trials, but has unfortunately showed no benefit in preserving visual function compared with IOP reduction alone (Allergan Press Release, 2008). One of the main challenges in designing effective and clinically relevant therapies for glaucoma is the multifactorial nature of the pathogenesis of the disease. Targeting specific molecular pathways may be beneficial for small subsets of patients, but not for all. Furthermore, multiple pathways may be involved in the progression of the disease in the same patient simultaneously or at different times. Therefore, targeted therapies may prove to be effective in personalized medicine tailored to the individual patient based on detailed information about the mechanisms involved in the pathogenesis of his/her disease. Until this information is available, enhancing the overall cell defense mechanisms, rather than targeting specific factors or pathways, may be a more viable strategy to protect RGCs. We believe that HSP70's antiapoptotic and cytoprotective characteristics, its ability to protect cells from a variety of stressful stimuli, and the possibility of its pharmacological induction in cells experiencing stress make this protein an attractive therapeutic target for this disease. Several studies briefly described here support the idea that stimulation of HSP70 expression may be beneficial to the survival of RGCs.
4.2. HSP70 protects RGCs from ocular hypertension- and optic nerve crush-induced injury
A rat model of experimental glaucoma to evaluate the protective effects of HSP70 induced by heat shock, zinc and geranylgeranylacetone (GGA) was generated by trabecular laser photocoagulation. A moderate elevation of IOP was sustained over 8 weeks (Ueda et al., 1998; Park et al., 2001b). Glaucomatous changes in this model have been characterized by evaluating RGC density with retrograde labeling, grading optic nerve injury, cell counting in the ganglion cell layer (GCL) of cresyl-violet-stained retinas, and counting terminal deoxynucleotidyl transferase dUTP nick end labeling-positive apoptotic cells in the GCL (Ueda et al., 1998; Park et al., 2001b; Ishii et al., 2003). The histopathological studies were performed 1 day, 3 days, and 1, 2, 4, 5 and 8 weeks after IOP elevation. The earliest statistically significant RGC loss of 8%-13% in this model was observed two weeks after IOP elevation. Reduction in RGC numbers reached 20%-27% and approximately 45% by five and eight weeks after IOP elevation, respectively, compared to control eyes (Park et al., 2001b; Ishii et al., 2003; Piri et al., 2007). One week after IOP elevation, a mild drop-out of RGCs in the GCL was observed and irregularity of astrocytic columns and axons in optic nerve head was noted. Electron microscopic examination revealed degeneration of RGC bodies and their axons with various features at the level of lamina cribrosa in the optic nerve head such as accumulation of organelles in scattered axons starting at 3 days, and swollen axoplasm, demyelination and swollen mitochondria (Fig. 4).
Figure 4.
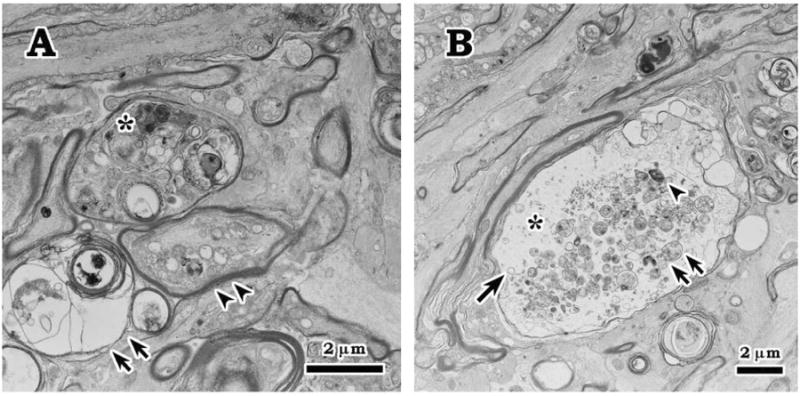
Axonal degeneration at 7 days after IOP elevation. A. Electron microscopic examination revealed degenerating axons with various features close to the level of lamina cribrosa in the optic nerve head such as accumulation of condensed (*) and swollen (double arrowheads) intra-axonal organelles, and myelin degeneration (double arrows). B. Scattered swollen axon with expanded axoplasm (*) and accumulation of myellin materials (arrowhead), swollen mitochondria (double arrows), and dilated membrane-bounded organelle (arrow).
RGCs' susceptibility to damage in glaucoma is almost certainly associated with more than one risk factor. Risk factors sufficient for or contributing to the development of the disease may vary by glaucoma type and from patient to patient within the same subtype of the disease, as well as in the same individual over time. These factors determine not only the likelihood of developing the disease, but also the severity and the rate of its progression. The multifactorial nature and a limited knowledge about the causative and contributing factors in human glaucoma present a major challenge in designing and generating appropriate animal models, which in turn creates an enormous obstacle for studying the mechanisms of glaucomatous neurodegeneration as well as developing new therapeutic strategies for this disease. The elevated IOP model described above is traditionally used as a model for glaucoma. However, this model represents only one, although important, risk factor associated with glaucoma and even if an effective therapy is developed in this model, such therapy may not target mechanisms unrelated to IOP elevation. From the standpoint of designing new neuroprotective strategies to control the progression of glaucomatous damage, it is important to target non-IOP components of glaucoma pathophysiology since the desired IOP levels can often be achieved by an array of currently available medications and surgical procedures. Since the HSP70-based RGC protective strategy is predicted to be effective against various types of stress or cellular damage, the role of this protein in stimulation of cell survival was assessed not only in animals with ocular hypertension (OH) but also in animals with optic nerve crush (ONC)-induced RGC degeneration. Although the ONC model is not typically considered a model for glaucoma, the mechanisms involved in the RGC degeneration after axonal injury may be relevant to both IOP-dependent and IOP-independent components of glaucomatous pathophysiology. Results of the studies on the effect of pharmacological (17-allylamino-17-demethoxygeldanamycin, 17-AAG) and viral-mediated induction of HSP70 on survival of RGC with axonal injury are briefly discussed here.
4.2.1. HSP70 induction by hyperthermia
A brief period of hyperthermia (heat stress) in cultured cells as well as in the whole animal correlates with enhanced cell survival upon further stress. For instance, heat stress effectively induces HSP70 expression in the retina and significantly decreases photoreceptor degeneration in animals exposed to bright light compared to normothermic animals (Barbe et al., 1988; Tytell et al., 1994). This cell resistance to damage was associated with hyperthermia-induced synthesis and accumulation of HSPs. To test the effect of hyperthermia on survival of RGCs injured by elevated IOP, anesthetized rats were treated with heat stress by placing them in a water bath at a constant temperature of 42°C (Park et al., 2001b). Their body temperatures were continuously monitored and upon reaching 40°C animals were kept in the bath for 15 minutes. The average RGC densities at four weeks after IOP elevation were 890 cells/mm2 for the control group, 1318 cells/mm2 for the heat stress group and 1069 cells/mm2 for the heat stress plus quercetin injection group. An increase in the immunoreactivity of HSP70 was present in RGCs purified from heat-stressed rats compared with control animals. The expression of HSP70 in RGCs from heat-stressed rats was inhibited when these rats were pretreated with quercetin (400 mg/kg; an inhibitor of HSP synthesis) before heat stress. These data indicate that hyperthermia supports the survival of RGCs damaged by OH and that hyperthermia-mediated cell protection depends on the upregulation of HSP70 expression.
4.2.2. HSP70 induction by zinc sulfate
Zinc is a ubiquitous element in the cell and is the second (after calcium) most abundant divalent cation in organisms. As a structural or catalytic component of more than 300 enzymes, zinc plays an important role in regulating cellular processes, including communication, proliferation, differentiation, and survival. Zinc deficiency can lead to various pathological conditions while its excess is toxic. Zinc concentration is tightly regulated by zinc transporters: ZnTs [members of the solute-linked carrier 30 transporter family] reduce cytosolic zinc concentration and ZIPs [members of the solute-linked carrier 39 family] increase it. In the brain, zinc is concentrated in synaptic vesicles of glutamatergic neurons (Paoletti et al., 2009; Marger et al., 2014). During active neurotransmission, zinc release into the synaptic cleft is increased. Zinc has been reported to be a potent inducer of HSP70 expression and although the exact mechanism of zinc induction of HSP70 is unknown, it has been proposed that translocation of synaptic zinc to postsynaptic neurons in the hippocampus induces HSP70 expression (Lee et al., 2000). The ability to induce Hsp70 expression together with its low toxicity compared with other transition metals were essential factors in choosing zinc to evaluate HSP70-mediated RGC protection from glaucomatous damage (Choi and Koh, 1998). To stimulate HSP70 expression, animals with experimental glaucoma were treated with intraperitoneal injection of 10 mg/kg of zinc sulfate twice a week, which, as expected, did not show any systemic side effects during the 4-week study period. This regimen for zinc treatment was chosen based on a previously published study showing no noticeable pathologic changes in rats receiving 16 mg/kg of zinc daily for 32 weeks (Klosterhalfen et al., 1997). In a human clinical study, patients with macular degeneration were treated with an oral dose of 200 mg of zinc sulfate daily for 2 years with no significant adverse effects (Newsome et al., 1988). The average density of RGCs was increased from about 890 cells/mm2 in a control group to 1600 cells/mm2 in the zinc-injected group 4 weeks after IOP elevation (Park et al., 2001b). This approximately 80% increase in RGC survival was associated with the induction of HSP70 expression in zinc-treated animals. The involvement of HSP70 in RGC protection was further demonstrated in animals injected with quercetin, which diminished the HSP70 expression and significantly decreased the number of surviving RGCs in the zinc-treated group (Park et al., 2001b). Zinc sulfate-induced upregulation of HSP72 was associated with blockage of the stress-activated protein kinase SAPK/JNK apoptotic pathway, and consequently with enhanced RGC and lateral geniculate nucleus neuron survival in rat glaucoma model (Li et al., 2014).
4.2.3. HSP70 induction by GGA
GGA (or Teprenone) is an acylic polyisoprenoid used in Japan and other Asian countries for the treatment of gastric ulcers under the brand name Selbex since 1984. In multiple animal models of ischemia and reperfusion, GGA prevents oxidative stress in the liver, heart, brain, kidney, and retina (Ohkawara et al., 2006; Tanito et al., 2005; Suzuki et al., 2005; Zhao et al., 2013; He et al., 2015). GGA's cytoprotective effect was associated primarily with induction of HSP70, as well as stimulation of the thioredoxin system (Patury et al., 2009; Hirota et al., 2002). The effect of GGA on induction of HSP70 expression and its correlation with RGC survival was evaluated in rats with induced glaucoma (Ishii et al., 2003). Animals in this study were treated with daily intraperitoneal injections of GGA at a dose of 200 mg/kg. HSP70 expression levels were analyzed at 1, 3 and 7 days after GGA administration. An increase in HSP70 expression in RGCs isolated from GGA-treated animals was detected as early as one day after drug administration and was significantly higher at days 3 and 7. The survival of RGCs 5 weeks after IOP elevation was increased by almost 60% in the GGA-injected group compared to controls. A possible correlation between HSP70 induction and RGC survival was evaluated by coadministration of quercetin, an inhibitor of HSP70 expression. Inhibition of HSP70 expression abolished the RGC protective effect of GGA against OH. Systemic administration of GGA protects retina from chronic IOP elevation by regulating the expression of HSP70. GGA-induced upregulation of HSP70 and RGC protection in chronic IOP elevation was also observed by Liu et al. (2010). Results of these experiments suggest that although GGA may promote cell survival by modulation of different pathways, the mechanisms involved in its protection of RGCs from elevated IOP strongly depend on the induction of HSP70 expression.
4.2.4. HSP70 induction by 17-AAG
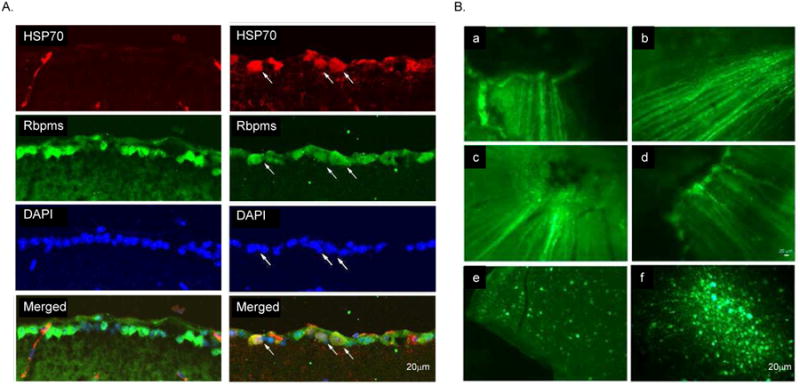
17-AAG (a derivative of the antibiotic geldanamycin) is a potent Hsp90 inhibitor that specifically binds to Hsp90's ATP-binding site and modulates its function (Whitesell et al., 1994; Prodromou et al., 1997; Pratt and Toft, 2003; Giubellino et al., 2013). The binding of these drugs to HSP90 facilitates the dissociation and activation of HSF1, which forms a complex with HSP90 in the resting cells (Figs. 3 and 5A). Upon its activation, which involves trimerization, phosphorylation, and nuclear translocalization, HSF1 binds to the heat shock response elements in HSP promoters and induces transcription of various HSPs (Neef et al., 2011). The ability of HSP90 inhibitors (including 17-AAG) to imitate the cellular heat shock response and induce the expression of HSPs (HSP70 in particular) has been associated with their cell protective effects in various models of degenerative diseases (McLean et al., 2004; Waza et al., 2005; Batulan et al., 2006; Fujikake et al., 2008; Putcha et al., 2010; Tam et al., 2010). This was the main rationale in our study to evaluate the effect of 17-AAG on the survival of RGCs after axonal injury. Another important factor in choosing this drug is that 17-AAG, as a potent HSP90 inhibitor, has been extensively studied in cancer research as a new strategy for the proteosomal degradation of HSP90 client oncoproteins and has been tested in several Phase II clinical trials in patients with various types of advanced or metastatic cancer (Heath et al., 2008; Gartner et al., 2012; Pacey et al., 2012; Ronnen et al., 2006). These studies provide important information about the maximum tolerated dose, potential toxicity, and bioavailability of the drugs used to design our experiments. Recently, we have evaluated the effect of 17-AAG-mediated upregulation of HSP70 on the survival of RGCs injured by ONC (Kwong et al., 2015). The extent of RGC loss in these animals was evaluated by counting RGCs labeled with Rbpms (RNA-binding protein with multiple splicing), an RGC marker identified and characterized in our laboratory (Fig. 6; Kwong et al., 2010). A single intravitreal injection of 17-AAG (4 ul; 0.2 ug/ul) increased survival of ONC-injured RGCs by approximately 50% compared to the vehicle-treated animals. We believe that the effect of 17-AAG on RGC survival could be even stronger with more than a single dose administration. An intravitreal injection was chosen in this study to increase the exposure of retinal cells to the drug. However, the treatment was limited to one injection in order to minimize potential injury to the eye associated with the procedure. Immunoblot analysis showed an upregulation of HSP70 expression in retinas of 17-AAG-treated animals with and without optic nerve injury by about 2.2-fold compared to the vehicle-injected uninjured and ONC animals, respectively (Figs. 5B and C; Kwong et al., 2015). An increase in the HSP70 expression level in retinas of 17-AAG-injected animals was also observed by immunohistochemistry. The expression of HSP70 in this study was evaluated 2 weeks after intravitreal injection of 17-AAG, and it was somewhat unexpected to observe sustained upregulation of HSP70. To our knowledge, there is only one published report that assessed the effect of intravitreal injection of 17-AAG on cell survival. In this report, 17-AAG was used to rescue photoreceptors in a murine model of autosomal dominant retinitis pigmentosa caused by a mutation in the inosine-5′-monophosphate dehydrogenase 1 gene (Tam et al., 2010). HSP70 expression in experimental retinas was shown to be upregulated by about 1.8 fold at 24 hours, but not at 48 or 72 hours after 17-AAG injection compared to the control retinas.
Figure 5.
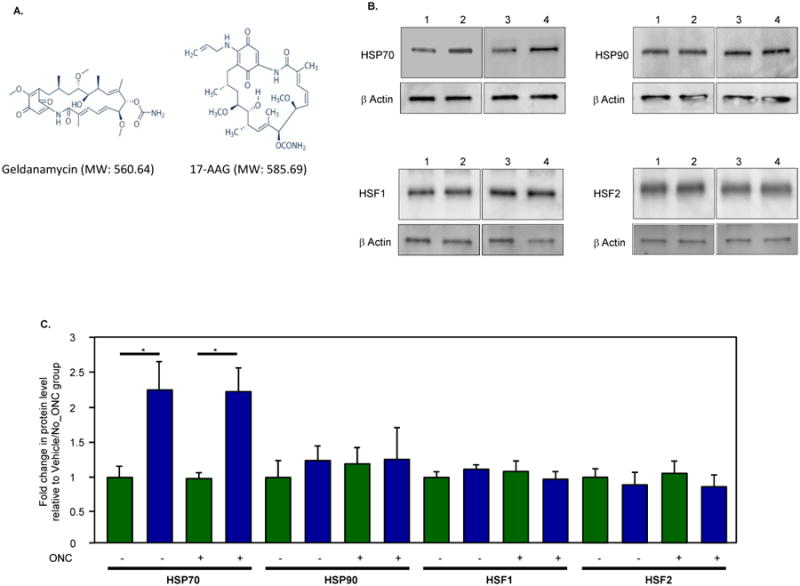
17-AAG (17-N-allylamino-17-demethoxygeldanamycin; Tanespimycin), an HSP90 inhibitor, induces expression of HSP70. A. Structures of geldanamycin and its derivative 17-AAG used as HSP90 inhibitors. B. Western blot analysis of HSP70 in retinal extracts from ONC and uninjured rats treated with vehicle or 17-AAG showed a significant change in HSP70 expression level associated with the administration of 17-AAG. Protein were quantified 2 weeks after ONC. A single intravitreal injection of 17-AAG or vehicle was given on the day of ONC. Lanes: 1, vehicle; 2, 17-AAG; 3, ONC/vehicle; 4, ONC/17-AAG. C. Quantitative analysis of HSP70 expression in retinas of animals treated with 17-AAG. In 17-AAG-treated animals with and without optic nerve injury (blue bars), HSP70 expression was upregulated about 2.2 fold (*P=0.01; n=6) compared to vehicle-injected uninjured and ONC animals (green bars). 17-AAG had no significant effect on expression levels of HSP90, HSF1 or HSF2.
Figure 6.
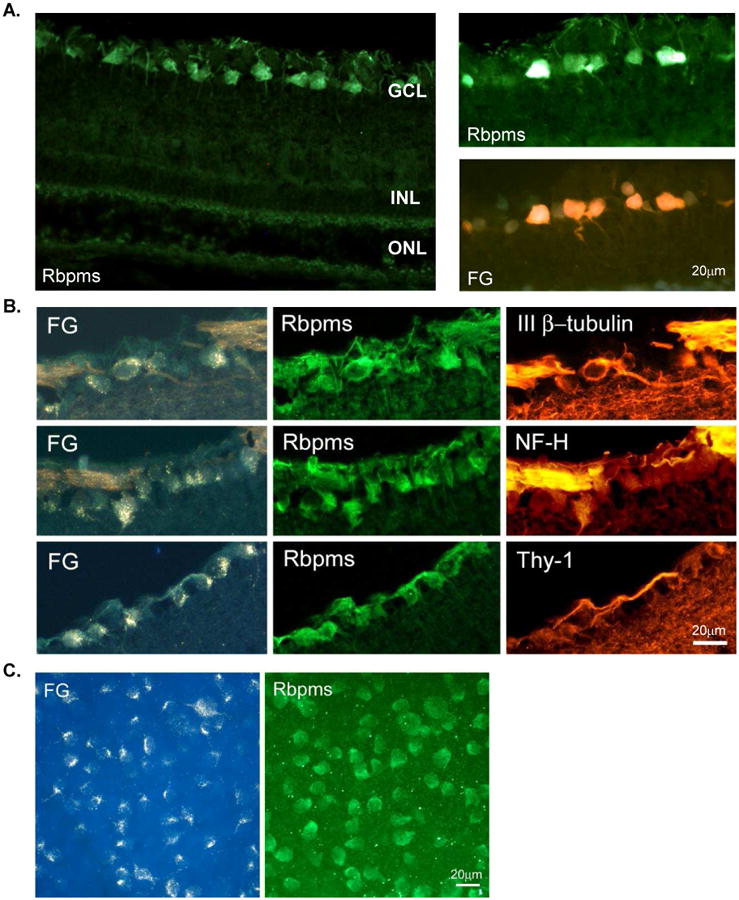
RBPMS is an RGC marker. A. Rbpms immunohistochemistry in the retina sections and its co-localization with retrogradely labeled RGCs. B. Immunohistochemical co-localization of Rbpms expression with commonly used RGC markers, Thy-1 and neurofilament H (NF-H) and neuronal marker, III β-tubulin. Approximately 97%, 95% and 96% of Rbpms-positive cells were also stained with III β-tubulin, NF-H or Thy-1, respectively. C. Rbpms immunohistochemistry in the whole mount retina and its co-localization with retrogradely labeled RGCs. Large irregularly shaped cells, as well as cells with smaller somas were among the labeled cells. Rbpms staining is present in the nucleus but is predominantly localized in the cytoplasm. Almost 100% (over 99.5%) of cells labeled by FG were also Rbpms positive irrespective of their location relative to the optic nerve head. FG, Fluorogold; ONL outer nuclear layer; INL, inner nuclear layer; and GCL, ganglion cell layer.
