

Abstract
Histone acetylation and deacetylation are key epigenetic gene regulatory mechanisms that play critical roles in eukaryotes. Acetylation of histone 4 lysine 16 (H4K16ac) is implicated in many cellular processes. However, its biological function and relationship with transcription are largely unexplored in plants. We generated first genome-wide high-resolution maps of H4K16ac in Arabidopsis thaliana and Oryza sativa. We showed that H4K16ac is preferentially enriched around the transcription start sites and positively correlates with gene expression levels. Co-existence of H4K16ac and H3K23ac is correlated with high gene expression levels, suggesting a potentially combinatorial effect of H4K16ac and H3K23ac histone 3 lysine 23 acetylation on gene expression. Our data further revealed that while genes enriched with both H4K16ac and H3K23ac are ubiquitously expressed, genes enriched with only H4K16ac or H3K23ac showed significantly distinct expression patterns in association with particular developmental stages. Unexpectedly, and unlike in Arabidopsis, there are significant levels of both H4K16ac and H3K23ac in the lowly expressed genes in rice. Furthermore, we found that H4K16ac-enriched genes are associated with different biological processes in Arabidopsis and rice, suggesting a potentially species-specific role of H4K16ac in plants. Together, our genome-wide profiling reveals the conserved and unique distribution patterns of H4K16ac and H3K23ac in Arabidopsis and rice and provides a foundation for further understanding their function in plants.
Keywords: Arabidopsis, epigenome, gene regulation, histone acetylation, H4K16ac, plant development, rice
Abbreviations
- ChIP
Chromatin-immunoprecipitation
- H4K16ac
Histone H4 lysine 16 acetylation
- H3K23ac
Histone H3 lysine 23 acetylation
- TSS
transcription start site
- TTS
transcription termination site.
Introduction
Eukaryotic DNA is packed inside the nucleus by wrapping around a histone octamer consisting of 2 copies of each core histone H2A, H2B, H3, and H4,1,2 forming a nucleosome that is the basic unit of chromatin. The unstructured N-termini of histone tails are subject to a plethora of chemical modifications, including acetylation, methylation, ubiquitylation, phosphorylation, and sumoylation.3 Histone modifications have been extensively demonstrated to be essential gene regulatory mechanisms.4-7 Histone acetylation homeostasis plays a vital role in a diverse array of biological processes, including DNA replication, DNA repair, gene silencing, genome defense, genome organization, normal growth and development, physiology, aging, and numerous diseases.8-13 In plants, recent advances in high throughput sequencing technology have enabled the large-scale profiling of histone acetylation marks and have greatly improved our understanding of the function of histone acetylation and its relationship with transcription.14-18 Besides its role in gene expression, plant histone acetylation plays crucial roles in the crosstalk between genomes and the environment during plant responses to diverse stresses at the cellular and organismal levels.19-24
Acetylation occurs at many histone lysine residues and regulates chromatin activity by at least 2 different mechanisms. On the one hand, individual acetylation marks have precise function on a specific chromatin-based process independent of other acetylated residues. On the other hand, there is a functional redundancy between multiple acetylation marks. Many acetylated residues act coordinately and the combined effect ultimately dictates the functional outcome.25 For example, acetylation at K5, K8, and K12 of H4 are redundant with each other for nucleosome assembly and transcription.26 Only the combined mutations in H4 K5, K8, and K12 residues exhibit accumulative impacts on gene expression levels, whereas the individual mutation has a negligible effect on transcription.27 In contrast, acetylation of lysine 16 of histone H4 (H4K16ac) appears to have unique and distinct functions from other acetylation marks.28,29 In budding yeast, hypoacetylated H4K16 is critical for maintaining the proper heterochromatin boundaries30,31 and is implicated as a central switch for high-order chromatin compaction.29,32-34 Yeast hyperacetylation of H4K16 at specific subtelomeric regions is also correlated with lifespan.35 In Drosophila, global acetylation of H4K16 on the male X chromosome plays important roles in reducing chromatin compaction for active transcription and accounts significantly for dosage compensation.36 Mammalian H4K16ac appears to have a cell type-specific role in transcription. In mouse embryonic stem cells, H4K16ac is critical in releasing compacted chromatin for transcriptional activation.37 However, H4K16ac has little impact on transcription in the differentiated HEK293 cells.38
Compared to the large amount of studies on H4K16ac in fungi and animals, its biological function in plants is completely unknown. Considering the critical roles of H4K16ac in chromatin dynamics and transcription in various organisms, understanding its global distribution pattern and its relationship to transcription is the key step toward uncovering its function in plants. In this study, we generated first genome-wide high-resolution maps of H4K16ac in Arabidopsis thaliana and rice (Oryza sativa), one of the most important food crops in the world. Our data showed that H4K16ac was preferentially associated with euchromatic regions particularly around transcription start sites and colocalized with H3K23ac in plants. The abundance of H4K16ac was positively correlated with the levels of gene expression. Interestingly, genes enriched in both H4K16ac and H3K23ac had significantly higher expression levels than those associated with only H4K16ac. Finally, our data revealed that while Arabidopsis H4K16ac and H3K23ac were depleted in the silent genes, significant levels of these 2 marks were found in the silent genes in rice.
Results
H4K16ac is highly enriched around transcription start sites and positively correlates with gene expression levels
To begin to understand the function of H4K16ac in plants, we determined its genome-wide distribution pattern by using chromatin immunoprecipitation coupled with sequencing (ChIP-seq). We first evaluated the specificity of the antibody against H4K16ac by dot blot using modified and unmodified peptides (Fig. S1A). Next, we generated global enrichment map of H4K16ac across the 5 Arabidopsis chromosomes by calculating their read density normalized to the reads derived from H3. The resulting map showed that H4K16ac was highly enriched in the gene-rich euchromatic arms and was absent from pericentromeric regions, where transposons and other repetitive elements cluster (Figs. 1A; Fig. S1B). We then determined its peak distribution over different regions of protein-coding genes [i.e., transcription start site (TSS), gene body, promoter, and intergenic region]. Unlike in yeast, fly, or human,39,40 Arabidopsis H4K16ac peaked 200 bp downstream of the TSS (Figs. 1B; Fig. S1A) and was depleted in transposable elements (Fig. S2B). To determine whether gene length may contribute to these distribution patterns, we divided genes into 4 groups based on their sizes and found a similar pattern among all genes (Fig. S2C), suggesting that enrichment of H4K16ac around the TSS is independent of gene size.
Figure 1.
