

Abstract
Background and Purpose
Aristolochic acid (AristA) is found in plants used in traditional medicines to treat pain. We investigated the action of AristA on TREK and TRESK, potassium (K2P) channels, which are potential therapeutic targets in pain. Balkan endemic nephropathy (BEN) is a renal disease associated with AristA consumption. A mutation of TASK‐2 (K2P5.1) channels (T108P) is seen in some patients susceptible to BEN, so we investigated how both this mutation and AristA affected TASK‐2 channels.
Experimental Approach
Currents through wild‐type and mutated human K2P channels expressed in tsA201 cells were measured using whole‐cell patch‐clamp recordings in the presence and absence of AristA.
Key Results
TREK‐1‐ and TREK‐2‐mediated currents were enhanced by AristA (100 μM), whereas TRESK was inhibited. Inhibition of TRESK did not depend on the phosphorylation of key intracellular serines but was completely blocked by mutation of bulky residues in the inner pore (F145A_F352A). The TASK‐2_T108P mutation markedly reduced both current density and ion selectivity. A related mutation (T108C) had similar but less marked effects. External alkalization and application of flufenamic acid enhanced TASK‐2 and TASK‐2_T108C current but did not affect TASK‐2_T108P current. AristA (300 μM) produced a modest enhancement of TASK‐2 current.
Conclusions and Implications
Enhancement of TREK‐1 and TREK‐2 and inhibition of TRESK by AristA may contribute to therapeutically useful effects of this compound in pain. Whilst AristA is unlikely to interact directly with TASK‐2 channels in BEN, loss of functional TASK‐2 channels may indirectly increase susceptibility to AristA toxicity.
Abbreviations
- AristaA
aristolochic acid
- BEN
Balkan endemic nephropathy
- FFA
flufenamic acid
Tables of Links
| TARGETS | |
|---|---|
| TASK‐2 (K2P5.1) channel | TREK‐1 (K2P2.1) channel |
| TASK‐3 (K2P9.1) channel | TREK‐2 (K2P10.1) channel |
| THIK‐1 (K2P13.1) channel | TRESK (K2P18.1) channel |
| TRAAK (K2P4.1) channel | TRPA1 channel |
| LIGANDS |
|---|
| Flufenamic acid (FFA) |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Introduction
Aristolochic acid (AristA) is found in plants of the Aristolochiaceae family, which have been used widely in traditional medicine for thousands of years. These plants are mentioned in early first‐century Roman texts as components of frequently ingested medicines to treat a variety of conditions including asthma, hiccups, spasms, pains and expulsion of afterbirth, and they were described as components of certain Chinese herbal medicines in the fifth century AD (Scarborough, 2011). Although these herbal remedies were used for a variety of conditions, a recurring theme was their use in many cultures for the treatment of pain. For example, Aristolochia clematitis (birthwort) has been used globally in childbirth, chronic pain and arthritis (Debelle et al., 2008), and Asarum caudatum (wild ginger) has been used in Chinese and Native American herbal remedies for migraine, intestinal pain and knee pain (Smith, 1929). The use of extracts of this family of plants in the treatment of pain was particularly widespread in Chinese herbal medicine. Aristolochia debilis has been used for headache, abdominal pain and epigastric pain, Aristolochia manshuriensis, as part of complex prescriptions, as an anti‐inflammatory treatment and Aristolochia fangchi, also in complex prescriptions, as an analgesic (IARC, 2002, 2012).
AristA has, however, been associated with nephropathy leading to end‐stage renal disease and also with urological malignancies. This first came to light when cases of nephritis and kidney failure were seen in a group of women in Belgium who had all taken a particular weight‐loss supplement, which, inadvertently, contained AristA (Vanherweghem et al., 1993; Debelle et al., 2008). A recent pair of studies identified an AristA mutational signature in upper urinary tract cancer patients from Taiwan (Hoang et al., 2013; Poon et al., 2013) and showed that this carcinogenic effect of AristA is amongst the most potent yet described. In animal studies, either brief exposure to high concentrations (e.g. 50 mg·kg−1 for 3 days) or extended exposure to low concentrations (e.g. 0.1 mg·kg−1 for 12 months) causes a variety of malignant tumours (IARC, 2012). As a result, AristA is now banned as a component of herbal medicines in many countries, including the UK and USA (Debelle et al., 2008). Despite this, its use is still widespread (Debelle et al., 2008). For example, fruits from a variety of plants of the Aristolochiaceae family are freely available in Iran and used to treat headache and back pain (Ardalan et al., 2015).
Balkan endemic nephropathy (BEN) is a renal disease restricted to rural areas of the Balkans and characterized by insidious onset, chronic renal failure and strong association with urothelial carcinoma of the upper urinary tract (Grollman et al., 2007). It is widely believed that BEN is caused by low‐level AristA exposure, probably from contamination of wheat flour seeds by A. clematitis (Grollman et al., 2007; De Broe, 2012; Bui‐Klimke and Wu, 2014). Because, like the diseases associated with AristA‐containing herbal medicines, BEN is linked to AristA, this has prompted the use of the term aristolochic acid nephropathy to describe all related renal diseases associated with consumption of this compound (e.g. De Broe, 2012). It is of interest that a number of patients predisposed to BEN have been shown to carry a mutation of the physiological important renal (Sepúlveda et al., 2015) two‐pore‐domain potassium (K2P) channel, TASK‐2 (K2P5.1) (Toncheva et al., 2014).
Despite the profound toxicity issues associated with AristA, we were intrigued by the widespread use of this compound in traditional medicine in the treatment of pain. A role for K2P channels as therapeutic targets for the treatment of pain has been proposed (e.g. Alloui et al., 2006; Mathie, 2010; Devilliers et al., 2013; Mathie and Veale, 2015), in particular the TREK (K2P2.1, K2P4.1, K2P10.1) and TRESK (K2P18.1) families of K2P channels (as reviewed by Mathie and Veale, 2015). The first aim of this study, therefore, was to investigate the action of AristA on these K2P channels to determine whether this may contribute to its apparent therapeutic usefulness in the treatment of pain. The second aim of this study was to describe how the mutation of TASK‐2 channels seen in patients susceptible to BEN influenced the functional properties of TASK‐2 channels and to determine whether AristA altered the activity of this K2P channel and thus whether this may contribute to the putative action of AristA in BEN.
Methods
Group sizes
The exact group size (n) for each experimental group/condition is provided, and n refers to independent values, not replicates. Data subjected to statistical analysis have an n of at least five per group.
Randomization
When comparisons are made between different recording conditions or different, mutated, forms of a channel, recordings were alternated between one condition and the other on a given experimental day.
Blinding
No blinding was undertaken in this study. It is not a usual procedure for this form of study and cannot be applied retrospectively.
Normalization
No normalization of primary data was performed.
Statistical comparison
Group mean values and statistical analysis use independent values. When comparing groups, a level of probability (P < 0.05) was deemed to constitute the threshold for statistical significance.
Cell culture
tsA201 cells (ECACC; Sigma‐Aldrich, Gillingham, Dorset, UK), modified human embryonic kidney 293 cells, were grown in a monolayer tissue culture flask maintained in a growth medium, which was composed of 88% minimum essential media with Earle's salts and 2 mM l‐glutamine, 10% of heat‐inactivated fetal bovine serum, 1% penicillin (10 000 U·mL−1) and streptomycin (10 mg·mL−1) and 1% non‐essential amino acids. The cells were placed in an incubator at 37°C with a humidified atmosphere of 95% oxygen and 5% carbon dioxide. After 2 or 3 days, when the cells were 70 to 90% confluent, they were split and resuspended in a four‐well plate containing 13‐mm‐diameter cover slips (poly‐d‐lysine coated) in 0.5 mL of media, ready to be transfected the next day.
