

Abstract
Prenatal stress has been widely associated with a number of short- and long-term pathological outcomes. Epigenetic mechanisms are thought to partially mediate these environmental insults into the fetal physiology. One of the main targets of developmental programming is the hypothalamic-pituitary-adrenal (HPA) axis as it is the main regulator of the stress response. Accordingly, an increasing number of researchers have recently focused on the putative association between DNA methylation at the glucocorticoid receptor gene (NR3C1) and prenatal stress, among other types of psychosocial stress. The current study aims to systematically review and meta-analyze the existing evidence linking several forms of prenatal stress with DNA methylation at the region 1F of the NR3C1 gene. The inclusion of relevant articles allowed combining empirical evidence from 977 individuals by meta-analytic techniques, whose methylation assessments showed overlap across 5 consecutive CpG sites (GRCh37/hg19 chr5:142,783,607-142,783,639). From this information, methylation levels at CpG site 36 displayed a significant correlation to prenatal stress (r = 0.14, 95% CI: 0.05–0.23, P = 0.002). This result supports the proposed association between a specific CpG site located at the NR3C1 promoter and prenatal stress. Several confounders, such as gender, methylation at other glucocorticoid-related genes, and adjustment for pharmacological treatments during pregnancy, should be taken into account in further studies.
Keywords: epigenetics, DNA methylation, glucocorticoid receptor, HPA axis, maternal stress, NR3C1 gene
Introduction
The prenatal period constitutes a crucial ontogenic developmental window where environmental influences have a preponderant effect on shaping fetal physiology by means of epigenetic mechanisms.1 One of the most complex organs to rapidly develop is the brain, which presents its fastest rate of expression change during the fetal period.2,3 Furthermore, the intrauterine environment is thought to be essential not only for fetal development itself but also for long-term health and disease, as prenatal environment risk factors, such as fetal nutrition, have been associated with a number of adult conditions, such as cardiovascular disease, which has encouraged the notion of a certain programming.4,5 Although early plasticity allows a proper adaptation to the environment that enables survival until reproductive age, it might be deleterious for both mental and physical long-term health.
Maternal stressors experienced during pregnancy that may influence offspring health include: i) physical stressors, such as malnutrition and toxins6 like alcohol, nicotine, or polychlorinated biphenyls; ii) psychosocial chronic stressors,7 such as suffering from a psychiatric disorder, taking care of terminally ill relatives,8 or being exposed to continuous violence (e.g., domestic violence or living in a war zone) or poverty (e.g., famine9 or low socioeconomic status10); and iii) being exposed to severe incontrollable acute trauma (e.g., natural disaster,11 terrorism,12 or genocide13), which may result in post-traumatic stress disorder (PTSD) development. It is important to note that these prenatal stressors do not present isolated but rather tend to converge. Intriguingly, the experience of chronic stress together with the development of anxious-depressive spectrum disorders have been associated with the onset of glucocorticoid resistance and higher circulating levels of cortisol,14,15 whereas PTSD has been consistently described to involve a higher glucocorticoid sensitivity and decreased cortisol levels.16 Due to this dual nature of cortisol effects after exposure to psychosocial stressors, we will focus on chronic conditions that have been described to increase circulating cortisol levels of pregnant women.
The human fetal hypothalamic-pituitary-adrenal (HPA) axis is developed and functioning at week 22 of pregnancy, although its plasticity is maintained during the first 2 years of life.17,18 Thus, maternal experiences of stress during pregnancy have the potential to permanently alter the physiology of their offspring, especially to program the HPA axis functioning. As the HPA axis regulates a myriad of biological processes, such as metabolism, blood pressure, and the immune response, these alterations can predispose prenatally stressed individuals to suffer metabolic, cardiovascular, and mental disorders in adulthood.19
Typically, after exposure to an acute stressor, the hypothalamus releases corticotropin-releasing hormone (CRH), which promotes the release of adrenocorticotropic hormone (ACTH) from the pituitary gland. Once in the adrenal glands, ACTH triggers the release of cortisol. This system is self-regulated by a negative feedback in which cortisol inhibits CRH and ACTH release from the central nervous system. Nevertheless, and as discussed above, during the exposure to chronic stress, this feedback may be impaired by the development of glucocorticoid resistance mediated by glucocorticoid receptor (GR) desensitization.20 This situation leads to an overactivation of the HPA axis that has been suggested to underpin the development of depressive symptoms at least in a subset of patients showing alterations of the HPA axis functioning.21
Hence, the GR is essential in the modulation and functioning of the HPA axis. The NR3C1 gene, which codes for the GR, has extensively been studied regarding its role in several psychiatric disorders characterized by HPA axis dysfunction and in subjects exposed to several forms of early stress by means of both genetic and epigenetic approaches.22,23
In this regard, several authors have analyzed DNA methylation at a CpG island located at the promoter region of this gene in different paradigms of early stress exposure. First, the discovery of a CpG-specific hypermethylation as found in exon 17 of postnatally neglected rat pups when compared to pups reared by mothers exhibiting high licking, grooming, and arched-back nursing (LG-ABN) behavior, prompted a new research field that has extended to human populations in the last decade.22,24 Special attention has been paid to exon 1F due to its homology with rat exon 17. Nevertheless, there is still no consensus about the effects of early stress on NR3C1 methylation due to the heterogeneity of assessed stress variables, regions analyzed, and subjects included in human studies.22 Although animal studies have largely focused in the study of postnatal stress as a modulator of epigenetic patterns, prenatal stages arise as a key ontogenic window of development where stress sensitization can also occur and become embedded in the epigenome; in fact, there is one study developed in mice that reported an increase in methylation in males exposed to early prenatal stress, which was restricted to an NGF1A binding region within GR promoter exon 17, thus paralleling prior results obtained in postnatal paradigms.25
By reviewing and meta-analyzing all previously published empirical reports in human populations, the current work aims to determine whether there is enough evidence to support a link between maternal chronic psychosocial stress (as experienced during pregnancy) and site-specific NR3C1 promoter methylation (as assessed in their offspring).
Results
Eligibility of studies and specific CpG sites to be meta-analyzed
Twenty-five scientific papers concurrently covering for the 3 topics of interest as described in the methodology section were retrieved by our defined search strategy for further assessment of eligibility. As can be seen in Figure 1, 15 articles were excluded due to the lack of new human data reported (reviews, animal studies, and human reports where there was no prenatal stress assessment); a total of 10 papers analyzed diverse prenatal stressors in relation to NR3C1 promoter methylation. In order to homogenize the final sample, 3 additional papers were excluded from the meta-analysis: one of them since smoking during pregnancy was not considered as a psychosocial stressor,26 and the remaining 2 since, as discussed in the introduction, PTSD status during pregnancy has been described to upregulate cortisol inactivation and decrease cortisol circulating levels, and not the other way around.12,13 The resulting pool consisted of 7 selected papers for the meta-analysis. Five of them analyzed maternal anxious-depressive status during pregnancy.27-31 The remaining 2 papers focused on the effect of maternal exposure to violence (either intimate partner violence or war-related events) during pregnancy.32,33
Figure 1.