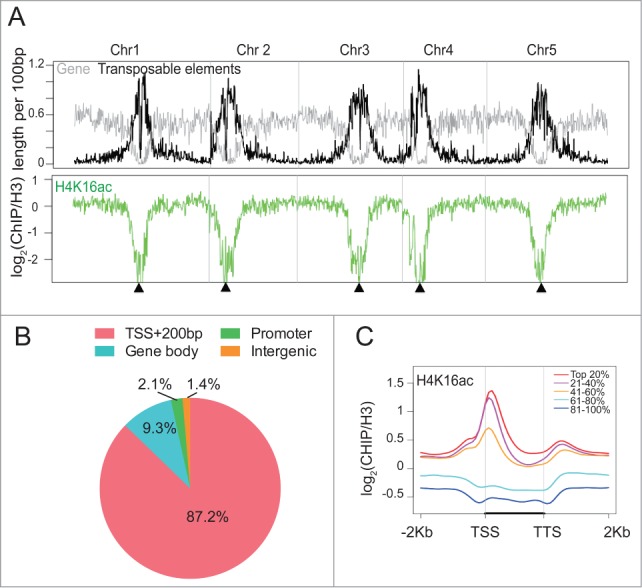
H4K16ac is highly enriched around transcription start sites (TSS) and positively correlates with transcription levels in Arabidopsis. (A) Chromosomal views show that H4K16ac are highly enriched in euchromatic regions. Y-axis represents the Log2 value of ChIP reads normalized to H3 reads. Transposable elements (black) and gene (gray) density were plotted to indicate the locations of pericentromeric heterochromatin. Chr1, Chr2, Chr3, Chr4, and Chr5 represent the 5 Arabidopsis chromosomes. Black triangle indicates the location of the centromere. (B) H4K16ac peak distribution at the TSS and 200 bp downstream of the TSS (TSS+200bp), gene body, promoter, and intergenic region. (C) Metaplot shows that H4K16ac is positively correlated with gene expression levels. “Top 20%” represents the gene group with the highest expression. The black bar in the X-axis represents genes from the TSS to the transcription termination site (TTS). Y-axis represents the log2 value of ChIP reads normalized to H3 reads.
Next, we asked whether H4K16ac-enrichment at the TSS was correlated with transcription. We classified a total of ~30,000 genes in Arabidopsis genome (annotated in TAIR10) into 5 groups based on their expression levels.41 We found that actively expressed genes had significantly higher levels of H4K16ac compared to moderately expressed genes (41-60% group, Fig. 1C; Fig. S1B, P < 1 × 10−20). Lowly expressed genes showed depletion of H4K16ac across the entire gene-body (Fig. 1C). This result revealed a positive correlation between transcript abundance and H4K16ac enrichment levels.
Combined enrichment of H4K16ac and H3K23ac is correlated with high gene expression levels
To further examine the relationship between H4K16ac and chromatin states, we performed a pairwise association analysis between H4K16ac and other epigenetic marks. H4K16ac is positively correlated with active marks (H3K9ac, H3K18ac, H3K4me2/3 and H3K36me2/315,42-44) and anti-correlated with repressive modifications (H3K27me1 and H3K9me215,45) and histone variants H3.1 and H2A.W, which are required for heterochromatin condensation46.,47.,48.,49.,50. (Fig. 2A).46-50 Interestingly, our results showed a significant positive correlation between H4K16ac and H3K23ac (Pearson's correlation coefficient = 0.73, P < 2.2 × 10−16), previously shown to be associated with transcriptional activation. 18 It is to be noted that the published H3K23ac ChIP-seq was performed using 3-week-old leaves,18 whereas, in our study, H4K16ac was profiled from 2-week-old seedlings. To rule out the possibility that H3K23ac may have distinct distribution patterns in different developmental stages, we determined the genome-wide profile of H3K23ac in parallel with that of H4K16ac from 2-week-old seedlings and found a significant correlation (Pearson's correlation coefficient = 0.52, P < 2.2 × 10−16). We next calculated the acetylation level of H4K16 and H3K23 for each gene in the Arabidopsis genome and revealed a positive correlation between these 2 marks (Pearson's correlation coefficient = 0.92, P < 2.2 × 10−16) (Fig. 2B). We further divided all genes into 4 clusters by calculating the log2 value of H4K16ac or H3K23ac reads normalized to H3 reads. Only genes with log2 value above 1 or below 0 were used for cluster classification. Approximately 15,800 genes (56%) containing both H4K16ac and H3K23ac were defined as belonging to the “Both” cluster (Figs. 2B; Fig. S3A). The genes only enriched with H4K16ac (but not H3K23ac) and H3K23ac (but not H4K16ac) were defined as “H4K16ac only” and “H3K23ac only”, respectively (Figs. 2B; Fig. S3A). A ChIP-quantitative PCR in 2 randomly chosen genes from “H4K16ac only” or “H3K23ac only” clusters further confirmed this classification (Fig. S3B). Around 6,700 genes (24%) that have none of H4K16ac and H3K23ac marks were defined as “None” cluster (Figs. 2B; Fig. S3A, and Table S1). We next compared the transcript abundance of genes in different clusters and found that genes enriched with both H4K16ac and H3K23ac have significantly higher expression levels than the other 3 clusters (Fig. 2C, P < 0.0001). Notably, the expression level of “H4K16ac only” (green box) genes is similar to that of “None” cluster (purple box) (Fig. 2C), suggesting that combined enrichment of H4K16ac and H3K23ac is correlated with high gene expression.
Figure 2.
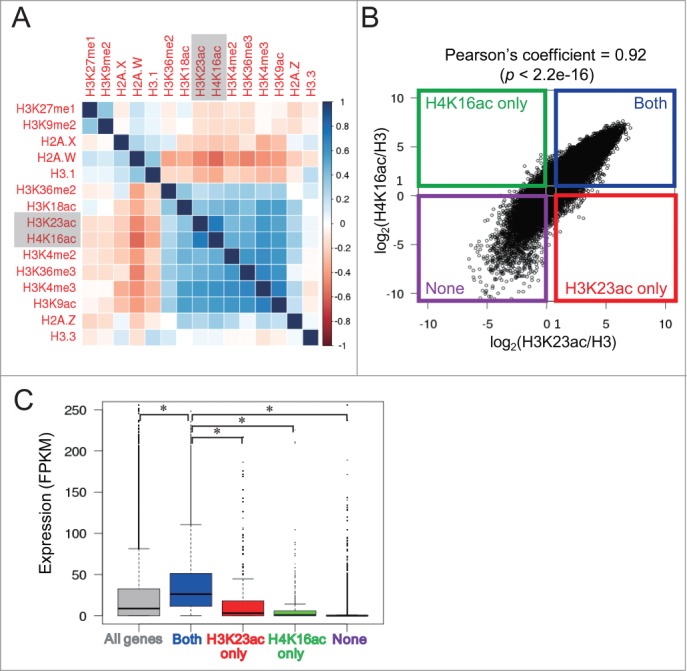
Co-existence of H4K16ac and H3K23ac is correlated with high gene expression. (A) Pearson correlation analysis shows the positive correlations of H4K16ac with active epigenetic marks and the negative correlations with silent marks. The coefficient is indicated by the color of each square. (B) Positive correlation between H4K16ac and H3K23ac in Arabidopsis genome. Pearson correlation was tested between H4K16ac and H3K23ac levels of each gene (Pearson correlation coefficient = 0.92, P < 2.2 × 10−16). Four gene clusters were divided based on their H4K16ac and H3K23ac levels. (C) Boxplot shows the expression level (FPKM) of genes in 4 clusters as defined in (B). All genes in the Arabidopsis genome were plotted as an average expression level (gray). The * indicates P < 0.0001 by t-test.
