

abstract
Pancreatic ductal adenocarcinoma originates from acinar cells that undergo acinar-to-ductal metaplasia (ADM). ADM is initiated in response to growth factors, inflammation, and oncogene activation and leads to a de-differentiated, duct-like phenotype. Our recent publication demonstrated a transforming growth factor α-KrasG12D-protein kinase D1-Notch1 signaling axis driving the induction of ADM and further progression to pancreatic intraepithelial neoplasia. This suggests that protein kinase D1 might be an early marker for tumor development and a potential target for drug development.
Keywords: Acinar-to-ductal metaplasia, mechanisms of oncogenesis and tumor progression, mouse model, oncogenic Kras, pancreatic cancer, protein kinase D1
Pancreatic cancer is one of the most lethal types of cancer with an extremely low 5-year survival rate. The high mortality of pancreatic cancer has persisted over decades because of its early metastasis to other organs and the inadequacy of methods for early detection and clinical diagnosis. Among the various types of pancreatic cancer, pancreatic ductal adenocarcinoma (PDA) accounts for 95% of all cases. More than 90% of PDA patients harbor oncogenic Kras mutations, indicating that pancreatic cancer is a genetic disorder. Permanent transgene expression of oncogenic versions of Kras or of transforming growth factor β (TGFβ), a ligand for the epidermal growth factor receptor (EGFR), in mouse pancreas recapitulates processes that lead to initiation of human pancreatic cancer. However, although mutant Kras expression leads to formation of precancerous lesions, in the absence of additional stimuli (i.e., growth factor signaling, inflammation) it is not sufficient to drive further progression to PDA in mice. In addition to initiating tumor formation, Kras has important functions in the maintenance of pancreatic cancer. This was demonstrated with a KrasG12D-inducible transgenic mouse model, in which established pancreatic cancer regressed when KrasG12D expression was abolished.1
A large body of evidence from transgenic and knockout mouse models indicates that PDA originates from acini of the pancreas. Under certain cell environments such as inflammation or growth factor- or cytokine-enriched conditions, pancreatic acinar cells can transdifferentiate into duct-like progenitor cells, a process known as acinar-to-ductal metaplasia (ADM). In the presence of oncogenic Kras mutations and upon activation of oncogenes and inactivation of tumor suppressor genes in a specific sequential order, these duct-like cells progress into pancreatic intraepithelial neoplasia (PanIN) lesions and eventually to PDA.
Protein kinase D (PKD) enzymes regulate various fundamental biological processes such as proliferation, differentiation, membrane trafficking, secretion, apoptosis, inflammation, cell migration, angiogenesis, cardiac hypertrophy, neuron plasticity, diabetes, and cancer. In mammals, PKD family members include PKD1, PKD2, and PKD3, which share similar domain homology and arrangements but have certain distinct physiologic functions. In the pancreas, activation of PKD3 in response to stimulation of gastrointestinal hormones in pancreatic acini increases amylase secretion to facilitate food digestion, whereas PKD1 has a role in insulin secretion in β-islets. Under pathologic conditions, dysregulation of PKD1 has been associated with diabetes, acute pancreatitis, and pancreatic cancer. In our very recently publication, we describe an in vivo role for PKD1 in the initiation of PDA, and use 3D organoid explant culture to delineate the mechanisms by which it contributes to this process.2 We demonstrate that increased expression and activity of PKD1, but not of PKD2 nor PKD3, was detected in the regions of ADM in pancreata of both MT-TGFα mice and p48cre;LSL-KrasG12D mice. Importantly, exogenous expression of constitutively active PKD1 in primary pancreatic acinar cells induced their metaplasia to duct-like cells with progenitor properties. Blockade of PKD1 activity by PKD kinase inhibitors or knockdown of PKD1 expression by PKD1-shRNA reduced ADM induced by TGFα with wild-type Kras and by oncogenic Kras, demonstrating that PKD1 functions downstream of both pathways (Fig. 1). Further investigation of the role of PKD1 in the initiation of PDA in vivo by specifically knocking out PKD1 in the pancreas of p48cre;LSL-KrasG12D mice revealed that PKD1 has a role not only in the initiation of precancerous lesions, but also in advancing PanIN progression to higher grades. We have identified the activation of Notch and its target genes as a major signaling event that drives ADM downstream of PKD1.