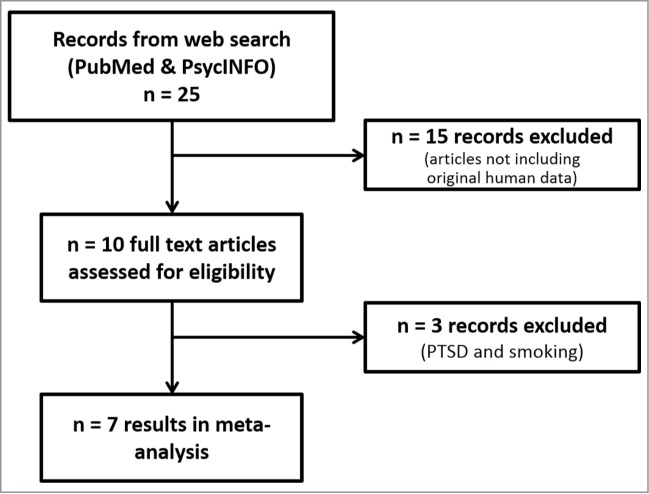
Flowchart of study selection and inclusion of results. Data from 7 papers were included in the meta-analysis.
As depicted in Figure 2, there is an overlap of 5 analyzed CpG sites (35 to 39) at the promoter of the exon 1F among 6 of the 7 papers meta-analyzed herein (see also Figure 3 for specific location in the sequence). Thus, 5 meta-analyses were carried out individually for each CpG site (Fig. 4) since previous literature supports the association between distinct early stressors and specific CpG sites methylation rather than the mean methylation of a region containing several CpG sites.24,34
Figure 2.
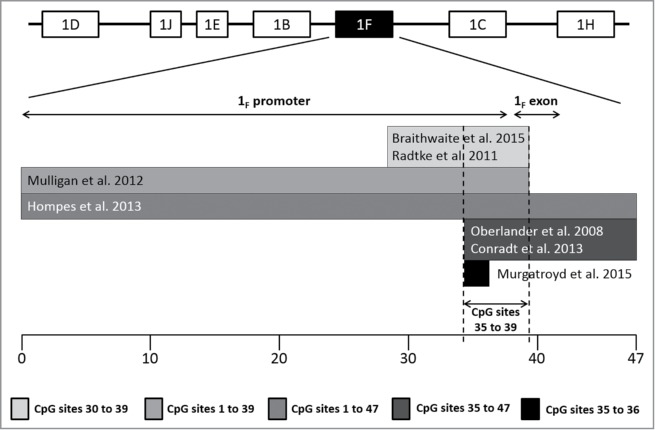
CpG site distribution and overlap among included papers in the meta-analysis. Research groups focused on distinct regions of promoter and exon 1F. Six out of the 7 papers included in the meta-analysis assessed the DNA methylation of a small cluster of CpG sites located immediately preceding exon 1F, from 35 to 39; additionally, Murgatroyd et al. limited their analyses to CpG sites 35 and 36 as they had been already pointed out as informative in the previous papers. The whole sequence of this region is displayed in Figure 3.
Figure 3.

Exon 1F (NR3C1 gene) structure. Exon is underlined. All CpG sites (CG) are numbered, remarked in bold and capital letters. Reviewed CpG sites herein (35 to 39) are highlighted in dark red.
Figure 4.
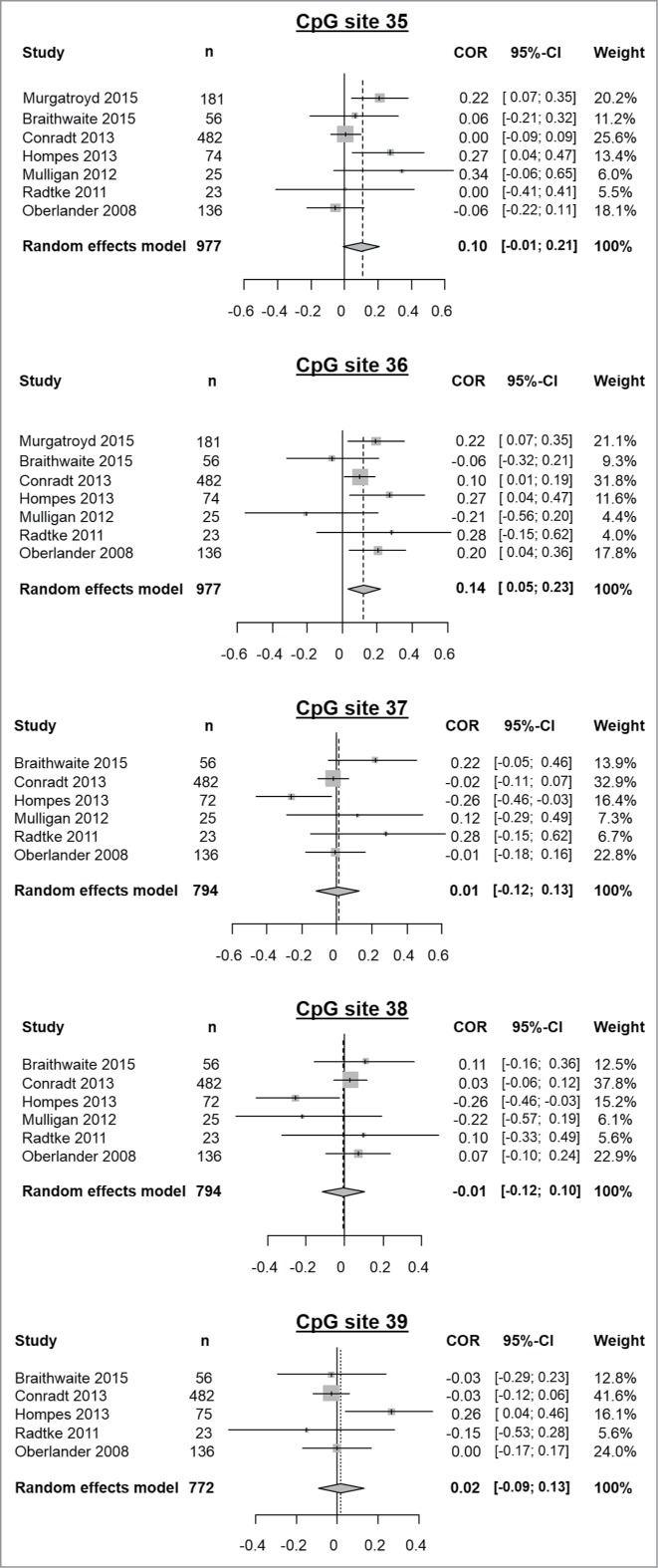
Results of meta-analyses of prenatal stress exposure influence on methylation of the CpG sites 35 to 39. From left to right, displayed variables consist of: study, sample size (n), a graphical representation of the computed correlations, correlation values between methylation and the assessed prenatal stress variable, their corresponding confidence intervals and the weight of each study in the meta-analysis as determined by a random effects model.
Since all of the reviewed articles included several CpG sites in their analyses, which led to a mixture of positive and negative findings, we expected publication bias to be avoided. Indeed, this hypothesis has been statistically assessed by funnel plot asymmetry tests for each individual CpG site included in the meta-analysis, further confirming the lack of publication bias (data not shown). In addition, presence of heterogeneity was statistically tested by means of 4 different estimators as reported below and described in the methodology.
NR3C1 exon 1F CpG site 35 methylation
DNA methylation at CpG site 35 of the NR3C1 gene was examined in 977 subjects across the 7 studies. We detected no significant correlation between offspring methylation and prenatal stress (r = 0.10, 95% CI: −0.01–0.21, pr = 0.08; τ2: 0.01, I2: 54.7%, H2 = 1.49, Q = 13.23, pQ = 0.04).