H4K16ac- and H3K23ac-enriched genes are ubiquitously expressed
Besides the combinatorial action, we asked whether there are any unique patterns of H4K16ac and H3K23ac in plants. To this end, we examined the transcript levels of genes in 4 clusters in various tissues from different developmental stages using previously published microarray data.51 Approximately 66% of genes (10,522 out of 15,810) containing both H4K16ac and H3K23ac showed a globally high expression level (average gcRMA expression levels > 6) across all tissue types except for pollen (Fig. 3A). In contrast, genes depleted in both H4K16ac and H3K23ac (“None” cluster) had generally low expression in all tissues (Fig. 3A). Interestingly, a certain fraction of “H3K23ac only” genes showed a slightly higher expression level in leaves compared to flowers. Also, a small number of “H4K16ac only” genes exhibited a moderate increase in transcript abundance in floral tissues compared to leaves (Fig. 3A). To further confirm the tissue-specific expression patterns of “H4K16ac only” and “H3K23ac only” genes, we calculated an entropy value for each gene cluster in all developmental stages and tissue types. The entropy value is a measure of the extent of tissue specificity, with a small value indicating high tissue specificity and a large value indicating low tissue specificity.52 Previous studies established that H3K27me3-enriched genes showed strong developmental regulation and had a significant smaller entropy value than genes in “Both” cluster (P < 1 × 10−20).53 Interestingly, genes enriched with both H4K16ac and H3K23ac had the largest entropy value among the 4 clusters (Fig. 3B; Fig. S4A), suggesting that they are ubiquitously expressed. In contrast, genes depleted of both H4K16ac and H3K23ac (“None” cluster) had a significantly smaller value compared to “Both” genes (Fig. 3B ;Fig. S4A, P < 1 × 10−20), indicating their high tissue-specific expression. Consistent with Fig. 3A, “H3K23ac only” and “H4K16ac only” enriched genes have significantly smaller entropy values compared to “Both” genes (P = 4.6 × 10−8 for “H3K23ac only” and P = 1.2 × 10−9 for “H4K16ac only”), suggesting that these genes, like those enriched in H3K27me3, have distinct expression patterns in association with particular development stages (Fig. 3B; Fig. S4A). We further determined the entropy values of the 4 gene clusters under stress, light, and pathogen responses. Interestingly, only “H4K16ac only” enriched genes showed a significantly smaller entropy value compared to “Both” genes in stress response (Fig. 3C; Fig. S4B, P = 2.2 × 10−7). None of the 4 clusters exhibited a significant difference under light and pathogen conditions (Figs. S4C-D).
Figure 3.
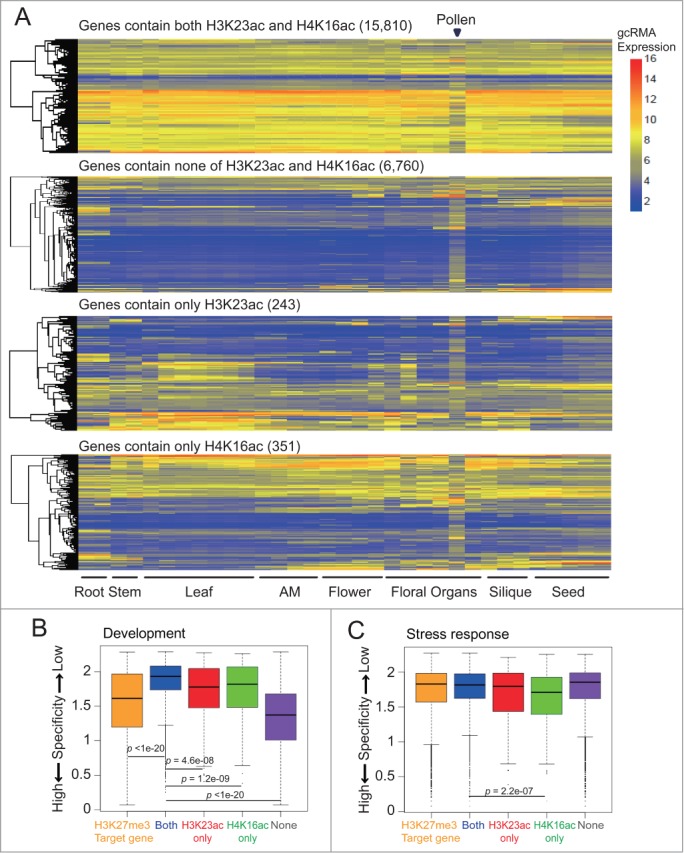
Genes enriched with both H4K16ac and H3K23ac are ubiquitously expressed. (A) Heatmaps show the expression patterns of 4 gene clusters in tissues from different developmental stages. Four gene clusters were defined in Figure 2B. (B-C) Tissue specificity of “Both”, “None”, “H3K23ac only”, and “H4K16ac only” genes in development (B) and abiotic stress (C) measured by entropy values. H3K27me3 target genes were included as a control for high expression specificity in development.
Together, these results suggest that the presence of both H4K16ac and H3K23ac is associated with highly and ubiquitously expressed genes, but these 2 marks when present in isolation likely have tissue-specific expression.
Higher order interaction between H4K16ac, H3K23ac, and other histone modifications
Extensive studies have established that multiple histone modifications act in a combinatorial fashion to specify distinct chromatin states.14,17 To understand the relationship between H4K16ac and H3K23ac with other histone methylation marks, we examined the correlation of these 2 marks with previously reported histone modifications in Arabidopsis. Consistent with the expression difference among the 4 clusters, we found that active marks (H3K4me2/3 and H3K36me2/3) are significantly enriched at genes in the “Both” cluster (P < 2.2 × 10−16) and are depleted in the “None” cluster (Fig. 4A-D). The situation is opposite for repressive marks. We found that H3K27me3 is depleted at genes in the “Both” cluster (Fig. 4E). Unexpectedly, “H3K23ac only” cluster had a significantly higher level of H3K27me3 than those of “Both” or “H4K16ac only” (Fig. 4E, P < 2.2 × 10−16 for “Both” and P = 3 × 10−3 for “H4K16ac only”). Combined with the expression patterns, these results suggest that the “Both” cluster is associated with multiple active histone modifications in seedlings.
Figure 4.