Figure 1.
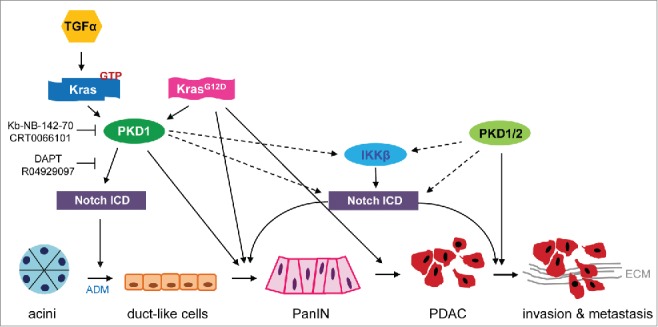
PKD1-dependent signaling pathways involved in pancreatic cancer initiation and progression. Our recent work has demonstrated the role of an axis of TGFα-Kras-PKD1-Notch1 in potentiation of ADM, which initiates pancreatic cancer.2 Moreover, knockout of PKD1 impeded KrasG12D-driven PanIN formation and progression. The figure puts our data in the context of published literature, summarizing mechanisms of how PKD1 signaling may contribute to each stage of cancer initiation and progression to a metastatic phenotype. Solid lines indicate published targets. Dashed lines indicate potential downstream signaling targets. ADM, acinar-to-ductal metaplasia; ECM, extracellular matrix; IKKβ, inhibitor of nuclear factor kappa-B subunit β; Notch ICD, Notch intercellular domain; PanIN, pancreatic intraepithelial neoplasia; PDAC, pancreatic ductal adenocarcinoma cell; PKD, protein kinase D; TGFα, transforming growth factor α.
Notch1 is an important regulator of pancreas development during embryogenesis, and its re-activation is crucial not only for tumor initiation through ADM but also for the formation and progression of KrasG12D-induced PanIN and cancer. For example, more ductal structures are present in mouse pancreas tissues after dual expression of Notch1 and KrasG12D, suggesting an important role of Notch1 in reprogramming acinar cells to a duct-like phenotype. Moreover, overexpression of Notch1 accelerates KrasG12D-induced PanIN formation and progression.3 Knockout of Notch2 in KrasG12D mice interrupts PanIN progression, prolongs survival, and causes a phenotypic switch toward anaplastic pancreatic cancer.4 Our data now indicate that one mechanism by which PKD1 drives PanIN progression is through activation of the Notch signaling pathway.2 Another PKD1-regulated transcription factor is nuclear factor κ-B (NF-κB), and NF-κB and Notch can cooperate to promote pancreatic cancer progression. For example, knockout of the Ikbkb gene in mouse has been reported to hinder KrasG12D-driven PanIN progression through downregulation of Notch signaling.5 Therefore, it is plausible that PKD1 potentiates the generation of high-grade PanIN through both of its downstream targets, NF-κB and Notch. Interestingly, Notch signaling also maintains pancreatic cancer stem cells, in which it mediates multidrug resistance and epithelial-mesenchymal transition (EMT).6 Moreover, gemcitabine-resistant pancreatic cancer cells express high levels of Notch and its ligands, Jagged and Dil,6 and inhibition of Notch signal by γ-secretase inhibitors resensitizes pancreatic cancer cells to gemcitabine-induced cell death through reduction of a cancer stem cell phenotype.7,8
Although our recent data show that PKD1 has a role in the initiation of PDA,2 little is known about its in vivo role in the progression of pancreatic cancer. In cancer cell lines, ectopically-expressed PKD1 or PKD2 increases anchorage-independent growth and invasiveness, as well as tumor growth and angiogenesis.9,10 However, these studies are hampered by the exclusive use of human pancreatic cell lines or xenografted mice, instead of genetically engineered mouse models. Given the importance of the tumor microenvironment and the contribution of different aspects of the innate and acquired immune system to the development and progression of pancreatic cancer, genetic animal models provide a great advantage over the above methods for understanding PKD function in PDA.