Transfection
For the electrophysiological experiments, the pcDNA3.1 vector was cloned with the gene of interest [hTREK‐1 (or K2P2.1, as designated by Alexander et al., 2015), hTREK‐2 (K2P10.1), hTRAAK (K2P4.1), hTRESK (K2P18.1), hTASK‐2 (K2P5.1) and hTASK‐3 (K2P9.1) wild type (WT) and mutated]. The hTHIK‐1 (K2P13.1) gene was in the vector pcDNA3.1_NEO_N8. These vectors and a similar vector containing GFP were incorporated into the tsA201 cells (0.5 μg per well) using the calcium phosphate method. The cells were incubated for 6–12 h at 37°C in 95% oxygen and 5% carbon dioxide. Cells were then washed using a PBS solution, and new media added to each well. The cells were used for experiments after 24 h.
Mutations
Point mutations were introduced by site‐directed mutagenesis into the K2P channel clones using the Quikchange kit (Stratagene, Amsterdam, the Netherlands). A pair of short (25–35 bases) complementary oligonucleotide primers, incorporating the intended mutation, were synthesized (Eurofins MWG Operon, Ebersberg, Germany). Mutant DNA constructs were sequenced (Eurofins MWG Operon) to confirm the introduction of the correct mutated bases.
Whole‐cell patch‐clamp electrophysiology
Currents were recorded using the whole‐cell patch clamp in a voltage clamp configuration in tsA201 cells transiently transfected with the channel of interest. The cover slip with the cells was placed in a recording chamber filled with an external medium composed of 145 mM NaCl, 2.5 mM KCl, 3 mM MgCl2, 1 mM CaCl2 and 10 mM HEPES (pH to 7.4, using NaOH). In experiments where external [K] was varied, NaCl was replaced with KCl to give the appropriate concentration of K. The internal medium used in the glass pipette comprised 150 mM KCl, 3 mM MgCl2, 5 mM (or 0.1 mM) EGTA and 10 mM HEPES (pH to 7.4, using KOH). Modulatory compounds were applied by bath perfusion at a rate of 4–5 mL·min−1. Complete exchange of bath solution usually occurred within 100–120 s; as such, any effect of these compounds on current amplitude was only measured once the response had reached steady state.
All the data presented have been collected at room temperature (19–22°C). The transfected cells were detected using a fluorescent microscope with UV light. Cells were voltage clamped using an Axopatch 1D or Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA, USA) and low‐pass filtered at 5 kHz before sampling (2–10 kHz) and online capture.
In order to study the potassium leak current, a ‘step‐ramp’ voltage protocol was used. For the step component of the protocol, cells were hyperpolarized from a holding voltage of −60 to −80 mV for 100 ms then stepped to −40 mV for 500 ms. For the ramp, cells were then stepped to −120 mV for 100 ms, followed by a 500 ms voltage ramp to +20 mV and a step back to −80 mV for another 100 ms, before being returned to the holding voltage of −60 mV. This protocol was composed of sweeps that lasted 1.5 s (including sampling at the holding voltage) and was repeated once every 5 s (see also Veale et al., 2014b).
Data analysis
For analysis of outward current, we measured the current difference between the −80 and −40 mV steps. The current–voltage graphs were obtained from the ramp change in voltage between −120 and +20 mV. The currents obtained with the imposed voltage protocol were recorded and analysed using pclamp 10.2 software and Microsoft Excel. For each cell, the current amplitude (pA) was normalized to the cell capacitance (pF).
Data are expressed as mean ± SEM, and n represents the number of cells used for the experiment. The statistical analyses used either Student's t‐test (both paired and unpaired) or a one‐way ANOVA with post hoc Dunnett's multiple comparisons test, using graphpad prism 6.02 (GraphPad Software, Inc., La Jolla, CA, USA). For the t‐test, the differences between means were considered as significant for P < 0.05. For the Dunnett's test, data were considered significantly different at the <0.05 level (confidence interval > 95% for the difference between the two compared means). Concentration–response data were fitted using the Hill equation. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Chemicals
All fine chemicals were purchased from Sigma‐Aldrich. Flufenamic acid (FFA) stock (10 mM) was made up in ethanol and diluted fresh in external solution before use (pH adjusted to 7.4). Aristolochic acid I (AristA) stock (50 mM) was made up in water and diluted in fresh external before use (pH adjusted to 7.4). Extracts of Aristolochia species comprise, primarily, of a mixture of aristolochic acid I and its demethoxylated derivative, aristolochic acid II. In this study, we used purified aristolochic acid 1 (Figure 1), and the term aristolochic acid (AristA) is used to denote this.
Figure 1.
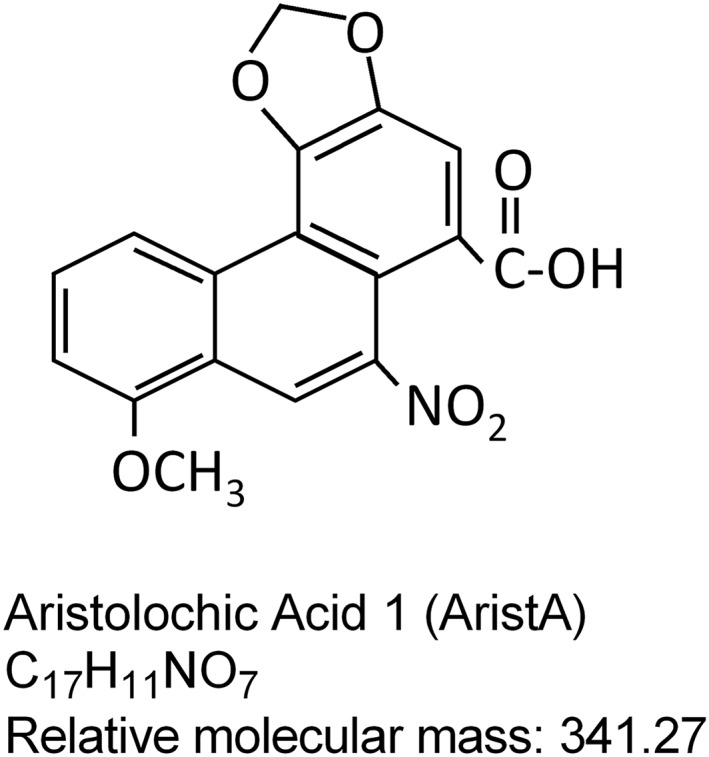
Structure of AristA. Adapted from IARC (2012).
Results
AristA enhances TREK‐1 and TREK‐2 channel currents
Application of AristA (aristolochic acid I, see Section on Methods; 100 μM) resulted in an enhancement of current through TREK‐1 and TREK‐2 channels of 26 ± 6% (mean ± SEM, n = 6) and 44 ± 11% (n = 6), respectively, when current is measured as the difference current between that seen at −40 mV and that at −80 mV (see Section on Methods). Enhancement of both currents was rapid and easily reversible (Figure 2A–D). There was some voltage‐dependence to the enhancement for both currents. This is illustrated for exemplar cells in the range −60 to +20 mV for either current (Figure 2 insets). Enhancement of TREK‐1 was 42 ± 9% (n = 6) at −60 mV, which was significantly larger than the 17 ± 6% (n = 6) enhancement at +20 mV in the same cells. For TREK‐2, enhancement was 50 ± 14% (n = 5) at −60 mV compared with 17 ± 3% (n = 5) at +20 mV in the same cells. Thus, enhancement was greater near the reversal potential of the currents and therefore near the resting membrane potential of cells in which they are expressed. In some recordings for both TREK‐1 and TREK‐2 (Figure 2A and C), there does appear to be a biphasic response to AristA (100 μM) with an ‘over‐recovery’ on washout leading to a sustained reduced current amplitude compared with that measured before application of the drug. However, a lower concentration of the compound (1 μM) had no detectable effect on either TREK‐1 or TREK‐2.