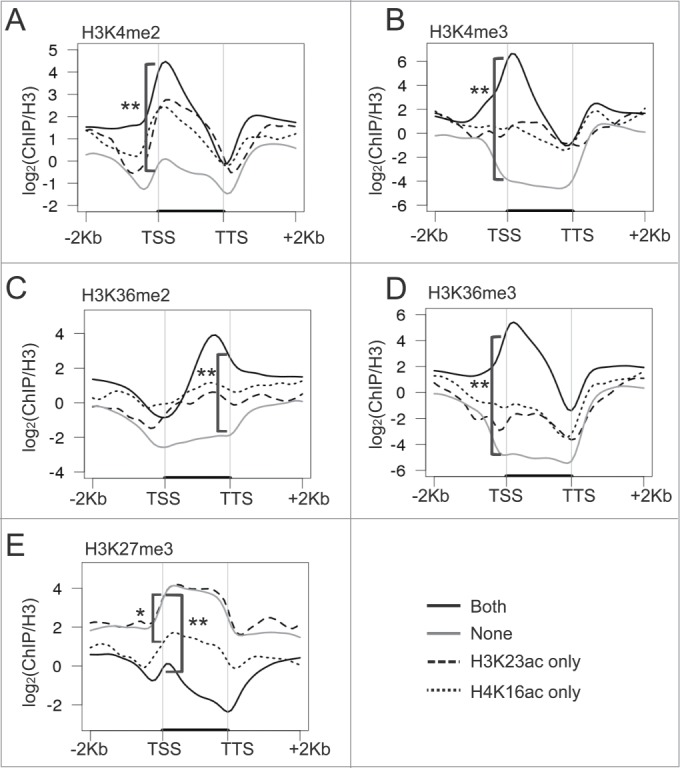
Correlation of histone methylation marks with H4K16ac and H3K23ac. Metaplots show the correlation of genes in the 4 clusters with H3K4me2 (A), H3K4me3 (B), H3K36me2 (C), H3K36me3 (D), and H3K27me3 (E). Four gene clusters were defined in Figure 2B. The * indicates P < 0.01 and ** indicates P < 2.2 × 10−16.
Conserved and unique distribution pattern of H4K16ac and H3K23ac in rice
Although the overall distribution patterns of H3K23ac across genes are similar in Arabidopsis, yeast and mammals,39 there are some notable species-specific H4K16ac patterns. H4K16ac is found mostly at the TSS of expressed genes in Arabidopsis (Fig. 1B), but is enriched at promoters and transcribed regions of active genes in human.37,38,40 To explore the conservation of H4K16ac and H3K23ac distribution patterns in plants, we determined their genome-wide occupancy in rice by using ChIP-seq.
Both H4K16ac and H3K23ac were enriched at euchromatic arms and depleted from pericentromeric regions (Figs. 5A; Fig. S5). We next performed a Pearson correlation analysis and found a significantly positive correlation of H3K23ac and H4K16ac with gene density (Fig. S6A, Pearson's correlation coefficient = 0.27, P< 2.2 × 10−16 for H3K23ac and coefficient = 0.57, P < 2.2 × 10−16 for H4K16ac). We also found a significantly negative correlation of both H3K23ac and H4K16ac with transposon density (Fig. S6A, Pearson's correlation coefficient = − 0.24, P < 2.2 × 10−16 for H3K23ac and coefficient = − 0.39, P < 2.2 × 10−16 for H4K16ac). To investigate the relationship of H3K23ac and H4K16ac with gene activity, we classified a total of ~40,000 rice genes into 5 groups based on their expression levels in rice seedlings.54 While 87.2% of H4K16ac and 88.2% of H3K23ac peaks are enriched at the TSS in Arabidopsis, only 56.5% of H4K16ac and 52.6% of H3K23ac peaks are at the TSS in rice (Fig. 5B). We further noticed a considerably increased fraction of these 2 marks in the gene body (22.5% for H4K16ac and 18.2% for H3K23ac) and intergenic regions (15.4% for H4K16ac and 22.2% for H3K23ac) in rice compared to 9% and 1.3% of both H4K16ac and H3K23ac in gene body and intergenic regions, respectively, in Arabidopsis (Fig. 5B). Unlike Arabidopsis, rice H4K16ac and H3K23ac have no significant correlation with transcript abundance of the top 2 groups of actively expressed genes (Figs. 5C; Fig. S6B, P = 0.7 for H3K23ac and P = 0.07 for H4K16ac). More interestingly, we found a considerable level of these 2 marks in the coding regions of many lowly expressed genes (Fig. 5C and 5D). To further understand the relationship of H4K16ac and H3K23ac with gene expression, we compared the expression levels of genes that have H4K16ac and H3K23ac (“Both”), “H3K23ac only”, “H4K16ac only”, and none of these 2 marks (“None”) (Fig. S7A). Like in Arabidopsis, genes enriched with both H4K16ac and H3K23ac showed significantly higher expression levels than the genes in the other 3 clusters (Fig. 5E, P < 0.001), suggesting that the co-existence of both H4K16ac and H3K23ac is also correlated with gene expression levels in rice.
Figure 5.

H4K16ac and H3K23ac are highly enriched around transcription start sites (TSS) and positively correlate with transcription levels in rice. (A) Chromosomal views of H4K16ac and H3K23ac distribution patterns in rice chromosome 1. Transposable elements and gene density were plotted to indicate the locations of the pericentromeric heterochromatin. (B) H4K16ac and H3K23ac peak distribution at the TSS and 200 bp downstream of the TSS (TSS+200bp), gene body, promoter, and intergenic region in Arabidopsis and rice. (C) Metaplots showing the H4K16ac and H3K23ac levels over 5 gene groups divided based on their expression levels. (D) Representative snapshots of H4K16ac and H3K23ac in an active gene and a silent gene in rice. (E) Boxplot showing the expression levels of 4 gene clusters. The * indicates P < 0.0001 by a Student's t-test. (F) Heatmaps showing distribution patterns of H4K16ac and H3K23ac in the top 20% and bottom 81–100% expressed genes in Arabidopsis and rice. TTS, transcription termination site; −2K, 2 kb upstream of the TSS; +2K, 2 kb downstream of the TTS.
To investigate the potential functional roles of H4K16ac-enriched genes, we performed Gene Ontology analysis. We found that, in terms of biological processes, rice H4K16ac-containing genes were mostly enriched in functions related to photosynthesis (P = 2.4 × 10−44), generation of precursor for metabolites and energy (P = 1.0 × 10−28), and metabolic processes (P = 7.8 × 10−22) (Fig. S7B).In contrast, Arabidopsis H4K16ac-enriched genes were mostly involved in development (P = 6.1 × 10−7), responses to stimuli (P = 3.7 × 10−6), and signal transduction (P = 1.4 × 10−5) (Fig. S7B).
Together, these results suggest that the distribution patterns of H4K16ac and H3K23ac are generally conserved between rice and Arabidopsis with the exception of their enrichment across the lowly expressed genes (Fig. 5F) and their association with genes involved in different biological processes.
Discussion
Our study generated the first genome-wide high-resolution maps of H4K16ac in the model plants A. thaliana and O. sativa. By comparative analyses, we showed that H4K16ac exhibited both conserved and unique distribution patterns in plants and acted in a combinatorial manner with H3K23ac on gene expression. We have also found some potential plant-specific roles of H4K16ac in Arabidopsis and rice.