Our Prkd1 knockout animal model provides the first in vivo proof for an essential role of PKD1 in the initiation and progression of PDA. This supports the development and clinical use of specific kinase inhibitors, which not only may prevent cancer progression in pancreatic cancer patients, but might also be effective for high-risk populations such as patients with familial pancreatitis.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by grants from the NIH (CA135102 and CA140182) and AACR (08–20–25-STOR) to PS; a grant from the Norwegian Research Council (grant: 197261) to M.L.; and a Postdoctoral Fellowship in Regenerative Medicine from the Center for Regenerative Medicine at Mayo Clinic to G-Y.L.
Author Contribution Statement
The content of this manuscript was discussed with all authors. The manuscript was written by G-Y.L. and edited by M.L. and P.S.
References
- 1.Collins MA, Bednar F, Zhang Y, Brisset JC, Galban S, Galban CJ, Rakshit S, Flannagan KS, Adsay NV, Pasca di Magliano M. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest 2012; 122:639-53; PMID:22232209; http://dx.doi.org/ 10.1172/JCI59227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liou GY, Doppler H, Braun UB, Panayiotou R, Scotti Buzhardt M, Radisky DC, Crawford HC, Fields AP, Murray NR, Wang QJ, et al.. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat Commun 2015; 6:6200; PMID:25698580; http://dx.doi.org/ 10.1038/ncomms7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.De La OJ, Emerson LL, Goodman JL, Froebe SC, Illum BE, Curtis AB, Murtaugh LC. Notch and Kras reprogram pancreatic acinar cells to ductal intraepithelial neoplasia. Proc Natl Acad Sci U S A 2008; 105:18907-12; PMID:19028876; http://dx.doi.org/ 10.1073/pnas.0810111105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mazur PK, Einwachter H, Lee M, Sipos B, Nakhai H, Rad R, Zimber-Strobl U, Strobl LJ, Radtke F, Kloppel G, et al.. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc Natl Acad Sci U S A 2010; 107:13438-43; PMID:20624967; http://dx.doi.org/ 10.1073/pnas.1002423107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maniati E, Bossard M, Cook N, Candido JB, Emami-Shahri N, Nedospasov SA, Balkwill FR, Tuveson DA, Hagemann T. Crosstalk between the canonical NF-kappaB and Notch signaling pathways inhibits Ppargamma expression and promotes pancreatic cancer progression in mice. J Clin Invest 2011; 121:4685-99; PMID:22056382; http://dx.doi.org/ 10.1172/JCI45797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sarkar FH, Li Y, Wang Z, Kong D. Pancreatic cancer stem cells and EMT in drug resistance and metastasis. Minerva Chir 2009; 64:489-500; PMID:19859039. [PMC free article] [PubMed] [Google Scholar]
- 7.Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad A, Banerjee S, Azmi AS, Miele L, Sarkar FH. Notch-1 induces epithelial-mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett 2011; 307:26-36; PMID:21463919; http://dx.doi.org/ 10.1016/j.canlet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 8.Kang M, Jiang B, Xu B, Lu W, Guo Q, Xie Q, Zhang B, Dong X, Chen D, Wu Y. Delta like ligand 4 induces impaired chemo-drug delivery and enhanced chemoresistance in pancreatic cancer. Cancer Lett 2013; 330:11-21; PMID:23200678; http://dx.doi.org/ 10.1016/j.canlet.2012.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Ochi N, Tanasanvimon S, Matsuo Y, Tong Z, Sung B, Aggarwal BB, Sinnett-Smith J, Rozengurt E, Guha S. Protein kinase D1 promotes anchorage-independent growth, invasion, and angiogenesis by human pancreatic cancer cells. J Cell Physiol 2011; 226:1074-81; PMID:20857418; http://dx.doi.org/ 10.1002/jcp.22421. [DOI] [PubMed] [Google Scholar]
- 10.Wille C, Kohler C, Armacki M, Jamali A, Gossele U, Pfizenmaier K, Seufferlein T, Eiseler T. Protein kinase D2 induces invasion of pancreatic cancer cells by regulating matrix metalloproteinases. Mol Biol Cell 2014; 25:324-36; PMID:24336522; http://dx.doi.org/ 10.1091/mbc.E13-06-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]