NR3C1 exon 1F CpG site 36 methylation
DNA methylation at CpG site 36 of the NR3C1 gene was examined in 977 subjects across the 7 studies. We detected a significant correlation between offspring's methylation and prenatal stress (r = 0.14, 95% CI: 0.05–0.23, pr = 0.002; τ2: 0, I2: 34.7%, H2 = 1.24, Q = 9.18, pQ = 0.16) pointing to an increased methylation of CpG site 36 after prenatal stress exposure. Final correlation values obtained by means of both random effects model and fixed effects model are the same (data not shown).
NR3C1 exon 1F CpG site 37 methylation
DNA methylation at CpG site 37 of the NR3C1 gene was examined in 794 subjects across the 6 studies; Murgatroyd et al. did not provide methylation percentages for this CpG site. We detected no significant correlation between offspring methylation and prenatal stress (r = 0.01, 95% CI: −0.12–0.13, pr = 0.91; τ2: 0.01, I2: 47.6%, H2 = 1.38, Q = 9.54, pQ = 0.09).
NR3C1 exon 1F CpG site 38 methylation
DNA methylation at CpG site 38 of the NR3C1 gene was examined in 794 subjects across 6 studies; Murgatroyd et al. did not provide methylation percentages for this CpG site. We detected no significant correlation between offspring methylation and prenatal stress (r = −0.01, 95% CI: −0.12–0.10, pr = 0.91; τ2: 0, I2: 35.7%, H2 = 1.25, Q = 7.78, pQ = 0.17).
NR3C1 exon 1F CpG site 39 methylation
DNA methylation at CpG site 39 of the NR3C1 gene was examined in 769 subjects across 5 studies; Murgatroyd et al. and Mulligan et al. did not provide methylation percentages for this CpG site. We detected no significant correlation between offspring methylation and prenatal stress (r = 0.02, 95% CI: −0.09–0.13, pr = 0.54; τ2: 0, I2: 35.9%, H2 = 1.25, Q = 6.24, pQ = 0.18).
Discussion
By meta-analyzing data available in the literature we found a significant correlation between psychosocial maternal stress and offspring methylation at a specific CpG site located in the exon 1F of the human glucocorticoid receptor gene NR3C1. Interestingly, we report a significant correlation between DNA methylation and presence of prenatal stress only in CpG site 36 but not in its adjacent positions 35, 37, 38, or 39. These results provide further evidence of the previously suggested biological relevance of CpG site-specific methylation when compared with average methylation of a region spanning several CpG sites.22
Interest in the methylation signature of NR3C1 exon 1F followed the publication of the seminal work developed in rats by Weaver and collaborators.24 They described a maternally mediated change in DNA methylation in the rat homolog of human CpG site 37, which is located at a transcription factor (NGFI-A)-binding region, thus suggesting the functional relevance of this modification. Although most of the human studies have focused on this specific CpG site due to its biological relevance, it is interesting to note that, in their animal model, Weaver et al. also found significant methylation differences in the adjacent CpG sites, including those who are homologous to human CpG sites 35 and 36. These unprecedented results in rats support human findings reported herein as other CpG sites located within the promoter of exon 1F could alter its transcription efficacy.
Of note, there was only one human study that paralleled the experimental conditions tested in rats. Murgatroyd and colleagues31 took into account both early postnatal depression and maternal tactile stimulation when analyzing newborn methylation. This design allowed the researchers to describe a mediating effect of both variables in altering DNA methylation at CpG sites 35 and 36 where postnatal depression in newborns non-exposed to prenatal depression increased methylation while maternal stroking decreased it. These results are in line with previous research from this group where maternal stroking has been reported to lessen the influence of prenatal depression upon infant behavioral and physiological outcomes.35
The evidence of a replicated correlation between prenatal stress and NR3C1 methylation is particularly relevant in the context of 3 infant neurobehavioral and neuroendocrine studies developed by Marsit and Lester's research groups, whose findings point to a significant effect of placental NR3C1 methylation in both newborn neurobehavior and cortisol reactivity after a newborn-adjusted stress procedure (the still-face paradigm).36-38 It is worth noting that the statistically significant correlations between NR3C1 methylation and neurobehavioral parameters were restricted to either (i) CpG sites 39 to 47, which are located within the exon 1F and beyond (instead of localizing to a promoter region as CpG sites 35 to 38), or (ii) average methylation across 13 CpG sites (35 – 47) instead of providing CpG specific associations. Anyhow, longitudinal follow-up of these infants will be crucial in order to assess the long-term impact of these methylation patterns and whether they last into adulthood. Interestingly, an independent study measuring HPA axis functioning in adults as associated with NR3C1 methylation also reported a correlation between methylation at 1F CpG sites 41 to 47 and cortisol levels after the Dex/CRH challenge, further supporting the role of epigenetic modifications at this region in neuroendocrine systems.39 Additionally, it would be interesting to assess DNA methylation in prenatally stressed newborns in CpG sites 40 to 47, as it seems to be associated with several neurobehavioral and neuroendocrine outcomes.36-38 Nevertheless, meta-analyzed papers which already assessed methylation in these extended regions did not report any correlation between pregnant anxious-depressive symptomatology and methylation at CpG sites 40–47, thus implying that prenatal stress may not modulate neurobehavior and neuroendocrine response as mediated by NR3C1 methylation.27-29
Prenatal stress-induced methylation changes are thought to partially arise due to increased fetal circulating cortisol levels. Recent evidence points to the essential role of the placental enzyme 11 β-hydroxysteroid dehydrogenase type 2 (11β-HSD2) in regulating the cortisol flux from the mother to the fetus. Maternal cortisol levels increase progressively during pregnancy. Since high concentrations of cortisol are deleterious to fetal development, 11β-HSD2 is required to protect the fetus by oxidizing cortisol into its less active metabolite cortisone.40 Accordingly, evidence supports a critical role of this enzyme in preventing cortisol damaging effects; lower 11β-HSD2 activity has been associated with lower birth weight in both humans and rodents.41,42 Generally, the experience of psychosocial stress entails the overproduction of cortisol. As previously stated, 11β-HSD2 protects the fetus from the increased cortisol levels present in maternal blood; nevertheless, it has been described in rats that, in the presence of chronic maternal stress exposure, 11β-HSD2 activity gets downregulated;43 furthermore, a posterior study determined that the placental effects of this enzyme come from the fetus and not the mother.44 Interestingly, a recent study reported similar effects due to exposure to maternal prenatal anxiety in humans, prenatal trait anxiety scores negatively correlated to placental 11β-HSD2 mRNA expression.45 This decrease on 11β-HSD2 expression is thought to be mediated, at least partially, by epigenetic mechanisms such as DNA methylation.46 This evidence points to a fetal overexposure to cortisol in stressed pregnant mothers, which may induce fetal epigenetic changes in glucocorticoid-related genes as the one reported herein. Thus, further studies analyzing the interaction between HSD11B2 and NR3C1 methylation are needed to disentangle the role of each gene in mediating prenatal experiences into adulthood.