H4K16ac and H3K23ac are preferentially enriched at actively expressed genes in Arabidopsis, consistent with the fact that acetylation marks are generally associated with open chromatin and actively transcribed regions.14,22,23,55-58 This distribution pattern is also in agreement with an anti-correlation of H3K23ac with DNA methylation.18,59 Surprisingly, we found that H4K16ac and H3K23ac are also enriched across the gene-body of many lowly expressed genes in rice (Fig. 5C and 5D). Consistent with this observation, H3K23ac is also associated with the coding regions of silent genes in fly.60 H4K16ac is also present at the silent genes in yeast.56 The biological significance of the enrichment of H4K16ac or H3K23ac in the silent genes is unknown. Their unexpected enrichment in the silent genes in rice, but not Arabidopsis, implies that there may be a species-specific difference in the organization of plant genomes. One possible explanation is that these silent genes may have relatively open chromatin in the gene body to make it accessible for acetylation, but their expression is strictly controlled by promoter chromatin configuration. This is consistent with a model in which the chromatin configuration of promoter acts on top of that of the gene body to regulate gene expression.61,62 Besides distribution pattern difference, our Gene Ontology analysis revealed that H4K16ac-enriched genes are associated with different biological processes in Arabidopsis and rice (Fig. S7B), further supporting a potentially species-specific role of H4K16ac in plants.
Another intriguing finding was a species-specific distribution pattern of H4K16ac and H3K23ac between plants and animals. H3K23ac was enriched at the promoters in worm63 and across the entire gene bodies in fly.60 Interestingly, the distribution pattern of H4K16ac is very dynamic in different organisms. Fly H4K16ac is mostly enriched around the TSS,60 whereas the majority of H4K16ac is associated with promoters and a relatively small fraction near the TSS in worm.63 In human, the highest enriched peaks of H4K16ac fall within the promoters and the transcribed regions of actively expressed genes.40 In plants, H4K16ac is mostly enriched around the TSS with slight differences in Arabidopsis and rice. Approximately 87% of H4K16ac peaks are around the TSS in Arabidopsis (Fig. 1B), but only 56% are present near the TSS in rice (Fig. 5B). This is not surprising given that a moderate fraction (38%) of H4K16ac is found in the body or intergenic regions in rice.
The distribution pattern of H3K23ac and H4K16ac is also in agreement with their potential roles in transcriptional activation. We found that 56% of the Arabidopsis genes modified by H4K16ac are also enriched with H3K23ac. A similar co-localization between these 2 marks was also found in human.40 Interestingly, the genes with both marks have the greatest transcript abundance and their expression levels are significantly higher than those of genes with only H4K16ac or H3K23ac (Figs. 2C and 5E). This result is consistent with the observation that genes associated with multiple active marks have high expression.40 Thus, our findings support a cooperative model in which multiple epigenetic modifications act in a combinatorial manner and the combined effect ultimately determines the function.40,64
Acetylation of H4K16 has been well documented to have many unique features in different organisms.29,58 Our systemic analysis of the expression patterns of H4K16ac-enriched genes in different developmental stages of 8 tissues revealed a correlation with plant development. As expected, genes enriched with both H4K16ac and H3K23ac are highly and ubiquitously expressed (Fig. 3A). Unexpectedly, these genes appear to be lowly expressed in the pollen. More surprisingly, the genes containing none of H4K16ac and H3K23ac are relatively highly expressed in the pollen. It is possible that H4K16ac and H3K23ac may have a tissue-specific role in transcriptional regulation in plants. Consistent with this hypothesis, H4K16ac appears to have cell-type dependent function in transcription in human cells.38 Furthermore, H4K16ac has been reported to be lost in many cancer cells and is frequently used as a hallmark for certain types of cancer.65 The mechanism regarding the differential expression pattern of H4K16ac-enriched genes in pollen is unclear. It is possible that, like in human HEK293 cells,37 H4K16ac has distinct effects on transcription in pollen. In the future, it will be important to directly examine the roles of H4K16ac in regulating transcriptional activity in the different stages of the development.
Materials and Methods
Plant material
Arabidopsis thaliana (accession Columbia-0) plants were grown in soil at 21°C under continuous light for 2 weeks. Oryza sativa (Nipponbare) was grown on MS medium at 28°C under short day condition (8 h light and 16 h dark) for 2 weeks. The aerial sections of the seedlings were harvested for the following experiments.
Dot blot
PVDF membrane (GE Healthcare, Hybond-P, RPN303F) was submerged in methanol for 2–3 s and then washed with TBS (50 mM Tris pH7.5, 150 mM NaCl). The membrane was placed on a wet filter paper. Peptides with dilution series of 250 pmol and 50 pmol were spotted on the membrane respectively and air-dried. The membrane with peptide was soaked with methanol and washed with TBS, followed by blocking for 1 h at room temperature (RT) with blocking buffer (3% BSA in TBS plus 0.1% Tween-20). The membrane was further incubated with antibodies against H3K23ac (07-355, Millipore) and H4K16ac (07-329, Millipore) for 2 h at H4K16ac (07-329, Millipore) for 2 h at room temperature.
Chromatin-immunoprecipitation (ChIP) sequencing
ChIP was performed based on the previously described paper with slight modifications.41 Two grams of plant tissues were ground into a fine powder with liquid nitrogen and re-suspended in 25 mL of nuclear isolation buffer (10 mM Hepes pH 8.0, 1 M sucrose, 5 mM KCl, 5 mM EDTA, 0.6% Triton X-100, 0.4 mM PMSF, 1 ug/ul pepstain, and protease inhibitor complete mini (Roche Applied Science)). The antibodies were purchased from Millipore (07-329 for H4K16ac and 07-355 for H3K23ac). ChIP libraries were constructed by using the Ovation Ultralow Library kit (NuGEN, Part No. 0330), following the manufacturer instructions. The libraries were sequenced using the HiSeq 2000 Sequencing System (Illumina) in UW-Madison biotechnology center.
Real-time PCR
Primers for Arabidopsis ChIP quantitative PCR were designed within the 400 bp region following TSS of each gene (Table S2). Quantitative real-time PCR analysis was performed using the SYBR Taq Kit (Life technologies, Catalog: 4309155) on the Applied Biosystems 7500 system. For each primer set, the ΔCT value was calculated using the comparative CT method: ΔCT = CT (ChIP isolated DNA) - CT (input DNA). Relative enrichment was calculated as 2-ΔΔCT± standard deviation (SD), where ΔΔCT=ΔCT (ChIP isolated DNA) – CT (H3 ChIP isolated DNA). Three technical replicates were performed.
ChIP-seq data analysis
Sequenced reads were mapped to TAIR10 genome for Arabidopsis and MSU7.0 genome for Oryza sativa with Bowtie2 (v2.1.0)66 using default parameters. Reads mapping to identical positions in the genome were collapsed into one read. The total reads obtained for each sample were listed in Table S3. Enrichment regions of H4K16ac and H3K23ac were defined using SICER package67 with a histone H3 ChIP as control. Rice ChIP was normalized to Micrococcal Nuclease digested sequencing reads (GSM1228041)68 for nucleosome density control.
