

Abstract
PIWI-interacting RNAs (piRNAs) are responsible for maintaining the genome stability by silencing retrotransposons in germline tissues– where piRNAs were first discovered and thought to be restricted. Recently, novel functions were reported for piRNAs in germline and somatic cells. Using deep sequencing of small RNAs and CAGE of postnatal development of mouse brain, we identified piRNAs only in adult mouse brain. These piRNAs have similar sequence length as those of MILI-bound piRNAs. In addition, we predicted novel candidate regulators and putative targets of adult brain piRNAs.
PIWI-interacting RNAs (piRNAs) are short non-coding RNAs that protect the genome integrity by silencing transposable elements (TEs)1,2,3,4,5,6. They are scattered in genomic clusters that can span up to hundreds of kilobases, and sometimes overlap7. These piRNA clusters are transcribed as long single-stranded transcripts (piRNA precursors) by RNA POL II8. In postnatal testes, these precursors undergo primary processing9 in which they are exported from the nucleus into the cytoplasm where they are finally made into mature piRNAs with Uracil predominantly at their 5′ end7,10. Mature piRNAs then associate in a length-dependent manner with PIWI-like family proteins, such as MILI, MIWI and MIWI2 to create a mature piRNA-induced silencing complex (piRISC)7. Guided by the piRNA, piRISC seeks out complementary target sequences and effectively silences them through either post-transcriptional gene silencing11,12,13 or by inducing DNA methylation14,15.
In mouse, piRNAs can be subdivided into two groups according to the time of their peak expression; pre-pachytene and pachytene piRNAs. Pre-pachytene piRNAs are most abundant prior to the pachytene stage in meiosis but maintain a basal expression level even during later stages8. They are known to target TEs and cleave them using the slicer activity of the PIWI domain16. Whereas pachytene piRNAs reach their peak expression during the pachytene stage of meiosis and are more abundant8. They are primarily derived from intergenic regions and are reported to control expression of protein-coding genes17. However, their functions are yet to be fully determined.
Although piRNAs were previously thought to be exclusively found in germ cells, recent studies reported them in somatic cells such as follicle cells in fruit fly ovaries, principal/basal cells in macaque epididymis and tissues such as sea slug central nervous system, and rat cerebral cortex14,18,19,20,21. A recent effort to identify piRNAs in the mouse brain fell short when the reported sequences were compared with known gene annotation. It has been revealed that snoRNAs and other abundant RNAs were misclassified as piRNAs14,22. Saxena and colleagues observed a 1.9 increase in total piRNA of Mecp2 knockout mouse cerebellum when compared to wild type mouse. It also suggested that this increase might cause gene-specific mis-regulation in Rett syndrome, which is often associated with mutations in Mecp2 gene. However, the authors admitted that more in-depth analysis was required to corroborate their preliminary results23. Here we identified piRNAs in adult mouse brain that exhibit the hallmarks of piRNA such as length (24–31 bp) and 1U bias. Moreover, we demonstrate that these piRNAs are similar to MILI-bound piRNAs with regards to their length. Finally, we predicted novel candidate regulators and potential targets of piRNAs in adult mouse brain.
Results
piRNAs are expressed in mouse brain
To investigate whether piRNAs were present in mouse brain, we deep-sequenced small RNAs (enriching for piRNAs based on their size) from brain and testes tissues. The samples were taken at 10 days post-partum (dpp), 14 dpp, and adult stages. We obtained 199,003; 457,461; 529,081 and 223,341; 1,177,846; 8,359,064 reads for 10 dpp; 14 dpp; adult stages in brain and testes, respectively. Of these reads 161,869 (81%); 390,929 (85%); 453,456 (86%) and 213,740 (96%); 1,132,436 (96%); 8,302,184 (99%) mapped to the UCSC mouse genome release 9 (mm9) for brain and testes, respectively (Fig. 1a). We focused on previously defined 214 piRNA clusters8. Out of the mapped reads, 741 (0.46%) and 1,106 (0.28%) reads mapped within piRNA clusters at 10 dpp and 14 dpp in brain, respectively, in contrast to 31,398 (7%) reads in adult brain (Fig. 1a). As for testes samples, 58,835 (28%); 340,181 (30%) and 8,234,258 (99%) mapped within piRNA clusters for 10 dpp; 14 dpp and adult stages, respectively. This suggests that piRNAs are unlikely to be present at 10 and 14 dpp in brain. In fact, only reads from these two stages lack significant 1U bias characteristic of primary piRNAs (Fig. 1b).
Figure 1. Sequencing of piRNA in brain and testes.

(a) Bar plot shows the number of sequenced reads; reads that mapped to genome; and reads mapped within annotated piRNA clusters. (b) Sequence bias of reads obtained in each stage and tissue; unlike reads at adult stage in brain, reads at 10 dpp and 14 dpp lack significant 1U bias, all reads from testes samples showed significant 1U bias. Sequence logo was obtained using Weblogo68.
The expression of piRNAs in the central nervous system of adult mouse has been reported by Lee and colleagues22. However, other reports14 noted that those piRNAs were partial snoRNA. Saxena and colleagues23 also identified piRNA in mouse cerebellum. However, many of their most abundant piRNAs were previously reported by Lee et al.22. To avoid such a misclassification, we compared adult brain reads that mapped within piRNA clusters against annotated non-coding RNAs24,25,26 using BLAST27. Only 1.1% of the reads had perfect matches. This result, coupled with the facts that adult brain reads map within annotated piRNA clusters and show significant 1U bias, suggests that those reads are bona fide piRNAs.
piRNA clusters expressed in brain are not tissue specific
piRNA clusters exhibit distinct expression profiles across developmental stages in both tissues (Fig. 2a). For instance, 47 piRNA clusters were expressed (see Materials and Methods and Fig. S1) in the adult stage in brain and 139, 154, and 115 piRNA clusters were expressed in testes at 10 dpp, 14 dpp and adult stages, respectively.