Figure 2.
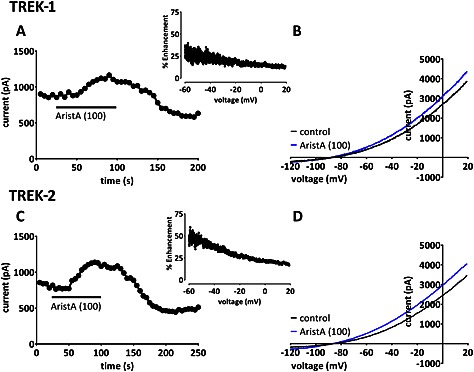
Aristolochic acid (AristA) enhances TREK‐1 and TREK‐2 currents, with apparent voltage‐dependence. (A) Time course plot showing an enhancement of human TREK‐1 current transiently expressed in tsA201 cells by AristA (100 μM). Each point is a 5 ms average of the difference current between that at −40 mV and that at −80 mV (see Methods for detailed description of voltage ramp protocol). (B) TREK‐1 currents evoked by ramp changes in voltage from −120 to +20 mV in control conditions and in the presence of 100 μM AristA. Inset shows percentage enhancement of TREK‐1 current by 100 μM AristA plotted as a function of voltage. (C) Time course plot showing an enhancement of human TREK‐2 current by AristA (100 μM). Inset shows percentage enhancement of TREK‐2 current by 100 μM AristA plotted as a function of voltage. (D) TREK‐2 currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA.
By contrast, AristA (100 μM) had little effect on currents mediated through TRAAK channels (Figure 3A and B) with an enhancement of 8 ± 5% (n = 5), or on the short form of TREK‐1 channels (TREK‐1_ΔN, Figure 3C and D), formed by alternative translation initiation (Thomas et al., 2008; Veale et al., 2010; 2014a), with an enhancement of −4 ± 5% (n = 5). This was despite the fact that both of these channels could be substantially enhanced by flufenamic acid (Veale et al., 2014a) in recordings from the same cells (Figure 3A and C).
Figure 3.
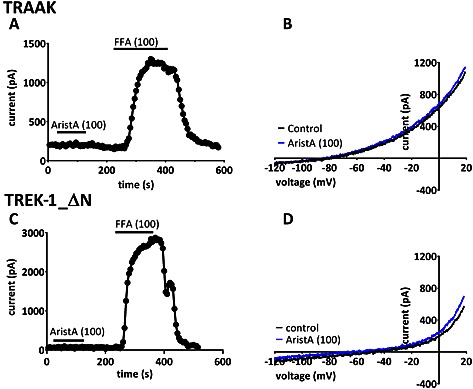
Aristolochic acid (AristA) has no effect on the closely related two‐pore‐domain potassium channel, TRAAK and the N‐terminally truncated TREK‐1 isoform (TREK‐1_ΔN). (A) Time course plot showing lack of effect of AristA (100 μM) on human TRAAK isoform 2 (the sequence of this isoform differs from the canonical sequence by the addition of 26 amino acids, preceding the start codon of isoform 1). (B) TRAAK currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA. (C) Time course plot showing lack of effect of AristA (100 μM) on the alternative translational initiation isoform of TREK‐1 (TREK‐1_ΔN), where the first 41 amino acids of wild‐type TREK‐1 are missing. (D) TREK‐1_ΔN currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA.
TREK channel activity is subject to convergent, polymodal regulation by a variety of factors including temperature, pressure and the phosphorylation state of the channel, and this may influence the response of the channels to other regulatory compounds (Bagriantsev et al., 2012). Recent experiments by Bagriantsev et al. (2012) showed that a triple glycine (3G) mutant, which decouples the intracellular C terminal tail of the channels from the pore‐forming core, rendered the channels insensitive to polymodal regulation by the aforementioned factors. Bagriantsev et al. (2013) found that the TREK channel activator, ML67‐33, still activated these mutated channels. We have made the corresponding 3G mutant of TREK‐2 (TREK‐2_I318G_G319_D320G). These channels were still enhanced by 100 μM AristA (26 ± 5%, n = 5), and this enhancement was not significantly different to that seen for WT TREK‐2 channels (44 ± 11%, n = 6). This suggests that enhancement of TREK‐2 channels by AristA occurs independently from polymodal regulation through the intracellular C terminal tail of the channel.
AristA inhibits TRESK channel currents
In contrast to the enhancement seen at high concentrations for TREK‐1 and TREK‐2 channels, we observed that AristA was an inhibitor of TRESK channels (Figure 4A and B). Inhibition had a fast onset but was only slowly reversible, particularly at higher concentrations such as 100 μM (Figure 4A), with a calculated 50% effective concentration of 13 ± 2 μM for AristA on TRESK and a Hill slope of 0.56 ± 0.08 (Figure 4C). As for enhancement of TREK channels, the effect of AristA was voltage‐dependent. This is illustrated for an exemplar cell in Figure 4D. Inhibition of TRESK by 100 μM AristA was 96 ± 6% (n = 6) at −60 mV, which was significantly larger than the 56 ± 5% (n = 6) inhibition seen at +20 mV in the same cells. At +20 mV, AristA had a calculated 50% effective concentration of 86 ± 3 μM with a Hill slope of 0.34 ± 0.11. Thus, as with enhancement of TREK‐1 and TREK‐2 channels, inhibition of TRESK was greater around the reversal potential of the currents and therefore around the resting membrane potential of cells in which they are expressed.
Figure 4.
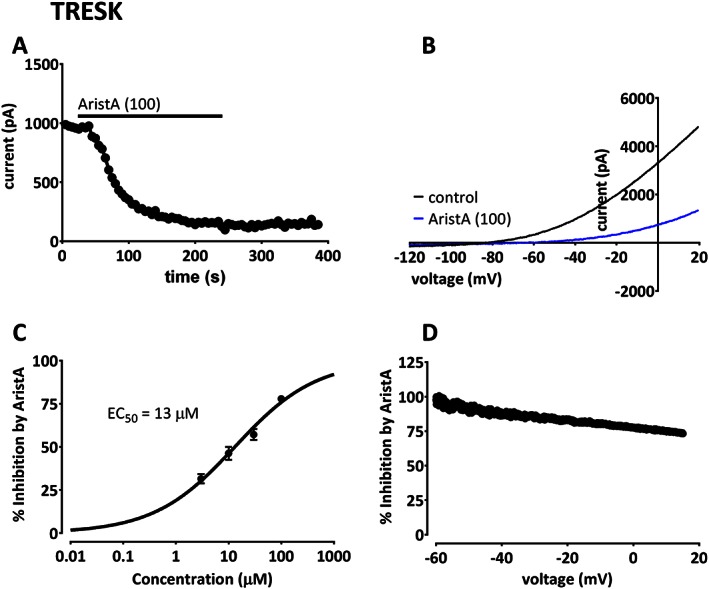
Aristolochic acid (AristA) inhibits TRESK‐mediated currents with apparent voltage‐dependence. (A) Time course plot showing an inhibition of human TRESK current by AristA (100 μM). (B) TRESK currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA. (C) Concentration–response curve for AristA inhibition of TRESK current. (D) Percentage inhibition of TRESK current by 100 μM AristA plotted as a function of voltage.
AristA and TRESK channel mutants
TRESK channels expressed in oocytes have been shown to be directly regulated by increases in intracellular calcium through activation of the calcium/calmodulin‐dependent phosphatase calcineurin (e.g. Czirják and Enyedi, 2010). We, therefore, carried out a number of experiments to investigate the potential role of AristA in calcium‐dependent processes. In the first set of experiments, we altered the internal calcium buffer in the recording solution between 0.1 mM (‘low’ EGTA) and 5 mM (‘high’ EGTA). This had no significant effect on inhibition by AristA (100 μM) with 73 ± 4% (n = 6) inhibition in low EGTA and 79 ± 2% (n = 6) inhibition in high EGTA. Thus, inhibition was unaffected by the degree of intracellular calcium buffering.