In genome-wide profiling of Arabidopsis H4K16ac, log2 value of the ChIP reads divided by H3 reads was calculated and binned in 100 kb increments. In rice, normalized ChIP read density for each 100 kb bin was calculated and plotted. For plots of acetylation level over genes or transposable elements, each gene was divided into 20 intervals (5% each interval) separately for the body of the gene, 2 kb upstream of the TSS, and 2 kb downstream of the TTS. Gene Ontology analysis was performed based on agriGO.69
Pairwise association analysis
We divided the Arabidopsis genome into 100 bp sections and calculated the log2 value of ChIP reads normalized to input reads for each histone modification. The histone modification ChIP-sequencing datasets were obtained from previously published papers.43,46,47,70 Pairwise association analysis between histone modification marks was calculated by Pearson correlation method. The P values was calculated by a Student t-test.
Classification of gene clusters
Based on our findings that H4K16ac and H3K23ac mostly enrich at 400 bp downstream of the TSS (Fig. S2A), we assign the acetylation score for each gene by taking a log2 value of ChIP reads normalized to H3 reads from the TSS to 400 bp downstream of the TSS. Genes with log2 value above 1 in both H4K16ac and H3K23ac were defined as “Both” cluster. Genes with log2 value below 0 in both acetylation marks were defined as “None” cluster. Genes with log2 value above 1 in H4K16ac but below 0 in H3K23ac were defined as “H4K16ac only”. Genes with log2 value above 1 in H3K23ac but below 0 in H4K16ac were defined as “H3K23ac only”. Gene lists of these 4 clusters were provided in Table S1. In Oryza sativa, “Both”, “H4K16ac only”, “H3K23ac only”, and “None” gene clusters were assigned to genes that had at least 1 bp overlapping between the genic regions and the defined peaks. Gene lists for 4 rice clusters were provided in Table S4.
Expression pattern analysis
In defining gene expression clusters, expression level of Arabidopsis seedling was obtained from published data.41 Rice gene expression profile was obtained from seedlings of “Nipponbare” (http://rice.plantbiology.msu.edu/; data set SRX016110). Expression profiles for a broad range of developmental stages throughout the plant life cycle were downloaded from a previously published paper.51 Triplicate data for each developmental time point were averaged and the sample list used in this study are listed in Table S5. Entropy was calculated by using the “entropy” package in R.52 All plots were normalized for their sequencing coverage.
Data access
The ChIP Sequencing data sets have been submitted to the National Center for Biotechnology Information database (GSE69426).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Authors' Contributions
LL, XC, DS and SQ performed the experiments. LL performed all statistical data analyses and drafted the manuscript. XZ designed the experiments, supervised all data analyses, and wrote the final version of manuscript. All authors read and approved the final manuscript.
Acknowledgments
We apologize for many important works that were not cited due to space limitations. We thank Ray Scheid, Rachael Fieweger and anonymous reviewers for critical comments on this manuscript. We also thank UW-Madison's HTCondor high throughput computing for computational resource.
Funding
This work is supported by startup fund from UW-Madison and grant from the US Department of Agriculture & National Institute of Food and Agriculture (Hatch 1002874 to XZ).
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
References
- 1.Kornberg RD. Chromatin structure: a repeating unit of histones and DNA. Science 1974; 184:868-71; PMID:4825889; http://dx.doi.org/ 10.1126/science.184.4139.868 [DOI] [PubMed] [Google Scholar]
- 2.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 1997; 389:251-60; PMID:9305837; http://dx.doi.org/ 10.1038/38444 [DOI] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin modifications and their function. Cell 2007; 128:693-705; PMID:17320507; http://dx.doi.org/ 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature 2000; 403:41-5; PMID:10638745; http://dx.doi.org/ 10.1038/47412 [DOI] [PubMed] [Google Scholar]
- 5.Jenuwein T, Allis CD. Translating the histone code. Science 2001; 293:1074-80; PMID:11498575; http://dx.doi.org/ 10.1126/science.1063127 [DOI] [PubMed] [Google Scholar]
- 6.Latham JA, Dent SY. Cross-regulation of histone modifications. Nat Struct Mol Biol 2007; 14:1017-24; PMID:17984964; http://dx.doi.org/ 10.1038/nsmb1307 [DOI] [PubMed] [Google Scholar]
- 7.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 2011; 12:7-18; PMID:21116306; http://dx.doi.org/ 10.1038/nrg2905 [DOI] [PubMed] [Google Scholar]
- 8.Unnikrishnan A, Gafken PR, Tsukiyama T. Dynamic changes in histone acetylation regulate origins of DNA replication. Nat Struct Mol Biol 2010; 17:430-7; PMID:20228802; http://dx.doi.org/ 10.1038/nsmb.1780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gong F, Miller KM. Mammalian DNA repair: HATs and HDACs make their mark through histone acetylation. Mutat Res 2013; 750:23-30; PMID:23927873; http://dx.doi.org/ 10.1016/j.mrfmmm.2013.07.002 [DOI] [PubMed] [Google Scholar]
- 10.Timmermann S, Lehrmann H, Polesskaya A, Harel-Bellan A. Histone acetylation and disease. Cell Mol Life Sci 2001; 58:728-36; PMID:11437234; http://dx.doi.org/ 10.1007/PL00000896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Archer SY, Hodin RA. Histone acetylation and cancer. Curr Opin Genet Dev 1999; 9:171-4; PMID:10322142; http://dx.doi.org/ 10.1016/S0959-437X(99)80026-4 [DOI] [PubMed] [Google Scholar]
- 12.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997; 389:349-52; PMID:9311776; http://dx.doi.org/ 10.1038/38664 [DOI] [PubMed] [Google Scholar]
- 13.Zentner GE, Henikoff S. Regulation of nucleosome dynamics by histone modifications. Nat Struct Mol Biol 2013; 20:259-66; PMID:23463310; http://dx.doi.org/ 10.1038/nsmb.2470 [DOI] [PubMed] [Google Scholar]
- 14.Roudier F, Ahmed I, Berard C, Sarazin A, Mary-Huard T, Cortijo S, Bouyer D, Caillieux E, Duvernois-Berthet E, Al-Shikhley L, et al.. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J 2011; 30:1928-38; PMID:21487388; http://dx.doi.org/ 10.1038/emboj.2011.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou J, Wang X, He K, Charron JB, Elling AA, Deng XW. Genome-wide profiling of histone H3 lysine 9 acetylation and dimethylation in Arabidopsis reveals correlation between multiple histone marks and gene expression. Plant Mol Biol 2010; 72:585-95; PMID:20054610; http://dx.doi.org/ 10.1007/s11103-009-9594-7 [DOI] [PubMed] [Google Scholar]