Figure 2. Expression profile of piRNA clusters in brain and testes clusters.

(a) Heatmap showing expression profiles of piRNA clusters (rows); brain adult sample cluster with testes adult sample (based on Pearson correlation), suggesting they share expressed piRNA clusters; annotation at the left of heatmap shows that most piRNA clusters expressed in adult brain (BT clusters) are intergenic (b) boxplot showing distributions of coverage correlation that was computed along the length of expressed piRNA between all samples; high correlation indicate that piRNAs production along the length of piRNAS clusters is similar across all conditions.
Adult brain piRNAs were mostly produced by intergenic piRNA clusters (p-value < 0.002, one tailed fisher exact test) which dominate piRNA production in pachytene stage of spermatogenesis8 (Fig. 2a). Since all piRNA clusters expressed in adult brain (henceforth denoted as BT clusters) were expressed in adult testes, we sought to determine whether there were any tissue-specific piRNA-producing regions within the clusters. We examined the coverage of piRNAs along the length of BT clusters in both tissues at adult stage and did not find tissue-specific piRNA-producing regions (median Pearson correlation 0.87; Fig. 2b). Therefore, we expected BT clusters to produce similar piRNA populations in both tissues. Indeed, out of 14,978 unique piRNA sequences mapped within BT clusters 13,253 (88.5%) were also present in adult testes (exact sequence match). Whereas only 2,692 (18.0%) and 6,417 (42.8%) were present in testes at 10 dpp and 14 dpp. Overall, this suggests that BT clusters produce a subpopulation of adult testes piRNAs.
piRNAs in adult brain are similar to MILI-bound piRNAs
Adult testes piRNAs are composed of both MIWI- and MILI-bound piRNAs28, each of those proteins binds to a distinct sequence length of piRNAs7. Since adult brain piRNAs are a subpopulation of adult testes piRNAs, we sought to determine which of those two proteins might bind adult brain piRNAs. The length distribution of adult brain piRNAs peaks at 26~27 bps, which is the nominal length for MILI-bound piRNAs7. Similarly to the length distribution of piRNAs in the adult brain, the length distribution of testes piRNAs at 14 dpp also peaks at 26~27 bps. This has been attributed to the presence of MILI-bound piRNAs29. Whereas, in adult testes, the distribution peaks at 26~27 and 29~30 bps; the first peak corresponds to MILI-bound piRNAs and the second peak corresponds to MIWI-bound piRNAs7 (Fig. 3). These peaks and their associations to PIWI-like proteins are consistent with publicly available immuno-precipitation (IP) data (Supplementary Fig. S2). Although adult brain piRNAs are similar to MILI-bound piRNAs with regards to their length, we could not detect an appreciable Mili expression (normalized expression ≤0.5) in adult brain based on our CAGE expression data nor through independent sources30,31.
Figure 3. piRNAs in adult brain are similar be MILI-bound.
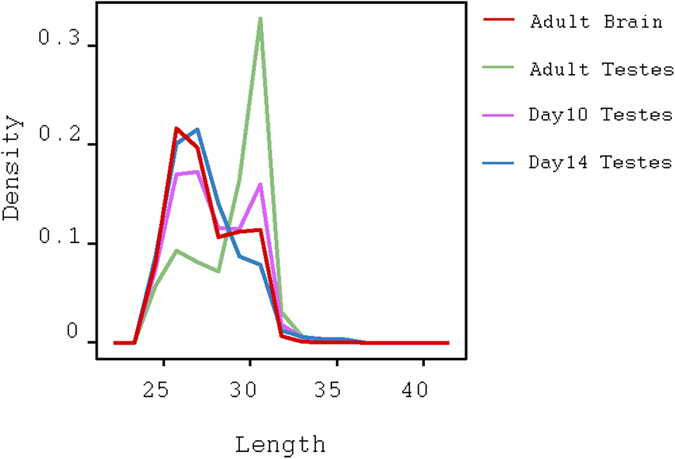
(a) Length distribution of piRNAs at adult brain reveals a peak at 26~27 bases which is commonly associated with MILI-binding. Consistent with current knowledge, A high peak at 29~31 bases in adult testes and a smaller peak at 26~27 bases are associated with MIWI- and MILI-binding, respectively.
ZIC2 and MEIS1 may regulate transcription of piRNA clusters in adult brain
MYBL1 is necessary for the transcription of pachytene piRNA clusters in mouse testes8. In fact, MYBL1 binds to the promoters of 106 piRNA clusters in adult testes, including 46 BT piRNA clusters. However, our CAGE expression data shows that Mybl1 is lowly expressed (see Materials and Methods) in adult brain (Fig. 4a). This is consistent with data obtained from other studies31 as well as Allen Brain Atlas30. Moreover, only a subset of its target piRNA clusters was expressed in adult brain. Thus, we suspect that other factors, alongside MYBL1, control the expression of this subset. Here, we considered two possible scenarios: either a transcription factor (TF) is activating only BT clusters (Fig. 4b) or a TF repressing piRNA clusters not expressed in adult brain but expressed in adult testes (non-BT clusters) (Fig. 4c).