Besides, as pointed out by Braithwaite and colleagues,30 newborn gender may be a key factor in order to assimilate prenatal stress into the epigenome. As they discuss, only male subjects exhibited higher NR3C1 methylation after prenatal exposure to depressive maternal status during pregnancy whereas females tended to be hypomethylated when exposed to prenatal depression. In this sense, the sex-specific programming effects of prenatal stress have been widely discussed in a number of reviews, as they have been repeatedly reported during the last decade in both rodents and humans.25,47-50 Interestingly, the placenta behaves differently depending on the fetal sex,51 which could be one of the mediating mechanisms of the increased male mortality after exposure to stressful experiences during pregnancy.52 This sexual dimorphism is currently under study, as its underlying causal mechanisms remain unknown. This topic is of major relevance in the psychiatric field, as most of the more prevalent mental disorders are sex-biased and this different sensitivity to maternal prenatal stress could be one of the involved mechanisms. From an evolutionary point of view, females have adapted in order to protect their offspring under a threat, which can be promoted by anxious behavior (tend and befriend hypothesis), while males have adapted to abandon their offspring and look for new mates, which may involve aggression (flight or fight hypothesis).53 Nevertheless, these hypotheses cannot be tested in this meta-analysis as Braithwaite and colleagues30 were the only group to study methylation in separate analyses depending on newborn gender. As a consequence, when averaging male and female methylation values, the significance of their findings is lost (see Fig. 4). Thus, gender-specific effects should be further examined in future studies assessing NR3C1 methylation.
Notoriously, methylation measures reported by Oberlander and colleagues27 differ considerably from the ones collected in this meta-analysis. This is due to a new calculation of unadjusted correlation coefficients, provided by Oberlander for proper meta-analysis calculation, instead of analyzing the previously reported adjusted regressions. As a consequence, exposure to serotonin reuptake inhibitors during pregnancy was neglected from the meta-analysis in order to compare data from the different papers meta-analyzed herein. Combining adjusted and unadjusted findings in mixed research synthesis can lead to artificial associations; given the nature of meta-analyzed studies, inclusion of exclusively unadjusted results appeared more appropriate.54 Nevertheless, this divergence is intriguing as it could suggest a protective role for serotonin reuptake inhibitor treatment during pregnancy, which may be minimizing the anxious-depressed-mediated methylation observed in offspring. Therefore, antidepressant treatment should be thoroughly investigated as a potential protective factor in further studies.
In the frame of epigenome-wide association studies (EWAS) examining prenatal stress, it is surprising to find no reference to either exon 1F of the NR3C1 gene or other related genes such as HSD11B2. On the contrary, these studies detected non-replicated significant hits in a number of genes such as MORC1, LTA, SGC5, TNFRSF21, CHRNA2, or COL7A1, among others.55-58 Interestingly, antidepressant treatment during pregnancy seems to be a major influencing factor, which would be in line with the discussion above. In this regard, Non and colleagues reported several changes in methylation only in the non-medicated depressed group when compared to healthy controls.57 On the other hand, Schroeder and colleagues found a significant methylation change at 2 CpG sites only in newborns exposed to antidepressants, while no global effect of maternal depression could be found. One of the limitations of this study is that the comparison group was recruited at a psychiatric center and, therefore, all controls had a lifetime history of at least one psychiatric disorder.58 Of note, 2 EWAS detected associations between NR3C1 CpG-specific methylation and prenatal environment: Non et al.57 found an association between methylation at an exon 1C located CpG site and non-medicated anxious/depressed group; Teh et al.59 found a gene-environment effect explaining a variably methylated CpG site located at an intronic region of NR3C1 gene; one the environmental variables assessed in this study was maternal depression during pregnancy as measured by EDPS.
As briefly stated in the introduction, we excluded 2 papers analyzing NR3C1 methylation in the offspring of mothers suffering from PTSD; their results were not eligible to be meta-analyzed herein due to a number of issues. On the one hand, Perroud et al.13 studied CpG site-specific methylation in the offspring of mothers exposed and non-exposed to the Rwandan genocide. Nevertheless, they analyzed a posterior region of exon 1F that exceeds its coding region, starting in CpG site 38. Although they reported a clear hypermethylation in this region associated with maternal PTSD status, there was no effect in CpG sites 38 and 39. Furthermore, this hypermethylation appeared coupled with lower cortisol levels, which cannot be explained by lower GR expression; the authors discussed a preponderant role of the mineralocorticoid receptor rather than GR in the pathology of PTSD. On the other hand, Yehuda et al.12 assessed the effects of transgenerational PTSD; however, they did not study prenatal stress as their included subjects were born after the World War II and the transversal nature of the study does not allow knowing whether parents were suffering from PTSD specifically before, during, or after pregnancy. In any case, maternal PTSD was associated with lower NR3C1 methylation, thus supporting opposite directions of the effect of maternal depression- and PTSD-derived epigenetic modifications affecting HPA axis functioning. Intriguingly, paternal rather than maternal PTSD status was associated with their offspring's higher methylation thus suggesting that the sex-specific effects discussed above are not limited to the assessed subjects but also affect their progenitors. Finally, both studies analyzed peripheral blood of either children or adult subjects, thus including a potential confounder due to immeasurable effects of life events experienced during later stages of life.
Interestingly, the own nature of the meta-analyzed data points to a major homogenous effect of anxious-depressive spectrum disorders in increasing CpG-specific methylation at the NR3C1 exon 1F promoter. Both war stress and intimate partner violence may be too different from diagnoses of either anxiety or depression and include a number of additional confusing factors into the analysis such as ethnicity (since Mulligan et al. study was performed in the Democratic Republic of Congo and Radtke et al. assessed a very reduced sample with a wide ethnic heterogeneity, further detailed in Table 1) and assessment timing (since Radtke et al. analyzed methylation in children instead of newborns).
Table 1.
Summary of data considered for meta-analysis and potential confounders.