- 16.Du Z, Li H, Wei Q, Zhao X, Wang C, Zhu Q, Yi X, Xu W, Liu XS, Jin W, et al.. Genome-wide analysis of histone modifications: H3K4me2, H3K4me3, H3K9ac, and H3K27ac in Oryza sativa L. Japonica. Mol Plant 2013; 6:1463-72; PMID:23355544; http://dx.doi.org/ 10.1093/mp/sst018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang C, Liu C, Roqueiro D, Grimm D, Schwab R, Becker C, Lanz C, Weigel D. Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res 2015; 25:246-56; PMID:25367294; http://dx.doi.org/ 10.1101/gr.170332.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stroud H, Do T, Du J, Zhong X, Feng S, Johnson L, Patel DJ, Jacobsen SE. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol 2014; 21:64-72; PMID:24336224; http://dx.doi.org/ 10.1038/nsmb.2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollender C, Liu Z. Histone deacetylase genes in Arabidopsis development. J Integr Plant Biol 2008; 50:875-85; PMID:18713398; http://dx.doi.org/ 10.1111/j.1744-7909.2008.00704.x [DOI] [PubMed] [Google Scholar]
- 20.Li Y, Shin D, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J 2013; 280:775-93; PMID:23181831 [DOI] [PubMed] [Google Scholar]
- 21.Liu X, Yang S, Zhao M, Luo M, Yu CW, Chen CY, Tai R, Wu K. Transcriptional repression by histone deacetylases in plants. Mol Plant 2014; 7:764-72; PMID:24658416; http://dx.doi.org/ 10.1093/mp/ssu033 [DOI] [PubMed] [Google Scholar]
- 22.Ma X, Lv S, Zhang C, Yang C. Histone deacetylases and their functions in plants. Plant Cell Rep 2013; 32:465-78; PMID:23408190; http://dx.doi.org/ 10.1007/s00299-013-1393-6 [DOI] [PubMed] [Google Scholar]
- 23.Wang Z, Cao H, Chen F, Liu Y. The roles of histone acetylation in seed performance and plant development. Plant Physiol Biochem 2014; 84C:125-33; http://dx.doi.org/ 10.1016/j.plaphy.2014.09.010 [DOI] [PubMed] [Google Scholar]
- 24.Kim JM, To TK, Seki M. An epigenetic integrator: new insights into genome regulation, environmental stress responses and developmental controls by histone deacetylase 6. Plant & cell physiology 2012; 53:794-800; PMID:22253092; http://dx.doi.org/ 10.1093/pcp/pcs004 [DOI] [PubMed] [Google Scholar]
- 25.Kim JA, Hsu JY, Smith MM, Allis CD. Mutagenesis of pairwise combinations of histone amino-terminal tails reveals functional redundancy in budding yeast. Proc Natl Acad Sci U S A 2012; 109:5779-84; PMID:22451923; http://dx.doi.org/ 10.1073/pnas.1203453109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma XJ, Wu J, Altheim BA, Schultz MC, Grunstein M. Deposition-related sites K5/K12 in histone H4 are not required for nucleosome deposition in yeast. Proc Natl Acad Sci U S A 1998; 95:6693-8; PMID:9618474; http://dx.doi.org/ 10.1073/pnas.95.12.6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A 2005; 102:5501-6; PMID:15795371; http://dx.doi.org/ 10.1073/pnas.0500136102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vaquero A, Sternglanz R, Reinberg D. NAD+-dependent deacetylation of H4 lysine 16 by class III HDACs. Oncogene 2007; 26:5505-20; PMID:17694090; http://dx.doi.org/ 10.1038/sj.onc.1210617 [DOI] [PubMed] [Google Scholar]
- 29.Shia WJ, Pattenden SG, Workman JL. Histone H4 lysine 16 acetylation breaks the genome's silence. Genome Biol 2006; 7:217; PMID:16689998; http://dx.doi.org/ 10.1186/gb-2006-7-5-217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suka N, Luo K, Grunstein M. Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin. Nat Genet 2002; 32:378-83; PMID:12379856; http://dx.doi.org/ 10.1038/ng1017 [DOI] [PubMed] [Google Scholar]
- 31.Kimura A, Umehara T, Horikoshi M. Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet 2002; 32:370-7; PMID:12410229; http://dx.doi.org/ 10.1038/ng993 [DOI] [PubMed] [Google Scholar]
- 32.Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, Peterson CL. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 2006; 311:844-7; PMID:16469925; http://dx.doi.org/ 10.1126/science.1124000 [DOI] [PubMed] [Google Scholar]
- 33.Johnson LM, Kayne PS, Kahn ES, Grunstein M. Genetic evidence for an interaction between SIR3 and histone H4 in the repression of the silent mating loci in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A 1990; 87:6286-90; PMID:2201024; http://dx.doi.org/ 10.1073/pnas.87.16.6286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shogren-Knaak M, Peterson CL. Switching on chromatin: mechanistic role of histone H4-K16 acetylation. Cell Cycle 2006; 5:1361-5; PMID:16855380; http://dx.doi.org/ 10.4161/cc.5.13.2891 [DOI] [PubMed] [Google Scholar]
- 35.Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, Berger SL. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 2009; 459:802-7; PMID:19516333; http://dx.doi.org/ 10.1038/nature08085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akhtar A, Becker PB. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol Cell 2000; 5:367-75; PMID:10882077; http://dx.doi.org/ 10.1016/S1097-2765(00)80431-1 [DOI] [PubMed] [Google Scholar]
- 37.Taylor GC, Eskeland R, Hekimoglu-Balkan B, Pradeepa MM, Bickmore WA. H4K16 acetylation marks active genes and enhancers of embryonic stem cells, but does not alter chromatin compaction. Genome Res 2013; 23:2053-65; PMID:23990607; http://dx.doi.org/ 10.1101/gr.155028.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Horikoshi N, Kumar P, Sharma GG, Chen M, Hunt CR, Westover K, Chowdhury S, Pandita TK. Genome-wide distribution of histone H4 Lysine 16 acetylation sites and their relationship to gene expression. Genome Integr 2013; 4:3; PMID:23587301; http://dx.doi.org/ 10.1186/2041-9414-4-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, et al.. Comparative analysis of metazoan chromatin organization. Nature 2014; 512:449-52; PMID:25164756; http://dx.doi.org/ 10.1038/nature13415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, et al.. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet 2008; 40:897-903; PMID:18552846; http://dx.doi.org/ 10.1038/ng.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhong X, Hale CJ, Law JA, Johnson LM, Feng S, Tu A, Jacobsen SE. DDR complex facilitates global association of RNA polymerase V to promoters and evolutionarily young transposons. Nat Struct Mol Biol 2012; 19:870-5; PMID:22864289; http://dx.doi.org/ 10.1038/nsmb.2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang X, Bernatavichute YV, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol 2009; 10:R62; PMID:19508735; http://dx.doi.org/ 10.1186/gb-2009-10-6-r62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luo C, Sidote DJ, Zhang Y, Kerstetter RA, Michael TP, Lam E. Integrative analysis of chromatin states in Arabidopsis identified potential regulatory mechanisms for natural antisense transcript production. Plant J 2013; 73:77-90; PMID:22962860; http://dx.doi.org/ 10.1111/tpj.12017 [DOI] [PubMed] [Google Scholar]