Figure 4. Identification of candidate regulators of BT piRNA clusters.

(a) Expression profile of Mybl1. (b) Diagram depicting the first possible scenarios in which BT clusters are expressed due to an activator TF. (c) Similar to (b) but for second scenario, in which non-BT clusters are silenced in adult brain due to a repressor TF. (d) Expression profile of candidate TFs that fit first scenario. (e) Expression profile of candidate TFs that fit the second scenario. (f) Co-expression network obtained from GeneMANIA63 showing that many candidates co-express with Mybl1; TFs were classified as activator or a repressor based on our suggested scenarios as their assumed function in the scenarios does not contradict with known literature about their function.
To determine the likely scenario, we used DREME32 in conjunction with TOMTOM33 to identify discriminatory transcription factor binding sites (TFBFs) between the promoters of BT and non-BT clusters. Initially, we identified 162 discriminatory TFs. Using CAGE to determine their expression profiles and UniProt34 to determine their functional annotation (repression or activation), we filtered them to five candidate TFs (see Materials and Methods and Supplementary Table S1): two were unique to BT clusters, namely, MEIS1 and SOX4; three were unique to non-BT clusters, namely, ZIC1, ZIC2 and EOMES (Fig. 4d,e). All TFs were implicated in neuron differentiation and development35,36,37,38.
Although ZIC2 can act as both repressor and activator, as a repressor it fits the second scenario. Indeed, based on public Chromatin Immunoprecipitation Sequencing (ChIP-Seq) data, ZIC2 binds the promoters of non-BT clusters in adult cerebellum39. Unfortunately, we did not find data for the other TFs related to their binding in adult brain. Interestingly, Zic2, Eomes and Meis1 co-express with Mybl1 (Fig. 4f). Therefore, we predict that MEIS1, EOMES and specifically ZIC2 are likely candidates for the regulation of piRNA cluster transcription.
Prediction of targets of adult brain piRNAs
In addition to the well-established role of piRNAs in TE-silencing, several studies have suggested that piRNAs may be involved in the regulation of genes in several species14,17,40,41. In order to investigate whether adult brain piRNAs were involved in mRNA deadenylation, we adapted the methodology described by17 and took into account only the top-scoring predicted gene target for each piRNA according to miRanda42 (Supplementary Table S2). Considering that putative gene targets should be down-regulated when compared to non-target genes if adult brain piRNAs were involved in mRNA deadenylation, we performed Mann Whitney test on the non-zero fold change, as calculated by GFOLD43, of the genes between 14 dpp and adult stages in brain (see Materials and Methods). Although there was no significant divergence in differential expression between predicted target genes and non-target genes (p-value 0.5122) (Supplementary Fig. S3), we cannot deny the possibility that a few adult brain piRNAs may be involved in small-scale regulation of target mRNAs.
A more recent study reported an alternative approach to identify piRNA targets44. This approach relied on a specific signature of partial complementarity of piRNAs to their putative targets (see Materials and Methods). Using this specific signature on our adult brain piRNAs, we were able to identify 41 potential mRNA targets of piRNAs (Supplementary Table S3). Using CPDB45, Gene Ontology (GO) enrichment for the putative mRNA targets revealed they were significantly (p-value ≤ 0.001) associated with pigmentation. Moreover, these putative targets were significantly (p-value ≤ 0.001) associated with Cholinergic synapse pathway. Applying the same signature on repeat elements, we identified 7,565 putative targets (Supplementary Table S4). Of which, 3,081 were short interspersed nucleotide elements (SINEs); 1,151 were long terminal repeats (LTRs); 450 were long interspersed nucleotide elements (LINEs). GO enrichment for repeat element targets revealed their significant (p-value ≤ 0.001) association with cardiac neural crest cell development involved in outflow tract morphogenesis based on GREAT46. In conclusion, these in silico predictions of putative targets should help guide future in vitro experimental validation.
Discussion
Here, we characterize piRNA in mouse brain throughout postnatal development and show that piRNAs are only present in the adult stage of brain development. These piRNAs display significant 1U bias and are found in an intergenic subset of previously annotated piRNA clusters8.
piRNAs bind to PIWI-like proteins according to their sequence length7. The sequence length of adult brain piRNAs peaks at 26~27 bases, which is a characteristic of piRNAs that bind MILI. However, the expression of PIWI-like genes– including Mili– were absent in adult brain. Since piRNAs function as part of a complex with PIWI-like proteins, it is unclear how these piRNAs function in the adult brain. One hypothesis is that piRNAs associate with a different protein; another is that Mili is expressed in a unique cell type that is hard to detect using whole brain sequencing.
MYBL1 is a key regulator of adult testes piRNA clusters8. In the brain, we showed that Mybl1 was solely expressed in adult stage based on our CAGE expression data. However, despite its expression, only a subset of adult testes piRNA clusters was expressed. This suggests that TFs other than MYBL1 may be implicated in the regulation of piRNA clusters in the brain. Consequently, we identified five TFs that may regulate piRNA clusters’ expression alongside MYBL1. Three of them co-express with Mybl1. One candidate is ZIC2, which is involved in neurogenesis36,47. Zic2 is widely expressed in adult brain48 and binds promoters of non-BT piRNA clusters in adult cerebellum39. Thus, we suspect that ZIC2 may repress those piRNA clusters. Another candidate is MEIS1, an activator TFs, which has binding sites only in promoters of piRNA clusters expressed in adult brain. In conclusion, we predicted novel candidate regulators of piRNA clusters for future validation.