| Authors | Main stress outcome | Timing of the stressor | n | Tissue | Methylation assessment | Ethnicity | Observations |
|---|---|---|---|---|---|---|---|
| Oberlander et al. 200827 | Maternal depression or anxiety during pregnancy, as assessed by HAM-D, HAM-A and EPDS | Psychiatric assessments at 2nd and 3rd trimesters | 136 | Cord blood | Pyrosequencing | Canadian recruited sample | This is the first study to explore NR3C1 methylation in humans in relation to early stress. Loss of significance due to unadjusted nature of the meta-analyzed results. |
| Radtke et al. 201133 | Prenatal exposure to intimate partner violence, as assessed by CAS | During pregnancy (all trimesters) | 23 | Whole blood (at 14.1 ± 0.5 years) | Sodium bisulfite mapping, cloning and Sanger sequencing | Mixed origin of the sample including Turkey, Iraq and Kosovo, among others | This is the only study that studied pre-adolescent and adolescent subjects instead of newborns (or infants). |
| Mulligan et al. 201232 | War stress, as assessed by trauma surveys | During pregnancy (all trimesters) | 25 | Cord blood | Sodium bisulfite mapping, cloning and Sanger sequencing | 100% of the sample was recruited at the Democratic Republic of Congo | Although Mulligan et al. reported an overall significant effect of war stress upon NR3C1 methylation, they found no significant effect of individual CpG sites. |
| Hompes et al. 201329 | Maternal depression or anxiety during pregnancy, as assessed by EPDS and PRAQ | Psychiatric assessments at 1st, 2nd and 3rd trimesters | 74 | Cord blood | MALDI-TOF mass spectrometry | 93% of the sample was Belgian | Conflicting results may be due to the study of methylation in CpG units rather than in individual CpG sites. |
| Conradt et al. 201328 | Maternal depression or anxiety during pregnancy, as assessed retrospectively from clinical records and interviews | Retrospective psychiatric assessment (all trimesters) | 482 | Placenta(fetal origin) | Pyrosequencing | 73% of the sample was Caucasian | Since the included sample in Conradt et al. study is several folds higher than the others, their reported associations drive the meta-analysis results, even in a random effects model as can be seen by the accounted weight. |
| Braithwaite et al. 201530 | Maternal depression during pregnancy, as assessed by EPDS | 2nd or 3rd trimester of pregnancy | 56 | Buccal swabs (collected at 53.6 ± 9.99 days) | Pyrosequencing | 89% of the sample was Caucasian | They successfully replicated Weaver and colleagues' findings in rats in male subjects but not in the females.As DNA methylation was examined in buccal swabs instead of cord or whole blood, replication of results suggests inter-reliability between tissues. |
| Murgatroyd et al. 201531 | Maternal depression during pregnancy, as assessed by EPDS | 2nd trimester of pregnancy (20 weeks) | 181 | Saliva (collected at 14 months) | MALDI-TOF mass spectrometry | 96% of the sample identified themselves as white British | This is the only study to date to examine both prenatal and postnatal depression and their interaction on offspring methylation. They also described a moderating role of maternal stroking in final methylation values.As DNA methylation was examined in saliva instead of cord or whole blood, replication of results suggests inter-reliability between tissues. |
Abbreviations: CAS, composite abuse scale; EPDS, Edinburgh Postnatal Depression Scale; HAM-A, Hamilton Rating Scale for Anxiety; HAM-D, Hamilton Rating Scale for Depression; PRAQ, pregnancy related anxiety questionnaire.
There are some limitations to be noted regarding the reviewed papers herein. First, each paper assessed DNA methylation by means of distinct technologies including pyrosequencing, mass spectrometry, and Sanger sequencing of cloned vectors. This variability might be biasing the meta-analysis results as each technique has its own limitations. As a matter of fact, both Murgatroyd et al.31 and Hompes et al.29 reported methylation values as for pairs of CpG sites due to the reduced resolution of mass spectrometry analyses thus compromising the comparison. Second, none of the studies explored paired GR expression in association with methylation; therefore, the phenotypic effect of CpG site 36 methylation remains unknown. Third, the timing of the psychiatric assessment is fundamental since the developmental effects of a single stressor fluctuate substantially according to different windows of susceptibility.19 Accordingly, the epigenetic modulation caused by anxious-depressive diagnosis during pregnancy seems to impact harder the fetuses when it takes place during the early stages of pregnancy as shown both in animal and human studies.25,27,29 Thus, the restricted correlations reported by Conradt et al.28 could be due to inclusion of women who experienced either depression or anxiety at any time during pregnancy without specifying the trimester. Fourth, given the length of the NR3C1 promoter area and the preliminary results correlating DNA methylation at additional CpG sites to distinct early stressors, other regions besides exon 1F could have been investigated. Fifth, all the methylation analyses were performed in peripheral tissues such as cord blood or saliva (see Table 1 for further details), which calls into question the comparability with brain measures; nevertheless, similar NR3C1 methylation findings have been found in both peripheral and brain tissues in relation with early adversity thus suggesting the tissue unspecificity of the signatures elicited by psychosocial stress in this gene.22 Furthermore, NR3C1 peripheral methylation and its correlated GR differential expression may be informative on their own as a substantial component of cortisol's actions occurs at a peripheral level. Finally, the overall methylation differences reported by all the reviewed papers, when specified, are modest (below 5 percent) and significance should be interpreted with caution.
There are also several limitations to be noted in this meta-analysis. To begin with, prenatal anxious-depressive disorder and exposure to violence may exert differential effects upon fetal methylation; we have tried to avoid this confounder by excluding certain prenatal stressors such as smoking or PTSD status as their long-term effects on offspring's health are unclear. Furthermore, the limited number of papers on this topic restricts the statistical power of this meta-analysis; nevertheless, the relatively large final sample size (977 subjects), together with the finding of replicated and significant results, strengthens the notion of epigenetically-driven changes modulated by very early environment. Finally, these results should be interpreted with caution since the correlation value is low (0.14). Further studies assessing newborn methylation with a CpG-specific level resolution controlling for prenatal maternal psychiatric diagnosis at specific time points of pregnancy are needed to replicate this correlation, whereas longitudinal studies are required to disentangle the long-term effects of this methylation in adult health.
Materials and Methods
Search strategy and exclusion criteria
Research was conducted via a systematic literature search in the PubMed and PsycINFO databases. The search terms were the following: «“NR3C1 or glucocorticoid receptor” and “methylation” and “prenatal stress or maternal psychiatric disorder”».
When necessary, corresponding authors of the selected papers were contacted in order to obtain data required for the meta-analysis. Selection of specific CpG sites to be studied was in accordance with the reviewed papers coverage.
NR3C1 gene and CpG island structure
NR3C1 gene contains 9 exons. The first of them includes 9 alternate untranslated variants (1A to 1J, excluding 1G) that are thought to influence GR tissue distribution, mRNA stability and translational efficiency. 60 Seven of these 9 alternate first exons are located within a CpG island spanning 3 kb that has been extensively studied regarding its methylation.
Statistical analysis
All statistical analyses were performed in R.61 Since both psychosocial stress and methylation levels are typically measured in continuous scales and, thus, authors typically report correlation coefficients, meta-analyses of correlations were performed. However, as not all manuscripts provide the same effect size measure (for instance, authors may report mean differences or t-statistics), correlation coefficients were estimated when necessary using R's compute.es package.62 This package allows statistics from one study to be converted to many other effect size estimates, using standard procedures on meta-analysis methodology.63
The meta-analyses were conducted following conventional protocols as implemented in R's meta package.64 The inverse variance weighting method was used for pooling. For comparison, sensitivity analyses were conducted using both fixed and random effects. Since there were no large differences between them, and as random effects models are especially suitable for sets of studies with different methods and samples, the Results section only shows the outcome of random effects models.65
Considering that sample sizes of the studies in the meta-analyses are relatively small, the Fisher's z transformation of correlations was conducted, and residual heterogeneity was accounted for through the DerSimonian-Laird estimator, in accordance with ordinary guidelines on meta-analysis methods.63,64
To assess between-study differences, the next measures of heterogeneity and variability were computed: τ2 (estimated amount of total heterogeneity), I2 (total heterogeneity / total variability), H (square root of the χ2 heterogeneity statistic / degrees of freedom), and Cochran's Q (evaluation of whether the outcome variability is greater than expected based on sampling variability).66
Publication bias was assessed by funnel plot asymmetry tests. In the present context, these tests should be regarded as exploratory due to the small number of studies in each meta-analysis.64,67
Due to the presence of several stress/psychopathology assessment scales in most of the studies, selection of one informative scale per study was performed if necessary. For Oberlander et al. data, Edinburgh Postnatal Depression Scale (EPDS) measurements at 2nd trimester of pregnancy were selected (so Ham-A and EPDS scores at 3rd trimester were dismissed) in order to reduce heterogeneity between studies given (i) the major force of Conradt et al. results driving the meta-analyses' final results, which were obtained in depressed pregnant mothers, (ii) the timing of Hompes et al. and Murgatroyd et al. results, which were restricted to the 2 first trimesters of pregnancy, and (iii) the timing of the effects reported in animal models, which also are restricted to early stages of pregnancy.25 For Hompes et al. study, available data was extracted from their published paper thus selecting average PRAQ scores for CpG sites 35–36 analyses and EPDS scores for CpG sites 37–39, following the aforementioned criteria. For Mulligan et al. paper, war stress was selected as the variable of interest as it had the largest effect on birth weight.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are indebted to David Allen Hughes Jr., Ph.D. (Universitat Pompeu Fabra), Elisabeth Conradt, Ph.D. (University of Utah), Jonathan Hill (University of Reading), Karl Radtke (Universität Konstanz) and Tim Oberlander, Ph.D. (University of British Columbia), for gently providing us with specific data of their corresponding works in order to run the meta-analyses.