The cell protective effects of the HSP90 inhibitors, including 17-AAG, are generally associated with an induction of HSPs, and of HSP70 in particular (Fig. 3). For instance, in the drosophila model of SCA, it has been shown that the therapeutic effect of 17-AAG on photoreceptor degeneration depends on the activation of HSF1, which leads to the induction of HSP70, HSP40, and HSP90 expression (Fujikake et al., 2008). As expected, the HSF1 knockdown abolished the effect of this drug on HSP expression and consequently no photoreceptor protection was observed. In our study, we observed 17-AAG-induced upregulation of HSP70, but the level of HSP90 was similar to that of vehicle-treated control retinas (Kwong et al., 2015). Although we favor the idea of 17-AAG-mediated RGC protection through HSF1-induced upregulation of HSP70, recent studies suggest a non-canonical, HSP-independent mechanism for HSF1 stimulation of cell survival, which involves association of HSF1 with sirtuin 1 (Sirt1). Sirt1 is a member of the sirtuin family of nicotinamide adenine dinucleotide-dependent histone deacetylases (Verma et al., 2014; Mimura et al., 2013). By deacetylating multiple histone and nonhistone proteins, including FOXO transcription factors family, p53, Ku-70, PGC1α, and p65/RelA NF-κB, Sirt 1 regulates a wide array of cellular processes crucial to cell survival, apoptosis, cell senescence and metabolism (Finkel et al., 2009). Furthermore, Sirt1 has been shown to increase the heat shock response by maintaining HSF1 in a deacetylated, DNA-binding competent state and by prolonging HSF1 binding to the HSP70 promoter (Liu et al., 2014; Westerheide et al., 2009).
4.2.5. AAV2-mediated expression of HSP70
The neuroprotective effect of hyperthermia, systemic administration of zinc and GGA, as well as intravitreal treatment with 17-AAG were associated with HSP70 induction. However, such treatments in addition to induction of HSP70 expression, will most certainly affect the expression of a host of proteins (including but not limited to other members of the HSP family) that may contribute to boosting cell defense mechanisms. This complicates the interpretation of the role of HSP70 in this process. To determine the ability of HSP70 to stimulate the survival of injured RGCs, an AAV2-HSP70 vector was used to express this protein in the retina of animals with ONC injury (Kwong et al., 2015). We found that RGC survival was increased by an average of 110% two weeks after axonal injury compared to the control. In the inferior retina, more than 300% increase in RGC numbers was observed. This suggests that more efficient cell transfection with AAV2-HSP70 in this portion of the retina is responsible for more efficient RGC protection. Relatively higher transfection efficiency of cells in the inferior retina was also observed after intravitreal injection of AAV2-GFP (Fig. 7; Kwong et al., 2015). The results of this study clearly indicate that HSP70-based therapy can be applied to protect RGCs from axonal injury. This therapy has the potential to be developed into a clinically relevant treatment for optic neuropathies. AAV-based gene therapy appears to be an attractive strategy for the treatment of different ocular diseases, including glaucoma, and has been evaluated in clinical trials in patients with Leber's congenital amaurosis and choroideremia (Hauswirth et al., 2008; Maguire et al., 2008; MacLaren et al., 2014).
Figure 7.
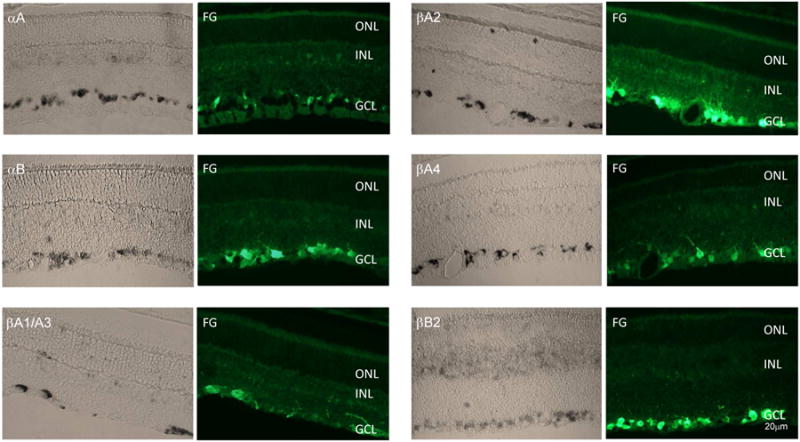
AAV2-mediated GFP expression in the retina. (B) GFP fluorescence was observed in RGC axons and somas 4 weeks after intravitreal administration of AAV2-GFP. Regional GFP expression was present in the inferior (a), temporal (b), inferior-temporal (c) and inferior-nasal (d) retina. GFP-positive cells were detected primarily in superior (e) and inferior (f) retina. A few GFP-positive axons were noted in the temporal and nasal retina.
5. Small HSP (sHSP) superfamily: structure, function and expression
5.1. sHSPs contain signature α-crystallin domain and form oligomers
The sHSP (HSPB) superfamily includes proteins with a molecular mass of the monomer between 12 and 43 kDa. sHSPs are characterized by the presence of a conserved signature sequence of 80–100 amino acids termed the “α-crystallin domain” (Kampinga et al., 2009). The α-crystallin domains are enriched in beta-strands (6–8 strands) organized in a beta-sheet sandwich (Bagnéris et al, 2009; Langanowsky et al, 2010; Baranova et al, 2011). They are involved in various intra- and inter-molecular interactions resulting in formation of dimers, building blocks for higher order oligomers that may consist of one or more sHSP family members (Van Montfort et al., 2001a, Van Montfort et al., 2001b and Kim et al., 1998). The flanking N- and C-terminal regions, although highly divergent among sHSPs, are involved in the stabilization of the oligomers (Kim et al., 1998, Lambert et al., 1999, Van Montfort et al., 2001a and Van Montfort et al., 2001b). Sequence alignment of the C-terminal regions of sHSPs shows a presence of a highly conserved IXI/V motif in all but two members (HSP20/HSPB6 and HSP22/HSPB8) of the family that contributes to the oligomer stabilization (Caspers et al., 1995). The interactions between the C-terminal region and the α-crystallin domain involve three α-crystallin domain regions: the β3 strand, the β4/β8 groove on the edge of the α-crystallin domain structure and the surface formed by the dimer interface (Kim et al, 1998; van Montfort et al, 2001; Bagnéris et al, 2009; Langanowsky et al, 2010). The most frequently detected interaction is the association of the IxI/V motif in the C-terminal region with the α-crystallin domain β4/β8 groove. This interaction involves neighboring dimers, enabling the formation of oligomer structure. The IxI/V-β4/β8 interactions have been observed in crystal structures of αB-crystallin, Methanococcus jannaschii HSP16.5, and wheat HSP16.9 homo-oligomers, as well as in α-crystallin domain crystals, where the interactions take place among neighboring dimers in the crystal lattice (Kim et al, 1998; van Montfort et al, 2001; Langanowsky et al, 2010). Heteromeric IxI/V-β4/(38 interactions between αB and αA-crystallins have been identified in solution by fluorescence resonance energy transfer experiments (Pasta et al, 2004). Mutations or truncations of the region containing this motif cause disassembly of the high molecular weight oligomer complexes and loss of chaperone-like activity (Pasta et al, 2004; Kim et al., 2003; Studer et al., 2002; Fu et al., 2005).
sHSP oligomer formation, as well as their substrate binding ability and consequently, chaperone activities, also depend on the integrity of the sHSP's N-terminal regions (Studer et al., 2002; Stromer et al., 2004; Smith et al., 1996; Fu et al., 2005; Basha et al., 2006). This critical relationship was clearly demonstrated for several sHSPs including HSP26, class A and class B α-HSPs, HSP16.9 and HSP18.1. Studer et al. (2002) investigated the contribution of the N-terminal end in oligomerization of class A and class B α-HSP proteins of B. japonicum, which normally form a complex of about 24 subunits with a molecular mass of 400–500 kDa. Deletion of a few amino-acid residues [HspH(Δ3N) and HspF(Δ5N)] from the N-terminus had no effect on the chaperone activity and oligomeric state of these proteins. However, further deletion of HspH N-terminal amino acids [HspH(Δ9N), HspH(Δ15N) and HspH(Δ20N)] resulted in assembly of non- functional α-Hsp complexes consisting of approximately eight subunits (Stromer et al., 2004). Extended truncations in the N-terminal region of HspF [HspF(Δ30N) and HspF(Δ40N)] reduced the formation of oligomer complexes to dimers and abolish the protein's chaperone activity. The importance of the N-terminal in the assembly of the higher order oligomer complexes was evaluated in two closely related dodecameric wheat Hsp16.9 and pea (Pisum sativum) Hsp18.1 (Bash et al., 2006). Variable N-terminal ends of these otherwise highly conserved proteins on one hand and significant differences in their chaperone activities on the other, suggests the importance of the N-terminus in substrate interactions. The N-terminal regions of Hsp18.1 and Hsp16.9 are only 41% identical (50% similar), and the N terminus of Hsp18.1 has an additional 6-residue insertion (residues 5–10). Chimeric proteins were produced by switching the entire N-terminal arms or the first 10 residues between Hsp18.1 and Hsp16.9. The chimeric proteins with the Hsp18.1 N terminus were similar to native Hsp18.1 with respect to their oligomeric structures and chaperone activities. However, both chimeras containing the Hsp16.9 N terminal ends and especially the one with the entire Hsp16.9 N terminus, displayed impaired oligomerization and were present as dimer or unstable oligomers. The sHSPs assembly and disassembly into mono- and poly-disperse oligomers can be influenced by a number of factors including phosphorylation, temperature, pH, protein concentration, oxidative stress, and age (reviewed in Sun and MacRae 2005). The dynamics of this process plays an essential role in the effectiveness of sHSP substrate binding and chaperoning function.
5.1.1. Crystallin superfamily of sHSPs
Mammalian crystallins are heterogeneous proteins classified into three major families: alpha, beta, and gamma crystallins. Crystallins were first identified in the ocular lens fiber cells and characterized as lens structural components responsible for its transparency. Alpha crystallin, consisting of alpha A and alpha B in approximately a 3:1 molar ratio, is the main type of lens crystallin. Alpha A and alpha B form large polydisperse oligomeric species with a molecular weight ranging from 300 to 1200 kDa that undergo dynamic subunit exchange (Bova et al., 1997). Extra-lenticular expression of crystallins has been well established: alpha A present in the retina, thymus and spleen (Srinivasan et al., 1992; Deretic et al., 1994) and alpha B were found in the retina, cornea, optic nerve, astrocytes, Muller cells, as well as in non-ocular tissues such as the brain, kidneys, lungs, liver, spleen, skin, cardiac and skeletal muscles (Bhat and Nagineni, 1989; Dubin et al., 1989; Iwaki et al., 1989, Kato et al., 1991). The beta crystallin family includes four acidic (A1-A4) and three basic (B1-B3) subunits with estimated 22-28 kDA of molecular weight. The beta A1 and beta A3 crystallins are encoded by the same gene; the synthesis of the beta A3 crystallin protein is initiated from a more upstream initiation codon than the beta A1 crystallin. The beta crystallins are oligomeric proteins. Gamma-crystallins are monomeric proteins represented by seven polypeptides: A, B, C, D, E, F and S. With the exception of gamma S, gamma crystallins are encoded by a tandemly repeated gene cluster and share high sequence homology, particularly in the N-terminal domain. The beta and gamma crystallins are homologous β-sheet proteins and often grouped into the beta/gamma crystallin superfamily. The structural unit of beta and gamma crystallins is the βγ-crystallin domain of 80–100 residues. Both proteins have two domains, each comprised of four β-strands arranged in two Greek key motifs (Blundell et al., 1981; Wistow et al., 1983).
With respect to their function, alpha A and alpha B are the best-studied members of the crystallin superfamily. Their chaperone-like activity and the ability to increase cell resistance to stress-inducible apoptosis are well established (Piatigorsky, J., 1998; Alge et al., 2002; Horwitz 1992, 2003; Xi et al., 2003a; Liu et al., 2005). The mechanisms for alpha A and alpha B crystallins' cell protective effect have been associated with the regulation of several antiapoptotic pathways (see section 5.3. sHSP function).
5.2. Expression profiles of sHSPs
The mammalian sHSP superfamily is represented by eleven proteins with different expression profiles (Kampinga et al., 2009). It is commonly accepted that HSP27 (HSPB1), alpha B crystallin (HSPB5), HSP20 (HSPB6), HSP22 (HSPB8) and HSPB11 are mostly ubiquitously expressed, HSPB2, HSPB3 and HSPB7 are restricted to skeletal and cardiac muscles, alpha A crystallin (HSPB4) is lens-specific and HSPB9 and HSPB10 are both testis-specific (Garrido et al 2012; Loones et al., 2000, Tallot et al., 2003, Verschuure et al., 2003 and Zhu et al., 2010). It is important to note however, that large number of studies including those from our laboratory demonstrate extralenticular expression of alpha A crystallin in various tissues including the retina (Egwuagu and Chepelinsky, 1997; Piri et al., 2007). Kirbach and Golenhofen (2011) analyzed the expression of all 11 sHSPs in various regions of the rat brain at developmental stages P3, P9, P15, P21, and adult by in situ hybridization and real-time PCR. Expression of only alpha B crystallin, HSP20, and HSPB11 was detected by in situ hybridization. Alpha B crystallin expression in the adult brain was primarily localized to the cerebellum white matter and granular cell layer, telencephalon white matter, and thalamic nuclei. HSP20 staining was observed in the cerebellar cortex and the choroid plexus of the ventricles. HSPB11 mRNA expression was observed in hippocampus and choroid plexus. With respect to developmental changes, expression of both alpha B crystallin and HSP20 was markedly increased during brain maturation, whereas HSPB11 was increased from P3 to P15 with almost no change thereafter. These findings are in agreement with the original reports on alpha B crystallin expression in non-lenticular tissues including brain (Bhat and Nagineni, 1989; Dubin et al., 1989; Iwaki et al., 1989), as well as with number of other studies on expression of this gene in healthy tissues and during various pathologies.
5.2.1. Crystallin expression in the retina
We have analyzed expression patterns of several alpha and beta crystallins, including alpha A, alpha B, beta A1/A3, beta A2, beta A4, and beta B2 in the rat retina. These crystallin genes were selected since their expression levels were reduced in a OH animal model for glaucoma (Piri et al., 2007; 2013). Regulation of these genes at mRNA and protein levels in response to IOP elevation is described in the following section. In situ hybridization of control rat retinal sections with riboprobes corresponding to each of these crystallins showed similar distribution of alpha A, alpha B, beta A1/A3, beta A2, beta A4, and beta B2 mRNAs with strong presence in the GCL and relatively weak staining in the inner nuclear layer (INL, Fig. 8; Piri et al., 2007). In order to determine the identity of the crystallin positive cells in the GCL, which in rodents contain both RGCs and non-RGCs, such as displaced amacrine cells in a nearly 1:1 ratio, retinas with retrogradely labeled RGCs were used for in situ hybridizations (Fig. 8). Signals for both alpha (alpha A and alpha B) and beta (beta A2, beta A4, and beta B2) crystallin genes in the GCL were clearly co-localized with RGCs. Expression of alpha A, alpha B and beta H crystallins in the mouse retina was earlier analyzed by immunofluorescence (Xi et al., 2003b): alpha A was shown to be distributed in the GCL, INL and ONL; alpha B and beta H crystallins had a similar distribution pattern as alpha A, but were also detected in the photoreceptor inner segments.
Figure 8.
In situ analysis of the alpha and beta crystallin expression in the retina. The expression of crystallins alpha A (αA), alpha B (αB), beta A1/A3 (βA1/A3), beta A2 (βA2), beta A4 (βA4), and beta B2 (βB2) was primarily observed in the ganglion cell layer (GCL). Relatively weak staining can also be seen in the inner nuclear layer (INL) and, to a much lesser degree, in the outer nuclear layer (ONL). Crystallin positive cells in the GCL were colocalized with RGCs retrogradely labeled with Fluorogold (FG).
A similar pattern of expression of alpha and beta crystallin genes in the retina and coordinated change in their expression level in response to IOP elevation suggest a common mechanism for transcriptional regulation of these genes. Regulation of temporal and spatial expression of crystallin genes during lens differentiation may involve various arraignments of developmentally regulated TFs, including Pax6, c-Maf, MafA/L-Maf, MafB, NRL, Sox2, Sox1, RARβ/RXRβ, RORα, Prox1, Six3, γFBP-B, HSF2, and HSF4, with ubiquitously expressed factors AP-1, CREB, pRb, TFIID, and USF (Cvekl and Piatigorsky, 1996; Somasundaram and Bhat, 2004; Cvekl et al., 2004; Cvekl and Duncan, 2007). Crystallin transcription in the retina may involve some of the TFs that control lenticular expression of these genes, although it is very likely that the TF ensembles have significant tissue as well as cell specific differences. For instance, a 148 kb genomic fragment encompassing the alpha A crystallin gene contains all regulatory regions required for expression of this gene in the lens, but not in the retina, thymus or spleen (Wolf et al., 2008). From the list of non-ubiquitous factors involved in crystallin transcription in the lens, only Pax6 and RORα are known to be localized in the GCL of the differentiated retinas and therefore, can serve as candidate TFs for regulation of crystallin expression in RGCs [Cvekl et al., 2004; Jones et al., 1998; Bhat et al., 2004; Steinmayr et al., 1998). Pax6 is a “master control” gene of eye and brain development. Mutations in this gene have been associated with CNS defects, anophthalmia and nasal hypoplasia (Glaser et al., 1994). Pax6 conditional knockout in the developing retina results in a failure in the specification of all cell types except amacrine cells (Marquardt et al., 2001), whereas Pax6 haploinsufficiency causes aniridia, often accompanied by cataract, corneal opacification and glaucoma (Ton et al., 1991; Glaser et al., 1992). RORα plays a crucial role in the development of the CNS (Hamilton et al., 1996; Dussault et al., 1998; Steinmayr et al., 1998). RORα deficient mice have no morphological effect on the retina, but causes dramatic changes in cerebellum development: animals suffer from impaired motor coordination, hanging and equilibrium deficits.
5.2.2. Stress/injury-induced upregulation of crystallin expression
Crystallin upregulation in response to stress or injury is frequently observed and considered as an activation of the cell defense mechanism. Both alpha A and alpha B crystallins are upregulated in the retinas of animal models for diabetes, including the genetic model of spontaneous obesity-induced type 2 diabetes, high fat diet- and alloxan-induced diabetes (Kumar et al., 2005; Kim et al., 2007; Wang et al., 2007; Fort et al., 2009). Alpha A expression was also upregulated in human diabetic retinas (Kase, et al., 2011). A tissue expression analysis of crystallins in a streptozotocin-induced rat model for diabetes showed increased levels of alpha A in the retina and alpha B upregulation in the lens, retina, heart, muscle, and brain (Kumar et al., 2005). Upregulation of alpha A, but not of alpha B or other HSPs, was observed in the photoreceptor inner segments of animals with experimental autoimmune uveitis and was suggested to suppress mitochondrial oxidative stress-mediated apoptosis (Rao et al., 2008). Intravenous administration of alpha A but not alpha B crystallin preserved retinal architecture and prevented photoreceptor damage in animals with experimental autoimmune uveitis. Administration of alpha A led to a reduction of Th1 cytokines (TNF-α, IL-12 and IFN-γ) in the retina and in the spleen and IL-17 only in the retina, as well as in expression of Toll-like receptors and their associated adaptors. Based on these observations, the authors suggested that alpha A crystallin-mediated photoreceptor protection in experimental autoimmune uveitis is associated with suppression of both the adaptive and innate immune responses (Rao et al., 2011). Upregulation of alpha B crystallin in the retina was also described during S. aureus-induced endophthalmitis as a possible antiapoptotic cell response during immune clearance of the bacteria (Whiston et al., 2008). Several other studies of retinal gene expression profiles after various types of injury, such as ischemia-reperfusion injury, light injury and retinal tears, pointed to the elevation of alpha and beta/gamma members of crystallin family (Yoshimura et al., 2003; Sakaguchi et al., 2003; Vazquez-Chona et al., 2004).
Upregulation of alpha B crystallin have also been associated with multiple neurologic disorders such as tauopathies, AD, amyotrophic lateral sclerosis, PD, PD dementia, and prion disorders (Braak et al., 2001, Dabir et al., 2004, Head et al., 1993, Iwaki et al., 1989, 1992, Renkawek et al., 1992, 1994, 1999, Shinohara et al., 1993 and Wang et al., 2013). Increased expression of alpha B crystallin was reported in a subset of astrocytic and oligodendrocytic tau inclusions of both sporadic and familial tauopathies (Dabir et al., 2004; Liu et al., 2015). The authors suggested that the increased alpha B level in glial tau inclusions represents a response by glia to the accumulation of misfolded or aggregated tau protein. Alpha B-positive neurons were also found in the cerebral cortex, amygdala, and hippocampus of PD brain (Braak et al., 2001). In AD patients' brains, alpha B crystallin immunoreactivity in astrocytes and microglia was limited to regions with senile plaques and neurofibrillary tangles. Considering crystallins' chaperone-like activity in preventing protein aggregation, it was hypothesized that alpha B upregulation and its possible association with amyloid deposition protect cells from amyloid-induced to toxicity (Renkawek et al., 1994).