To determine whether piRNAs in adult brain were involved in mRNA deadenylation, we used miRanda42 to identify piRNA targets as previously described17. However, these predicted targets were not more likely to be down-regulated when compared to non-target genes (Mann-Whitney test; p-value 0.5122).
A more recent study reported a specific targeting signature of adult testes piRNAs44. Based on this signature, we predicted 41 candidate mRNA targets and 7,565 repeat element targets. GO enrichment of candidate mRNA and repeat element targets were associated with pigmentation, and cardiac neural crest development, respectively. In mouse embryo, cardiac neural crest stem cells were shown to be able to differentiate into pigment cells49. Furthermore, the predicted mRNA targets were also associated with cholinergic synapse pathway.
In adult testes, L1 elements are repressed through multiple mechanisms, including piRNAs50. Furthermore, L1 elements are derepressed in mice with mutant MILI11. Although L1 elements are active during brain development51, how they are controlled in brain and whether piRNAs play a role in their regulation is still unclear52. Therefore, future functional analysis is required.
In conclusion, we described the developmental expression of piRNA in postnatal mouse brain. We showed that these piRNAs are similar to MILI-bound piRNAs in terms of their length and suggested new candidate regulators of those piRNAs. Although a deeper investigation into piRNAs in the brain is required, we believe the data and results described here provide new insights and a valuable resource for the small-RNA community.
Materials and Methods
RNA Extraction
Total RNA was extracted using RNA-Bee (AMS Biotechnology) according to the manufacturer’s instructions, from pools of whole brain and pools of testis of male C57Bl/6 mice, sacrificed through cervical displacement, at 10 dpp, 14 dpp and adult, in accordance with the approved guidelines. All experimental protocols were approved by the Roslin Institute. Quality and quantity of the total RNA was measured by Nanodrop spectrophotometer and Bioanalyzer RNA chips (Agilent). For sequencing library preparation, low molecular weight RNA (<40 nucleotides long) was isolated from the total RNA using a FlashPAGE fractionator (Life Technologies).
Preparation of RNA-Seq library
50 ng of fractionated small RNA from brain and testes at 10 dpp, 14 dpp and adult was tagged and used to generate cDNA libraries according to Kawano et al.53. Briefly, adenylated 3′ adapters were ligated to 3′ end of small RNAs using a truncated RNA ligase enzyme followed by 5′ adaptor ligation to 5′-monophosphate ends using RNA ligase enzyme, ensuring specific ligation of non-degraded small RNAs. cDNA was prepared using a primer specific to the 3′ adaptor in the presence of dimer eliminator and amplified for 14 PCR cycles using a special forward primer targeting the 5′ adaptor containing additional sequence for sequencing and a reverse primer targeting the 3′ adaptor. The amplified libraries which contained piRNA and sequencing linkers were run on a 6% polyacrylamide gel and then the 80–84 bp bands (which correspond to inserts of 26–32 nucleotides cDNAs) were extracted by gel extraction protocols (QIAGEN). Libraries were sequenced after quality check on a Bioanalyzer DNA 1,000 chip (Agilent).
Preprocessing of RNA-Seq tags
Multiplexed barcode sequencing for three pools during three postnatal developmental stages from two tissues was performed on RNA-Seq tags using Illumina GA-IIX (40 bp single end tags), where the barcode and the adapter were ligated to the 5′ end and 3′ end, respectively. In order to retain high quality RNA-Seq reads, we trimmed all bases, including bases for barcode and 3′ adapter, which had a quality score ≤15 as well as all subsequent bases for each read. In order to extract the endogenous sequences from the RNA-Seq reads, we stripped the four base 5′ barcodes as well as at least one base of 3′ adapter. Any read, which was not stripped, was discarded, resulting in endogenous sequences with a maximum length of 35 bases. Since piRNA size ranges from 26 to 31 bases1, we discarded all sequences whose length was ≤24 bases. Finally, for each developmental stage in each tissue we created a single dataset by concatenating all the sequences from the three pools together. We retained duplicate reads. Subsequently, we aligned the sequences on the non-repeat-masked UCSC release 9 of the mouse genome (MM9)54 using bowtie2 v2.2.555 using the sensitive preset option and allowed a maximum 100 alignments. All the reads that aligned to the genome were retained and used for subsequent analysis.
Comparison against non-coding RNA
We compared adult brain reads that mapped within annotated piRNA clusters to known non-coding RNAs. These non-coding RNAs include NONCODE v3.0 snoRNA24, UCSC tRNA25, miRBase v21 miRNA26 and the NCBI complete ribosomal DNA unit. The comparison was performed using NCBI BLASTN v2.2.31+27. Except for “-task blastn”, Default parameters were used.
piRNA Cluster expression
The expression of each piRNA cluster was based on the number of reads mapped within the piRNA cluster keeping multi-mapping reads. We used RPKM (Reads Per Kilobase per Million mapped reads) for normalization. In order to distinguish between expressed and non-expressed piRNA clusters, we observe the following criteria: a piRNA cluster must have at least 100 mapped reads; the RPKM expression level of piRNA cluster must be at least 10; the reads must map within the entire piRNA cluster and not concentrated in a small region, to reduce the effect of PCR duplicates, this was done by requiring that the maximum depth of the reads divided by the total number of mapped reads per piRNA cluster be less than 0.9. Based on these criteria, we designate clusters as expressed or not for each developmental stage in each tissue independently (Supplementary Fig. S1).