- 44.Xu L, Zhao Z, Dong A, Soubigou-Taconnat L, Renou JP, Steinmetz A, Shen WH. Di- and tri- but not monomethylation on histone H3 lysine 36 marks active transcription of genes involved in flowering time regulation and other processes in Arabidopsis thaliana. Mol Cell Biol 2008; 28:1348-60; PMID:18070919; http://dx.doi.org/ 10.1128/MCB.01607-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacob Y, Stroud H, Leblanc C, Feng S, Zhuo L, Caro E, Hassel C, Gutierrez C, Michaels SD, Jacobsen SE. Regulation of heterochromatic DNA replication by histone H3 lysine 27 methyltransferases. Nature 2010; 466:987-91; PMID:20631708; http://dx.doi.org/ 10.1038/nature09290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stroud H, Otero S, Desvoyes B, Ramirez-Parra E, Jacobsen SE, Gutierrez C. Genome-wide analysis of histone H3.1 and H3.3 variants in Arabidopsis thaliana. Proc Natl Acad Sci U S A 2012; 109:5370-5; PMID:22431625; http://dx.doi.org/ 10.1073/pnas.1203145109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yelagandula R, Stroud H, Holec S, Zhou K, Feng S, Zhong X, Muthurajan UM, Nie X, Kawashima T, Groth M, et al.. The histone variant H2A.W defines heterochromatin and promotes chromatin condensation in Arabidopsis. Cell 2014; 158:98-109; PMID:24995981; http://dx.doi.org/ 10.1016/j.cell.2014.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coleman-Derr D, Zilberman D. Deposition of histone variant H2A.Z within gene bodies regulates responsive genes. PLoS Genet 2012; 8:e1002988; PMID:23071449; http://dx.doi.org/ 10.1371/journal.pgen.1002988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kumar SV, Wigge PA. H2A.Z-containing nucleosomes mediate the thermosensory response in Arabidopsis. Cell 2010; 140:136-47; PMID:20079334; http://dx.doi.org/ 10.1016/j.cell.2009.11.006 [DOI] [PubMed] [Google Scholar]
- 50.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 2008; 456:125-9; PMID:18815594; http://dx.doi.org/ 10.1038/nature07324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schmid M, Davison TS, Henz SR, Pape UJ, Demar M, Vingron M, Scholkopf B, Weigel D, Lohmann JU. A gene expression map of Arabidopsis thaliana development. Nat Genet 2005; 37:501-6; PMID:15806101; http://dx.doi.org/ 10.1038/ng1543 [DOI] [PubMed] [Google Scholar]
- 52.Hausser J, Strimmer K. Entropy Inference and the James-Stein Estimator, with Application to Nonlinear Gene Association Networks. J Mach Learn Res 2009; 10:1469-84 [Google Scholar]
- 53.Luo C, Lam E. ANCORP: a high-resolution approach that generates distinct chromatin state models from multiple genome-wide datasets. Plant J 2010; 63:339-51; PMID:20444227; http://dx.doi.org/ 10.1111/j.1365-313X.2010.04236.x [DOI] [PubMed] [Google Scholar]
- 54.He G, Zhu X, Elling AA, Chen L, Wang X, Guo L, Liang M, He H, Zhang H, Chen F, et al.. Global epigenetic and transcriptional trends among two rice subspecies and their reciprocal hybrids. The Plant cell 2010; 22:17-33; PMID:20086188; http://dx.doi.org/ 10.1105/tpc.109.072041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev 1998; 12:599-606; PMID:9499396; http://dx.doi.org/ 10.1101/gad.12.5.599 [DOI] [PubMed] [Google Scholar]
- 56.Natsume-Kitatani Y, Shiga M, Mamitsuka H. Genome-wide integration on transcription factors, histone acetylation and gene expression reveals genes co-regulated by histone modification patterns. PloS one 2011; 6:e22281; PMID:21829453; http://dx.doi.org/ 10.1371/journal.pone.0022281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ren XY, Vorst O, Fiers MW, Stiekema WJ, Nap JP. In plants, highly expressed genes are the least compact. Trends Genet 2006; 22:528-32; PMID:16934358; http://dx.doi.org/ 10.1016/j.tig.2006.08.008 [DOI] [PubMed] [Google Scholar]
- 58.Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Ann Rev Biochem 2007; 76:75-100; PMID:17362198; http://dx.doi.org/ 10.1146/annurev.biochem.76.052705.162114 [DOI] [PubMed] [Google Scholar]
- 59.Li X, Qian W, Zhao Y, Wang C, Shen J, Zhu JK, Gong Z. Antisilencing role of the RNA-directed DNA methylation pathway and a histone acetyltransferase in Arabidopsis. Proc Natl Acad Sci U S A 2012; 109:11425-30; PMID:22733760; http://dx.doi.org/ 10.1073/pnas.1208557109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al.. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 2011; 471:480-5; PMID:21179089; http://dx.doi.org/ 10.1038/nature09725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krawetz SA, Kramer JA, McCarrey JR. Reprogramming the male gamete genome: a window to successful gene therapy. Gene 1999; 234:1-9; PMID:10393233; http://dx.doi.org/ 10.1016/S0378-1119(99)00147-X [DOI] [PubMed] [Google Scholar]
- 62.Milon B, Sun Y, Chang W, Creasy T, Mahurkar A, Shetty A, Nurminsky D, Nurminskaya M. Map of open and closed chromatin domains in Drosophila genome. BMC genomics 2014; 15:988; PMID:25407537; http://dx.doi.org/ 10.1186/1471-2164-15-988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, et al.. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science 2010; 330:1775-87; PMID:21177976; http://dx.doi.org/ 10.1126/science.1196914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell 2007; 129:823-37; PMID:17512414; http://dx.doi.org/ 10.1016/j.cell.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 65.Fraga MF, Ballestar E, Villar-Garea A, Boix-Chornet M, Espada J, Schotta G, Bonaldi T, Haydon C, Ropero S, Petrie K, et al.. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet 2005; 37:391-400; PMID:15765097; http://dx.doi.org/ 10.1038/ng1531 [DOI] [PubMed] [Google Scholar]
- 66.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature methods 2012; 9:357-9; PMID:22388286; http://dx.doi.org/ 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 2009; 25:1952-8; PMID:19505939; http://dx.doi.org/ 10.1093/bioinformatics/btp340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang T, Talbert PB, Zhang W, Wu Y, Yang Z, Henikoff JG, Henikoff S, Jiang J. The CentO satellite confers translational and rotational phasing on cenH3 nucleosomes in rice centromeres. Proceedings of the National Academy of Sciences of the United States of America 2013; 110:E4875-83; PMID:24191062; http://dx.doi.org/ 10.1073/pnas.1319548110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Du Z, Zhou X, Ling Y, Zhang Z, Su Z. agriGO: a GO analysis toolkit for the agricultural community. Nucleic acids research 2010; 38:W64-70; PMID:20435677; http://dx.doi.org/ 10.1093/nar/gkq310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Costas C, de la Paz Sanchez M, Stroud H, Yu Y, Oliveros JC, Feng S, Benguria A, Lopez-Vidriero I, Zhang X, Solano R, et al.. Genome-wide mapping of Arabidopsis thaliana origins of DNA replication and their associated epigenetic marks. Nat Struct Mol Biol 2011; 18:395-400; PMID:21297636; http://dx.doi.org/ 10.1038/nsmb.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.