Funding
Supported by the Spanish Ministry of Science and Innovation (grant number SAF2008–05674-C03–01), European Twins Study Network on Schizophrenia Research Training Network (grant number EUTwinsS; MRTN-CT-2006-035987), the Comissionat per a Universitats I Recerca, DIUE, Generalitat de Catalunya (grant number 2014SGR1636), the CIBER of Mental Health (CIBERSAM) and the Ministry of Science and Innovation (PIM2010ERN-00642) in frame of ERA-NET NEURON. Helena Palma-Gudiel was supported by the Secretaria d'Universitats i Recerca del Departament d'Economia i Coneixement de la Generalitat de Catalunya (2015 FI_B 01011). A. Córdova-Palomera was funded by CONACyT (Mexico).
References
- 1.Kofink D, Boks MPM, Timmers HTM, Kas MJ. Epigenetic dynamics in psychiatric disorders: environmental programming of neurodevelopmental processes. Neurosci Biobehav Rev 2013; 37:831-45; PMID:23567520; http://dx.doi.org/ 10.1016/j.neubiorev.2013.03.020 [DOI] [PubMed] [Google Scholar]
- 2.Naumova OY, Lee M, Rychkov SY, Vlasova NV, Grigorenko EL. Gene expression in the human brain: the current state of the study of specificity and spatiotemporal dynamics. Child Dev 2013; 84:76-88; PMID:23145569; http://dx.doi.org/ 10.1111/cdev.12014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colantuoni C, Lipska BK, Ye T, Hyde TM, Tao R, Leek JT, Colantuoni EA, Elkahloun AG, Herman MM, Weinberger DR, et al.. Temporal dynamics and genetic control of transcription in the human prefrontal cortex. Nature 2011; 478:519-23; PMID:22031444; http://dx.doi.org/ 10.1038/nature10524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanson M a, Gluckman PD. Early developmental conditioning of later health and disease: physiology or pathophysiology? Physiol Rev 2014; 94:1027-76; PMID:25287859; http://dx.doi.org/ 10.1152/physrev.00029.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delisle H. Programming of chronic disease by impaired fetal nutrition. Evidence and implications for policy and intervention strategies. World Health Organization 2001; Retrieved from: http://www.who.int/nutrition/publications/obesity/WHO_NHD_02.3/en/ [Google Scholar]
- 6.Williams JHG, Ross L. Consequences of prenatal toxin exposure for mental health in children and adolescents. Eur Child Adolesc Psychiatry 2007; 16:243-53; PMID:17200791; http://dx.doi.org/ 10.1007/s00787-006-0596-6 [DOI] [PubMed] [Google Scholar]
- 7.Glover V. Annual research review: prenatal stress and the origins of psychopathology: an evolutionary perspective. J Child Psychol Psychiatry 2011; 52:356-67; PMID:21250994; http://dx.doi.org/ 10.1111/j.1469-7610.2011.02371.x [DOI] [PubMed] [Google Scholar]
- 8.Miller GE, Murphy MLM, Cashman R, Ma R, Ma J, Arevalo JMG, Kobor MS, Cole SW. Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun 2014; 41:191-9; PMID:25242587; http://dx.doi.org/ 10.1016/j.bbi.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod Toxicol 2005; 20:345-52; PMID:15893910; http://dx.doi.org/ 10.1016/j.reprotox.2005.04.005 [DOI] [PubMed] [Google Scholar]
- 10.Miller GE, Chen E, Fok AK, Walker H, Lim A, Nicholls EF, Cole S, Kobor MS. Low early-life social class leaves a biological residue manifested by decreased glucocorticoid and increased proinflammatory signaling. 2009; 106:14716-21; PMID:19617551; http://dx.doi.org/ 10.1073/pnas.0902971106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.King S, Dancause K, Turcotte-Tremblay A-M, Veru F, Laplante DP. Using natural disasters to study the effects of prenatal maternal stress on child health and development. Birth Defects Res C Embryo Today 2012; 96:273-88; PMID:24203917; http://dx.doi.org/ 10.1002/bdrc.21026 [DOI] [PubMed] [Google Scholar]
- 12.Yehuda R, Daskalakis NP, Lehrner A, Desarnaud F, Bader HN, Makotkine I, Flory JD, Bierer LM, Meaney MJ. Influences of maternal and paternal PTSD on epigenetic regulation of the glucocorticoid receptor gene in holocaust survivor offspring. Am J Psychiat 2014; 171:872-80; PMID:24832930; http://dx.doi.org/ 10.1176/appi.ajp.2014.13121571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perroud N, Rutembesa E, Paoloni-Giacobino A, Mutabaruka J, Mutesa L, Stenz L, Malafosse A, Karege F. The Tutsi genocide and transgenerational transmission of maternal stress: epigenetics and biology of the HPA axis. World J Biol Psychiat 2014; 15:334-45; PMID:24690014; http://dx.doi.org/ 10.3109/15622975.2013.866693 [DOI] [PubMed] [Google Scholar]
- 14.Cohen S, Janicki-Deverts D, Doyle WJ, Miller GE, Frank E, Rabin BS, Turner RB. Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proc Natl Acad Sci U S A 2012; 109:5995-9; PMID:22474371; http://dx.doi.org/ 10.1073/pnas.1118355109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lopez-Duran NL, Kovacs M, George CJ. Hypothalamic-pituitary-adrenal axis dysregulation in depressed children and adolescents: a meta-analysis. Psychoneuroendocrinology 2009; 34:1272-83; PMID:19406581; http://dx.doi.org/ 10.1016/j.psyneuen.2009.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehrner A, Bierer LM, Passarelli V, Pratchett LC, Flory JD, Bader HN, Harris IR, Bedi A, Daskalakis NP, Makotkine I, et al.. Maternal PTSD associates with greater glucocorticoid sensitivity in offspring of Holocaust survivors. Psychoneuroendocrinology 2014; 40:213-20; PMID:24485493; http://dx.doi.org/ 10.1016/j.psyneuen.2013.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mesiano S, Jaffe RB. Developmental and functional biology of the primate fetal adrenal cortex. Endocr Rev 1997; 18:378-403; PMID:9183569 [DOI] [PubMed] [Google Scholar]
- 18.Tarullo AR, Gunnar MR. Child maltreatment and the developing HPA axis. Horm Behav 2006; 50:632-9; PMID:16876168; http://dx.doi.org/ 10.1016/j.yhbeh.2006.06.010 [DOI] [PubMed] [Google Scholar]
- 19.Reynolds RM. Glucocorticoid excess and the developmental origins of disease: two decades of testing the hypothesis–2012 Curt Richter Award Winner. Psychoneuroendocrinology 2013; 38:1-11; PMID:22998948; http://dx.doi.org/ 10.1016/j.psyneuen.2012.08.012 [DOI] [PubMed] [Google Scholar]
- 20.Yang N, Ray DW, Matthews LC. Current concepts in glucocorticoid resistance. Steroids 2012; 77:1041-9; PMID:22728894; http://dx.doi.org/ 10.1016/j.steroids.2012.05.007 [DOI] [PubMed] [Google Scholar]