For mouse TRESK channels, Czirják and Enyedi (2010) have shown that mutation of key serine residues, located in the large intracellular loop between transmembrane domains M2 and M3, can abolish the effects of phosphatase action. We thus created two mutant human TRESK channels, S252E_S264E to mimic the phosphorylated form of the channel and S252A_S264A to mimic the dephosphorylated form of the channel.
For S252A_S264A channels, current density was significantly larger than WT channels (48 ± 3 pA/pF, n = 31, for the mutated channels compared with 27 ± 2 pA/pF, n = 28, for WT TRESK channels); however, AristA (100 μM) still inhibited channel current by 80 ± 2% (n = 6, Figure 5A and B). For S252E_S264E channels, current density was not significantly different to WT channels (33 ± 3 pA/pF, n = 39). AristA (100 μM) was, again, able to inhibit current through these channels by 73 ± 5% (n = 5) (Figure 5C and D). The inhibition of either phosphorylation mutant by AristA was not significantly different to that of WT TRESK.
Figure 5.
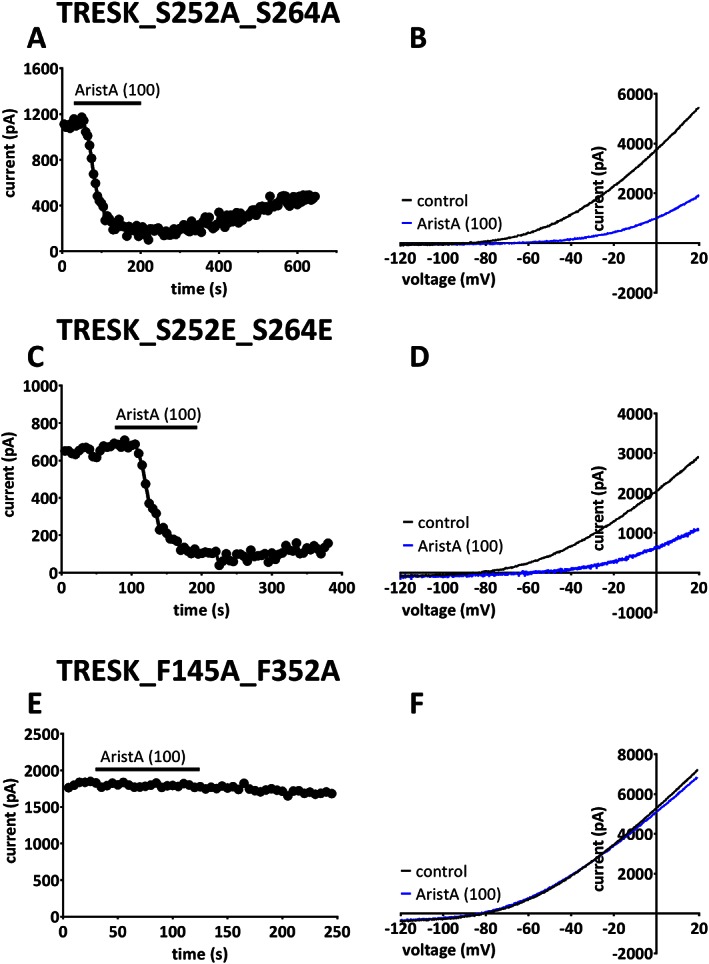
Aristolochic acid (AristA) inhibits serine phosphorylation mutants of TRESK, but its effect is significantly attenuated by the phenylalanine mutants in the inner pore. (A) Time course plot showing an inhibition of the phosphorylation mutant TRESK_S252A_S264A by AristA (100 μM). (B) TRESK_S252A_S264A currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA. (C) Time course plot showing an inhibition of the phosphorylation mutant TRESK_S252E_S264E by AristA (100 μM). (D) TRESK_S252E_S264E currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA. (E) Time course plot showing lack of effect on mutant TRESK_F145A_F352A by AristA (100 μM). (F) TRESK_F145A_F352A currents evoked by ramp changes in voltage in control conditions and in the presence of 100 μM AristA.
Recently, mutation of two amino acids in the M2 and M4 inner pore regions of mouse TRESK channels (equivalent to F145A_F352A in human TRESK) has been shown to occlude the action of a number of different blocking drugs of the channel, and it was proposed that this position forms a binding site for blockers targeting TRESK channels (Kim et al., 2013; Bruner et al., 2014). We investigated the effect of this double mutation on the action of AristA on TRESK channels. We found that AristA (100 μM) no longer inhibited current through these mutated channels (0 ± 5%, n = 7, Figure 5E and F).
TASK‐2, AristA and BEN
AristA has been implicated in BEN (see Introduction). A recent paper has suggested that a number of patients suffering from BEN have a mutation in the coding region of the K2P channel, TASK‐2, namely T108P, suggesting an important role of TASK‐2 channels in BEN predisposition (Toncheva et al., 2014). Given the potential importance of both AristA and TASK‐2 channels in BEN, we were interested, therefore, to determine whether AristA interacted with either (or both) of the WT or the T108P mutated form of TASK‐2. Initially, however, we characterized the properties of these T108P mutated TASK‐2 channels.
Figure 6A–C shows currents through WT TASK‐2 channels, TASK‐2_T108P channels and a more conservatively mutated TASK‐2 channel at the same residue, TASK‐2_T108C, in a variety of external K+ concentrations. In contrast to WT and TASK‐2_T108C channels, there is very little current through TASK‐2_T108P mutated channels regardless of the external K+ concentration. For example, in 2.5 mM external K+, current density for WT TASK‐2 was 52 ± 4 pA/pF (n = 27); for TASK‐2_T108C, it was 9 ± 1 pA/pF (n = 15), and for TASK‐2_T108P, it was just 2 ± 1 pA/pF (n = 26). Both mutations had significantly smaller current density than WT TASK‐2.
Figure 6.
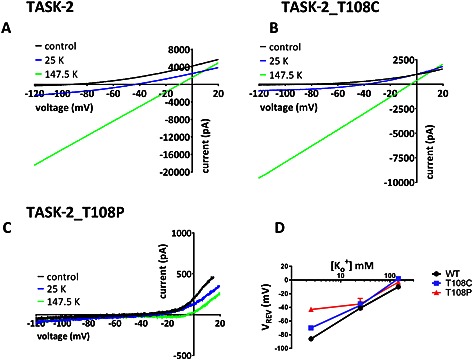
Characteristics of currents through WT TASK‐2, TASK‐2_T108C and TASK‐2_T108P channels in varying concentrations of extracellular potassium (K+). (A) Currents recorded through wild‐type TASK‐2 channels in 2.5 mM K+, 25 mM K+ and 147.5 mM K+. Currents evoked by ramp changes in voltage from −120 to +20 mV, as detailed in Methods. (B) Currents recorded through TASK‐2_T108C channels in 2.5 mM K+, 25 mM K+ and 147.5 mM K+. (C) Currents recorded through TASK‐2_T108P channels in 2.5 mM K+, 25 mM K+ and 147.5 mM K+. (D) Plot of reversal potential versus external K+ concentration for WT TASK‐2, TASK‐2_T108C and TASK‐2_T108P.