Preparation of CAGEscan library
CAGEscan is an enhancement upon CAGE (Cap Analysis of Gene Expression) which uses paired-end sequencing approach to sequence an additional random block in the 3′ direction of the same transcript. The preparation of the CAGEscan library was adapted from published protocol56 and modified to work with Illumina GA-IIX 36 cycles paired end CAGEscan read sequencer. The first-strand cDNAs were created using 500 μg brain and testes RNA of male C57Bl/6 mice, sacrificed through cervical displacement, with 2 μl of 0.66 M D-threalose, 3.3 M D-sorbitol57, 100 μM template switching oligonucleotide (5′-TAGTCGAACTGAAGGTCTCCAGCArGrGrG), 10 μM random reverse-transcription primer with a random pentadecamer tail (5′-GTACCAGCAGTAGTCGAACTGAAGGTCTCCTCTN15). The mixed solution was reduced in volume to 2 μl in a centrifugal evaporator at room temperature then heated for denaturizing at 65 °C for 10 min and transferred quickly on an ice-water mix. Reverse transcription was accomplished in a volume of 10 μl with the following components: 1.25 × first-strand buffer, 650 μM dNTPs, 1.3 mM DTT, 925 mM betain and 200 units SuperScript II, and the reaction was incubated at 22 °C for 10 min, 50 °C for 30 min, 75 °C for 15 min. Synthesized cDNAs were purified with Agencourt RNAClean XP kit following the manufacturer’s instructions. The purified cDNAs were eluted to 40 μl water then synthesized the second strand cDNA and the resulting product by semisuppressive PCR58. The determination of the optimal PCR cycle number in pilot reactions was used quantitative PCR: 0.15 × purified cDNA, 1 × SYBR Premix Ex Taq (TaKaRa), 100 mM semisuppressive forward primer (5′- TAGTCGAACTGAAGGTCTCCAGC) and 100 mM semisuppressive reverse primer (5′-TGACGTCGTCTAGTCGAACTGAAGGTCTCCGAACC) with a StepOne Real-Time PCR system. The thermal cycling program was: 5 min 95 °C and 40 × (15 s at 95 °C, 10 s at 65 °C, 2 min at 68 °C). After determination, PCR was performed on a large-scale hot-start PCR with 3 μl of purified first-strand cDNA as template in a final volume of 50 μl using a reaction mixture containing 1.25 U TaKaRa Ex Taq HS (TaKaRa), 200 μM each dNTP mixture (TaKaRa), 1 × Ex Taq buffer (TaKaRa), 100 mM semisuppressive forward primer and 100 mM semisuppressive reverse primer with the following thermal cycling program: 5 min 95 °C and 17–19 cycles × (15s at 95 °C, 10 s at 65 °C, 2 min at 68 °C). The amplified DNA was purified with Agencourt AMPure XP kit then measured the concentration by NanoDrop 1000.
To create adaptor sequences for Illumina GA-IIX sequencer, semisuppressive PCR cDNA was amplified using 20 ng DNA in a final volume of 50 μl with a mixture containing 1.25 U Ex Taq HS (TaKaRa), 200 μM each dNTP mixture (TaKaRa), 1 × Ex Taq buffer (TaKaRa), 200 mM forward primer (5′–AATGATACGGCGACCACCGAGATCTACACTAGTCGAACTGAAGG) and 200 mM reverse primer (5′–CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT) following thermal cycling program: 1 min 95 °C, 15 s at 95 °C, 10 s at 55 °C, 2 min at 68 °C and 6 cycles × (15 s at 95 °C, 10 s at 65 °C, 2 min at 68 °C). The amplified DNA was purified with Agencourt AMPure XP kit then measured the concentration by Agilent High Sensitivity DNA kit and adjusted 16 pM final concentration (from 200 bp–700 bp) on GAIIx. CAGEscan tags were amplified with Cluster generation kit with modified sequence primer (5′–TAGTCGAACTGAAGGTCTCCAGCA) following Cluster generation protocol (Illumina) then generated clusters were sequenced with 36 cycle paired end CAGEscan read sequencing kit by Illumina GA-IIX.
Preprocessing of CAGEscan tags
Multiplexed barcode sequencing for three pools during three postnatal developmental stages from two tissues was performed in CAGEscan tags using Illumina GA-IIX (105 bp paired end tags), where the adapter followed by six bases of barcode and three consecutive guanine bases were ligated to the 5′ end of the forward mate while only the adapter was ligated to the 5′ end of the reverse mate. After removing any partial 5′ and 3′ adapters for both mates, we trimmed all bases which had a quality score ≤15 as well as all subsequent bases. Afterwards, we removed the nine bases comprising the barcode and the three guanine bases from the 5′ end of the forward mate. We then discarded any read whose length was ≤15 bases.
We mapped the remaining paired-end sequences onto MM9 using bowtie2 v2.2.555, using the sensitive preset option, in addition to Phred64 quality scores. We considered dove-tail sequences to be discordant pairs and we retained non-discordant sequences.
Gene expression analysis
We used the R package CAGEr v1.4.159, allowing any mapping quality, to retrieve CTSSs. Then we normalized the expression of each of the CTSS according to power-law normalization60 in transcripts per million (tpm). To obtain expression value for each gene, we summed the tpm for each unique CTSS within the one Kb upstream and 0.5 Kb downstream of all its transcription starting sites. Any gene whose normalized expression value was ≥0.5 was designated as expressed. Differential gene expression was performed using GFOLD43 v1.1.2 using 0.01 as cutoff for the False Discovery Rate (FDR).