5.2.3. Modulation of alpha and beta crystallin genes in the retina in response to OH and optic nerve axotomy
Our interest in crystallin genes has emerged during the analysis of retinal gene expression profiles of the rat OH model of glaucoma, the most common form of optic neuropathy affecting more than 60 million people worldwide (Leske, 1983; Quigley and Broman, 2006). It is the second leading cause of blindness with an estimated 8.4 million people bilaterally blind in 2010, rising to 11.1 million by 2020 (Quigley and Broman, 2006). The degeneration of RGCs and their axons in the optic nerve is the cause of vision loss in glaucoma. Since the exact mechanisms underlying RGC damage and dysfunction in this disease are not well understood, several years ago we analyzed the changes in retinal expression profiles after elevation of IOP with the objective to identify genes that may be associated with glaucomatous neurodegeneration. The OH animal model used in this study was generated by trabecular laser photocoagulation and associated glaucomatous changes have been well characterized by the evaluation of extent of RGC loss that reached approximately 8% and 20% by two and five weeks after IOP elevation, respectively compared to the control eyes (Piri et al., 2007). Expression of six crystallin genes, including alpha A, alpha B, beta A1/A3, beta A2, beta A4, beta B1, beta B2, beta B3 and gamma 4 was downregulated two weeks after IOP elevation (Piri et al., 2007). Downregulation of crystallin expression in the retina in response to IOP elevation was unexpected since these genes are generally upregulated in stressed or damaged tissues. Interestingly, however, the downregulation of these genes was of transient nature as their mRNA levels returned to the control level at 5-weeks after IOP elevation. It is important to mention that the IOP-induced changes in crystallin expression are not specific to the animal model that was used in our experiments. Similar changes in the levels of crystallin mRNAs were noted in retinal gene profiles of several other animal models for glaucoma: in a rat glaucoma model generated by episcleral vein injection of hypertonic saline, alpha A, alpha B and beta B2 downregulation was reported in animals exposed to elevated IOP for 8 days, but not for 5 weeks (Ahmed et al., 2004); beta A1 level was reduced in a hereditary rat model of elevated IOP (Naskar and Thanos, 2006); and in DBA/2J mouse, a model for secondary angle closure glaucoma, the expression of 9 crystallin genes including alpha A, beta A1, beta A2, beta A4, beta B1, beta B3, gamma B, gamma D, and gamma N was downregulated at 8 months of age when IOP in these animals was elevated (Steel et al., 2006).
IOP-induced dynamic changes in crystallin transcription observed by gene profiling were further evaluated by quantitative analyses at both RNA and protein levels (Piri et al., 2007). The levels of alpha A, alpha B, beta A1/A3, beta A4, and beta B2 transcripts were reduced by ∼50% and beta A2 – 40% in experimental retinas two weeks after IOP elevation (8% RGC loss) compared to controls. In retinas of animals exposed to high IOP for 5 weeks (20% RGC loss), the mRNA levels of these crystallin genes were recovered to the levels of control or were even 5% to 10% higher. This dynamic change in crystallin expression suggest: a) at an early stage of the glaucomatous process (2 weeks after IOP elevation), high IOP suppresses the transcriptional activity of crystallin genes in RGCs and possibly other retinal cells by yet unknown mechanism; b) a recovery in crystallin levels at 5 weeks after IOP elevation is due to the activation of cell defense mechanisms in response to significant RGC degeneration that leads to stimulation of crystallin gene transcription in the remaining RGCs and/or other retinal cells. The hypothesis of crystallin upregulation in the retina by non-RGC cells after extensive RGC loss is supported by the data showing an increase in crystallin levels in retinas of the optic nerve transection (ONT) model of RGC degeneration. Despite the loss of approximately 90% of RGCs by 2 weeks after axotomy, alpha A and alpha B expression was 1.4 and 1.2 fold higher compared to control retinas, respectively. The analysis of crystallin expression at the protein levels showed reduction in alpha A, alpha B, beta A1/A3, and beta B2 crystallin levels in the retina both 2 and 5-week post IOP elevation compared to controls (Piri et al., 2007). Alpha A, alpha B, beta A1/A3 and beta B2 were about 2, 2.7, 3.3 and 1.6 fold lower at two weeks after IOP elevation, respectively, compared to controls. In animals exposed to elevated IOP for five weeks, the levels of alpha A and alpha B were decreased 1.6 fold and of beta A1/A3 and beta B2 almost twofold. We were not able to detect beta A2 and beta A4 crystallin proteins, which suggests their low expression level in the retina. In ONT retinas, a reduction of 1.6 fold in alpha A and alpha B crystallin protein levels were observed (Munemasa et al., 2009b). The discordance between mRNA and protein levels at five weeks post IOP elevation and after ONT can be explained by post-transcriptional regulation of crystallin expression or by an increased turnover rate of crystallins. Proteomic analysis of rat retina in a steroid-induced OH model also detected a 2.7-fold downregulation of alpha A crystallin (Miyara et al., 2008). Since crystallins, particularly alpha crystallins are known to have antiapoptotic and cell protective effects and generally upregulated in response to stress or injury, their downregulation after IOP elevation could undermine the survival properties of RGCs, and consequently be associated with RGC death in glaucoma.
5.3. sHSP function
sHSPs are implicated in a wide range of physiological cellular processes with three interrelated key functions that are attributed to all or some members of the sHSP family: chaperone-like activity, stabilization of the cytoskeleton and antiapoptotic activity. sHSPs are classified as ATP-independent molecular chaperones based on their ability to prevent protein aggregation and may cooperate with ATP-dependent chaperones of the HSP70 family to assist the correct folding of misfolded polypeptides (Horwitz, 1992; Jakob et al., 1993). The following two sections overview the roles of sHSPs in cytoskeleton dynamics and in regulation of pathways associated with apoptotic cell death.
5.3.1. sHSPs and the cellular cytoskeleton
The cellular cytoskeleton is a complex, dynamic structure composed of an interconnected network of filaments that provides the framework for cellular shape, organelle movement and cell division. It consists of microtubules (MTs; tubular polymers of tubulin), microfilaments (MFs or actin filaments), and intermediate filaments (IFs). sHSPs, particularly HSP27 and alpha B crystallin, contribute to maintaining the integrity of IFs and actin filaments, stabilization of IFs assembly, modulation of IF interactions within their networks and reorganization of the IF network (Liang and MacRae, 1997, Perng et al., 1999). sHSPs can interact with IF monomers and whole IFs in both normal and stress conditions, albeit the number of sHSP/IF interactions in stress conditions is increased to prevent IF aggregation. sHSP mutations or the absence of sHSPs may lead to IF aggregation. IF aggregation phenotypes due to sHSP mutations mimic diseases associated with IF proteins such as desmin, vimentin and neuro-filament. For instance, in desmin-related myopathy, an inherited neuromuscular disorder, mutations in closely interacting proteins, desmin and alpha B crystallin, lead to accumulation of desmin/alpha B aggregates, which consequently alter the mechanical properties of IFs (Vicart et al., 1998, Goldfarb et al., 2008). In Alexander disease, mutations in IF protein, glial fibrillary acidic protein, lead to the formation of aggregates known as Rosenthal fibers that contain glial fibrillary acidic protein, vimentin, and several sHSP such as HSP27 and alpha B crystallin. Overexpression of alpha B crystallin may induce the disaggregation of glial fibrillary acidic protein filaments in a mouse model of Alexander disease and improve survival rates (Tang et al., 2010). IF proteins, including lens-specific beaded filament structural proteins 1 and 2, and vimentin, as well as their association with alpha A and alpha B crystallins are essential for the optical properties of the lens (Quinlan et al., 1996; Nicholl and Quinlan, 1994; Sandilands et al. 2003). Mutations in these proteins are associated cataract formation. Mutations in alpha B-crystallin can cause both cataract and cardiomyopathy (R120G), only cataract (450delA) or myofibrillar myopathy (464delCT; Q151X) (Vicart, P., et al. 1998; Berry, V., et al. 2001; Selcen and Engel, 2003.).
HSP27, Hsp20 and alpha B crystallin are also involved in dynamics of actin filaments under normal conditions, and more importantly in their stabilization during stress, which can induce actin fibers depolymerization and aggregation and disruption of the cytoskeleton (Wieske et al., Wettstein et al., 2012; Seit-Nebi et al., 2012). For instance, in H9C2 cells, under normal conditions about 20% of alpha B crystallin is colocalized with actin fibers (Singh et al., 2007). However, upon heat stress, colocalization of alpha B crystallin with actin fibers was increased to 86%. Furthermore, evenly distributed in the cytosol alpha B crystallin was shown to translocate into the nucleus at the onset of heat stress suggesting its role in the stabilization of the nucleoskeletal assembly (Adhikari et al., 2004). The interaction of alpha B crystallin with actin is tightly regulated by alpha B phosphorylation on both Ser45 and Ser59 residues. HSP27 interaction with the actin cytoskeleton has been associated with inhibition of actin polymerization. Overexpression of HSP27 was reported to accelerate rebuilding of actin filaments after stress, whereas downregulation of HSP27 expression leads to actin filament disorganization (Huot et al., 1996; Horman et al., 1999; Mairesse et al., 1996). The mechanisms underlying this function of HSP27 remain controversial. Wettstein et al. (2012) explain HSP27 inhibition of actin polymerization by its ability to cap the plus end of actin filaments, thus preventing the fixation of a new actin monomer. However, this model has been challenged by Seit-Nebi et al., (2012), who claimed that the results of studies by Miron et al., (1988 and 1991) that were used by Wettstein et al. (2012) to create this model, were not supported by later studies (Panasenko et al., 2003; Butt et al., 2001). HSP27 interaction with actin and its activity appears to be regulated by phosphorylation: unphosphorylated but not phosphorylated HSP27 inhibits actin polymerization. The importance of phosphorylation in protection of the microfilament network from stress induced by hyperthermia (Lavoie et al., 1993b, Schneider et al., 1998), cytochalasin D (Lavoie et al., 1993b), free oxygen radicals (Huot et al., 1996) and cholecystokinin (Schaefer et al., 1999) was demonstrated by expression of wild-type or nonphophorylatable HSP27. Furthermore, it has been reported that protein phosphatase inhibitors prevent HSP25 dephosphorylation and F-actin disruption (Loktionova and Kabakov, 1998). Clarke and Mearow (2013) showed that in control neuroendocrine PC12 cells, HSP27 interacts with F-actin as a predominantly nonphosphorylated protein. However, the association of phosphorylated HSP27 with F-actin was significantly increased after stress.
Hsp27 and alpha B crystallin are also implicated in regulation of the dynamics of MT structural network. The spatial and temporal regulation of microtubule cycles involves nucleation, growing and shrinking. In most cell types, centrosomes serve as the main MT organizing centers; however, MTs can be nucleated from other sites as well. HSP27 has been shown to bind to newly formed non-centrosomal MTs, assist their formation, and thus, increase the probability of successful de novo MT formation (Almeida-Souza et al., 2013). Mutations in HSP27 are associated with Charcot-Marie-Tooth neuropathy. A subset of these mutations displays enhanced affinity to tubulin and MTs resulting in MT stabilization (Almeida-Souza et al., 2011). The authors suggested that the neuron-specific phenotype of Charcot-Marie-Tooth neuropathy caused by HSP27 mutations is due to particular vulnerability of neuronal cells to disturbances in MT dynamics. Several lines of evidence implicate alpha B crystallin in MT preservation and dynamics. Houck and Clark (2010) proposed a model for regulation of tubulin assembly by alpha B:crystallin, which is based on the molar ratio of alpha B:tubulin: a) low molar ratios of alpha B:tubulin are favorable for MT assembly and alpha B crystallin monomers stabilize assembled MTs; b) high molar ratios of αB crystallin:tubulin favor assembly of mixed αB crystallin-tubulin complexes, decreasing the tubulin available for MT assembly. The assembly of MTs was maximal at alpha B to tubulin molar ratios between 1:4 and 2:1, while molar ratios >2:1 inhibited MT assembly (Ghosh et al., 2007). The work by this group has also identified interactive sequences in the human alpha B crystallin involved in the assembly/disassembly of MT and aggregation of tubulin (Ghosh et al., 2007). The sequence 113FISREFHR120 on the surface of alpha B crystallin decreased MT assembly by ∼45%, whereas the sequences 131LTITSSLSSDGV142 and 156ERTIPITRE164, corresponding to the β8 strand and the C-terminal respectively, increased MT assembly by ∼34–45%. Furthermore, the alpha B crystallin peptides 113FISREFHR120 and 131LTITSSLSSDGV142 decreased the thermal aggregation of tubulin by more than 40%. Alpha B crystallin has been shown to bind and prevent tubulin self-aggregation at the onset of the temperature induced denaturation process (Arai and Atomi, 1997). Prevention of MT aggregation may involve the interaction of crystallin with MT-associated proteins (Xi et al., 2006).
5.3.2. sHSPs and the apoptotic cell death
Hsp27 and alpha crystallins have a cell protective effect against a wide range of stress inducers and pro-apoptotic factors by modulating several cell death pathways. Mechanisms involved in HSP27- and alpha crystallin-mediated cell protection vary depending on the cell type and nature of injury or stress. HSP27 antiapoptotic effects have been associated with its ability to interact with pro-caspase 3 and inhibit its activation, suppress oligomerization and translocation of Bax to the mitochondria, block the release of cytochrome C and Smac from mitochondria, reduce activation of pro-caspase 9 by inhibiting interaction with cytochrome c and formation of the apoptosome, prevent Fas/APO-1 receptor from inducing cell death, inactivate the pro-death JNK pathway and activate the pro-survival Akt/PKB pathway (Mehlen et al., 1996; Machado et al., 2011; Stetler et al., 2009; Arrigo, 2007; Acunzo et al., 2012). Hsp27 has been demonstrated to modulate a caspase-independent apoptosis by interacting with a mediator of Fas-induced apoptosis, Daxx (death domain-associated protein 6), and consequently preventing its binding with Ask1 (apoptosis signal-regulated kinase 1) and Fas (Charette et al., 2000). The activity of HSP27 in this study was shown to be regulated by phosphorylation that affects the supramolecular organization of the protein from oligomers to dimers. HSP27 phosphorylation mutant expressed as oligomers only failed to block Daxx-mediated apoptosis (Charette et al., 2000). Nonphosphorylatable HSP27 form was also insufficient to suppress ASK1-mediated cell death induced by cerebral ischemia (Stetler et al., 2012). Protein kinase D (PKD)-dependent HSP27 phosphorylation at Ser15 and Ser82 was identified as a critical upstream step for the subsequent interaction between HSP27 and ASK1 and suppression of downstream cell death signaling (Stetler et al., 2012). Alpha B crystallin cell protective effect has been related to its ability to interact with proapoptotic regulators Bax and Bcl-X(S) and inhibit their translocation from the cytosol to the mitochondria (Mao et al., 2004), inhibit activation of caspase-3 and PARP (poly (ADP-ribose) polymerase) (Sreekumar et al., 2010), inhibit the cytochrome c- and caspase-8-dependent activation of caspase-3 (Kamradt et al., 2001), interact with p53 and prevent its translocation from the cytoplasm to mitochondria (Liu et al., 2007), prevent RAS activation and inhibit the RAF/MEK/ERK pathway (Fig. 2; Cortese et al., 2005). Similar to HSP27, phosphorylation of alpha B crystallin appears to be essential for its cells protective effect. Using alpha B crystallin constructs with serine (S) to alanine (A) [block phosphorylation] or S to glutamate (E) [mimic phosphorylation] mutations at positions 19, 45, and 59, Morrison et al. (2003) showed that expression of the alpha B with S to A substitutions at all three positions (AAA) resulted in 3-fold more stress-induced apoptosis of cardiac myocytes compared with αB-crystallin (AAE) or (EEE), indicating that phosphorylation of S59 is important for alpha B crystallin-mediated inhibition of apoptosis. Alpha B crystallin phosphorylation at S59 as well as S45 but not at S19 was also reported to be critical for protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death (Li and Reiser 2011). The cytoprotective effect of alpha A crystallin involves its interactions with cytochrome c or caspase-6 (Fig. 2; Rao et al., 2008; Morozov and Wawrousek, 2006). Furthermore, alpha A has been shown to display similar affinity to Bax and Bcl-X(S) as alpha B. Its interaction with these proapoptotic factors prevents their translocation from cytosol into mitochondria thus preserving mitochondrial integrity and subsequently reducing cytochrome c release and inhibiting caspase-3 activation (Mao et al., 2004).
6. Crystallins support RGC survival and axon regeneration
6.1. Alpha crystallins protect RGCs from ONT and ONC
As stated above, crystallins, particularly alpha A and alpha B, as members of a sHSP superfamily, possess chaperone-like and antiapoptotic activities and generally upregulated in stressed or injured tissues. Therefore, the observed downregulation of crystallin genes in retinas of animals with OH could weaken the survival properties of RGCs, and consequently lead to their degeneration. To test this hypothesis, we evaluated the roles of alpha A and alpha B crystallin in survival of RGCs in a rat optic nerve axotomy model (Munemasa et al., 2009b). ONT causes fast and specific degeneration of RGCs with approximately 50% and 90% of RGC loss by 7 and 14 days after axotomy, respectively. As expected, the loss of RGCs was correlated with the reduced number of crystallin-positive cells in the GCL (Fig. 9; Munemasa et al., 2009b). The effect of alpha A and alpha B crystallin overexpression on RGC survival was evaluated 7 and 14 days after axotomy (Fig. 10; Munemasa et al., 2009b). At day 7 after ONT, 1426±70 and 1418±81 RGCs/mm2 were present in retinas electroporated with alpha A and alpha B expression constructs, respectively, compared with 1010±121 RGCs/mm2 in vector-transfected or 1016±88 RGCs/mm2 in nontransfected retinas. At 14 days after ONT, numbers of surviving RGCs were 389±57 and 353.57±60 cells/mm2 after alpha A and alpha B transfection, respectively, compared with 198±29 cells/mm2 after transfection with the vector alone or 206±60 cells/mm2 in nontransfected retinas. Thus, overexpression of alpha A and alpha B crystallins increases the number of survived RGCs by about 40% 1 week after axotomy and by 95% and 75%, respectively, 2 weeks after ONT. The cell protective effect of crystallin genes could be even higher with more efficient transfection. In this study, the total number of ELP-mediated alpha A- and alpha B-transfected RGCs in adult rat retinas was around 26%. RGCs constituted 85% and 80% of all alpha A- and alpha B–transfected cells in the GCL, respectively.
Figure 9.
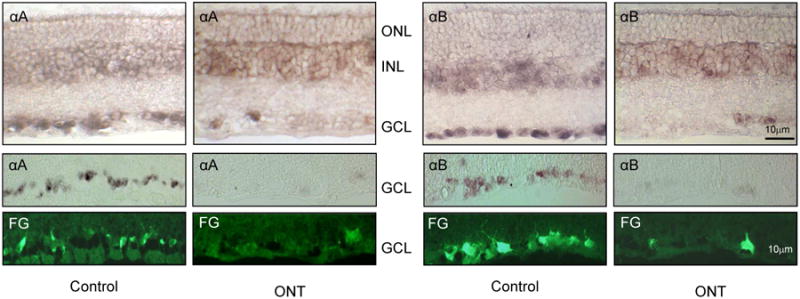
In situ analysis of alpha A and alpha B crystallin expression in control and axotomized retinas. In control retinas, alpha A (αA) and alpha B (αB) crystallin-positive cells were observed primarily in the GCL and INL. In the GCL, crystallin-positive were colocalized with RGCs retrogradely labeled with FG. Very few crystallin-positive cells and retrogradely labeled RGCs were present in the GCL two weeks after optic nerve axotomy.
Figure 10.
Effect of alpha A and alpha B crystallin overexpression on RGC survival injured by ONT. A. At day 7, RGC protective effect of alpha A (αA) and alpha B (αB) crystallin overexpression was about 40%. ONT (n=5) vs. ONT+αA (n=7), *P=0.004; ONT+EGFP (n=6) vs. ONT+αA (n=7), **P=0.003; ONT (n=5) vs. ONT+αB (n=6), *P=0.006; and ONT+EGFP (n = 6) vs. ONT+αB (n=6), **P=0.004. B. Approximately a 95% and 75% increase in the RGC survival mediated by αA and αB overexpression, respectively, was observed two weeks after ONT. ONT (n=9) vs. ONT+αA (n=8), *P=0.001; ONT+EGFP (n=5) vs. ONT+αA (n=8), **P=0.003; ONT (n=9) vs. ONT+αB (n=8), *P=0.001; and ONT+EGFP (n=5) vs. ONT+αB (n=8), **P = 0.003. C. FG-labeled RGCs in nontransfected, EGFP, αA and αB transfected retinas two weeks after ONT.
The effects of alpha crystallin on RGC survival after optic nerve injury was also evaluated in rats that received intravenous injections of 0.05 g/kg, 0.5 g/kg and 5 g/kg of alpha crystallin protein (Wu et al., 2014). Two weeks after optic nerve injury RGC numbers were reduced to 85±15 cells/mm2 from 2074±150 cells/mm2 counted in uninjured control retinas. In crystallin treated animals, RGC numbers in animals injured by optic nerve crush were significantly increased compared to that in saline-injected group: 124±26 cells/mm2, 128±31 cells/mm2, and 164±20 cells/mm2 were survived in animals treated with 0.05 g/kg, 0.5 g/kg and 5 g/kg of alpha crystallin, respectively. The cell protective effect of alpha crystallin was aassociated with the inhibition of microglial activation and TNF-α/iNOS release (Wu et al., 2009; 2014).