Furthermore, unlike WT TASK‐2 channels, the reversal potential of TASK‐2_T108P channels measured in different external [K+] are not consistent with the presence of a K+‐selective current (Figure 6D). For example, in 2.5 mM external K+, reversal of current for WT TASK‐2 was −86 ± 1 mV (n = 27), but for TASK‐2_T108P, this was −43 ± 3 mV (n = 26). It is of interest that the TASK‐2_T108C channel also showed a reduced K+ selectivity with a reversal potential in 2.5 mM external K+ of −70 ± 3 mV (n = 15).
Activators of TASK‐2 channels
We next considered whether activators of TASK‐2 channels might induce current through T108P mutated channels, as has been seen previously for other non‐functional or reduced‐function, mutated K2P channels (Ma et al., 2013; Veale et al., 2014a, 2014b). Changes in external pH are known to regulate TASK‐2 channels (Reyes et al., 1998). Figure 7A and B illustrates that very similar regulation by external pH for TASK‐2_T108C channels to that known for WT TASK‐2 channels occurs, with clear enhancement of current at pH 8.4 and inhibition at pH 6.4 compared with pH 7.4. We found a 120 ± 17% (n = 9) enhancement at pH 8.4 for WT TASK‐2 and a 143 ± 22% (n = 9) enhancement for TASK‐2_T108C channels. The reversal potential at pH 8.4 (in 2.5 mM external K+) was −91 ± 3 mV (n = 5) for WT TASK‐2 and −76 ± 3 mV (n = 7) for TASK‐2_T108C channels. However, in contrast, pH 8.4 had no effect on TASK‐2_T108P channels (Figure 7C and D) with 10 ± 12% (n = 7) enhancement of current and a reversal potential of −52 ± 5 mV (n = 7) at pH 8.4.
Figure 7.
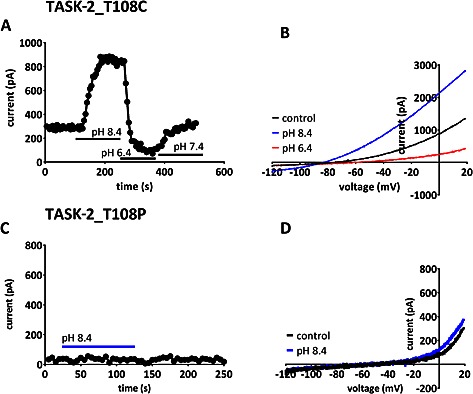
Effect of changes in extracellular pH on mutant TASK‐2 channels. (A) Representative time course for enhancement and inhibition of outward current by extracellular alkalization and acidification, respectively, through TASK‐2_T108C channels. (B) TASK‐2_T108C currents evoked by ramp changes in voltage in control solution (pH 7.4), pH 8.4 and pH 6.4. (C) Representative time course for changes of outward current by extracellular alkalization through TASK‐2_T108P channels. (D) TASK‐2_T108P currents evoked by ramp changes in voltage in control solution (pH 7.4) and in alkaline solution (pH 8.4).
FFA (100 μM) is a known enhancer of several K2P channels (Veale et al., 2014a, 2014b; see also Figure 2). We showed that this compound is also able to enhance current through both WT TASK‐2 channels (Figure 8A and B) and TASK‐2_T108C channels (Figure 8C and D). We found an 80 ± 31% (n = 7) enhancement for WT TASK‐2 by FFA and a 97 ± 25% (n = 6) enhancement for TASK‐2_T108C channels. The reversal potential in FFA (in 2.5 mM external K) was −90 ± 3 mV (n = 7) for WT TASK‐2 and −72 ± 7 mV (n = 6) for TASK‐2_T108C channels. However, FFA (100 μM) produced no enhancement of TASK‐2_T108P channels (Figure 8E and F) with 15 ± 14% (n = 9) inhibition of current and a reversal potential of −44 ± 4 mV (n = 9) in FFA.
Figure 8.
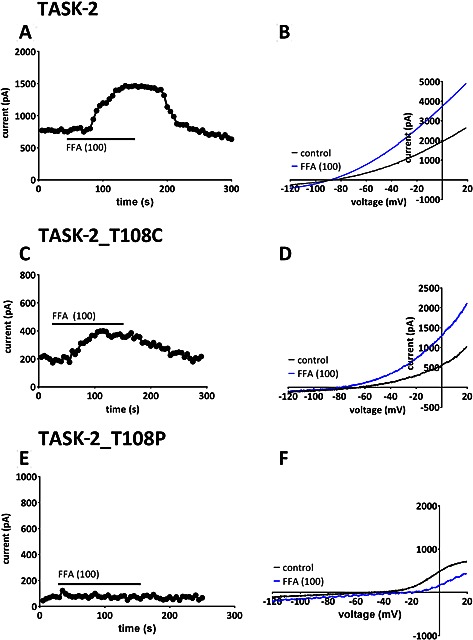
Regulation of WT TASK‐2, TASK‐2_T108C and TASK‐2_T108P mutant channels by flufenamic acid (FFA). (A) Representative time course for enhancement of outward current by FFA (100 μM) for current through WT TASK‐2 channels. (B) Current–voltage relationships for WT TASK‐2 channels in the absence and presence of FFA (100 μM). (C) Representative time course for enhancement of outward current by FFA (100 μM) for current through TASK‐2_T108C mutant channels. (D) Current–voltage relationships for TASK‐2_T108C mutant channels in the absence and presence of FFA (100 μM). (E) Representative time course demonstrating lack of effect on the outward current by FFA (100 μM) on current through TASK‐2_T108P mutant channels. (F) Current–voltage relationships for TASK‐2_T108P mutant channels in the absence and presence of FFA (100 μM).
AristA and TASK‐2 channels
At 100 μM, AristA has no effect on WT TASK‐2, TASK‐2_T108C or TASK‐2_T108P channels. However, at a concentration of 300 μM, AristA was able to induce a very modest enhancement of WT TASK‐2 channels (Figure 9A and B, 21 ± 13%, n = 5). At this high concentration, AristA also produced a very small enhancement of TASK‐2_T108P channels (Figure 9C and D, 12 ± 6%, n = 5) although current density of these channels remained unaltered compared with the absence of AristA at 2 ± 1 pA/pF (n = 5). Furthermore, the reversal potential of these currents through TASK‐2_T108P in the presence of AristA in 2.5 mM external [K+] was −40 ± 4 mV (n = 5), still inconsistent with the presence of a K+ selective current.
Figure 9.
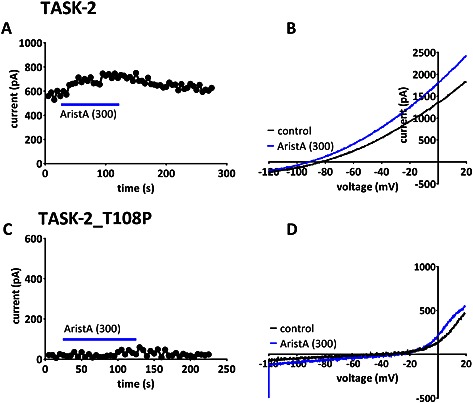
Regulation of WT TASK‐2 and TASK‐2_T108P mutant channels by aristolochic acid (AristA). (A) Representative time course for enhancement of outward current by AA (300 μM) for current through WT TASK‐2 channels. (B) Current–voltage relationships for WT TASK‐2 channels in the absence and presence of AristA (300 μM). (C) Representative time course for the outward current of TASK‐2_T108P channels in the presence and absence of AristA (300 μM). (D) Current–voltage relationships for TASK‐2_T108P channels in the absence and presence of AristA (300 μM).
Discussion
In this study, we have shown that AristA has a range of actions on K2P channels. This is summarized for all K2P channels tested for a single concentration of AristA (100 μM) in Figure 10, where the percentage change in current in the presence of AristA compared with that in the absence of the compound is measured. The TREK family members, TREK‐1 and TREK‐2, are the only K2P channels to show substantive enhancement by AristA, whereas TRESK channels, which are the most distant from TREK channels in terms of sequence homology (e.g. Enyedi and Czirják, 2010; Feliciangeli et al., 2015), are the only channels that are inhibited by AristA. There is no substantive effect of AristA, at this concentration, on TRAAK, TASK‐3, THIK‐1 or TASK‐2 channels.