Prediction of discriminatory regulatory TFs
In order to investigate the TFs that may be regulating the intergenic piRNA clusters expressed in adult brain, we used DREME v4.10.132, using default parameters, to find significantly overrepresented (E-value ≤0.05) discriminatory motifs that are unique to piRNA clusters expressed in adult brain and motifs that are unique to piRNA clusters expressed in adult testes excluding those expressed in adult brain. Then we compared the motifs against mouse UniPROBE61 and JASPAR62 vertebrate using TOMTOM33, with default parameters, to identify the TFs with binding sites most similar to those specific motifs. Using a normalized expression cutoff of 0.5 tpm, we required that all TFs be higher than this threshold in adult brain. Furthermore, TFs whose binding sites were overrepresented in promoters of adult brain piRNA clusters were required to be expressed above this threshold for all testes developmental stages. Whereas, TFs whose binding sites are overrepresented in the subset of piRNA clusters expressed in adult testes but not in adult brain were required to be expressed no more than that threshold on all testes developmental stages. Finally, we retained only mouse TFs whose expression patterns are consistent with the two proposed scenarios as mentioned above. Finally, we used UniProt34 to determine whether a TF was an activator or a repressor. We used GeneMANIA63 database to visualize co-expression between the candidate TFs.
piRNA Target Prediction
In order to determine whether piRNAs in adult brain were involved in mRNA deadenylation as shown previously in testes, we adopted a similar methodology to that described by Gou et al.17. Briefly, we considered all expressed (normalized CAGE expression ≥0.5) genes at either 14 dpp or adult stages in brain as potential gene targets of any adult brain piRNA. Using miRanda42 v3.3a to match piRNAs to their targets, we set a more stringent score threshold, 160, for reporting hits in contrast to 150 used originally by Gou et al.17. Then for each piRNA, we considered only the top-scoring gene target. The Differential expression was obtained from the generalized fold change as given by GFOLD43 for each gene between 14 dpp and adult stages in brain. To determine whether predicted target genes were more likely to be down-regulated than non-target genes, we selected all the genes that exhibited any differential expression, up or down. The distribution of non-zero GFOLD values of target genes was then compared to the distribution of non-zero GFOLD values of non-target genes using Mann-Whitney test.
Alternatively, we also used the method introduced by Goh et al.44 that takes advantage of unique signatures of piRNA targeting. This signature relies on the divergent partial complementarity of sense piRNAs and guide piRNAs. Sense piRNAs were aligned to the genome using bowtie v1.1.164 with parameters “–a –v 0”. Guide piRNAs were mapped also using bowtie v1.1.164 with parameters “-a -n 0 -l 10 -e 160”. We used SAMtools65 to ensure partial complementarity. Since this method applies to both mRNA and repeats, we also investigated any transposable element, whose length was 75% of the consensus length as determined by HOMER66. Using BEDTools67, any mRNA or repeat element which overlapped the piRNA targeting signature was designated as potential target of piRNAs.
Coverage of piRNAs along the length of piRNA clusters
Each expressed piRNA cluster was divided into non-overlapping windows of 100 bases. Coverage was determined based on the percentage of total reads that mapped within each window for each piRNA cluster to the total reads that mapped within the entire piRNA cluster. This was done for all expressed piRNA clusters in each developmental stage for each tissue independently. We then tested the Pearson correlation of the coverage, as previously defined, of the same expressed piRNA cluster across two samples for each of the expressed piRNA clusters common to both samples. For the correlation distribution, we only retained significant (p-value ≤ 0.001) correlations.
Additional Information
Accession Codes: The data is available under the Bioproject ID SRP073223.
How to cite this article: Ghosheh, Y. et al. Characterization of piRNAs across postnatal development in mouse brain. Sci. Rep. 6, 25039; doi: 10.1038/srep25039 (2016).
Supplementary Material
Acknowledgments
Y.G., L.S., T.R.1, V.O. and T.R.2 are supported by King Abdullah University of Science and Technology. Research Grant from the Japanese Ministry of Education, Culture, Sports, Science and Technology (MEXT) to the RIKEN Center for Life Science Technologies. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Author Contributions Conceived and designed the study: T.R.2 (Timothy Ravasi), P.C. and V.O. Performed the wet lab experiments: H.T. Preprocessed the data: Y.G. Analyzed the data: Y.G., L.S. and T.R.1 (Taewoo Ryu). Prepared the figures: Y.G. and L.S. Wrote the manuscript: Y.G., L.S. and H.T. All authors reviewed the manuscript.