- 21.Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 2000; 23:477-501; PMID:11027914; http://dx.doi.org/ 10.1016/S0893-133X(00)00159-7 [DOI] [PubMed] [Google Scholar]
- 22.Palma-Gudiel H, Córdova-Palomera A, Leza JC, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: a critical review. Neurosci Biobehav Rev 2015; 55:520-35; PMID:26073068; http://dx.doi.org/ 10.1016/j.neubiorev.2015.05.016 [DOI] [PubMed] [Google Scholar]
- 23.Van Rossum EFC, Binder EB, Majer M, Koper JW, Ising M, Modell S, Salyakina D, Lamberts SWJ, Holsboer F. Polymorphisms of the glucocorticoid receptor gene and major depression. Biol Psychiatry 2006; 59:681-8; PMID:16580345; http://dx.doi.org/ 10.1016/j.biopsych.2006.02.007 [DOI] [PubMed] [Google Scholar]
- 24.Weaver ICG, Cervoni N, Champagne F a, D'Alessio AC, Sharma S, Seckl JR, Dymov S, Szyf M, Meaney MJ. Epigenetic programming by maternal behavior. Nat Neurosci 2004; 7:847-54; PMID:15220929; http://dx.doi.org/ 10.1038/nn1276 [DOI] [PubMed] [Google Scholar]
- 25.Mueller BR, Bale TL. Sex-specific programming of offspring emotionality after stress early in pregnancy. J Neurosci 2008; 28:9055-65; PMID:18768700; http://dx.doi.org/ 10.1523/JNEUROSCI.1424-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stroud LR, Papandonatos GD, Rodriguez D, Mccallum M, Salisbury AL, Phipps MG, Lester B, Huestis MA, Niaura R, Padbury JF, et al.. Maternal smoking during pregnancy and infant stress response: test of a prenatal programming hypothesis. Psychoneuroendocrinology 2014; 48:29-40; PMID:24999830; http://dx.doi.org/ 10.1016/j.psyneuen.2014.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics 2008; 3:97-106; PMID:18536531; http://dx.doi.org/ 10.4161/epi.3.2.6034 [DOI] [PubMed] [Google Scholar]
- 28.Conradt E, Lester BM, Appleton AA, Armstrong DA, Marsit CJ. The roles of DNA methylation of NR3C1 and 11β-HSD2 and exposure to maternal mood disorder in utero on newborn neurobehavior. Epigenetics 2013; 8:1321-9; PMID:24135662; http://dx.doi.org/ 10.4161/epi.26634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hompes T, Izzi B, Gellens E, Morreels M, Fieuws S, Pexsters A, Schops G, Dom M, Van Bree R, Freson K, et al.. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiatr Res 2013; 47:880-91; PMID:23566423; http://dx.doi.org/ 10.1016/j.jpsychires.2013.03.009 [DOI] [PubMed] [Google Scholar]
- 30.Braithwaite EC, Kundakovic M, Ramchandani PG, Murphy SE, Champagne F a. Maternal prenatal depressive symptoms predict infant NR3C1 1F and BDNF IV DNA methylation. Epigenetics 2015; 10:408-17; PMID:25875334; http://dx.doi.org/ 10.1080/15592294.2015.1039221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murgatroyd C, Quinn JP, Sharp HM, Pickles A, Hill J. Effects of prenatal and postnatal depression, and maternal stroking, at the glucocorticoid receptor gene. Transl Psychiatry 2015; 5:e560; PMID:25942041; http://dx.doi.org/ 10.1038/tp.2014.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mulligan CJ, D'Errico NC, Stees J, Hughes DA. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics 2012; 7:853-7; PMID:22810058; http://dx.doi.org/ 10.4161/epi.21180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radtke KM, Ruf M, Gunter HM, Dohrmann K, Schauer M, Meyer A, Elbert T. Transgenerational impact of intimate partner violence on methylation in the promoter of the glucocorticoid receptor. Transl Psychiat 2011; 1:e21; PMID:22832523; http://dx.doi.org/ 10.1038/tp.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McGowan PO, Sasaki A, D'Alessio AC, Dymov S, Labonté B, Szyf M, Turecki G, Meaney MJ. Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat Neurosci 2009; 12:342-8; PMID:19234457; http://dx.doi.org/ 10.1038/nn.2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharp H, Pickles A, Meaney M, Marshall K, Tibu F, Hill J. Frequency of infant stroking reported by mothers moderates the effect of prenatal depression on infant behavioural and physiological outcomes. PLoS One 2012; 7:e45446; PMID:23091594; http://dx.doi.org/ 10.1371/journal.pone.0045446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bromer C, Marsit CJ, Armstrong D a, Padbury JF, Lester B. Genetic and epigenetic variation of the glucocorticoid receptor (NR3C1) in placenta and infant neurobehavior. Dev Psychobiol 2013; 55:673-83; PMID:22714792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Appleton AA, Lester BM, Armstrong DA, Lesseur C, Marsit CJ. Examining the joint contribution of placental NR3C1 and HSD11B2 methylation for infant neurobehavior. Psychoneuroendocrinology 2015; 52:32-42; PMID:25459891; http://dx.doi.org/ 10.1016/j.psyneuen.2014.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Conradt E, Fei M, LaGasse L, Tronick E, Guerin D, Gorman D, Marsit CJ, Lester BM. Prenatal predictors of infant self-regulation: the contributions of placental DNA methylation of NR3C1 and neuroendocrine activity. Front Behav Neurosci 2015; 9:130; PMID:26074794; http://dx.doi.org/ 10.3389/fnbeh.2015.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tyrka AR, Price LH, Marsit C, Walters OC, Carpenter LL. Childhood adversity and epigenetic modulation of the leukocyte glucocorticoid receptor: preliminary findings in healthy adults. PLoS One 2012; 7:e30148; PMID:22295073; http://dx.doi.org/ 10.1371/journal.pone.0030148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Murphy B, Clark S, Donald I, Pinsky M, Velady D. Conversion of maternal cortisol to cortisone during placental transfer to the human fetus. Am J Obstet Gynecol 1974; 118:538-41; PMID:4812574 [DOI] [PubMed] [Google Scholar]
- 41.Benediktsson R, Lindsay R, Noble J, Seckl JR, Edwards C. Glucocorticoid exposure in utero: New model for adult hypertension. Lancet 1993; 341:339-41; PMID:8094115; http://dx.doi.org/ 10.1016/0140-6736(93)90138-7 [DOI] [PubMed] [Google Scholar]
- 42.Murphy VE, Zakar T, Smith R, Giles WB, Gibson PG, Clifton VL. Reduced 11β-hydroxysteroid dehydrogenase type 2 activity is associated with decreased birth weight centile in pregnancies complicated by asthma. J Clin Endocrinol Metab 2002; 87:1660-8; PMID:11932298 [DOI] [PubMed] [Google Scholar]