Figure 10.
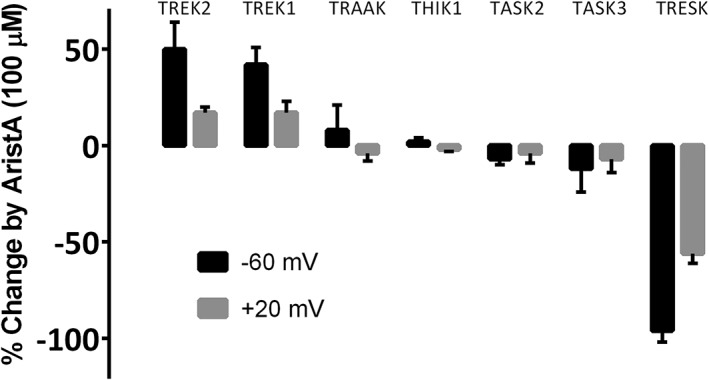
Effect of aristolochic acid (AristA) at 100 μM on representative human two‐pore‐domain potassium (K2P) channels. The effet on each channel is shown at two different voltages (−60 and +20 mV). Representative time courses for THIK‐1 and TASK‐3 channels are given in the Supporting Information.
Mechanism of action of AristA
AristA is a non‐selective agent that has been suggested to regulate the activity of a number of different ion channels. For example, TRPA1−/− flies have been shown to have a reduced avoidance to AristA, and AristA induces action potential firing in these flies (Kim et al., 2010). However, this effect of AristA is not linked to direct effect on TRPA1 channels (Kim et al., 2010). AristA has also been demonstrated to influence the activity of certain K+ channels (Lopes et al., 1998; Roch et al., 2006), but again, these are supposed to be indirect actions. In these studies, AristA was proposed to work through block of the enzyme phospholipase A2 (Soderquist et al., 2014). Phospholipase A2 is a calcium‐dependent enzyme that generates arachidonic acid. It is unlikely that block of phospholipase A2 underlies the substantive effects on TREK and TRESK channels seen here, because arachidonic acid enhances TREK (and TRAAK) channels (Fink et al., 1996; Enyedi and Czirják, 2010) and blocks TRESK channels (Sano et al., 2003; Kang et al., 2004). Thus, AristA should produce the opposite effects to those observed here if it were working through block of phospholipase A2, namely, an inhibition of TREK (and TRAAK) channels and an enhancement of TRESK channels. Furthermore, we have shown that inhibition of TRESK does not depend on the intracellular calcium concentration determined by varying the concentration of intracellular calcium buffer, EGTA.
Inhibition of TRESK channels by AristA also does not work through calcium‐mediated stimulation of phosphatase activity, nor does it depend on the phosphorylation state of the channel on key serine residues in the intracellular loop between M2 and M3, known to be regulated by phosphatase activity (Czirják et al., 2004; Czirják and Enyedi, 2006, 2010; Enyedi and Czirják, 2015), because it still caused substantive inhibition of both the permanently ‘phosphorylated’ form of the channel (S252E_S264E) and the permanently ‘dephosphorylated’ form of the channel (S252A_S264A). This can be contrasted with the block of TRESK channels by benzocaine, which is dependent on the phosphorylation state of these serine residues on the channel (Czirják and Enyedi, 2006).
Mutation of two bulky amino acids in the M2 and M4 inner pore regions of mTRESK channels (equivalent to F145A_F352A in hTRESK) has been shown to occlude the action of a number of different blocking drugs of the channel, and it was proposed that this position forms a binding site for blockers targeting TRESK channels (Kim et al., 2013; Bruner et al., 2014). These mutations also occlude the action of AristA and increase TRESK current density. Inhibition by AristA is voltage‐dependent, consistent with the idea that these amino acids, in the inner pore, may contribute to the binding site for AristA. However, we cannot exclude the possibility that these amino acids are important in regulating gating of the channel rather than forming a binding site, per se.
Enhancement of TREK‐1 and TREK‐2 channels in the treatment of pain
TREK‐1−/− mice have been found to be more sensitive than WT mice to painful heat sensations (Alloui et al., 2006). Furthermore, more recent work has shown that morphine, acting through μ opioid receptors, enhances TREK‐1 current directly. In the same study, TREK‐1−/− animals showed significantly less morphine‐induced analgesia than WT animals (Devilliers et al., 2013). In both small‐ and medium‐sized dorsal root ganglion neurons, single‐channel and whole‐cell patch recordings suggested that TREK‐2 channels were most likely to underlie a substantial portion of background current present in these cells (Kang and Kim, 2006; Marsh et al., 2012). More recently, TREK‐2 channels have been shown to be selectively expressed in IB4 binding C nociceptors (Acosta et al., 2014) and contribute to the resting membrane potential of these neurons. As such, it was proposed that TREK‐2 expression might act to limit pathological pain (Acosta et al., 2014). Enhancement of both TREK‐1 and TREK‐2 channel activity by AristA would contribute to a therapeutically useful effect of this compound in pain and may help to explain the persistent use of plant extracts containing this compound in herbal remedies for pain (see Introduction; IARC, 2002, 2012).
Inhibition of TRESK channels and pain
The potential importance of TRESK channels in pain associated with migraine was highlighted by the observation that familial migraine with aura is associated with a dominant‐negative mutation in TRESK channels (Lafrenière et al., 2010; Enyedi and Czirják, 2015). This suggests that decreased TRESK channel activity might exacerbate migraine pain and that activators of TRESK channels might provide a useful therapeutic approach in pain, particularly given their rather restricted expression in certain central neurons, spinal cord and nociceptive sensory neurons (Kang and Kim, 2006; Dobler et al., 2007; Lafrenière et al., 2010; Marsh et al., 2012).
Hydroxy‐α‐sanshool, a primary active ingredient of Szechuan peppers, has been shown to block TRESK channels, and this action on TRESK channels has been proposed to underlie the distinctive numbing effect induced by this natural, widely used analgesic (Bautista et al., 2008). This is a rather paradoxical observation given that TRESK channel activity is inhibited rather than enhanced by hydroxy‐α‐sanshool. However, it is possible that the tingling paraesthesia (Lennertz et al., 2010) induced by hydroxy‐α‐sanshool eventually leads to desensitization of the excited neurons and a numbing of the sensation of pain in a manner analogous to that seen for the nociceptive agent, capsaicin, albeit through a different molecular mechanism (Mathie, 2010; Mathie and Veale, 2015). Because AristA is a potent inhibitor of TRESK channels, it might be predicted to act in a similar manner to hydroxy‐α‐sanshool to produce analgesia.
The voltage dependence of the effect of AristA on TREK‐1, TREK‐2 and TRESK channels shows that the effect of the compound is most prominent around the resting membrane potential of cells. This suggests that, in sensory neurons, AristA is most effective at membrane potentials that regulate the excitability and firing frequency of these neurons either dampening (for TREK‐1 and TREK‐2) or increasing (for TRESK) excitability. However, given that different K2P channels contribute differently to the resting membrane potential in different neuronal subpopulations, this is likely to be a complex, non‐linear mechanism.