References
- Aravin A. et al. A novel class of small RNAs bind to MILI protein in mouse testes. Nature 442, 203–207, 10.1038/nature04916 (2006). [DOI] [PubMed] [Google Scholar]
- Betel D., Sheridan R., Marks D. S. & Sander C. Computational analysis of mouse piRNA sequence and biogenesis. PLoS Comput Biol 3, e222, 10.1371/journal.pcbi.0030222 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennecke J. et al. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell 128, 1089–1103, 10.1016/j.cell.2007.01.043 (2007). [DOI] [PubMed] [Google Scholar]
- Girard A., Sachidanandam R., Hannon G. J. & Carmell M. A. A germline-specific class of small RNAs binds mammalian Piwi proteins. Nature 442, 199–202, 10.1038/nature04917 (2006). [DOI] [PubMed] [Google Scholar]
- Grivna S. T., Beyret E., Wang Z. & Lin H. A novel class of small RNAs in mouse spermatogenic cells. Genes Dev 20, 1709–1714, 10.1101/gad.1434406 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T. et al. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev 20, 1732–1743, 10.1101/gad.1425706 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siomi M. C., Sato K., Pezic D. & Aravin A. A. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol 12, 246–258, 10.1038/nrm3089 (2011). [DOI] [PubMed] [Google Scholar]
- Li X. Z. et al. An ancient transcription factor initiates the burst of piRNA production during early meiosis in mouse testes. Mol Cell 50, 67–81, 10.1016/j.molcel.2013.02.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyret E., Liu N. & Lin H. piRNA biogenesis during adult spermatogenesis in mice is independent of the ping-pong mechanism. Cell Res 22, 1429–1439, 10.1038/cr.2012.120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto Y. et al. Targeted gene silencing in mouse germ cells by insertion of a homologous DNA into a piRNA generating locus. Genome Res. 10.1101/gr.137224.112 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Fazio S. et al. The endonuclease activity of Mili fuels piRNA amplification that silences LINE1 elements. Nature 480, 259–263, 10.1038/nature10547 (2011). [DOI] [PubMed] [Google Scholar]
- Lim A. K., Tao L. & Kai T. piRNAs mediate posttranscriptional retroelement silencing and localization to pi-bodies in the Drosophila germline. J Cell Biol 186, 333–342, 10.1083/jcb.200904063 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M. et al. Miwi catalysis is required for piRNA amplification-independent LINE1 transposon silencing. Nature 480, 264–267, 10.1038/nature10672 (2011). [DOI] [PubMed] [Google Scholar]
- Rajasethupathy P. et al. A role for neuronal piRNAs in the epigenetic control of memory-related synaptic plasticity. Cell 149, 693–707, 10.1016/j.cell.2012.02.057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T. et al. Role for piRNAs and noncoding RNA in de novo DNA methylation of the imprinted mouse Rasgrf1 locus. Science 332, 848–852, 10.1126/science.1203919 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M. & Wu Y. Fighting an old war with a new weapon–silencing transposons by Piwi-interacting RNA. IUBMB life 65, 739–747, 10.1002/iub.1192 (2013). [DOI] [PubMed] [Google Scholar]
- Gou L. T. et al. Pachytene piRNAs instruct massive mRNA elimination during late spermiogenesis. Cell Res 24, 680–700, 10.1038/cr.2014.41 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A., Nakka V. P. & Vemuganti R. Altered expression of PIWI RNA in the rat brain after transient focal ischemia. Stroke 42, 1105–1109, 10.1161/STROKEAHA.110.598391 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. et al. Collapse of germline piRNAs in the absence of Argonaute3 reveals somatic piRNAs in flies. Cell 137, 509–521, 10.1016/j.cell.2009.04.027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malone C. D. et al. Specialized piRNA pathways act in germline and somatic tissues of the Drosophila ovary. Cell 137, 522–535, 10.1016/j.cell.2009.03.040 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z. et al. Widespread expression of piRNA-like molecules in somatic tissues. Nucl Acids Res 39, 6596–6607, 10.1093/nar/gkr298 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E. J. et al. Identification of piRNAs in the central nervous system. RNA 17, 1090–1099, 10.1261/rna.2565011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena A., Tang D. & Carninci P. piRNAs warrant investigation in Rett Syndrome: an omics perspective. Disease markers 33, 261–275, 10.3233/DMA-2012-0932 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu D. et al. NONCODE v3.0: integrative annotation of long noncoding RNAs. Nucl Acids Res 40, D210–215, 10.1093/nar/gkr1175 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D. et al. The UCSC Table Browser data retrieval tool. Nucl Acids Res 32, D493–496, 10.1093/nar/gkh103 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A. & Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucl Acids Res 42, D68–73, 10.1093/nar/gkt1181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W. & Lipman D. J. Basic local alignment search tool. J Mol Biol 215, 403–410, 10.1016/S0022-2836(05)80360-2 (1990). [DOI] [PubMed] [Google Scholar]
- Vourekas A. et al. Mili and Miwi target RNA repertoire reveals piRNA biogenesis and function of Miwi in spermiogenesis. Nat Struct Mol Biol 19, 773–781, 10.1038/nsmb.2347 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S. et al. piRNA-triggered MIWI ubiquitination and removal by APC/C in late spermatogenesis. Developmental cell 24, 13–25, 10.1016/j.devcel.2012.12.006 (2013). [DOI] [PubMed] [Google Scholar]
- Sunkin S. M. et al. Allen Brain Atlas: an integrated spatio-temporal portal for exploring the central nervous system. Nucl Acids Res 41, D996–D1008, 10.1093/nar/gks1042 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Lu A. & Sharp F. R. Regional genome transcriptional response of adult mouse brain to hypoxia. BMC genomics 12, 499, 10.1186/1471-2164-12-499 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey T. L. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27, 1653–1659, 10.1093/bioinformatics/btr261 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S., Stamatoyannopoulos J. A., Bailey T. L. & Noble W. S. Quantifying similarity between motifs. Genome Biol 8, R24, 10.1186/gb-2007-8-2-r24 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. Activities at the Universal Protein Resource (UniProt). Nucl Acids Res 42, D191–198, 10.1093/nar/gkt1140 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsen G. E. et al. The protomap is propagated to cortical plate neurons through an Eomes-dependent intermediate map. Proc Natl Acad Sci USA 110, 4081–4086, 10.1073/pnas.1209076110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J., Inoue T., Hoshino J. & Mikoshiba K. Zic2 controls cerebellar development in cooperation with Zic1. J Neuroscience 22, 218–225 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Zhou Y., Barcarse E. A. & O’Gorman S. Altered neuronal lineages in the facial ganglia of Hoxa2 mutant mice. Dev Biol 314, 171–188, 10.1016/j.ydbio.2007.11.032 (2008). [DOI] [PubMed] [Google Scholar]