A single intravitreal administration of alpha crystallin protein protected RGC axons from optic nerve crush: RGC axonal survival was significantly greater in those crystallin treated than in control animals 2 weeks after injection: 16.0% vs 12%, 21% vs 11%, and 28% vs 17% at 0.5 mm, 2 mm, and 5 mm distal to the injury site, respectively (Ying et al., 2008). The protective effect of alpha crystallin declined by 4 weeks after injection but remained greater than in controls. In a rat model of acute OH, intravitreal injection of alpha B crystallin increased the survival of RGCs. The cell protecive effect of alpha B crystallin was associated with reduced caspase 3 expression (Wu et al., 2012).
6.2. Crystallins and neuroprotective effect of wolfberry
Up-regulation of alpha and beta crystallins was also associated with the RGC protective effect of wolfberry (fruit of Lycium barbarum) in a rat OH model. Wolfberry has been known in Asia for more than 2,500 years as a remedy that among other things improves visual acuity. An active compound in wolfberry, L. barbarum polysaccharide (LBP), has been shown to stimulate anti-oxidative effects and inhibit several pro-apoptotic signaling pathways (Li, 2007; Yu et al., 2005, 2007). Daily feeding of LBP (1 mg/kg) through a nasogastric tube, prior to IOP elevation reduced the extent of RGC loss from about 18% to about 1% in animals with experimental glaucoma (Chan et al., 2007). LBP treatment increased the levels of both alpha (A and B) and beta (A4 and B2) crystallins more than 10 times compared to control 2 days after IOP elevation, suggesting their roles in mediating RGC protection rat OH model (Chiu et al., 2010; Chan et al., 2007). The neuroprotective effects of LBP are associated with its ability to modulate several pathways that are activated after injury including anti-apoptotic, anti-oxidative and immune response. LBP modulates microglia activation in chronic OH model (Chiu et al., 2009), down-regulate RAGE (the receptor for advanced glycation end-products (AGEs), AGE, Aβ and endothelin-1 in the retinas of an acute OH model (Mi et al., 2012), activate antioxidant pathway by increasing Nrf2 (nuclear factor erythroid 2-related factor) nuclear accumulation and heme oxygenase-1 expression in the retina after ischemia/reperfusion injury (Li et al., 2011; He et al., 2014), reduces oxidative stress and inhibit the JNK pathway after ONT (Li et al., 2013, 2015), decrease the phosphorylation of double-stranded RNA-dependent protein kinase and inhibits the activity of caspases 3 and 2 in neurons stressed by beta-amyloid peptide induced neurotoxicity (Yu et al., 2007). LBP's capacity to modulate various pathways to increase cell resistance to stress- and injury-induced damage could be especialy benefecial in the treatment of multifactorial disorders such as glaucoma.
6.3. Beta B2 crystallin stimulates RGC regeneration
Members of the crystallin superfamily are implicated not only in enhancing the survival of RGCs injured by elevated IOP, ONC and ONT but have been also shown to contribute to the egeneration of RGC axons. Mature CNS neurons including RGCs, gradually loose their capacity to regenerate damaged axons and reestablish connections. This has been attributed to the decline in intrinsic neuronal growth ability (Teng and Tang, 2006; Park et al., 2008; Moore et al., 2009; Smith et al., 2009), as well as extrinsic inhibition by the adult CNS glial microenvironment that includes proteoglycans associated with astroglial scarring and myelin-associated inhibitors such as Nogo-A, myelin-associated glycoprotein and oligodendrocyte myelin glycoprotein (Wong et al., 2003; Silver and Miller, 2004; Yiu and He, 2006). Crystallin-associated support for the regenerative growth of axotomized RGC axons was described both in vitro and in vivo. Liedtke et al. (2007) reported up-regulation of beta B2 crystallin during retinal regeneration in vitro and its localization within RGCs and their axons, including growth cones and filopodia. Overexpression of beta B2 crystallin in dissociated retinal and hippocampal neurons increased axonogenesis (Liedtke et al., 2007). The axon growth promoting effect of crystallins was further confirmed after intravitreal injections of beta or gamma crystallins, but not of alpha crystallins (Fisher et al., 2008). Stimulation of CNTF (ciliary neurotrophic factor) expression in retinal astrocytes and activation of CNTF's major downstream JAK/STAT3 (Janus kinase/signal transducers and activators of transcription) signaling pathway by beta crystallin suggests that the effect of this protein in axonal regeneration is mediated by astrocyte-derived CNTF (Fisher et al., 2008; Müller et al., 2007). The observation of the lack of alpha crystallin's stimulatory effect on axon regeneration, however was challenged by Wang et al. (2012), who showed that alpha crystallin promotes neurite initiation and stimulates neurite outgrowth. In this study, the effect of alpha crystallin in a neurite outgrowth assay was evaluated on retinal cells isolated from newborns (postnatal day 0 to day 2) and adult rats and cultured on myelin-coated dishes to mimic the in vivo inhibitory microenvironment. Alpha crystallin's stimulation of neurite growth in the presence of myelin inhibitory factors was attributed to its ability to regulate the RhoA/Rock signaling pathway (Wang et al., 2012). Inconsistency of alpha crystallin's effect on axon regeneration was explained by different concentrations of alpha crystallin tested in these studies. The optimum concentration of alpha crystallins was determined to be 10−4 g/l; treatment with higher concentrations of this protein weakens its effect on neurite outgrowth (Wang et al., 2011).
7. HSP27 and HSP60 in RGC survival, regeneration and autoimmunity-mediated degeneration
Upregulation of HSP60 and HSP27 have been reported in patients with glaucoma as well as in animal models of RGC degeneration induced by elevated IOP, optic nerve axotomy and ischemic injury. In patients with primary open-angle glaucoma and normal-pressure glaucoma, increased expression of HSP60 was observed in RGCs and photoreceptors, whereas HSP27 upregulation was present in the nerve fiber layer, RGCs and in optic nerve heads where it was associated with astroglial cells in the lamina cribrosa (Tezel et al., 2000). The increased expression of proteins was suggested to contribute to a cellular defense mechanism against stress or injury in glaucoma (Tezel et al., 2000). The levels of HSP60 and HSP27 were also elevated in the inner retinal layers, especially in the ganglion cell and nerve fiber layer of monkeys with laser-induced glaucoma (Sakai et al., 2003). The expression of HSP27 was increased in RGCs and retinal astrocytes in rat OH model (Kalesnykas et al., 2007). A significant increase in total HSP27 (∼4-fold) and pHSP27 (∼2-fold) was reported in the experimental rat glaucoma model and in DBA/2J mice with elevated IOP (∼3-fold and ∼2-fold for HSP27 and pHSP27, respectively; Huang et al., 2007). HSP27 level was also elevated after acute pressure-induced ischemia (Windisch et al., 2009). Upregulation of HSP27 in RGCs, glial cells of the optic tract, and astrocytes in the optic layer of the superior colliculus was observed after optic nerve axotomy (Krueger-Naug et al., 2002). Induction of HSP27 expression in axotomized RGC by intravitreal injection of the 3-Hydroxy-3-methylglutaryl-CoA reductase inhibitor simvastatin improved RGC survival 7 and 14 days after optic nerve axotomy by approximately 90% and 20%, respectively (Kretz et al., 2006). Specific upregulation of HSP27 after ischemic preconditioning was strongly correlated with the protection of retinal function from ischemic injury (Li et al., 2003).
The mechanisms that initiate upregulation of HSPs in these models of RGC degeneration are most likely associated with proteotoxic stress. Axonal damage caused by ONC, ONT or OH severely alters the integrity and function of several vital cellular structures such as cytoskeleton, mitochondria and ER with resulting devastating consequences on protein homeostasis. The protein homeostasis (or proteostasis) is supported by a network of pathways that include: degradation - the ubiquitin proteasome and the ER–associated degradation systems; posttranslational modification -phosphorylation, acylation, and oxidation; and protein synthesis/folding/unfolding - ribosomes, the unfolded protein response (UPR) and HSF1 (Roth and Balch, 2011; Li et al., 2013; Díaz-Villanueva JF, Díaz-Molina, 2015). ER stress appears to be a common neuronal response to axonal injury: ER stress and activation of UPR has been implicated in RGC degeneration caused by ONT (Pernet et al., 2012), ONC (Hu et al., 2012; Jiang et al., 2013; Yasuda et al., 2014; Lindsey et al., 2015) and OH (Doh et al., 2010; Shimazawa et al., 2007). HSF1, as one of the important sensors of proteotoxic stress, plays a critical role in the heat shock response network that regulates the expression of molecular chaperones, such as HSPs, as well as proteinases and other inducible genes for protection and recovery from diverse forms of environmental and physiological stress. Activation of HSF1 in response to disbalance of proteostasis (Fig. 3) can upregulate expression of various HSPs, including HSP60 and HSP27 in animals models of optic nerve injury. The involvement of alternative to classical HSF1-mediated transcriptional activation of HSP27 during retinal ischemic preconditioning was investigated since the expression levels of other HSPs were not affected by this experimental condition (Whitlock et al., 2005). It has been shown that CoCl2, an inducer of hypoxia-inducible factor-1 (HIF-1), upregulates HSP27 expression in cultured neurons and in vivo, supporting the idea of HIF1 regulation of HSP27 expression during ischemic preconditioning; HIF-1 binding sites in the promoter region and in the first intron of the HSP27 gene were involved in gene expression in the presence of the CoCl2 (Whitlock et al., 2005). CoCl2-induced upregulation of HSP27 as well as electroporation-mediated delivery of this protein to retinal cells protected RGCs from ischemia-reperfusion injury (Whitlock et al., 2005; Yokoyama et al., 2001).
HSP60 and HSP27 are also involved in regeneration of injured RGCs. Upregulation of HSP60 was associated with RGC axon regeneration in retinal explants of rats with hereditary buphthalmos characterized by IOP elevation (Lasseck et al., 2007). The presence of antibodies against this protein in the culture medium prevented axonal growth, substantiating the functional relevance of HSP60 in RGC regeneration. HSP27 expression level was significantly correlated with axonal regeneration of mature RGCs after ONT; the number of HSP27-positive RGCs that extended new axons into the peripheral nerve autografts used in this study to stimulate regeneration of transected axons was five-fold higher compared to those that failed to regenerate (Hebb et al., 2006).
These studies directly or indirectly associate the stress- or injury-induced upregulation of HSP27 and HSP60 with activation of cellular defense mechanism, which can be explained by the well established functions of these molecular chaperones in the regulation of cytoskeleton dynamics, cellular redox state and apoptotic death pathways. However, contrary to their role in protecting and repairing damaged cells, the increased levels of HSPs were proposed to recruit immune responses that may contribute to the progression of the diseases such as glaucoma (Tezel et al., 2004). Several lines of evidence supporting this hypothesis are presented below. Patients with either normal-pressure or primary open-angle glaucoma had an elevated serum immunoreactivity to HSP60 (Wax et al., 1998; 2001), HSP27 and alpha crystallins (Tezel et al., 1998; Wax et al., 2001). Increased titers of circulating antibodies against these proteins were suggested to be pathogenic in patients with an autoimmune form of glaucomatous neuropathy (Tezel et al., 2004). Antibodies against sHSPs can enter living cells by an endocytic mechanism and trigger apoptosis in ex vivo and in vitro retina models (Tezel et al., 1998; Tezel and Wax, 2000). It has been also shown that after internalization, HSP27 antibody binds to the actin cytoskeleton and causes depolymerization and proteolytic cleavage of actin (Tezel and Wax, 2000). Actin cleavage was decreased in the presence of a nonselective caspase inhibitor, which suggests an involvement of caspase activation in the proteolytic process mediated by HSP27 antibody. A marked shortening and disorganization of actin microfilaments after incubation of human retinal cells with HSP27 antibody were detected by electron microscopy. Taken together, the data suggest that exogenous HSP27 antibody may obstruct the ability of HSP27 to stabilize the actin cytoskeleton and consequently induce cell death (Tezel and Wax, 2000). Further support for the involvement of an autoimmune component in a subset of glaucoma, was demonstrated by immunization of rats with HSP27 and HSP60 that resulted in RGC degeneration and axon loss (Wax et al., 2008; Joachim et al., 2009; Joachim et al., 2010). A recently published study, however, found no significant difference in the serum levels of antibodies against human HSP60 between 35 NTG and 34 POAG subjects and 36 controls and concluded that their results “do not confirm the hypothesis that normal-tension glaucoma is associated with elevated blood levels of antibodies against human HSP60” (Grabska-Liberek et al., 2015). This apparent disagreement about the role of antibodies against HSP60 in glaucoma pathogenesis can be resolved if we accept that the autoimmune factor as proposed by Tezel and Wax is associated with a certain subset of glaucoma, which may have been underrepresented among patients included in the study by Grabska-Liberek et al. (2015).
8. Conclusion and future directions
Optic neuropathies and glaucoma in particular affects millions of people worldwide and if left untreated could lead to debilitating visual impairments due to damage to RGCs and their axons in the optic nerve (3-5). Current glaucoma treatment is limited to lowering of IOP and although it has been proven to be effective in slowing the progression of the disease in most patients, it is common for visual function to deteriorate despite successful IOP management. This dictates the necessity of developing alternative or complementary therapies to IOP reduction for this disease. Since the mechanisms responsible for glaucomatous damage are not well understood, multiple neuroprotective strategies targeting different pathways that may be involved in this process were proposed and tested (8-15). Despite extensive studies in this direction during the last two decades and the accumulation of a wealth of information about RGC degeneration and neuroprotection in animal models, the research community is somewhat disappointed by the lack of substantial progress in developing new clinically relevant therapeutic approaches (16, 17). In our opinion, the major challenge for the development of clinically relevant therapy for preserving RGCs from glaucomatous neurodegeneration is the multifactorial nature of this disease. Risk factors sufficient for or contributing to the development of the disease may vary by glaucoma type and from patient to patient within the same subtype of the disease, as well as in the same individual during the course of the disease. These factors determine not only the likelihood of developing the disease, but also the severity and the rate of its progression. Some of the cellular factors that are commonly associated with the pathogenesis of glaucoma include biomechanical stress, oxidative stress, axonal transport failure, insufficient nutrient supply, autoimmunity, and glial cell dysfunction. The most recognizable risk factor, IOP, even when within a normal range, could be a sufficient or contributing factor associated with RGC damage.
The multifactorial nature of glaucoma pathogenesis could be addressed by the stimulation of cellular defense mechanisms such as heat shock response that will involve the induction of HSPs including HSP70 and alpha crystallins in RGCs as well as in other ocular and non-ocular tissues involved in glaucoma pathogenesis. HSP70 and alpha crystallins have been recognized for their ability to protect cells from an array of stress stimuli by inhibiting multiple apoptotic cell death pathways, stabilizing the cellular cytoskeleton, preventing possible protein misfolding/unfolding/aggregation, and assisting in the disposal of damaged or defective proteins. Results of our studies and studies from other researchers provide strong support for HSP70's and alpha crystallins' RGC protective effects after systemic or ocular induction of these proteins in various animal models of RGC degeneration. With respect to potential clinical applications of an HSP-based strategy, both gene therapy and pharmacological approaches can provide an enduring neuroprotection, which is essential in chronic neurodegenerative diseases such as glaucoma. That being said, the pharmacological induction of HSPs appears to be more appropriate than a viral-mediated approach since it can be administered non-invasively (orally for instance vs intravitreal injection); the treatment regimen can easily be modified or interrupted if necessary; and the systemic induction of HSP70 or crystallin expression may exert their benefits by targeting both ocular and non-ocular tissues including trabecular meshwork and glial cells in the retina, as well as neurons in the central visual system, lateral geniculate nucleus and primary visual cortex, that are known to contribute to RGC dysfunction and death in glaucoma (He et al., 2008; Sacca and Izzotti, 2008; Yücel et al., 2001; Yücel, 2013; Chen et al., 2013). Alternatively, to address possible concerns related to exposure of vital organs to HSP-inducing agents, sustained delivery of HSP inducers to ocular tissues could be achieved by intravitreal implants. The ocular delivery of these drugs would make even more sense in patients with no involvement of CNS neurons in pathogenesis of their disease.
In conclusion, we believe that an approach that targets various mechanisms which support the integrity and normal function of RGCs, as well as other cells that are known to participate in glaucomatous neurodegeneration, HSP-mediated neuroprotection could be an effective therapeutic strategy regardless of the diverse molecular pathways which are likely associated with glaucoma pathogeneses. Since the functional loss of RGCs may precede the RGCs' physical loss by years (Ventura et al., 2006; Banitt et al., 2013), we have an opportunity not only to preserve the existing RGC population but also rescue injured cells and restore their function.
Acknowledgments
This work was supported by the National Institutes of Health/National Eye Institute Grant EY018644 (NP) and the Research to Prevent Blindness (JC).
Abbreviations
- 17-AAG
17-allylamino-17-demethoxygeldanamycin
- AAV
adeno-associated virus
- AD
Alzheimer disease
- Aβ
amyloid-β
- ER
endoplasmic reticulum
- GCL
ganglion cell layer
- GFP
green fluorescent protein
- GGA
geranylgeranylacetone
- HD
Huntington disease
- HSF
heat shock factor
- HSP
heat shock protein
- HSV
Herpes simplex virus
- IF
intermediate filament
- IOP
intraocular pressure
- MF
microfilaments
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MT
microtubule
- OH
ocular hypertension
- ONC
optic nerve crush
- ONT
optic nerve transection
- PD
Parkinson disease
- polyQ
polyglutamine
- RGC
retinal ganglion cell
- SBMA
spinobulbar muscular atrophy
- SCA
spinocerebellar ataxia
- Sirt
sirtuin
- TF
transcription factor
Footnotes
Conflict of interest: The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abravaya K, Phillips B, Morimoto RI. Heat shock-induced interactions of heat shock transcription factor and the human hsp70 promoter examined by in vivo footprinting. Mol Cell Biol. 1991;11:586–592. doi: 10.1128/mcb.11.1.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acunzo J, Katsogiannou M, Rocchi P. Small heat shock proteins HSP27 (HspB1), αB-crystallin (HspB5) and HSP22 (HspB8) as regulators of cell death. Int J Biochem Cell Biol. 2012;44:1622–1631. doi: 10.1016/j.biocel.2012.04.002. [DOI] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adhikari AS, Sridhar Rao K, Rangaraj N, Parnaik VK, Rao ChM. Heat stress-induced localization of small heat shock proteins in mouse myoblasts: intranuclear lamin A/C speckles as target for alphaB-crystallin and Hsp25. Expt Cell Res. 2004;299:393–403. doi: 10.1016/j.yexcr.2004.05.032. [DOI] [PubMed] [Google Scholar]
- Ahmed F, Brown KM, Stephan DA, Morrison JC, Johnson EC, Tomarev SI. Microarray analysis of changes in mRNA levels in the rat retina after experimental elevation of intraocular pressure. Invest Ophthalm Vis Sci. 2004;45:1247–1258. doi: 10.1167/iovs.03-1123. [DOI] [PubMed] [Google Scholar]
- Alge CS, Priglinger SG, Neubauer AS, Kampik A, Zillig M, Bloemendal H, Welge-Lussen U. Retinal pigment epithelium is protected against apoptosis by alphaB-crystallin. Invest Ophthalmol Vis Sci. 2002;43:3575–3582. [PubMed] [Google Scholar]
- Ali A, Bharadwaj S, O'Carroll R, Ovsenek N. HSP90 interacts with and regulates the activity of heat shock factor 1 in Xenopus oocytes. Mol Cell Biol. 1998;18:4949–4960. doi: 10.1128/mcb.18.9.4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allergan Press Release: Trials, Fourth Quarter Operating Results. 2008 Jan 30; http://agn.client.shareholder.com/releasedetail.cfm?ReleaseID=290764.