- 43.Welberg L a M, Thrivikraman KV, Plotsky PM. Chronic maternal stress inhibits the capacity to up-regulate placental 11beta-hydroxysteroid dehydrogenase type 2 activity. J Endocrinol 2005; 186:R7-R12; PMID:16135661; http://dx.doi.org/ 10.1677/joe.1.06374 [DOI] [PubMed] [Google Scholar]
- 44.Holmes MC, Abrahamsen CT, French KL, Paterson JM, Mullins JJ, Seckl JR. The mother or the fetus? 11beta-hydroxysteroid dehydrogenase type 2 null mice provide evidence for direct fetal programming of behavior by endogenous glucocorticoids. J Neurosci 2006; 26:3840-4; PMID:16597738; http://dx.doi.org/ 10.1523/JNEUROSCI.4464-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Donnell KJ, Bugge A, Freeman L, Khalife N, Connor TGO, Glover V. Maternal prenatal anxiety and downregulation of placental 11 b -HSD2. Psychoneuroendocrinology 2012; 37:818-26; PMID:22001010; http://dx.doi.org/ 10.1016/j.psyneuen.2011.09.014 [DOI] [PubMed] [Google Scholar]
- 46.Jensen Peña C, Monk C, Champagne F a. Epigenetic effects of prenatal stress on 11β-hydroxysteroid dehydrogenase-2 in the placenta and fetal brain. PLoS One 2012; 7:e39791; PMID:22761903; http://dx.doi.org/ 10.1371/journal.pone.0039791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bale TL. Sex differences in prenatal epigenetic programming of stress pathways. Stress 2011; 14:348-56; PMID:21663536 [DOI] [PubMed] [Google Scholar]
- 48.Goel N, Bale TL. Examining the intersection of sex and stress in modelling neuropsychiatric disorders. J Neuroendocrinol 2009; 21:415-20; PMID:19187468; http://dx.doi.org/ 10.1111/j.1365-2826.2009.01843.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kundakovic M, Gudsnuk K, Franks B, Madrid J, Miller RL, Perera FP, Champagne F a. Sex-specific epigenetic disruption and behavioral changes following low-dose in utero bisphenol A exposure. Proc Natl Acad Sci U S A 2013; 110:9956-61; PMID:23716699; http://dx.doi.org/ 10.1073/pnas.1214056110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Van Os J, Selten JP. Prenatal exposure to maternal stress and subsequent schizophrenia. The May 1940 Invasion of The Netherlands. Br J Psychiat 1998; 172:324-6 [DOI] [PubMed] [Google Scholar]
- 51.Clifton VL. Review: sex and the human placenta: mediating differential strategies of fetal growth and survival. Placenta 2010; 31 Suppl:S33-9; PMID:20004469; http://dx.doi.org/ 10.1016/j.placenta.2009.11.010 [DOI] [PubMed] [Google Scholar]
- 52.Catalano R, Yorifuji T, Kawachi I. Natural selection in utero: evidence from the Great East Japan Earthquake. Am J Hum Biol 2013; 25:555-9; PMID:23754635; http://dx.doi.org/ 10.1002/ajhb.22414 [DOI] [PubMed] [Google Scholar]
- 53.Glover V, Hill J. Sex differences in the programming effects of prenatal stress on psychopathology and stress responses: an evolutionary perspective. Physiol Behav 2012; 106:736-40; PMID:22353310; http://dx.doi.org/ 10.1016/j.physbeh.2012.02.011 [DOI] [PubMed] [Google Scholar]
- 54.Voils CI, Crandell JL, Chang Y, Leeman J, Sandelowski M. Combining adjusted and unadjusted findings in mixed research synthesis. J Eval Clin Pract 2011; 17:429-34; PMID:21040243; http://dx.doi.org/ 10.1111/j.1365-2753.2010.01444.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao-Lei L, Massart R, Suderman MJ, Machnes Z, Elgbeili G, Laplante DP, Szyf M, King S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: project ice storm. PLoS One 2014; 9:e107653; PMID:25238154; http://dx.doi.org/ 10.1371/journal.pone.0107653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nieratschker V, Massart R, Gilles M, Luoni A, Suderman MJ, Krumm B, Meier S, Witt SH, Nöthen MM, Suomi SJ, et al.. MORC1 exhibits cross-species differential methylation in association with early life stress as well as genome-wide association with MDD. Transl Psychiat 2014; 4:e429; PMID:25158004; http://dx.doi.org/ 10.1038/tp.2014.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Non AL, Binder AM, Kubzansky LD, Michels KB. Genome-wide DNA methylation in neonates exposed to maternal depression, anxiety, or SSRI medication during pregnancy. Epigenetics 2014; 9:964-72; PMID:24751725; http://dx.doi.org/ 10.4161/epi.28853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schroeder JW, Smith AK, Brennan P a, Conneely KN, Kilaru V, Knight BT, Newport DJ, Cubells JF, Stowe ZN. DNA methylation in neonates born to women receiving psychiatric care. Epigenetics 2012; 7:409-14; PMID:22419064; http://dx.doi.org/ 10.4161/epi.19551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Teh AL, Pan H, Chen L, Ong M, Dogra S, Wong J, Macisaac JL, Mah SM, Mcewen LM, Saw S, et al.. The effect of genotype and in utero environment on interindividual variation in neonate DNA methylomes. Genome Res 2014; 24:1064-74; PMID:24709820; http://dx.doi.org/ 10.1101/gr.171439.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Turner JD, Vernocchi S, Schmitz S, Muller CP. Role of the 5′-untranslated regions in post-transcriptional regulation of the human glucocorticoid receptor. Biochim Biophys Acta 2014; 1839:1051-61; PMID:25150144; http://dx.doi.org/ 10.1016/j.bbagrm.2014.08.010 [DOI] [PubMed] [Google Scholar]
- 61.R Core Team R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2014. Retrived from: http://www.r-project.org. [Google Scholar]
- 62.Del Re AC Compute.es: compute effect sizes. 2014; Retrieved from: http://cran.r-project.org/web/packages/compute.es. [Google Scholar]
- 63.Cooper H, Hedges LV, Valentine JC. The Handbook of Research Synthesis and Meta-Analysis. Second New York: Russell Sage Foundation; 2009. [Google Scholar]
- 64.Schwarzer G. Meta: general package for meta-analysis. 2015; Retrieved from: https://cran.r-project.org/web/packages/meta/ [Google Scholar]
- 65.Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw 2010; 36:1-48. [Google Scholar]
- 66.Cochran W. The combination of estimates from different experiments. Biometrics 1954; 10:101-29; http://dx.doi.org/ 10.2307/3001666 [DOI] [Google Scholar]
- 67.Sterne JAC, Sutton AJ, Ioannidis JPA, Terrin N, Jones DR, Lau J, Carpenter J, Rücker G, Harbord RM, Schmid CH, et al.. Recommendations for examining and interpreting funnel plot asymmetry in meta-analyses of randomised controlled trials. BMJ 2011; 343:d4002; PMID:21784880; http://dx.doi.org/ 10.1136/bmj.d4002 [DOI] [PubMed] [Google Scholar]