TASK‐2 channels and BEN
The mutation T108P in TASK‐2 channels is found in a number of patients diagnosed with BEN. This suggests that this mutation predisposes these patients to the disease because the proportion of BEN patients with this mutation is much larger than that seen in the general population (Toncheva et al., 2014). We have found that this mutation markedly reduces TASK‐2 channel current density and alters the ion selectivity of the current, so that it is no longer potassium selective. Because we do not have a functional, tagged form of TASK‐2_T108P at present, we cannot exclude the possibility that this channel gives a small current because it does not express well at the membrane. A more conservative mutation at the same site (T108C) also reduces current density but not to nearly the same degree and slightly alters ion selectivity. This threonine residue is located at the top of the M2 transmembrane domain of TASK‐2, close to the channel selectivity filter, and it is present in 13 out of the 15 mammalian K2P channels (Mathie et al., 2010). In other K2P channels, this threonine residue is known to influence channel gating (Zilberberg et al., 2001; Mathie et al., 2010). External alkalization (to pH 8.4) and application of FFA were shown to enhance current through both WT TASK‐2 channels and TASK‐2_T108C channels to around the same degree. These two treatments, however, caused no enhancement of TASK‐2_T108P channels and did not influence the ion selectivity of this mutated channel.
TASK‐2 has an important role in renal function (Cid et al., 2013). Indeed, it is highly expressed in renal tissue and was originally isolated and cloned from human kidney (Reyes et al., 1998). It is highly expressed in the basolateral membrane of proximal tubule cells (Warth et al., 2004). TASK‐2−/− mice have been shown to have a reduced capacity for HCO3 − transport, which leads to proximal renal tubular acidosis (Warth et al., 2004; Sepúlveda et al., 2015). Poor or non‐functioning TASK‐2 channels in patients carrying the T108P mutation would be anticipated to produce similar effects in these patients and compromise renal function. Furthermore, because TASK‐2 channels form functional dimers, it is likely that the T108P mutation will act as a dominant negative in these patients.
TASK‐2 channels expressed in the retrotrapezoid nucleus regulate the ventilator response to CO2 (Bayliss et al., 2015), but a parallel pathway mediated through the proton‐activated receptor GPR4 acts as a distinct central mediator of respiratory chemosensitivity (Kumar et al., 2015). In this way, independent molecular pH sensors provide redundancy for this vital physiological function. It is possible, therefore, that similar independent parallel processes may exist to compensate for TASK‐2 in the kidney, if required.
We have found that AristA has little effect on either WT TASK‐2 channels or on TASK‐2_T108P channels except to induce a very modest enhancement of current through these channels at a high concentration (300 μM). This suggests that AristA is unlikely to interact directly with TASK‐2 channels in contributing to its action in BEN. Rather, loss of functional TASK‐2 channels in BEN (such as in patients with the T108P mutation) may indirectly increase susceptibility to AristA toxicity (and thus to BEN) acting through independent molecular pathway(s).
Author contributions
A.M and E.L.V participated in research design, E.L.V conducted experiments, E.L.V and A.M performed data analysis, A.M and E.L.V wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S1 Aristolochic acid (AristA) has no effect on the two pore domain potassium channels, TASK3 and THIK1
Acknowledgements
Supported by the Biotechnology and Biological Sciences Research Council (UK) [BB/J000930/1] and Pfizer.
Veale, E. L. , and Mathie, A. (2016) Aristolochic acid, a plant extract used in the treatment of pain and linked to Balkan endemic nephropathy, is a regulator of K2P channels. British Journal of Pharmacology, 173: 1639–1652. doi: 10.1111/bph.13465.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acosta C, Djouhri L, Watkins R, Berry C, Bromage K, Lawson SN (2014). TREK2 expressed selectively in IB4‐binding C‐fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci 34: 1494–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noël J, Chemin J et al. (2006). TREK1, a K+ channel involved in polymodal pain perception. EMBO J 25: 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardalan MR, Khodaie L, Nasri H, Jouyban A (2015). Herbs and hazards: risk of aristolochic acid nephropathy in Iran. Iran J Kidney Dis 9: 14–17. [PubMed] [Google Scholar]
- Bagriantsev SN, Ang KH, Gallardo‐Godoy A, Clark KA, Arkin MR, Renslo AR et al. (2013). A high‐throughput functional screen identifies small molecule regulators of temperature‐ and mechano‐sensitive K2P channels. ACS Chem Biol 8: 1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev SN, Clark KA, Minor DL Jr (2012). Metabolic and thermal stimuli control K(2P)2.1 (TREK1) through modular sensory and gating domains. EMBO J 31: 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bautista DM, Sigal YM, Milstein AD, Garrison JL, Zorn JA, Tsuruda PR et al. (2008). Pungent agents from Szechuan peppers excite sensory neurons by inhibiting two‐pore potassium channels. Nat Neurosci 11: 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayliss DA, Barhanin J, Gestreau C, Guyenet PG (2015). The role of pH‐sensitive TASK channels in central respiratory chemoreception. Pflugers Arch 467: 917–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner JK, Zou B, Zhang H, Zhang Y, Schmidt K, Li M (2014). Identification of novel small molecule modulators of K2P18.1 two‐pore potassium channel. Eur J Pharmacol 740: 603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui‐Klimke T, Wu F (2014). Evaluating weight of evidence in the mystery of Balkan endemic nephropathy. Risk Anal 34: 1688–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cid LP, Roa‐Rojas HA, Niemeyer MI, González W, Araki M, Araki K et al. (2013). TASK‐2: a K2P K(+) channel with complex regulation and diverse physiological functions. Front Physiol 4: 198. doi: 10.3389/fphys.2013.00198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirják G, Tóth ZE, Enyedi P (2004). The two‐pore domain K+ channel, TRESK, is activated by the cytoplasmic calcium signal through calcineurin. J Biol Chem 279: 18550–18558. [DOI] [PubMed] [Google Scholar]
- Czirják G, Enyedi P (2006). Targeting of calcineurin to an NFAT‐like docking site is required for the calcium‐dependent activation of the background K+ channel, TRESK. J Biol Chem 281: 14677–14682. [DOI] [PubMed] [Google Scholar]
- Czirják G, Enyedi P (2010). TRESK background K(+) channel is inhibited by phosphorylation via two distinct pathways. J Biol Chem 285: 14549–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debelle FD, Vanherweghem JL, Nortier JL (2008). Aristolochic acid nephropathy: a worldwide problem. Kidney Int 74: 158–169. [DOI] [PubMed] [Google Scholar]
- De Broe ME (2012). Chinese herbs nephropathy and Balkan endemic nephropathy: toward a single entity, aristolochic acid nephropathy. Kidney Int 81: 513–515. [DOI] [PubMed] [Google Scholar]
- Devilliers M, Busserolles J, Lolignier S, Deval E, Pereira V, Alloui A et al. (2013). Activation of TREK‐1 by morphine results in analgesia without adverse side effects. Nat Commun 4: 2941. [DOI] [PubMed] [Google Scholar]
- Dobler T, Springauf A, Tovornik S, Weber M, Schmitt A, Sedlmeier R et al. (2007). TRESK two‐pore‐domain K+ channels constitute a significant component of background potassium currents in murine dorsal root ganglion neurones. J Physiol 585: 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi P, Czirják G (2010). Molecular background of leak K+ currents: two‐pore domain potassium channels. Physiol Rev 90: 559–605. [DOI] [PubMed] [Google Scholar]