- Almeida-Souza L, Asselbergh B, d'Ydewalle C, Moonens K, Goethals S, de Winter V, Azmi A, Irobi J, Timmermans JP, Gevaert K, Remaut H, Van Den Bosch L, Timmerman V, Janssens S. Small heat-shock protein HSPB1 mutants stabilize microtubules in charcot-marie-tooth neuropathy. J Neurosci. 2011;31:15320–15328. doi: 10.1523/JNEUROSCI.3266-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida-Souza L, Asselbergh B, De Winter V, Goethals S, Timmerman V, Janssens S. HSPB1 facilitates the formation of non-centrosomal microtubules. PLoS One. 2013;8:e66541. doi: 10.1371/journal.pone.0066541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson MG, Libby RT, Gould DB, Smith RS, John SW. High-dose radiation with bone marrow transfer prevents neurodegeneration in an inherited glaucoma. Proc Natl Acad Sci USA. 2005;102:4566–4571. doi: 10.1073/pnas.0407357102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andley UP, Song Z, Wawrousek EF, Fleming TP, Bassnett S. Differential protective activity of alpha A- and alphaB-crystallin in lens epithelial cells. J Biol Chem. 2000;275:36823–36831. doi: 10.1074/jbc.M004233200. [DOI] [PubMed] [Google Scholar]
- Arai H, Atomi Y. Chaperone activity of alpha B-crystallin suppresses tubulin aggregation through complex formation. Cell Struct Funct. 1997;22:539–544. doi: 10.1247/csf.22.539. [DOI] [PubMed] [Google Scholar]
- Auluck PK, Chan HY, Trojanowski JQ, Lee VM, Bonini NM. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson's disease. Science. 2002;295:865–868. doi: 10.1126/science.1067389. [DOI] [PubMed] [Google Scholar]
- Bagnéris C, Bateman OA, Naylor CE, Cronin N, Boelens WC, Keep NH, Slingsby C. Crystal structures of alpha-crystallin domain dimers of alphaB-crystallin and Hsp20. J Mol Biol. 2009;392:1242–1252. doi: 10.1016/j.jmb.2009.07.069. [DOI] [PubMed] [Google Scholar]
- Bakalash S, Kessler A, Mizrahi T, Nussenblatt R, Schwartz M. Antigenic specificity of immunoprotective therapeutic vaccination for glaucoma. Invest Ophthalmol Vis Sci. 2003;44:3374–3381. doi: 10.1167/iovs.03-0080. [DOI] [PubMed] [Google Scholar]
- Baler R, Dahl G, Voellmy R. Activation of human heat shock genes is accompanied by oligomerization, modification, and rapid translocation of heat shock transcription factor HSF1. Mol Cell Biol. 1993;13:2486–2496. doi: 10.1128/mcb.13.4.2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banitt MR, Ventura LM, Feuer WJ, Savatovsky E, Luna G, Shif O, Bosse B, Porciatti V. Progressive loss of retinal ganglion cell function precedes structural loss by several years in glaucoma suspects. Invest Ophthalmol Vis Sci. 2013;54:2346–2352. doi: 10.1167/iovs.12-11026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova EV, Weeks SD, Beelen S, Bukach OV, Gusev NB, Strelkov SV. Three-dimensional structure of α-crystallin domain dimers of human small heat shock proteins HSPB1 and HSPB6. J Mol Biol. 2011;411:110–122. doi: 10.1016/j.jmb.2011.05.024. [DOI] [PubMed] [Google Scholar]
- Barbe MF, Tytell M, Gower DJ, Welch WJ. Hyperthermia protects against light damage in the rat retina. Science. 1988;241:1817–1820. doi: 10.1126/science.3175623. [DOI] [PubMed] [Google Scholar]
- Basha E, Friedrich KL, Vierling E. The N-terminal arm of small heat shock proteins is important for both chaperone activity and substrate specificity. J Biol Chem. 2006;281:39943–39952. doi: 10.1074/jbc.M607677200. [DOI] [PubMed] [Google Scholar]
- Batulan Z, Taylor DM, Aarons RJ, Minotti S, Doroudchi MM, Nalbantoglu J, Durham HD. Induction of multiple heat shock proteins and neuroprotection in a primary culture model of familial amyotrophic lateral sclerosis. Neurobiol Dis. 2006;24:213–225. doi: 10.1016/j.nbd.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Beere HM, Wolf BB, Cain K, Mosser DD, Mahboubi A, Kuwana T, Tailor P, Morimoto RI, Cohen GM, Green DR. Heat-shock protein 70 inhibits apoptosis by preventing recruitment of procaspase-9 to the Apaf-1 apoptosome. Nat Cell Biol. 2000;2:469–475. doi: 10.1038/35019501. [DOI] [PubMed] [Google Scholar]
- Berry V, Francis P, Reddy MA, Collyer D, Vithana E, MacKay I, Dawson G, Carey AH, Moore A, Bhattacharya SS, Quinlan RA. Alpha-B crystallin gene (CRYAB) mutation causes dominant congenital posterior polar cataract in humans. Am J Hum Genet. 2001;69:1141–1145. doi: 10.1086/324158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat SP, Nagineni CN. alpha B subunit of lens-specific protein alpha-crystallin is present in other ocular and non-ocular tissues. Biochem Biophys Res Commun. 1989;158:319–325. doi: 10.1016/s0006-291x(89)80215-3. [DOI] [PubMed] [Google Scholar]
- Bhat SP, Rayner SA, Chau SC, Ariyasu RG. Pax-6 expression in posthatch chick retina during and recovery from form-deprivation myopia. Dev Neurosci. 2004;26:328–335. doi: 10.1159/000082274. [DOI] [PubMed] [Google Scholar]
- Bluhm WF, Martin JL, Mestril R, Dillmann WH. Specific heat shock proteins protect microtubules during simulated ischemia in cardiac myocytes. Am J Physiol. 1998;275:H2243–H2249. doi: 10.1152/ajpheart.1998.275.6.H2243. [DOI] [PubMed] [Google Scholar]
- Blundell T, Lindley P, Miller L, Moss D, Slingsby C, Tickle I, Turnell B, Wistow G. The molecular structure and stability of the eye lens: X-ray analysis of gamma-crystallin II. Nature. 1981;289:771–777. doi: 10.1038/289771a0. [DOI] [PubMed] [Google Scholar]
- Bova MP, Ding LL, Horwitz J, Fung BK. Subunit exchange of alphaA-crystallin. J Biol Chem. 1997;272:29511–29517. doi: 10.1074/jbc.272.47.29511. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Sandmann-Kiel D, Rub U, Schultz C. Nerve cells expressing heat-shock proteins in Parkinson's disease. Acta Neuropathol. 2001;102:449–454. doi: 10.1007/s004010100395. [DOI] [PubMed] [Google Scholar]
- Bu L, Jin Y, Shi Y, Chu R, Ban A, Eiberg H, Andres L, Jiang H, Zheng G, Qian M, Cui B, Xia Y, Liu J, Hu L, Zhao G, Hayden MR, Kong X. Mutant DNA-binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract. Nat Genet. 2002;31:276–278. doi: 10.1038/ng921. [DOI] [PubMed] [Google Scholar]
- Buccellato MA, Carsillo T, Traylor Z, Oglesbee M. Heat shock protein expression in brain: a protective role spanning intrinsic thermal resistance and defense against neurotropic viruses. Prog Brain Res. 2007;162:395–415. doi: 10.1016/S0079-6123(06)62019-0. [DOI] [PubMed] [Google Scholar]
- Butt E, Immler D, Meyer HE, Kotlyarov A, Laass K, Gaestel M. Heat shock protein 27 is a substrate of cGMP-dependent protein kinase in intact human platelets: phosphorylation-induced actin polymerization caused by HSP27 mutants. J Biol Chem. 2001;276:7108–7113. doi: 10.1074/jbc.m009234200. [DOI] [PubMed] [Google Scholar]
- Buzzard KA, Giaccia AJ, Killender M, Anderson RL. Heat shock protein 72 modulates pathways of stress-induced apoptosis. J Biol Chem. 1998;273:17147–17153. doi: 10.1074/jbc.273.27.17147. [DOI] [PubMed] [Google Scholar]
- Carmichael J, Chatellier J, Woolfson A, Milstein C, Fersht AR, Rubinsztein DC. Bacterial and yeast chaperones reduce both aggregate formation and cell death in mammalian cell models of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:9701–9705. doi: 10.1073/pnas.170280697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspers GJ, Leunissen JA, de Jong WW. The expanding small heat-shock protein family, and structure predictions of the conserved alpha-crystallin domain. J Mol Evol. 1995;40:238–248. doi: 10.1007/BF00163229. [DOI] [PubMed] [Google Scholar]
- Chan HC, Chang RC, Koon-Ching Ip A, Chiu K, Yuen WH, Zee SY, So KF. Neuroprotective effects of Lycium barbarum Lynn on protecting retinal ganglion cells in an ocular hypertension model of glaucoma. Exp Neurol. 2007;203:269–273. doi: 10.1016/j.expneurol.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Charette SJ, Lavoie JN, Lambert H, Landry J. Inhibition of Daxx-mediated apoptosis by heat shock protein 27. Mol Cell Biol. 2000;20:7602–7612. doi: 10.1128/mcb.20.20.7602-7612.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen WW, Wang N, Cai S, Fang Z, Yu M, Wu Q, Tang L, Guo B, Feng Y, Jonas JB, Chen X, Liu X, Gong Q. Structural brain abnormalities in patients with primary open-angle glaucoma: a study with 3T MR imaging. Invest Ophthalmol Vis Sci. 2013;54:545–554. doi: 10.1167/iovs.12-9893. [DOI] [PubMed] [Google Scholar]
- Cheng L, Sapieha P, Kittlerova P, Hauswirth WW, Di Polo A. TrkB gene transfer protects retinal ganglion cells from axotomy-induced death in vivo. J Neurosci. 2002;22:3977–3986. doi: 10.1523/JNEUROSCI.22-10-03977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu K, Chan HC, Yeung SC, Yuen WH, Zee SY, Chang RC, So KF. Modulation of microglia by Wolfberry on the survival of retinal ganglion cells in a rat ocular hypertension model. J Ocul Biol Dis Infor. 2009;2:47–56. doi: 10.1007/s12177-009-9023-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu K, Zhou Y, Yeung SC, Lok CK, Chan OO, Chang RC, So KF, Chiu JF. Up-regulation of crystallins is involved in the neuroprotective effect of wolfberry on survival of retinal ganglion cells in rat ocular hypertension model. J Cell Biochem. 2010;110:311–320. doi: 10.1002/jcb.22539. [DOI] [PubMed] [Google Scholar]
- Choi DW, Koh JY. Zinc and brain injury. Annu Rev Neurosci. 1998;21:347–375. doi: 10.1146/annurev.neuro.21.1.347. [DOI] [PubMed] [Google Scholar]
- Clarke JP, Mearow KM. Cell stress promotes the association of phosphorylated HspB1 with F-actin. PLoS One. 2013;8:e68978. doi: 10.1371/journal.pone.0068978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins P, Hightower LE. Newcastle disease virus stimulates the cellular accumulation of stress (heat shock) mRNAs and proteins. J Virol. 1982;44:703–707. doi: 10.1128/jvi.44.2.703-707.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese MS, Baird JP, Uversky VN, Dunker AK. Uncovering the unfoldome: enriching cell extracts for unstructured proteins by acid treatment. J Proteome Res. 2005;4:1610–1618. doi: 10.1021/pr050119c. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dillmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- Cullen KE, Sarge KD. Characterization of hypothermia-induced cellular stress response in mouse tissues. J Biol Chem. 1997;272:1742–1746. doi: 10.1074/jbc.272.3.1742. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Piatigorsky J. Lens development and crystallin gene expression: many roles for Pax-6. Bioessays. 1996;18:621–630. doi: 10.1002/bies.950180805. [DOI] [PubMed] [Google Scholar]
- Cvekl A, Yang Y, Chauhan BK, Cveklova K. Regulation of gene expression by Pax6 in ocular cells: a case of tissue-preferred expression of crystallins in lens. Int J Dev Biol. 2004;48:829–844. doi: 10.1387/ijdb.041866ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cvekl A, Duncan MK. Genetic and epigenetic mechanisms of gene regulation during lens development. Prog Retin Eye Res. 2007;26:555–597. doi: 10.1016/j.preteyeres.2007.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabir DV, Trojanowski JQ, Richter-Landsberg C, Lee VM, Forman MS. Expression of the small heat-shock protein alphaB-crystallin in tauopathies with glial pathology. Am J Pathol. 2004;164:155–166. doi: 10.1016/s0002-9440(10)63106-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JN, Antonawich FJ. Role of apoptotic proteins in ischemic hippocampal damage. Ann N Y Acad Sci. 1997;835:309–320. doi: 10.1111/j.1749-6632.1997.tb48638.x. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Ferrante RJ, Andreassen OA, Dillmann WH, Beal MF. Mice overexpressing 70-kDa heat shock protein show increased resistance to malonate and 3-nitropropionic acid. Exp Neurol. 2002;176:262–265. doi: 10.1006/exnr.2002.7933. [DOI] [PubMed] [Google Scholar]
- Deretic D, Aebersold RH, Morrison HD, Papermaster DS. Alpha A- and alpha B-crystallin in the retina. Association with the post-Golgi compartment of frog retinal photoreceptors. J Biol Chem. 1994;269:16853–16861. [PubMed] [Google Scholar]
- Díaz-Villanueva JF, Díaz-Molina R, García-González V. Protein folding and mechanisms of proteostasis. Int J Mol Sci. 2015;16:17193–17230. doi: 10.3390/ijms160817193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doh SH, Kim JH, Lee KM, Park HY, Park CK. Retinal ganglion cell death induced by endoplasmic reticulum stress in a chronic glaucoma model. Brain Res. 2010;1308:158–166. doi: 10.1016/j.brainres.2009.10.025. [DOI] [PubMed] [Google Scholar]
- Dong Z, Wolfer DP, Lipp HP, Bueler H. Hsp70 gene transfer by adeno-associated virus inhibits MPTP-induced nigrostriatal degeneration in the mouse model of parkinson disease. Mol Ther. 2005;11:80–88. doi: 10.1016/j.ymthe.2004.09.007. [DOI] [PubMed] [Google Scholar]
- Dubin RA, Wawrousek EF, Piatigorsky J. Expression of the murine alpha B-crystallin gene is not restricted to the lens. Mol Cell Biol. 1989;9:1083–1091. doi: 10.1128/mcb.9.3.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dussault I, Fawcett D, Matthyssen A, Bader JA, Giguère V. Orphan nuclear receptor RORα-deficient mice display the cerebellar defects of staggerer. Mech Dev. 1998;70:147–153. doi: 10.1016/s0925-4773(97)00187-1. [DOI] [PubMed] [Google Scholar]
- Egwuagu CE, Chepelinsky AB. Extralenticular expression of the alpha A-crystallin promoter/gamma interferon transgene. Exp Eye Res. 1997;64:491–495. doi: 10.1006/exer.1996.0202. [DOI] [PubMed] [Google Scholar]
- Finkel T, Deng C, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D, Hauk TG, Muller A, Thanos S. Crystallins of the beta/gamma-superfamily mimic the effects of lens injury and promote axon regeneration. Mol Cell Neurosci. 2008;37:471–479. doi: 10.1016/j.mcn.2007.11.002. [DOI] [PubMed] [Google Scholar]
- Fort PE, Freeman WM, Losiewicz MK, Singh RS, Gardner TW. The retinal proteome in experimental diabetic retinopathy: up-regulation of crystallins and reversal by systemic and periocular insulin. Mol Cell Proteomics. 2009;8:767–779. doi: 10.1074/mcp.M800326-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zhang H, Zhang X, Cao Y, Jiao W, Liu C, Song Y, Abulimiti A, Chang Z. A dual role for the N-terminal region of Mycobacterium tuberculosis Hsp16.3 in self-oligomerization and binding denaturing substrate proteins. J Biol Chem. 2005;280:6337–6348. doi: 10.1074/jbc.M406319200. [DOI] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26197. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikake N, Nagai Y, Popiel HA, Okamoto Y, Yamaguchi M, Toda T. Heat shock transcription factor 1-activating compounds suppress polyglutamine-induced neurodegeneration through induction of multiple molecular chaperones. J Biol Chem. 2008;283:26188–26189. doi: 10.1074/jbc.M710521200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto M, Izu H, Seki K, Fukuda K, Nishida T, Yamada S, Kato K, Yonemura S, Inouye S, Nakai A. HSF4 is required for normal cell growth and differentiation during mouse lens development. EMBO J. 2004;23:4297–4306. doi: 10.1038/sj.emboj.7600435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabai VL, Kabakov AE. Induction of heat-shock protein synthesis and thermotolerance in EL-4 ascites tumor cells by transient ATP depletion after ischemic stress. Exp Mol Pathol. 1994;60:88–99. doi: 10.1006/exmp.1994.1008. [DOI] [PubMed] [Google Scholar]
- Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, Sherman MY. Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance. J Biol Chem. 1997;272:18033–18037. doi: 10.1074/jbc.272.29.18033. [DOI] [PubMed] [Google Scholar]
- Garrido C, Paul C, Seigneuric R, Kampinga HH. The small heat shock proteins family: the long forgotten chaperones. Int J Biochem Cell Biol. 2012;44:1588–1592. doi: 10.1016/j.biocel.2012.02.022. [DOI] [PubMed] [Google Scholar]
- Gartner EM, Silverman P, Simon M, Flaherty L, Abrams J, Ivy P, Lorusso PM. A phase II study of 17-allylamino-17-demethoxygeldanamycin in metastatic or locally advanced, unresectable breast cancer. Breast Cancer Res Treat. 2012;131:933–937. doi: 10.1007/s10549-011-1866-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh JG, Houck SA, Clark JI. Interactive domains in the molecular chaperone human alphaB crystallin modulate microtubule assembly and disassembly. PLoS One. 2007;2:e498. doi: 10.1371/journal.pone.0000498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giubellino A, Sourbier C, Lee MJ, Scroggins B, Bullova P, Landau M, Ying W, Neckers L, Trepel JB, Pacak K. Targeting heat shock protein 90 for the treatment of malignant pheochromocytoma. PLoS One. 2013;8:e56083. doi: 10.1371/journal.pone.0056083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser T, Walton DS, Maas RL. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat Genet. 1992;2:232–238. doi: 10.1038/ng1192-232. [DOI] [PubMed] [Google Scholar]
- Glaser T, Jepeal L, Edwards JG, Young SR, Favor J, Maas RL. Pax6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat Genet. 1994;7:463–471. doi: 10.1038/ng0894-463. [DOI] [PubMed] [Google Scholar]
- Goldfarb LG, Olivé M, Vicart P, Goebel HH. Intermediate filament diseases: desminopathy. Adv Exp Med Biol. 2008;642:131–164. doi: 10.1007/978-0-387-84847-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman S. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol. 2003;19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- Grabska-Liberek I, Skonieczna K, Olesinska M, Terelak-Borys B, Kociecki J, Sikora M, Jamrozy-Witkowska A, Tesla P, Czarnocka B. Levels of antibodies against human heat shock protein (HSP) 60 in patients with glaucoma in Poland. Med Sci Monit. 2015;21:828–832. doi: 10.12659/MSM.893349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton BA, Frankel WN, Kerrebrock AW, Hawkins TL, FitzHugh W, Kusumi K, Russell LB, Mueller KL, van Berkel V, Birren BW, Kruglyak L, Lander ES. Disruption of the nuclear hormone receptor RORα in staggerer mice. Nature. 1996;379:736–739. doi: 10.1038/379736a0. [DOI] [PubMed] [Google Scholar]
- Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: Short-term results of a phase I trial. Human Gene Therapy. 2008;19:979–990. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- Hayward SA, Rinehart JP, Denlinger DL. Desiccation and rehydration elicit distinct heat shock protein transcript responses in flesh fly pupae. J Exp Biol. 2004;207:963–971. doi: 10.1242/jeb.00842. [DOI] [PubMed] [Google Scholar]
- Head MW, Corbin E, Goldman JE. Overexpression and abnormal modification of the stress proteins alpha B-crystallin and HSP27 in Alexander disease. Am J Pathol. 1993;143:1743–1753. [PMC free article] [PubMed] [Google Scholar]
- He D, Song X, Li L. Geranylgeranylacetone protects against cerebral ischemia and reperfusion injury: HSP90 and eNOS phosphorylation involved. Brain Res. 2015;1599:150–157. doi: 10.1016/j.brainres.2014.12.019. [DOI] [PubMed] [Google Scholar]
- He Y, Leung KW, Zhang YH, Duan S, Zhong XF, Jiang RZ, Peng Z, Tombran-Tink J, Ge J. Mitochondrial complex I defect induces ROS release and degeneration in trabecular meshwork cells of POAG patients: protection by antioxidants. Invest Ophthalmol Vis Sci. 2008;49:1447–1458. doi: 10.1167/iovs.07-1361. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Neuroprotection by sodium channel blockade with phenytoin in an experimental model of glaucoma. Invest Ophthalmol Vis Sci. 2005;46:4164–4169. doi: 10.1167/iovs.05-0618. [DOI] [PubMed] [Google Scholar]