- Cheung M., Abu-Elmagd M., Clevers H. & Scotting P. J. Roles of Sox4 in central nervous system development. Brain Research 79, 180–191 (2000). [DOI] [PubMed] [Google Scholar]
- Frank C. L. et al. Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat Neuroscience 18, 647–656, 10.1038/nn.3995 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiuchi T. et al. A single female-specific piRNA is the primary determiner of sex in the silkworm. Nature 509, 633–636, 10.1038/nature13315 (2014). [DOI] [PubMed] [Google Scholar]
- Rouget C. et al. Maternal mRNA deadenylation and decay by the piRNA pathway in the early Drosophila embryo. Nature 467, 1128–1132, 10.1038/nature09465 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enright A. J. et al. MicroRNA targets in Drosophila. Genome Biol 5, R1, 10.1186/gb-2003-5-1-r1 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J. et al. GFOLD: a generalized fold change for ranking differentially expressed genes from RNA-seq data. Bioinformatics 28, 2782–2788, 10.1093/bioinformatics/bts515 (2012). [DOI] [PubMed] [Google Scholar]
- Goh W. S. et al. piRNA-directed cleavage of meiotic transcripts regulates spermatogenesis. Genes Dev 29, 1032–1044, 10.1101/gad.260455.115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A., Stelzl U., Lehrach H. & Herwig R. The Consensus Path DB interaction database: 2013 update. Nucl Acids Res 41, D793–800, 10.1093/nar/gks1055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean C. Y. et al. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501, 10.1038/nbt.1630 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aruga J. The role of Zic genes in neural development. Mol Cell Neurosci 26, 205–221, 10.1016/j.mcn.2004.01.004 (2004). [DOI] [PubMed] [Google Scholar]
- Brown L. & Brown S. Zic2 is expressed in pluripotent cells in the blastocyst and adult brain expression overlaps with makers of neurogenesis. GEP 9, 43–49, 10.1016/j.gep.2008.08.002 (2009). [DOI] [PubMed] [Google Scholar]
- Youn Y. H., Feng J., Tessarollo L., Ito K. & Sieber-Blum M. Neural crest stem cell and cardiac endothelium defects in the TrkC null mouse. Mol Cell Neurosci 24, 160–170 (2003). [DOI] [PubMed] [Google Scholar]
- Di Giacomo M. et al. Multiple epigenetic mechanisms and the piRNA pathway enforce LINE1 silencing during adult spermatogenesis. Mol Cell 50, 601–608, 10.1016/j.molcel.2013.04.026 (2013). [DOI] [PubMed] [Google Scholar]
- Muotri A. R. et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature 468, 443–446, 10.1038/nature09544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyengar B. R. et al. Non-coding RNA interact to regulate neuronal development and function. Front Cell Neurosci 8, 47, 10.3389/fncel.2014.00047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawano M. et al. Reduction of non-insert sequence reads by dimer eliminator LNA oligonucleotide for small RNA deep sequencing. BioTechniques 49, 751–755, 10.2144/000113516 (2010). [DOI] [PubMed] [Google Scholar]
- Mouse Genome Sequencing, C. et al. Initial sequencing and comparative analysis of the mouse genome. Nature 420, 520–562, 10.1038/nature01262 (2002). [DOI] [PubMed] [Google Scholar]
- Langmead B. & Salzberg S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, 10.1038/nmeth.1923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salimullah M., Sakai M., Plessy C. & Carninci P. NanoCAGE: a high-resolution technique to discover and interrogate cell transcriptomes. Cold Spring Harb Protoc 2011, pdb prot5559, 10.1101/pdb.prot5559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carninci P., Shiraki T., Mizuno Y., Muramatsu M. & Hayashizaki Y. Extra-long first-strand cDNA synthesis. BioTechniques 32, 984–985 (2002). [DOI] [PubMed] [Google Scholar]
- Plessy C. et al. Linking promoters to functional transcripts in small samples with nanoCAGE and CAGEscan. Nat Methods 7, 528–534, 10.1038/nmeth.1470 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberle V., Forrest A. R., Hayashizaki Y., Carninci P. & Lenhard B. CAGEr: precise TSS data retrieval and high-resolution promoterome mining for integrative analyses. Nucl Acids Res 43, e51, 10.1093/nar/gkv054 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balwierz P. J. et al. Methods for analyzing deep sequencing expression data: constructing the human and mouse promoterome with deepCAGE data. Genome Biol 10, R79, 10.1186/gb-2009-10-7-r79 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newburger D. E. & Bulyk M. L. UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucl Acids Res 37, D77–82, 10.1093/nar/gkn660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A. et al. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucl Acids Res 42, D142–147, 10.1093/nar/gkt997 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warde-Farley D. et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic acids research 38, W214–220, 10.1093/nar/gkq537 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Trapnell C., Pop M. & Salzberg S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25, 10.1186/gb-2009-10-3-r25 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079, 10.1093/bioinformatics/btp352 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589, 10.1016/j.molcel.2010.05.004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan A. R. & Hall I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842, 10.1093/bioinformatics/btq033 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks G. E., Hon G., Chandonia J. M. & Brenner S. E. WebLogo: a sequence logo generator. Genome Res 14, 1188–1190, 10.1101/gr.849004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.