- Enyedi P, Czirják G (2015). Properties, regulation, pharmacology, and functions of the K₂p channel, TRESK. Pflugers Arch 467: 945–958. [DOI] [PubMed] [Google Scholar]
- Feliciangeli S, Chatelain FC, Bichet D, Lesage F (2015). The family of K2P channels: salient structural and functional properties. J Physiol 593: 2587–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink M, Duprat F, Lesage F, Reyes R, Romey G, Heurteaux C et al. (1996). Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15: 6854–6862. [PMC free article] [PubMed] [Google Scholar]
- Grollman AP, Shibutani S, Moriya M, Miller F, Wu L, Moll U et al. (2007). Aristolochic acid and the etiology of endemic (Balkan) nephropathy. Proc Natl Acad Sci U S A 104: 12129–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang ML, Chen C‐H, Sidorenko VS, He J, Dickman KG, Yun BH et al. (2013). Mutational signature of aristolochic acid exposure as revealed by whole‐exome sequencing. Sci Transl Med 5: 197ra102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IARC (2002). Some traditional herbal medicines, some mycotoxins, naphthalene and styrene. IARC Monogr Eval Carcinog Risks Hum 82: 1–556. [PMC free article] [PubMed] [Google Scholar]
- IARC (2012). Plants containing aristolochic acid. IARC Monogr Eval Carcinog Risks Hum 100a: 347–361. [Google Scholar]
- Kang D, Mariash E, Kim D (2004). Functional expression of TRESK‐2, a new member of the tandem‐pore K+ channel family. J Biol Chem 279: 28063–28070. [DOI] [PubMed] [Google Scholar]
- Kang D, Kim D (2006). TREK‐2 (K2P10.1) and TRESK (K2P18.1) are major background K+ channels in dorsal root ganglion neurons. Am J Physiol Cell Physiol 291: C138–C146. [DOI] [PubMed] [Google Scholar]
- Kim SH, Lee Y, Akitake B, Woodward OM, Guggino WB, Montell C (2010). Drosophila TRPA1 channel mediates chemical avoidance in gustatory receptor neurons. Proc Natl Acad Sci U S A 107: 8440–8445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee Y, Tak HM, Park HJ, Sohn YS, Hwang S et al. (2013). Identification of blocker binding site in mouse TRESK by molecular modeling and mutational studies. Biochim Biophys Acta 1828: 1131–1142. [DOI] [PubMed] [Google Scholar]
- Kumar NN, Velic A, Soliz J, Shi Y, Li K, Wang S et al. (2015). Regulation of breathing by CO₂ requires the proton‐activated receptor GPR4 in retrotrapezoid nucleus neurons. Science 348: 1255–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafrenière RG, Cader MZ, Poulin JF, Andres‐Enguix I, Simoneau M, Gupta N et al. (2010). A dominant‐negative mutation in the TRESK potassium channel is linked to familial migraine with aura. Nat Med 16: 1157–1160. [DOI] [PubMed] [Google Scholar]
- Lennertz RC, Tsunozaki M, Bautista DM, Stucky CL (2010). Physiological basis of tingling paresthesia evoked by hydroxy‐alpha‐sanshool. J Neurosci 30: 4353–4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes CM, Franks NP, Lieb WR (1998). Actions of general anaesthetics and arachidonic pathway inhibitors on K+ currents activated by volatile anaesthetics and FMRFamide in molluscan neurones. Br J Pharmacol 125: 309–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Roman‐Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F et al. (2013). A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 369: 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh B, Acosta C, Djouhri L, Lawson SN (2012). Leak K⁺ channel mRNAs in dorsal root ganglia: relation to inflammation and spontaneous pain behaviour. Mol Cell Neurosci 49: 375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A (2010). Ion channels as novel therapeutic targets in the treatment of pain. J Pharm Pharmacol 62: 1089–1095. [DOI] [PubMed] [Google Scholar]
- Mathie A, Al‐Moubarak E, Veale EL (2010). Gating of two pore domain potassium channels. J Physiol 588: 3149–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Veale EL (2015). Two‐pore domain potassium channels: potential therapeutic targets for the treatment of pain. Pflugers Arch 467: 931–943. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP et al. (2014). The IUPHAR/BPS guide to PHARMACOLOGY: an expert‐driven knowledge base of drug targets and their ligands. Nucleic Acids Res 42: D1098–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon SL, Pang S‐T, McPherson JR, Yu W, Huang KK, Guan P et al. (2013). Genome‐wide mutational signatures of aristolochic acid and its application as a screening tool. Sci Transl Med 5: 197ra101. [DOI] [PubMed] [Google Scholar]
- Reyes R, Duprat F, Lesage F, Fink M, Salinas M, Farman N et al. (1998). Cloning and expression of a novel pH‐sensitive two pore domain K+ channel from human kidney. J Biol Chem 273: 30863–30869. [DOI] [PubMed] [Google Scholar]
- Roch A, Shlyonsky V, Goolaerts A, Mies F, Sariban‐Sohraby S (2006). Halothane directly modifies Na+ and K+ channel activities in cultured human alveolar epithelial cells. Mol Pharmacol 69: 1755–1762. [DOI] [PubMed] [Google Scholar]
- Sano Y, Inamura K, Miyake A, Mochizuki S, Kitada C, Yokoi H et al. (2003). A novel two‐pore domain K+ channel, TRESK, is localized in the spinal cord. J Biol Chem 278: 27406–27412. [DOI] [PubMed] [Google Scholar]
- Scarborough J (2011). Ancient medicinal use of Aristolochia: birthwort's tradition and toxicity. Pharm Hist 53: 3–21. [PubMed] [Google Scholar]
- Sepúlveda FV, Pablo Cid L, Teulon J, Niemeyer MI (2015). Molecular aspects of structure, gating, and physiology of pH‐sensitive background K2P and Kir K+‐transport channels. Physiol Rev 95: 179–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith HI (1929). Materia medica of the Bella Coola and neighboring tribes of British Columbia. Natl Mus Can Bull 56: 47–68. [Google Scholar]
- Soderquist RS, Danilov AV, Eastman A (2014). Gossypol increases expression of the pro‐apoptotic BH3‐only protein NOXA through a novel mechanism involving phospholipase A2, cytoplasmic calcium, and endoplasmic reticulum stress. J Biol Chem 289: 16190–16199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas D, Plant LD, Wilkens CM, McCrossan ZA, Goldstein SA (2008). Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron 58: 859–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toncheva D, Mihailova‐Hristova M, Vazharova R, Staneva R, Karachanak S, Dimitrov P et al. (2014). NGS nominated CELA1, HSPG2, and KCNK5 as candidate genes for predisposition to Balkan endemic nephropathy. Biomed Res Int 2014: 920723. doi: 10.1155/2014/920723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanherweghem JL, Depierreux M, Tielemans C, Abramowicz D, Dratwa M, Jadoul M et al. (1993). Rapidly progressive interstitial renal fibrosis in young women: association with slimming regimen including Chinese herbs. Lancet 341: 387–391. [DOI] [PubMed] [Google Scholar]
- Veale EL, Al‐Moubarak E, Bajaria N, Omoto K, Cao L, Tucker SJ et al. (2014a). Influence of the N terminus on the biophysical properties and pharmacology of TREK1 potassium channels. Mol Pharmacol 85: 671–681. [DOI] [PubMed] [Google Scholar]
- Veale EL, Hassan M, Walsh Y, Al‐Moubarak E, Mathie A (2014b). Recovery of current through mutated TASK3 potassium channels underlying Birk Barel syndrome. Mol Pharmacol 85: 397–407. [DOI] [PubMed] [Google Scholar]
- Veale EL, Rees KA, Mathie A, Trapp S (2010). Dominant negative effects of a non‐conducting TREK1 splice variant expressed in brain. J Biol Chem 285: 29295–29304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warth R, Barrière H, Meneton P, Bloch M, Thomas J, Tauc M et al. (2004). Proximal renal tubular acidosis in TASK2 K+ channel‐deficient mice reveals a mechanism for stabilizing bicarbonate transport. Proc Natl Acad Sci U S A 101: 8215–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberberg N, Ilan N, Goldstein SAN (2001). KCNK0: opening and closing the 2‐P domain potassium leak channel entails “C‐type” gating of the outer pore. Neuron 32: 635–648. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Aristolochic acid (AristA) has no effect on the two pore domain potassium channels, TASK3 and THIK1