- Hare W, WoldeMussie E, Lai R, Ton H, Ruiz G, Feldmann B, Wijono M, Chun T, Wheeler L. Efficacy and safety of memantine, an NMDA-type open-channel blocker, for reduction of retinal injury associated with experimental glaucoma in rat and monkey. Surv Ophthalmol. 2001;45:S284–289. doi: 10.1016/s0039-6257(01)00200-4. [DOI] [PubMed] [Google Scholar]
- He M, Pan H, Chang RC, So KF, Brecha NC, Pu M. Activation of the Nrf2/HO-1 antioxidant pathway contributes to the protective effects of Lycium barbarum polysaccharides in the rodent retina after ischemia-reperfusion-induced damage. PLoS One. 2014;9:e84800. doi: 10.1371/journal.pone.0084800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath EI, Hillman DW, Vaishampayan U, Sheng S, Sarkar F, Harper F, Gaskins M, Pitot HC, Tan W, Ivy SP, Pili R, Carducci MA, Erlichman C, Liu G. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin Cancer Res. 2008;14:7940–7946. doi: 10.1158/1078-0432.CCR-08-0221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebb MO, Myers TL, Clarke DB. Enhanced expression of heat shock protein 27 is correlated with axonal regeneration in mature retinal ganglion cells. Brain Res. 2006;1073-1074:146–150. doi: 10.1016/j.brainres.2005.12.038. [DOI] [PubMed] [Google Scholar]
- Hirota K, Nakamura H, Masutani H, Yodoi J. Thioredoxin superfamily and thioredoxin-inducing agents. Ann N Y Acad Sci. 2002;957:189–199. doi: 10.1111/j.1749-6632.2002.tb02916.x. [DOI] [PubMed] [Google Scholar]
- Horwitz J. Alpha-crystallin can function as a molecular chaperone Proc. Natl Acad Sci USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horwitz J. Alpha-crystallin. Exp Eye Res. 2003;76:145–153. doi: 10.1016/s0014-4835(02)00278-6. [DOI] [PubMed] [Google Scholar]
- Houck SA, Clark JI. Dynamic subunit exchange and the regulation of microtubule assembly by the stress response protein human alphaB crystallin. PLoS One. 2010;5:e11795. doi: 10.1371/journal.pone.0011795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Park KK, Yang L, Wei X, Yang Q, Cho KS, Thielen P, Lee AH, Cartoni R, Glimcher LH, Chen DF, He Z. Differential effects of unfolded protein response pathways on axon injury-induced death of retinal ganglion cells. Neuron. 2012;73:445–452. doi: 10.1016/j.neuron.2011.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta JB, Dobberfuhl A, Filippopolous T, Guo Y, Kwon G, Grosskreutz CL. Calcineurin cleavage is triggered by elevated intraocular pressure, and calcineurin inhibition blocks retinal ganglion cell death in experimental glaucoma. Proc Natl Acad Sci USA. 2005;102:12242–12247. doi: 10.1073/pnas.0505138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Fileta JB, Filippopoulos T, Ray A, Dobberfuhl A, Grosskreutz CL. Hsp27 phosphorylation in experimental glaucoma. Invest Ophthalmol Vis Sci. 2007;48:4129–4135. doi: 10.1167/iovs.06-0606. [DOI] [PubMed] [Google Scholar]
- Huot J, Houle F, Spitz DR, Landry J. HSP27 phosphorylation-mediated resistance against actin fragmentation and cell death induced by oxidative stress. Cancer Res. 1996;56:273–279. [PubMed] [Google Scholar]
- Ishii Y, Kwong JM, Caprioli J. Retinal ganglion cell protection with geranylgeranylacetone, a heat shock protein inducer, in a rat glaucoma model. Invest Ophthalm Vis Sci. 2003;44:1982–1992. [PubMed] [Google Scholar]
- Iwaki T, Kume-Iwaki A, Liem RK, Goldman JE. Alpha B-crystallin is expressed in non-lenticular tissues and accumulates in Alexander's disease brain. Cell. 1989;57:71–78. doi: 10.1016/0092-8674(89)90173-6. [DOI] [PubMed] [Google Scholar]
- Iwaki T, Wisniewski T, Iwaki A, Corbin E, Tomokane N, Tateishi J, Goldman JE. Accumulation of alpha B-crystallin in central nervous system glia and neurons in pathologic conditions. Am J Pathol. 1992;140:345–356. [PMC free article] [PubMed] [Google Scholar]
- Jaattela M. Heat shock proteins as cellular lifeguards. Ann Med. 1999;31:261–271. doi: 10.3109/07853899908995889. [DOI] [PubMed] [Google Scholar]
- Jakob U, Gaestel M, Engel K, Buchner J. Small heat shock proteins are molecular chaperones. J Biol Chem. 1993;268:1517–1520. [PubMed] [Google Scholar]
- Jiang B, Zhang P, Zhou D, Zhang J, Xu X, Tang L. Intravitreal transplantation of human umbilical cord blood stem cells protects rats from traumatic optic neuropathy. PLoS One. 2013;8:e69938. doi: 10.1371/journal.pone.0069938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joachim SC, Grus FH, Kraft D, White-Farrar K, Barnes G, Barbeck M, Ghanaati S, Cao S, Li B, Wax MB. Complex antibody profile changes in an experimental autoimmune glaucoma animal model. Invest Ophthalmol Vis Sci. 2009;50:4734–4742. doi: 10.1167/iovs.08-3144. [DOI] [PubMed] [Google Scholar]
- Joachim SC, Wax MB, Seidel P, Pfeiffer N, Grus FH. Enhanced characterization of serum autoantibody reactivity following HSP 60 immunization in a rat model of experimental autoimmune glaucoma. Curr Eye Res. 2010;35:900–908. doi: 10.3109/02713683.2010.495829. [DOI] [PubMed] [Google Scholar]
- Jones SE, Jomary C, Grist J, Thomas MR, Neal MJ. Expression of Pax-6 mRNA in the retinal degeneration (rd) mouse. Biochem Biophys Res Commun. 1998;252:236–240. doi: 10.1006/bbrc.1998.9631. [DOI] [PubMed] [Google Scholar]
- Kalesnykas G, Niittykoski M, Rantala J, Miettinen R, Salminen A, Kaarniranta K, Uusitalo H. The expression of heat shock protein 27 in retinal ganglion and glial cells in a rat glaucoma model. Neuroscience. 2007;150:692–704. doi: 10.1016/j.neuroscience.2007.09.078. [DOI] [PubMed] [Google Scholar]
- Kamradt MC, Chen F, Cryns VL. The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–16063. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Hageman J, Vos MJ, Kubota H, Tanguay RM, Bruford EA, Cheetham ME, Chen B, Hightower LE. Guidelines for the nomenclature of the human heat shock proteins. Cell Stress Chaperones. 2009;14:105–111. doi: 10.1007/s12192-008-0068-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kase S, Ishida S, Rao NA. Increased expression of αA-crystallin in human diabetic eye. Int J Mol Med. 2011;28:505–511. doi: 10.3892/ijmm.2011.708. [DOI] [PubMed] [Google Scholar]
- Kato K, Shinohara H, Kurobe N, Inaguma Y, Shimizu K, Ohshima K. Tissue distribution and developmental profiles of immunoreactive αB crystallin in the rat determined with a sensitive immunoassay system. Biochim Biophys Acta. 1991;1074:201–208. doi: 10.1016/0304-4165(91)90062-l. [DOI] [PubMed] [Google Scholar]
- Kawazoe Y, Nakai A, Tanabe M, Nagata K. Proteasome inhibition leads to the activation of all members of the heat-shock-factor family. Eur J Biochem. 1998;255:356–362. doi: 10.1046/j.1432-1327.1998.2550356.x. [DOI] [PubMed] [Google Scholar]
- Kelly S, Uney JB, McCulloch J. Adenovirus HSP70 gene transfer ameliorates damage following global ischemia. J Cereb Blood Flow Metab. 2001;21:S23. [Google Scholar]
- Kelly S, Zhang ZJ, Zhao H, Xu L, Giffard RG, Sapolsky RM, Yenari MA, Steinberg GK. Gene transfer of HSP72 protects cornu ammonis 1 region of the hippocampus neurons from global ischemia: influence of Bcl-2. Ann Neurol. 2002;52:160–167. doi: 10.1002/ana.10264. [DOI] [PubMed] [Google Scholar]
- Kim HP, Morse D, Choi AM. Heat-shock proteins: new keys to the development of cytoprotective therapies. Expert Opin Ther Targets. 2006;10:7597–7569. doi: 10.1517/14728222.10.5.759. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kim R, Kim SH. Crystal structure of a small heat-shock protein. Nature. 1998;394:595–599. doi: 10.1038/29106. [DOI] [PubMed] [Google Scholar]
- Kim R, Lai L, Lee HH, Cheong GW, Kim KK, Wu Z, Yokota H, Marqusee S, Kim SH. On the mechanism of chaperone activity of the small heat-shock protein of Methanococcus jannaschii. Proc Natl Acad Sci USA. 2003;100:8151–8155. doi: 10.1073/pnas.1032940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YH, Choi MY, Kim YS, Han JM, Lee JH, Park CH, Kang SS, Choi WS, Cho GJ. Protein kinase C delta regulates anti-apoptotic alphaB-crystallin in the retina of type 2 diabetes. Neurobiol Dis. 2007;28:293–303. doi: 10.1016/j.nbd.2007.07.017. [DOI] [PubMed] [Google Scholar]
- Kirbach BB, Golenhofen N. Differential expression and induction of small heat shock proteins in rat brain and cultured hippocampal neurons. J Neurosci Res. 2011;89:162–175. doi: 10.1002/jnr.22536. [DOI] [PubMed] [Google Scholar]
- Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol. 1997;17:2107–2115. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klosterhalfen B, Hauptmann S, Offner FA, Amo-Takyi B, Töns C, Winkeltau G, Affify M, Küpper W, Kirkpatrick CJ, Mittermayer C. Induction of heat shock protein 70 by zinc-bis-(DL-hydrogenaspartate) reduces cytokine liberation, apoptosis, and mortality rate in a rat model of LD100 endotoxemia. Shock. 1997;7:254–262. doi: 10.1097/00024382-199704000-00003. [DOI] [PubMed] [Google Scholar]
- Klucken J, Shin Y, Masliah E, Hyman BT, McLean PJ. Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem. 2004;279:25497–25502. doi: 10.1074/jbc.M400255200. [DOI] [PubMed] [Google Scholar]
- Knauf U, Newton EM, Kyriakis J, Kingston RE. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996;10:2782–2793. doi: 10.1101/gad.10.21.2782. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kume A, Li M, Doyu M, Hata M, Ohtsuka K, Sobue G. Protective effect of chaperones on polyglutamine diseases. J Biol Chem. 2000;275:8772–8778. doi: 10.1074/jbc.275.12.8772. [DOI] [PubMed] [Google Scholar]
- Kollman JM, Merdes A, Mourey L, Agard DA. Microtubule nucleation by gamma-tubulin complexes. Nat Rev Mol Cell Biol. 2011;12:709–721. doi: 10.1038/nrm3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz A, Schmeer C, Tausch S, Isenmann S. Simvastatin promotes heat shock protein 27 expression and Akt activation in the rat retina and protects axotomized retinal ganglion cells in vivo. Neurobiol Dis. 2006;21:421–430. doi: 10.1016/j.nbd.2005.08.003. [DOI] [PubMed] [Google Scholar]
- Krueger-Naug AM, Emsley JG, Myers TL, Currie RW, Clarke DB. Injury to retinal ganglion cells induces expression of the small heat shock protein Hsp27 in the rat visual system. Neuroscience. 2002;110:653–665. doi: 10.1016/s0306-4522(01)00453-5. [DOI] [PubMed] [Google Scholar]
- Kukreja RC, Kontos MC, Loesser KE, Batra SK, Qian YZ, Gbur CJ, Jr, Naseem SA, Jesse RL, Hess ML. Oxidant stress increases heat shock protein 70 mRNA in isolated perfused rat heart. Am J Physiol. 1994;267:2213–2219. doi: 10.1152/ajpheart.1994.267.6.H2213. [DOI] [PubMed] [Google Scholar]
- Kumar PA, Haseeb A, Suryanarayana P, Ehtesham NZ, Reddy GB. Elevated expression of alphaA- and alphaB-crystallins in streptozotocin-induced diabetic rat. Arch Biochem Biophys. 2005;444:77–83. doi: 10.1016/j.abb.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Kwong JM, Caprioli J, Piri N. RNA binding protein with multiple splicing: a new marker for retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51:1052–1058. doi: 10.1167/iovs.09-4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JM, Gu L, Nassiri N, Bekerman V, Kumar-Singh R, Rhee KD, Yang XJ, Hauswirth WW, Caprioli J, Piri N. AAV-mediated and pharmacological induction of Hsp70 expression stimulates survival of retinal ganglion cells following axonal injury. Gene Ther. 2015;22:138–145. doi: 10.1038/gt.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert H, Charette SJ, Bernier AF, Guimond A, Landry J. HSP27 multimerization mediated by phosphorylation-sensitive intermolecular interactions at the amino terminus. J Biol Chem. 1999;274:9378–9385. doi: 10.1074/jbc.274.14.9378. [DOI] [PubMed] [Google Scholar]
- Langanowsky A, Benesch J, Landau M, Ding L, Sawaya M, Cascio D, Huang Q, Robinson C, Horwitz J, Eisenberg D. Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 2010;19:1031–1043. doi: 10.1002/pro.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasseck J, Schroer U, Koenig S, Thanos S. Regeneration of retinal ganglion cell axons in organ culture is increased in rats with hereditary buphthalmos. Exp Eye Res. 2007;85:90–104. doi: 10.1016/j.exer.2007.03.005. [DOI] [PubMed] [Google Scholar]
- Lavoie JN, Gingras-Breton G, Tanguay RM, Landry J. Induction of Chinese hamster HSP27 gene expression in mouse cells confers resistance to heat shock. HSP27 stabilization of the microfilament organization. J Biol Chem. 1993a;268:3420–3429. [PubMed] [Google Scholar]
- Lavoie JN, Hickey E, Weber LA, Landry J. Modulation of actin microfilament dynamics and fluid phase pinocytosis by phosphorylation of heat shock protein 27. Biol Chem. 1993b;268:24210–24214. [PubMed] [Google Scholar]
- Le DA, Lipton SA. Potential and current use of N-methyl-D-aspartate (NMDA) receptor antagonists in diseases of aging. Drugs Aging. 2001;18:717–724. doi: 10.2165/00002512-200118100-00001. [DOI] [PubMed] [Google Scholar]
- Lee JY, Park J, Kim YH, Kim DH, Kim CG, Koh JY. Induction by synaptic zinc of heat shock protein-70 in hippocampus after kainate seizures. Exp Neurol. 2000;161:433–441. doi: 10.1006/exnr.1999.7297. [DOI] [PubMed] [Google Scholar]
- Leske MC. The epidemiology of open-angle glaucoma: a review. Am J Epidemiol. 1983;118:166–191. doi: 10.1093/oxfordjournals.aje.a113626. [DOI] [PubMed] [Google Scholar]
- Li H, Liang Y, Chiu K, Yuan Q, Lin B, Chang RC, So KF. Lycium barbarum (wolfberry) reduces secondary degeneration and oxidative stress, and inhibits JNK pathway in retina after partial optic nerve transection. PLoS One. 2013;8:e68881. doi: 10.1371/journal.pone.0068881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998a;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- Li M, Nakagomi Y, Kobayashi Y, Merry DE, Tanaka F, Doyu M, Mitsuma T, Fischbeck KH, Sobue G. Nonneural nuclear inclusions of androgen receptor protein in spinal and bulbar muscular atrophy. Am J Pathol. 1998b;153:695–701. doi: 10.1016/S0002-9440(10)65612-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Li Y, Duan X. Heat shock protein 72 confers protection in retinal ganglion cells and lateral geniculate nucleus neurons via blockade of the SAPK/JNK pathway in a chronic ocular-hypertensive rat model. Neural Regen Res. 2014;9:1395–1401. doi: 10.4103/1673-5374.137595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Reiser G. Phosphorylation of Ser45 and Ser59 of alphaB-crystallin and p38/extracellular regulated kinase activity determine alphaB-crystallin-mediated protection of rat brain astrocytes from C2-ceramide- and staurosporine-induced cell death. J Neurochem. 2011;118:354–364. doi: 10.1111/j.1471-4159.2011.07317.x. [DOI] [PubMed] [Google Scholar]
- Li S, Yang L, Selzer ME, Hu Y. Neuronal ER stress in axon injury and neurodegeneration. Ann Neurol. 2013;74:768–777. doi: 10.1002/ana.24005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SY, Yang D, Yeung CM, Yu WY, Chang RC, So KF, Wong D, Lo AC. Lycium barbarum polysaccharides reduce neuronal damage, blood-retinal barrier disruption and oxidative stress in retinal ischemia/reperfusion injury. PLoS One. 2011;6:e16380. doi: 10.1371/journal.pone.0016380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XM. Protective effect of Lycium barbarum polysaccharides on streptozotocin-induced oxidative stress in rats. Int J Biol Macromol. 2007;40:461–465. doi: 10.1016/j.ijbiomac.2006.11.002. [DOI] [PubMed] [Google Scholar]
- Li Y, Roth S, Laser M, Ma JX, Crosson CE. Retinal preconditioning and the induction of heat-shock protein 27. Invest Ophthalmol Vis Sci. 2003;44:1299–1304. doi: 10.1167/iovs.02-0235. [DOI] [PubMed] [Google Scholar]
- Liang P, MacRae TH. Molecular chaperones and the cytoskeleton. J Cell Sci. 1997;110:1431–1440. doi: 10.1242/jcs.110.13.1431. [DOI] [PubMed] [Google Scholar]
- Liedtke T, Schwamborn JC, Schroer U, Thanos S. Elongation of axons during regeneration involves retinal crystallin beta b2 (crybb2) Mol Cell Proteomics. 2007;6:895–907. doi: 10.1074/mcp.M600245-MCP200. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Lindsey JD, Duong-Polk KX, Hammond D, Leung CK, Weinreb RN. Protection of injured retinal ganglion cell dendrites and unfolded protein response resolution after long-term dietary resveratrol. Neurobiol Aging. 2015;36:1969–1981. doi: 10.1016/j.neurobiolaging.2014.12.021. [DOI] [PubMed] [Google Scholar]
- Liu DJ, Hammer D, Komlos D, Chen KY, Firestein BL, Liu AY. SIRT1 knockdown promotes neural differentiation and attenuates the heat shock response. Cell Physiol. 2014;229:1224–1235. doi: 10.1002/jcp.24556. [DOI] [PubMed] [Google Scholar]
- Liu JP, Schlosser R, Ma WY, Dong Z, Feng H, Lui L, Huang XQ, Liu Y, Li DW. Human alphaA- and alphaB-crystallins prevent UVA-induced apoptosis through regulation of PKCalpha, RAF/MEK/ERK and AKT signaling pathways. Exp Eye Res. 2004;79:393–403. [PubMed] [Google Scholar]
- Liu S, Li J, Tao Y, Xiao X. Small heat shock protein alphaB-crystallin binds to p53 to sequester its translocation to mitochondria during hydrogen peroxide-induced apoptosis. Biochem Biophys Res Comm. 2007;354:109–114. doi: 10.1016/j.bbrc.2006.12.152. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhou Q, Tang M, Fu N, Shao W, Zhang S, Yin Y, Zeng R, Wang X, Hu G, Zhou J. Upregulation of alphaB-crystallin expression in the substantia nigra of patients with Parkinson's disease. Neurobiol Aging. 2015;36:1686–1691. doi: 10.1016/j.neurobiolaging.2015.01.015. [DOI] [PubMed] [Google Scholar]
- Liu ZL, Wang YR, Sha Q, Nie QZ. Influence of geranylgeranylacetone on the expression of HSP70 in retina of rats with chronic IOP elevation. Int J Ophthalmol. 2010;3:28–31. doi: 10.3980/j.issn.2222-3959.2010.01.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Freund R, Schweber M, Wensink PC, Meselson M. Sequence organization and transcription at two heat shock loci in Drosophila. Proc Nat Acad Sci USA. 1975;72:1117–1121. doi: 10.1073/pnas.75.11.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loktionova SA, Kabakov AE. Protein phosphatase inhibitors and heat preconditioning prevent Hsp27 dephosphorylation, F-actin disruption and deterioration of morphology in ATP-depleted endothelial cells. FEBS Lett. 1998;433:294–300. doi: 10.1016/s0014-5793(98)00920-x. [DOI] [PubMed] [Google Scholar]
- Loones MT, Chang Y, Morange M. The distribution of heat shock proteins in the nervous system of the unstressed mouse embryo suggests a role in neuronal and non-neuronal differentiation. Cell Stress Chaperones. 2000;5:291–305. doi: 10.1379/1466-1268(2000)005<0291:tdohsp>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu A, Ran R, Parmentier-Batteur S, Nee A, Sharp FR. Geldanamycin induces heat shock proteins in brain and protects against focal cerebral ischemia. J Neurochem. 2002;81:355–364. doi: 10.1046/j.1471-4159.2002.00835.x. [DOI] [PubMed] [Google Scholar]
- Machado P, Rostaing P, Guigonis JM, Renner M, Dumoulin A, Samson M, Vannier C, Triller A. Heat shock cognate protein 70 regulates gephyrin clustering. J Neurosci. 2011;31:3–14. doi: 10.1523/JNEUROSCI.2533-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLaren RE, Groppe M, Barnard AR, Cottriall CL, Tolmachova T, Seymour L, Clark KR, During MJ, Cremers FP, Black GC, Lotery AJ, Downes SM, Webster AR, Seabra MC. Retinal gene therapy in patients with choroideremia: Initial findings from a phase 1/2 clinical trial. Lancet. 2014;383:1129–1137. doi: 10.1016/S0140-6736(13)62117-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell'Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mairesse N, Horman S, Mosselmans R, Galand P. Antisense inhibition of the 27 kDa heat shock protein production affects growth rate and cytoskeletal organization in MCF-7 cells. Cell Biol Intern. 1996;20:205–212. doi: 10.1006/cbir.1996.0025. [DOI] [PubMed] [Google Scholar]
- Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci USA. 1994;91:1632–1636. doi: 10.1073/pnas.91.5.1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao YW, Liu JP, Xiang H, Li DW. Human alphaA- and alphaB-crystallins bind to Bax and Bcl-X(S) to sequester their translocation during staurosporine-induced apoptosis. Cell Death Differ. 2004;11:512–526. doi: 10.1038/sj.cdd.4401384. [DOI] [PubMed] [Google Scholar]
- Marger L, Schubert CR, Bertrand D. Zinc: an underappreciated modulatory factor of brain function. Biochem Pharmacol. 2014;91:426–435. doi: 10.1016/j.bcp.2014.08.002. [DOI] [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Martin KR, Quigley HA, Zack DJ, Levkovitch-Verbin H, Kielczewski J, Valenta D, Baumrind L, Pease ME, Klein RL, Hauswirth WW. Gene therapy with brain-derived neurotrophic factor as a protection: retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2003;44:4357–4365. doi: 10.1167/iovs.02-1332. [DOI] [PubMed] [Google Scholar]
- Martin LJ, Sieber FE, Traystman RJ. Apoptosis and necrosis occur in separate neuronal populations in hippocampus and cerebellum after ischemia and are associated with differential alterations in metabotropic glutamate receptor signaling pathways. J Cereb Blood Flow Metab. 2000;20:153–167. doi: 10.1097/00004647-200001000-00020. [DOI] [PubMed] [Google Scholar]
- Matsumori Y, Hong SM, Aoyama K, Fan Y, Kayama T, Sheldon RA, Vexler ZS, Ferriero DM, Weinstein PR, Liu J. Hsp70 overexpression sequester AIF and reduces neonatal hypoxic/ischemic brain injury. J Cereb Blood Flow Metab. 2005;25:899–910. doi: 10.1038/sj.jcbfm.9600080. [DOI] [PubMed] [Google Scholar]
- McKinnon SJ, Lehman DM, Tahzib NG, Ransom NL, Reitsamer HA, Liston P, LaCasse E, Li Q, Korneluk RG, Hauswirth WW. Baculoviral IAP repeat-containing-4 protects optic nerve axons in a rat glaucoma model. Mol Ther. 2002;5:780–787. doi: 10.1006/mthe.2002.0608. [DOI] [PubMed] [Google Scholar]
- McLean PJ KluckenJ, Shin Y HymanBT. Geldanamycin induces Hsp70 and prevents alpha-synuclein aggregation and toxicity in vitro. Biochem Biophys Res Commun. 2004;321:665–669. doi: 10.1016/j.bbrc.2004.07.021. [DOI] [PubMed] [Google Scholar]
- McMillan DR, Xiao X, Shao L, Graves K, Benjamin IJ. Targeted disruption of heat shock transcription factor 1 abolishes thermotolerance and protection against heat-inducible apoptosis. J Biol Chem. 1998;273:7523–7528. doi: 10.1074/jbc.273.13.7523. [DOI] [PubMed] [Google Scholar]
- Mehlen P, Schulze-Osthoff K, Arrigo AP. Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death. J Biol Chem. 1996;271:16510–16514. doi: 10.1074/jbc.271.28.16510. [DOI] [PubMed] [Google Scholar]
- Mi XS, Feng Q, Lo AC, Chang RC, Lin B, Chung SK, So KF. Protection of retinal ganglion cells and retinal vasculature by Lycium barbarum polysaccharides in a mouse model of acute ocular hypertension. PLoS One. 2012;7:e45469. doi: 10.1371/journal.pone.0045469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimura T, Kaji Y, Noma H, Funatsu H, Okamoto S. The role of SIRT1 in ocular aging. Exp Eye Res. 2013;116:17–26. doi: 10.1016/j.exer.2013.07.017. [DOI] [PubMed] [Google Scholar]
- Miron T, Wilchek M, Geiger B. Characterization of an inhibitor of actin polymerization in vinculin-rich fraction of turkey gizzard smooth muscle. Eur J Biochem. 1988;178:543–553. doi: 10.1111/j.1432-1033.1988.tb14481.x. [DOI] [PubMed] [Google Scholar]
- Miron T, Vancompernolle K, Vandekerckhove J, Wilchek M, Geiger B. A 25-kD inhibitor of actin polymerization is a low molecular mass heat shock protein. J Cell Biol. 1991;114:255–261. doi: 10.1083/jcb.114.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyara N, Shinzato M, Yamashiro Y, Iwamatsu A, Kariya K, Sawaguchi S. Proteomic analysis of rat retina in a steroid-induced ocular hypertension model: potential vulnerability to oxidative stress. Jpn J Ophthalm. 2008;52:84–90. doi: 10.1007/s10384-007-0507-5. [DOI] [PubMed] [Google Scholar]
- Moore DL, Blackmore MG, Hu Y, Kaestner KH, Bixby JL, Lemmon VP, Goldberg JL. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto R, Tissieres A, Georgopoulos C, editors. Stress Proteins in Biology and Medicine. Cold Spring Harbor, NY: Cold Spring Harbor Lab. Press; 1990. [DOI] [PubMed] [Google Scholar]
- Morozov V, Wawrousek EF. Caspase-dependent secondary lens fiber cell disintegration in {alpha}A-/{alpha}B-crystallin double-knockout mice. Development. 2006;133:813–821. doi: 10.1242/dev.02262. [DOI] [PubMed] [Google Scholar]
- Morrison LE, Hoover HE, Thuerauf DJ, Glembotski CC. Mimicking phosphorylation of alphaB-crystallin on serine-59 is necessary and sufficient to provide maximal protection of cardiac myocytes from apoptosis. Circul Res. 2003;92:203–211. doi: 10.1161/01.res.0000052989.83995.a5. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci USA. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Müller A, Hauk TG, Fischer D. Astrocyte-derived CNTF switches mature RGCs to a regenerative state following inflammatory stimulation. Brain. 2007;130:3308–3320. doi: 10.1093/brain/awm257. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Ahn JH, Kwong JM, Caprioli J, Piri N. Redox proteins thioredoxin 1 and thioredoxin 2 support retinal ganglion cell survival in experimental glaucoma. Gene Ther. 2009a;16:17–25. doi: 10.1038/gt.2008.126. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Kwong JM, Caprioli J, Piri N. The role of alphaA- and alphaB-crystallins in the survival of retinal ganglion cells after optic nerve axotomy. Invest Ophthalm Vis Sci. 2009b;50:3869–3875. doi: 10.1167/iovs.08-3138. [DOI] [PubMed] [Google Scholar]
- Naskar R, Thanos S. Retinal gene profiling in a hereditary rodent model of elevated intraocular pressure. Mol Vis. 2006;12:1199–1210. [PubMed] [Google Scholar]
- Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nat Rev Drug Discov. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neufeld AH, Das S, Vora S, Gachie E, Kawai S, Manning PT, Connor JR. A prodrug of a selective inhibitor of inducible nitric oxide synthase is neuroprotective in the rat model of glaucoma. J Glaucoma. 2002;11:221–225. doi: 10.1097/00061198-200206000-00010. [DOI] [PubMed] [Google Scholar]
- Newsome DA, Swartz M, Leone NC, Elston RC, Miller E. Oral zinc in macular degeneration. Arch Ophthalmol. 1988;106:192–198. doi: 10.1001/archopht.1988.01060130202026. [DOI] [PubMed] [Google Scholar]
- Nicholl ID, Quinlan RA. Chaperone activity of α-crystallins modulates intermediate filament assembly. EMBO J. 1994;13:945–953. doi: 10.1002/j.1460-2075.1994.tb06339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norton PM, Latchman DS. Levels of 90kd heat shock protein and resistance to glucocorticoid mediated cell killing in a range of human and murine lymphocyte cell lines. Genes Dev. 1989;33:149–154. doi: 10.1016/0022-4731(89)90288-4. [DOI] [PubMed] [Google Scholar]
- Nowak TS., Jr Protein synthesis and the heart shock/stress response after ischemia. Cerebrovasc Brain Metab Rev. 1990;2:345–366. [PubMed] [Google Scholar]
- Ohkawara T, Takeda H, Nishiwaki M, Nishihira J, Asaka M. Protective effects of heat shock protein 70 induced by geranylgeranylacetone on oxidative injury in rat intestinal epithelial cells. Scand J Gastroenterol. 2006;41:312–317. doi: 10.1080/00365520500319427. [DOI] [PubMed] [Google Scholar]
- Ohtsuka K, Hata M. Molecular chaperone function of mammalian Hsp70 and Hsp40 –a review. Int J Hyperthermia. 2000;16:231–45. doi: 10.1080/026567300285259. [DOI] [PubMed] [Google Scholar]
- Ostberg JR, Kaplan KC, Repasky EA. Induction of stress proteins in a panel of mouse tissues by fever-range whole body hyperthermia. Int J Hyperthermia. 2002;18:552–562. doi: 10.1080/02656730210166168. [DOI] [PubMed] [Google Scholar]
- Pacey S, Gore M, Chao D, Banerji U, Larkin J, Sarker S, Owen K, Asad Y, Raynaud F, Walton M, Judson I, Workman P, Eisen T. A Phase II trial of 17-allylamino, 17-demethoxygeldanamycin (17-AAG, tanespimycin) in patients with metastatic melanoma. Invest New Drugs. 2012;30:341–349. doi: 10.1007/s10637-010-9493-4. [DOI] [PubMed] [Google Scholar]
- Panasenko OO, Kim MV, Marston SB, Gusev NB. Interaction of the small heat shock protein with molecular mass 25 kDa (hsp25) with actin. Eur J Biochem. 2003;270:892–901. doi: 10.1046/j.1432-1033.2003.03449.x. [DOI] [PubMed] [Google Scholar]
- Paoletti P, Vergnano AM, Barbour B, Casado M. Zinc at glutamatergic synapses. Neuroscience. 2009;158:126–136. doi: 10.1016/j.neuroscience.2008.01.061. [DOI] [PubMed] [Google Scholar]
- Park HS, Lee JS, Huh SH, Seo JS, Choi EJ. Hsp72 functions as a natural inhibitory protein of c-Jun N-terminal kinase. EMBO J. 2001a;20:446–456. doi: 10.1093/emboj/20.3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park KH, Cozier F, Ong OC, Caprioli J. Induction of heat shock protein 72 protects retinal ganglion cells in a rat glaucoma model. Invest Ophthalmol Vis Sci. 2001b;42:1522–1530. [PubMed] [Google Scholar]
- Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–96.6. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasta SY, Raman B, Ramakrishna T, Rao CM. The IXI/V motif in the C-terminal extension of alpha-crystallins: alternative interactions and oligomeric assemblies. Mol Vis. 2004;10:655–662. [PubMed] [Google Scholar]
- Patel BA, Khaliq J, Evans J. Hypoxia induces hsp70 gene expression in human hepatoma (HEP G2) cells. Biochem Mol Biol Int. 1995;36:907–912. [PubMed] [Google Scholar]
- Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem. 2009;9:1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernet V, Joly S, Dalkara D, Schwarz O, Christ F, Schaffer D, Flannery JG, Schwab ME. Neuronal Nogo-A upregulation does not contribute to ER stress-associated apoptosis but participates in the regenerative response in the axotomized adult retina. Cell Death Differ. 2012;19:1096–1108. doi: 10.1038/cdd.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng MD, Cairns L, van den Ijssel P, Prescott A, Hutcheson AM, Quinlan RA. Intermediate filament interactions can be altered by HSP27 and alphaB-crystallin. J Cell Sci. 1999;112:2099–2112. doi: 10.1242/jcs.112.13.2099. [DOI] [PubMed] [Google Scholar]
- Piatigorsky J. Multifunctional lens crystallins and corneal enzymes: more than meets the eye. Ann N Y Acad Sci. 1998;842:7–15. doi: 10.1111/j.1749-6632.1998.tb09626.x. [DOI] [PubMed] [Google Scholar]
- Piri N, Song M, Kwong JM, Caprioli J. Modulation of alpha and beta crystallin expression in rat retinas with ocular hypertension-induced ganglion cell degeneration. Brain Res. 2007;1141:1–9. doi: 10.1016/j.brainres.2006.11.095. [DOI] [PubMed] [Google Scholar]
- Piri N, Kwong JM, Caprioli J. Crystallins in retinal ganglion cell survival and regeneration. Mol Neurobiol. 2013;48:819–828. doi: 10.1007/s12035-013-8470-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas AM, Soriano MA, Estrada A, Sanz O, Martin F, Ferrer I. The heat shock stress response after brain lesions: induction of 72 kDa heat shock protein (cell types involved, axonal transport, transcriptional regulation) and protein synthesis inhibition. Prog Neurobiol. 1997;51:607–636. doi: 10.1016/s0301-0082(97)00004-x. [DOI] [PubMed] [Google Scholar]
- Plesset J, Palm C, McLaughlin CS. Induction of heat shock proteins and thermotolerance by ethanol in Saccharomyces cerevisiae. Biochem Biophys Res Commun. 1982;108:1340–1345. doi: 10.1016/0006-291x(82)92147-7. [DOI] [PubMed] [Google Scholar]
- Plumier JC, Krueger AM, Currie RW, Kontoyiannis D, Kollias G, Pagoulatos GN. Transgenic mice expressing the human inducible Hsp70 have hippocampal neurons resistant to ischemic injury. Cell Stress Chaperones. 1997;2:162–167. doi: 10.1379/1466-1268(1997)002<0162:tmethi>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–360. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell. 1997;90:65–75. doi: 10.1016/s0092-8674(00)80314-1. [DOI] [PubMed] [Google Scholar]
- Putcha P, Danzer KM, Kranich LR, Scott A, Silinski M, Mabbett S, Hicks CD, Veal JM, Steed PM, Hyman BT, McLean PJ. Brain-permeable small-molecule inhibitors of Hsp90 prevent alpha-synuclein oligomer formation and rescue alpha-synuclein-induced toxicity. J Pharmacol Exp Ther. 2010;332:849–857. doi: 10.1124/jpet.109.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol. 2006;90:262–267. doi: 10.1136/bjo.2005.081224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan RA, Carter JM, Sandilands A, Prescott AR. The beaded filament of the eye lens: an unexpected key to intermediate filament structure and function. Trends Cell Biol. 1996;6:123–126. doi: 10.1016/0962-8924(96)20001-7. [DOI] [PubMed] [Google Scholar]
- Rajdev S, Hara K, Kokubo Y, Mestril R, Dillmann W, Weinstein PR, Sharp FR. Mice overexpressing rat heat shock protein 70 are protected against cerebral infarction. Ann Neurol. 2000;47:782–791. [PubMed] [Google Scholar]
- Rao NA, Saraswathy S, Wu GS, Katselis GS, Wawrousek EF, Bhat S. Elevated retina-specific expression of the small heat shock protein, alphaA-crystallin, is associated with photoreceptor protection in experimental uveitis. Invest Ophthalmol Vis Sci. 2008;49:1161–1171. doi: 10.1167/iovs.07-1259. [DOI] [PubMed] [Google Scholar]
- Rao NA, Saraswathy S, Pararajasegaram G, Bhat SP. Small heat shock protein αA-crystallin prevents photoreceptor degeneration in experimental autoimmune uveitis. PLoS One. 2012;7:e33582. doi: 10.1371/journal.pone.0033582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renkawek K, de Jong WW, Merck KB, Frenken CW, van Workum FP, Bosman GJ. Alpha B-crystallin is present in reactive glia in Creutzfeldt–Jakob disease. Acta Neuropathol. 1992;83:324–327. doi: 10.1007/BF00296796. [DOI] [PubMed] [Google Scholar]
- Renkawek K, Voorter CE, Bosman GJ, van Workum FP, de Jong WW. Expression of alpha B-crystallin in Alzheimer's disease. Acta Neuropathol. 1994;87:155–160. doi: 10.1007/BF00296185. [DOI] [PubMed] [Google Scholar]
- Renkawek K, Stege GJ, Bosman GJ. Dementia, gliosis and expression of the small heat shock proteins hsp27 and alpha B-crystallin in Parkinson's disease. Neuroreport. 1999;10:2273–2276. doi: 10.1097/00001756-199908020-00009. [DOI] [PubMed] [Google Scholar]
- Richard V, Kaeffer N, Thuillez C. Delayed protection of the ischemic heart from pathophysiology to therapeutic applications. Fundam Clin Pharmacol. 1996;10:409–415. doi: 10.1111/j.1472-8206.1996.tb00595.x. [DOI] [PubMed] [Google Scholar]
- Ritossa F. Discovery of the heat shock response. Cell Stress Chaperones. 1996;2:97–98. doi: 10.1379/1466-1268(1996)001<0097:dothsr>2.3.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronnen EA, Kondagunta GV, Ishill N, Sweeney SM, Deluca JK, Schwartz L, Bacik J, Motzer RJ. A phase II trial of 17-(Allylamino)-17-demethoxygeldanamycin in patients with papillary and clear cell renal cell carcinoma. Invest New Drugs. 2006;24:543–546. doi: 10.1007/s10637-006-9208-z. [DOI] [PubMed] [Google Scholar]
- Roth DM, Balch WE. Modeling general proteostasis: proteome balance in health and disease. Curr Opin Cell Biol. 2011;23:126–134. doi: 10.1016/j.ceb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacca SC, Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008;173:385–407. doi: 10.1016/S0079-6123(08)01127-8. [DOI] [PubMed] [Google Scholar]
- Saibil H. Molecular chaperones: containers and surfaces for folding, stabilizing or unfolding proteins. Curr Opin Struct Biol. 2000;10:251–258. doi: 10.1016/s0959-440x(00)00074-9. [DOI] [PubMed] [Google Scholar]
- Sakaguchi H, Miyagi M, Darrow RM, Crabb JS, Hollyfield JG, Organisciak DT, Crabb JW. Intense light exposure changes the crystallin content in retina. Exp Eye Res. 2003;76:131–133. doi: 10.1016/s0014-4835(02)00249-x. [DOI] [PubMed] [Google Scholar]
- Sakai M, Sakai H, Nakamura Y, Fukuchi T, Sawaguchi S. Immunolocalization of heat shock proteins in the retina of normal monkey eyes and monkey eyes with laser-induced glaucoma. Jpn J Ophthalmol. 2003;47:42–52. doi: 10.1016/s0021-5155(02)00627-5. [DOI] [PubMed] [Google Scholar]
- Sandilands A, Prescott AR, Wegener A, Zoltoski RK, Hutcheson AM, Masaki S, Kuszak JR, Quinlan RA. Knockout of the intermediate filament protein CP49 destabilises the lens fibre cell cytoskeleton and decreases lens optical quality, but does not induce cataract. Exp Eye Res. 2003;76:385–391. doi: 10.1016/s0014-4835(02)00330-5. [DOI] [PubMed] [Google Scholar]
- Sarge KD, Murphy SP, Morimoto RI. Activation of heat shock gene transcription by HSF1 involves oligomerization, acquisition of DNA binding activity, and nuclear localization and can occur in the absence of stress. Mol Cell Biol. 1993;13:1392–1407. doi: 10.1128/mcb.13.3.1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer C, Clapp P, Welsh MJ, Benndorf R, Williams JA. HSP27 expression regulates CCK-induced changes of the actin cytoskeleton in CHO-CCK-A cells. Am J Physiol. 1999;277:C1032–C1043. doi: 10.1152/ajpcell.1999.277.6.C1032. [DOI] [PubMed] [Google Scholar]
- Schneider GB, Hamano H, Cooper LF. In vivo evaluation of hsp27 as an inhibitor of actin polymerization: Hsp27 limits actin stress fiber and focal adhesion formation after heat shock. J Cell Physiol. 1998;177:575–584. doi: 10.1002/(SICI)1097-4652(199812)177:4<575::AID-JCP8>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Schori H, Kipnis J, Yoles E, WoldeMussie E, Ruiz G, Wheeler LA, Schwartz M. Vaccination for protection of retinal ganglion cells against death from glutamate cytotoxicity and ocular hypertension: implications for glaucoma. Proc Natl Acad Sci USA. 2001;98:3398–3403. doi: 10.1073/pnas.041609498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Selcen D, Engel AG. Myofibrillar myopathy caused by novel dominant negative alpha B-crystallin mutations. Ann Neurol. 2003;54:804–810. doi: 10.1002/ana.10767. [DOI] [PubMed] [Google Scholar]
- Seit-Nebi AS, Datskevich P, Gusev NB. Commentary on paper: Small heat shock proteins and the cytoskeleton: an essential interplay for cell integrity? (Wettstein et al.) Int J Biochem Cell Biol. 2013;45:344–346. doi: 10.1016/j.biocel.2012.11.011. [DOI] [PubMed] [Google Scholar]
- Sharp FR, Lowenstein D, Simon R, Hisanaga K. Heat shock protein hsp72 induction in cortical and striatal astrocytes and neurons following infarction. J Cereb Blood Flow Metab. 1991;11:621–627. doi: 10.1038/jcbfm.1991.113. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Inokuchi Y, Ito Y, Murata H, Aihara M, Miura M, Araie M, Hara H. Involvement of ER stress in retinal cell death. Mol Vis. 2007;13:578–587. [PMC free article] [PubMed] [Google Scholar]
- Silver J, Miller JH. Regeneration beyond the glial scar. Nat Rev Neurosci. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- Singh BN, Rao KS, Ramakrishna T, Rangaraj N, Rao ChM. Association of alphaB-crystallin, a small heat shock protein, with actin: role in modulating actin filament dynamics in vivo. Journal of Mol Biol. 2007;366:756–767. doi: 10.1016/j.jmb.2006.12.012. [DOI] [PubMed] [Google Scholar]
- Shinohara H, Inaguma Y, Goto S, Inagaki T, Kato K. Alpha B crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer's disease. J Neurol Sci. 1993;119:203–208. doi: 10.1016/0022-510x(93)90135-l. [DOI] [PubMed] [Google Scholar]
- Sittler A, Lurz R, Lueder G, Priller J, Lehrach H, Hayer-Hartl MK, Hartl FU, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- Smith JB, Liu Y, Smith DL. Identification of possible regions of chaperone activity in lens alpha-crystallin. Exp Eye Res. 1996;63:125–128. doi: 10.1006/exer.1996.0100. [DOI] [PubMed] [Google Scholar]
- Smith PD, Sun F, Park KK, Cai B, Wang C, Kuwako K, Martinez-Carrasco I, Connolly L, He Z. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram T, Bhat SP. Developmentally dictated expression of heat shock factors: exclusive expression of HSF4 in the postnatal lens and its specific interaction with alphaB-crystallin heat shock promoter. J Biol Chem. 2004;279:44497–44503. doi: 10.1074/jbc.M405813200. [DOI] [PubMed] [Google Scholar]
- Srinivasan AN, Nagineni CN, Bhat SP. Alpha A-crystallin is expressed in non-ocular tissues. J Biol Chem. 1992;267:23337–23341. [PubMed] [Google Scholar]
- Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest Ophthalm Vis Sci. 2006;47:977–985. doi: 10.1167/iovs.05-0865. [DOI] [PubMed] [Google Scholar]
- Steinmayr M, Andre E, Conquet F, Rondi-Reig L, Delhaye-Bouchaud N, Auclair N, Daniel H, Crepel F, Mariani J, Sotelo C. Becker-Andre M: staggerer phenotype in retinoid-related orphan receptor alpha-deficient mice. Proc Natl Acad Sci USA. 1998;95:3960–3965. doi: 10.1073/pnas.95.7.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins,proteasome components and SRC-1,and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- Stetler RA, Gao Y, Signore AP, Cao G, Chen J. HSP27: mechanisms of cellular protection against neuronal injury. Curr Mol Med. 2009;9:863–872. doi: 10.2174/156652409789105561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Gan Y, Zhang W, Liou AK, Gao Y, Cao G, Chen J. Heat shock proteins: cellular and molecular mechanisms in the central nervous system. Prog Neurobiol. 2010;92:184–211. doi: 10.1016/j.pneurobio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stromer T, Fischer E, Richter K, Haslbeck M, Buchner J. Analysis of the regulation of the molecular chaperone Hsp26 by temperature-induced dissociation: the N-terminal domail is important for oligomer assembly and the binding of unfolding proteins. J Biol Chem. 2004;279:11222–11228. doi: 10.1074/jbc.M310149200. [DOI] [PubMed] [Google Scholar]
- Sreekumar PG, Kannan R, Kitamura M, Spee C, Barron E, Ryan SJ, Hinton DR. AlphaB crystallin is apically secreted within exosomes by polarized human retinal pigment epithelium and provides neuroprotection to adjacent cells. PLoS One. 2010;5:e12578. doi: 10.1371/journal.pone.0012578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele MR, Inman DM, Calkins DJ, Horner PJ, Vetter ML. Microarray analysis of retinal gene expression in the DBA/2J model of glaucoma. Invest Ophthalmol Vis Sci. 2006;47:977–85. doi: 10.1167/iovs.05-0865. [DOI] [PubMed] [Google Scholar]
- Studer S, Obrist M, Lentze N, Narberhaus F. A critical motif for oligomerization and chaperone activity of bacterial alpha-heat shock proteins. Eur J Biochem. 2002;269:3578–3586. doi: 10.1046/j.1432-1033.2002.03049.x. [DOI] [PubMed] [Google Scholar]
- Sun Y, MacRae TH. Small heat shock proteins: molecular structure and chaperone function. Cell Mol Life Sci. 2005;62:2460–2476. doi: 10.1007/s00018-005-5190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Maruyama S, Sato W, Morita Y, Sato F, Miki Y, Kato S, Katsuno M, Sobue G, Yuzawa Y, Matsuo S. Geranylgeranylacetone ameliorates ischemic acute renal failure via induction of Hsp70. Kidney Int. 2005;67:2210–2220. doi: 10.1111/j.1523-1755.2005.00326.x. [DOI] [PubMed] [Google Scholar]
- Tam LC, Kiang AS, Campbell M, Keaney J, Farrar GJ, Humphries MM, Kenna PF, Humphries P. Prevention of autosomal dominant retinitis pigmentosa by systemic drug therapy targeting heat shock protein 90 (Hsp90) Hum Mol Genet. 2010;19:4421–4436. doi: 10.1093/hmg/ddq369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tallot P, Grongnet JF, David JC. Dual perinatal and developmental expression of the small heat shock proteins [FC12]aB-crystallin and Hsp27 in different tissues of the developing piglet. Biol Neonate. 2003;83:281–288. doi: 10.1159/000069488. [DOI] [PubMed] [Google Scholar]
- Tanabe M, Sasai N, Nagata K, Liu XD, Liu PC, Thiele DJ, Nakai A. The mammalian HSF4 gene generates both an activator and a repressor of heat shock genes by alternative splicing. J Biol Chem. 1999;274:27845–27856. doi: 10.1074/jbc.274.39.27845. [DOI] [PubMed] [Google Scholar]
- Tang G, Perng MD, Wilk S, Quinlan R, Goldman JE. Oligomers of mutant glial fibrillary acidic protein (GFAP) Inhibit the proteasome system in alexander disease astrocytes, and the small heat shock protein alphaB-crystallin reverses the inhibition. J Biol Chem. 2010;285:10527–10537. doi: 10.1074/jbc.M109.067975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanito M, Kwon YW, Kondo N, Bai J, Masutani H, Nakamura H, Fujii J, Ohira A, Yodoi J. Cytoprotective effects of geranylgeranylacetone against retinal photooxidative damage. J Neurosci. 2005;25:2396–2404. doi: 10.1523/JNEUROSCI.4866-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng FY, Tang BL. Axonal regeneration in adult CNS neurons–signaling molecules and pathways. J Neurochem. 2006;96:1501–1508. doi: 10.1111/j.1471-4159.2006.03663.x. [DOI] [PubMed] [Google Scholar]
- Tezel G, Seigel GM, Wax MB. Autoantibodies to small heat shock proteins in glaucoma. Invest Ophthalmol Vis Sci. 1998;39:2277–2287. [PubMed] [Google Scholar]
- Tezel G, Hernandez R, Wax MB. Immunostaining of heat shock proteins in the retina and optic nerve head of normal and glaucomatous eyes. Arch Ophthalmol. 2000;118:511–518. doi: 10.1001/archopht.118.4.511. [DOI] [PubMed] [Google Scholar]
- Tezel G, Wax MB. The mechanisms of hsp27 antibody-mediated apoptosis in retinal neuronal cells. J Neurosci. 2000;20:3552–3562. doi: 10.1523/JNEUROSCI.20-10-03552.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezel G, Yang J, Wax MB. Heat shock proteins, immunity and glaucoma. Brain Res Bull. 2004;62:473–480. doi: 10.1016/S0361-9230(03)00074-1. [DOI] [PubMed] [Google Scholar]
- Ton CC, Hirvonen H, Miwa H, Weil MM, Monaghan P, Jordan T, van Heyningen V, Hastie ND, Meijers-Heijboer H, Drechsler M, Royer-Pokora B, Collins F, Swaroop A, Strong LC, Saunders GF. Positional cloning and characterization of a paired box and homeobox containing gene from the aniridia region. Cell. 1991;67:1059–1074. doi: 10.1016/0092-8674(91)90284-6. [DOI] [PubMed] [Google Scholar]
- Tytell M, Barbe MF, Brown IR. Induction of heat shock (stress) protein 70 and its mRNA in the normal and light-damaged rat retina after whole body hyperthermia. J Neurosci Res. 1994;38:19–31. doi: 10.1002/jnr.490380105. [DOI] [PubMed] [Google Scholar]
- Ueda J, Sawaguchi S, Hanyu T, Yaoeda K, Fukuchi T, Abe H, Ozawa H. Experimental glaucoma model in the rat induced by laser trabecular photocoagulation after an intracameral injection of India ink. Jpn J Ophthalmol. 1998;42:337–344. doi: 10.1016/s0021-5155(98)00026-4. [DOI] [PubMed] [Google Scholar]
- Van Montfort RL, Slingsby C, Vierling E. Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Adv Protein Chem. 2001a;59:105–156. doi: 10.1016/s0065-3233(01)59004-x. [DOI] [PubMed] [Google Scholar]
- Van Montfort RL, Basha E, Friedrich KL, Slingsby C, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8:1025–1030. doi: 10.1038/nsb722. [DOI] [PubMed] [Google Scholar]
- Vazquez-Chona F, Song BK, Geisert EE. Temporal changes in gene expression after injury in the rat retina. Invest Ophthalm Vis Sci. 2004;45:2737–2746. doi: 10.1167/iovs.03-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura LM, Sorokac N, De Los Santos R, Feuer WJ, Porciatti V. The relationship between retinal ganglion cell function and retinal nerve fiber thickness in early glaucoma. Invest Ophthalmol Vis Sci. 2006;47:3904–3911. doi: 10.1167/iovs.06-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma P, Pfister JA, Mallick S, D'Mello SR. HSF1 protects neurons through a novel trimerization- and HSP-independent mechanism. J Neurosc. 2014;34:1599–1612. doi: 10.1523/JNEUROSCI.3039-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verschuure P, Tatard C, Boelens WC, Grongnet JF, David JC. Expression of small heat shock proteins HspB2, HspB8, Hsp20 and cvHsp in different tissues of the perinatal developing pig. Eur J Cell Biol. 2003;82:523–530. doi: 10.1078/0171-9335-00337. [DOI] [PubMed] [Google Scholar]
- Vicart P, Vicart P, Caron A, Guicheney P, Li Z, Prévost MC, Faure A, Chateau D, Chapon F, Tomé F, Dupret JM, Paulin D, Fardeau M. A missense mutation in the alphaB-crystallin chaperone gene causes a desmin-related myopathy. Nat Genet. 1998;20:92–95. doi: 10.1038/1765. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Spector A. alpha-crystallin stabilizes actin filaments and prevents cytochalasin-induced depolymerization in a phosphorylation-dependent manner. Eur J Biochem. 1996;242:56–66. doi: 10.1111/j.1432-1033.1996.0056r.x. [DOI] [PubMed] [Google Scholar]
- Wang K, Zhang J, Xu Y, Ren K, Xie WL, Yan YE, Zhang BY, Shi Q, Liu Y, Dong XP. Abnormally upregulated alphaB-crystallin was highly coincidental with the astrogliosis in the brains of scrapie-infected hamsters and human patients with prion diseases. J Mol Neurosci. 2013;51:734–748. doi: 10.1007/s12031-013-0057-x. [DOI] [PubMed] [Google Scholar]
- Wang YD, Wu JD, Jiang ZL, Wang YB, Wang XH, Liu C, Tong MQ. Comparative proteome analysis of neural retinas from type 2 diabetic rats by two-dimensional electrophoresis. Curr Eye Res. 2007;32:891–901. doi: 10.1080/02713680701593702. [DOI] [PubMed] [Google Scholar]
- Wang YH, Wang DW, Wu N, Wang Y, Yin ZQ. Alpha-crystallin promotes rat retinal neurite growth on myelin substrates in vitro. Ophthalmic Res. 2011;45:164–168. doi: 10.1159/000319944. [DOI] [PubMed] [Google Scholar]
- Wang YH, Wang DW, Wu N, Wang Y, Yin ZQ. α-Crystallin promotes rat axonal regeneration through regulation of RhoA/rock/cofilin/MLC signaling pathways. J Mol Neurosci. 2012;46:138–144. doi: 10.1007/s12031-011-9537-z. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G, Saito I, Gupta RS, Harley JB, Li Z, Romano C. Anti-Ro/SS-A positivity and heat shock protein antibodies in patients with normal-pressure glaucoma. Am J Ophthalmol. 1998;125:145–157. doi: 10.1016/s0002-9394(99)80084-1. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G, Kawase K, Kitazawa Y. Serum autoantibodies to heat shock proteins in glaucoma patients from Japan and the United States. Ophthalmology. 2001;108:296–302. doi: 10.1016/s0161-6420(00)00525-x. [DOI] [PubMed] [Google Scholar]
- Wax MB, Tezel G, Yang J, Peng G, Patil RV, Agarwal N, Sappington RM, Calkins DJ. Induced autoimmunity to heat shock proteins elicits glaucomatous loss of retinal ganglion cell neurons via activated T-cell-derived fas-ligand. J Neurosci. 2008;28:12085–12096. doi: 10.1523/JNEUROSCI.3200-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Raynes R, Powell C, Xue B, Uversky VN. HSF transcription factor family, heat shock response, and protein intrinsic disorder. Curr Protein Pept Sci. 2012;13:86–103. doi: 10.2174/138920312799277956. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Anckar J, Stevens SM, Jr, Sistonen L, Morimoto RI. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–1066. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettstein G, Bellaye PS, Micheau O, Bonniaud P. Small heat shock proteins and the cytoskeleton: an essential interplay for cell integrity? Int J Biochem Cell Biol. 2012;44:1680–1686. doi: 10.1016/j.biocel.2012.05.024. [DOI] [PubMed] [Google Scholar]
- Whiston EA, Sugi N, Kamradt MC, Sack C, Heimer SR, Engelbert M, Wawrousek EF, Gilmore MS, Ksander BR. Gregory MS: alphaB-crystallin protects retinal tissue during Staphylococcus aureus-induced endophthalmitis. Infect Immun. 2008;76:1781–1790. doi: 10.1128/IAI.01285-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc Natl Acad Sci USA. 1994;91:8324–8328. doi: 10.1073/pnas.91.18.8324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock NA, Agarwalm N, Mam JX, Crosson CE. Hsp27 upregulation by HIF-1 signaling offers protection against retinal ischemia in rats. Invest Ophthalmol Vis Sci. 2005;46:1092–1098. doi: 10.1167/iovs.04-0043. [DOI] [PubMed] [Google Scholar]
- Wieske M, Benndorf R, Behlke J, Dölling R, Grelle G, Bielka H, Lutsch G. Defined sequence segments of the small heat shock proteins HSP25 and αB-crystallin inhibit actin polymerization. Eur J Biochem. 2001;268:2083–2090. doi: 10.1046/j.1432-1327.2001.02082.x. [DOI] [PubMed] [Google Scholar]
- Windisch BK, LeVatte TL, Archibald ML, Chauhan BC. Induction of heat shock proteins 27 and 72 in retinal ganglion cells after acute pressure-induced ischaemia. Clin Experiment Ophthalmol. 2009;37:299–307. doi: 10.1111/j.1442-9071.2009.02032.x. [DOI] [PubMed] [Google Scholar]
- Wistow G, Turnell B, Summers L, Slingsby C, Moss D, Miller L, Lindley P, Blundell T. X-ray analysis of the eye lens protein gamma-II crystallin at 1.9 A resolution. J Mol Biol. 1983;170:175–202. doi: 10.1016/s0022-2836(83)80232-0. [DOI] [PubMed] [Google Scholar]
- Wolf L, Yang Y, Wawrousek E, Cvekl A. Transcriptional regulation of mouse alpha A-crystallin gene in a 148kb Cryaa BAC and its derivates. BMC Dev Biol. 2008;8:88. doi: 10.1186/1471-213X-8-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong EV, David S, Jacob MH, Jay DG. Inactivation of myelin-associated glycoprotein enhances optic nerve regeneration. J Neurosci. 2003;23:3112–3117. doi: 10.1523/JNEUROSCI.23-08-03112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JP, Schmidt KG, Melena J, Chidlow G, Allmeier H, Osborne NN. The beta-adrenoceptor antagonists metipranlol and timolol are retinal neuroprotectants: comparison with betaxolol. Exp Eye Res. 2003;76:505–516. doi: 10.1016/s0014-4835(02)00335-4. [DOI] [PubMed] [Google Scholar]
- Wu N, Wang YH, Zhao HS, Liu DN, Ying X, Yin ZQ, Wang Y. alpha-Crystallin downregulates the expression of TNF-alpha and iNOS by activated rat retinal microglia in vitro and in vivo. Ophthalmic Res. 2009;42:21–28. doi: 10.1159/000219681. [DOI] [PubMed] [Google Scholar]
- Wu Z, Wang L, Hou S. Alpha B-crystallin improved survival of retinal ganglion cells in a rat model of acute ocular hypertension. Neural Regen Res. 2012;7:1493–1497. doi: 10.3969/j.issn.1673-5374.2012.19.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Yu J, Chen S, Xu J, Ying X, Ye M, Li Y, Wang Y. α-Crystallin protects RGC survival and inhibits microglial activation after optic nerve crush. Life Sci. 2014;94:17–23. doi: 10.1016/j.lfs.2013.10.034. [DOI] [PubMed] [Google Scholar]
- Wyttenbach A, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:2898–2903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, Kato K, Rubinsztein DC. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet. 2001;10:1829–1845. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
- Yang XM, Baxter GF, Heads RJ, Yellon DM, Downey JM, Cohen MV. Infarct limitation of the second window of protection in a conscious rabbit model. Cardiovasc Res. 1996;31:777–783. doi: 10.1016/0008-6363(96)00026-0. [DOI] [PubMed] [Google Scholar]
- Yasuda M, Tanaka Y, Ryu M, Tsuda S, Nakazawa T. RNA sequence reveals mouse retinal transcriptome changes early after axonal injury. PLoS One. 2014;9:e93258. doi: 10.1371/journal.pone.0093258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yenari MA, Fink SL, Sun GH, Chang LK, Patel MK, Kunis DM, Onley D, Ho DY, Sapolsky RM, Steinberg GK. Gene therapy with HSP72 is neuroprotective in rat models of stroke and epilepsy. Ann Neurol. 1998;44:584–591. doi: 10.1002/ana.410440403. [DOI] [PubMed] [Google Scholar]
- Ying X, Zhang J, Wang Y, Wu N, Wang Y, Yew DT. Alpha-crystallin protected axons from optic nerve degeneration after crushing in rats. J Mol Neurosci. 2008;35:253–258. doi: 10.1007/s12031-007-9010-1. [DOI] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Oshitari T, Negishi H, Dezawa M, Mizota A, Adachi-Usami E. Protection of retinal ganglion cells from ischemia-reperfusion injury by electrically applied Hsp27. Invest Ophthalmol Vis Sci. 2001;42:3283–3286. [PubMed] [Google Scholar]
- Yoshimura N, Kikuchi T, Kuroiwa S, Gaun S. Differential temporal and spatial expression of immediate early genes in retinal neurons after ischemia-reperfusion injury. Invest Ophthalm Vis Sci. 2003;44:2211–2220. doi: 10.1167/iovs.02-0704. [DOI] [PubMed] [Google Scholar]
- Yu MS, Leung SKY, Lai SW, Che CM, Zee SY, So KF, Yuen WH, Chang RC. Neuroprotective effects of anti-aging oriental medicine Lycium barbarum against [beta]-amyloid peptide neurotoxicity. Exp Gerontol. 2005;40:716–727. doi: 10.1016/j.exger.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Yu MS, Lai CS, Ho YS, Zee SY, So KF, Yuen WH, Chang RC. Characterization of the effects of anti-aging medicine Fructus lycii on beta-amyloid peptide neurotoxicity. Int J Mol Med. 2007;20:261–268. [PubMed] [Google Scholar]
- Yücel YH, Zhang Q, Weinreb RN, Kaufman PL, Gupta N. Atrophy of relay neurons in magno- and parvocellular layers in the lateral geniculate nucleus in experimental glaucoma. Invest Ophthalmol Vis Sci. 2001;42:3216–3222. [PubMed] [Google Scholar]
- Yücel Y. Central nervous system changes in glaucoma. J Glaucoma. 2013;22:S24–25. doi: 10.1097/IJG.0b013e3182934a55. [DOI] [PubMed] [Google Scholar]
- Xi JH, Bai F, Andley UP. Reduced survival of lens epithelial cells in the alphaA-crystallin-knockout mouse. J Cell Sci. 2003a;116:1073–1085. doi: 10.1242/jcs.00325. [DOI] [PubMed] [Google Scholar]
- Xi J, Farjo R, Yoshida S, Kern TS, Swaroop A, Andley UP. A comprehensive analysis of the expression of crystallins in mouse retina. Mol Vis. 2003b;9:410–419. [PubMed] [Google Scholar]
- Xi JH, Bai F, McGaha R, Andley UP. Alpha-crystallin expression affects microtubule assembly and prevents their aggregation. FASEB J. 2006;20:846–857. doi: 10.1096/fj.05-5532com. [DOI] [PubMed] [Google Scholar]
- Zhang J, Wu SM, Gross RL. Effects of beta-adrenergic blockers on glutamate-induced calcium signals in adult mouse retinal ganglion cells. Brain Res. 2003;959:111–119. doi: 10.1016/s0006-8993(02)03735-6. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Sobel RA, Cheng D, Steinberg GK, Yenari MA. Mild hypothermia increases Bcl-2 protein expression following global cerebral ischemia. Brain Res Mol Brain Res. 2001;95:75–85. doi: 10.1016/s0169-328x(01)00247-9. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Faden AI, Loane DJ, Lipinski MM, Sabirzhanov B, Stoica BA. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. J Cereb Blood Flow Metab. 2013;33:1897–1908. doi: 10.1038/jcbfm.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Zhu J, Wan X, Zhang T. Gene expression of sHsps, Hsp40 and Hsp60 families in normal and abnormal embryonic development of mouse forelimbs. Toxicol Lett. 2010;193:242–251. doi: 10.1016/j.toxlet.2010.01.016. [DOI] [PubMed] [Google Scholar]
- Zimmerman SB, Minton AP. Macromolecular crowding: biochemical, biophysical and physiological consequences. Annu Rev Biophys Biomol Struct. 1993;22:27–65. doi: 10.1146/annurev.bb.22.060193.000331. [DOI] [PubMed] [Google Scholar]
- Zuo J, Rungger D, Voellmy R. Multiple layers of regulation of human heat shock transcription factor 1. Mol Cell Biol. 1995;15:4319–4330. doi: 10.1128/mcb.15.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]