

Replication of a specific site on the main chromosome of V. cholerae triggers the replication initiation of its secondary chromosome.
Keywords: Vibrio, replication, chromosome, secondary chromosome, plasmids, multipartite genome, replication initiation, pathogens, cholera, cell cycle
Abstract
Bacteria with multiple chromosomes represent up to 10% of all bacterial species. Unlike eukaryotes, these bacteria use chromosome-specific initiators for their replication. In all cases investigated, the machineries for secondary chromosome replication initiation are of plasmid origin. One of the important differences between plasmids and chromosomes is that the latter replicate during a defined period of the cell cycle, ensuring a single round of replication per cell. Vibrio cholerae carries two circular chromosomes, Chr1 and Chr2, which are replicated in a well-orchestrated manner with the cell cycle and coordinated in such a way that replication termination occurs at the same time. However, the mechanism coordinating this synchrony remains speculative. We investigated this mechanism and revealed that initiation of Chr2 replication is triggered by the replication of a 150-bp locus positioned on Chr1, called crtS. This crtS replication–mediated Chr2 replication initiation mechanism explains how the two chromosomes communicate to coordinate their replication. Our study reveals a new checkpoint control mechanism in bacteria, and highlights possible functional interactions mediated by contacts between two chromosomes, an unprecedented observation in bacteria.
INTRODUCTION
Bacteria with secondary chromosomes are frequent and have arisen independently in several taxa (1). This is the case for pathogens such as Vibrio or Burkholderia, symbionts such as Rhizobia, and others. Domestication of large plasmids, after transfer of essential genes, appears to explain the origin of secondary chromosomes (2). Evidence supporting this hypothesis includes the fact that all secondary chromosomes carry plasmid-like replication systems (2). Replication of bacterial chromosomes is regulated at initiation from a single well-conserved origin of replication (oriC) under the control of DnaA, the universal initiator of chromosome replication in bacteria (3), whereas plasmids have various types of replication origins. Usually, their replication is controlled by an initiator binding to directly repeated sequences (iterons) or by an antisense RNA (4).
All reported members of the Vibrionaceae family (Vibrio, Listonella, Aliivibrio, and Photobacterium) have two chromosomes of uneven size (5). Most of our knowledge on the replication control of secondary chromosomes comes from studies of Vibrio cholerae, the causative agent of cholera in humans. V. cholerae has two circular chromosomes, a main chromosome (Chr1) of 3 Mbp and a secondary chromosome (Chr2) of 1 Mbp (6). Chr1 replication is initiated at an oriC-like origin, ori1, by DnaA. Chr2 has a plasmid-like origin, ori2, where replication is regulated by a Vibrio-specific factor, RctB (7). RctB is a large protein [658 amino acids (AA)] that binds to DNA as a monomer or as a dimer (8). RctB binds and hydrolyzes adenosine 5′-triphosphate (ATP), but unlike DnaA, the ATP-bound form of RctB is inactive (9). Various regulatory mechanisms, such as initiator autoregulation, initiator titration, and origin handcuffing, control the level and activity of RctB [for review, see the study by Val et al. (10)]. RctB binds to iterons (12-mer sites) in ori2, which promotes replication initiation. Additionally, RctB binds other regulatory sites. Within ori2, RctB binds to 39-mer regulatory sites, which strongly inhibit ori2 initiation (11). Beyond the origin region of Chr2, chromatin immunoprecipitation with DNA microarray (ChIP-chip) analysis revealed that RctB binds to a locus on Chr2 (coordinates 956828-1030773) containing five iterons and one 39-mer, which inhibits the replication of an ori2-driven plasmid (mini-chr2) in Escherichia coli (12). RctB was also found to bind a locus on Chr1 sharing no homology with either iterons or 39-mers. This site was shown to act as a replication enhancer of ori2 by increasing RctB affinity for iterons and decreasing RctB affinity for 39-mers (12). Serial deletions of a DNA fragment containing the Chr1 RctB ChIP-chip binding peak (fragments chrI-2 to chrI-10) showed that the replication-enhancing activity of a mini-chr2 in E. coli could be narrowed down to a 70-bp chrI-9 fragment (coordinates 818000-818069). However, the larger (150 bp) chrI-4 fragment (coordinates 817947-818099) was more efficient in enhancing mini-chr2 replication in E. coli. The presence of such a site on Chr1 suggested that the two chromosomes communicate with each other during replication. However, the role of this locus in the replication coordination of Chr1 and Chr2 remains a subject of speculation.
Chromosomes replicate during a defined period of the cell cycle, ensuring a single round of replication per cell. Plasmids generally have no such constraint, replicating randomly during the bacterial cell cycle (13). Despite its plasmid origin, Chr2 replication occurs only once per cell cycle (14). Initiation of Chr2 replication is also delayed so that the two chromosomes terminate replication at nearly the same time (15). The mechanism responsible for triggering Chr2 replication at a specific time of the cell cycle remains unknown. Here, we provide new insights into this regulatory process, and we explain how Chr2 monitors the replication status of Chr1 to time its own replication. We show that the Chr1 RctB binding site (12), renamed crtS for Chr2 replication triggering site, is crucial for the activation of Chr2 replication. We demonstrate that the replication of crtS triggers the replication of Chr2. We also show that the crtS locus and ori2 localize to the same region of the cell during the entire cell cycle and display enhanced physical contacts, suggesting that the regulatory mechanisms may involve a structural interplay. This study reveals a new checkpoint control mechanism in bacteria.
RESULTS
Marker frequency analysis reveals the relative replication pattern of the two chromosomes of V. cholerae
Replication of bacterial chromosomes occurs bidirectionally and terminates in the region opposite to the origin, in the vicinity of the dimer resolution site (dif), forming two replicated halves called replichores (Fig. 1A, top) (16). Assuming that Chr1 and Chr2 replicate at the same speed, replication of Chr2 must be delayed so that replication termination of the two chromosomes can be synchronous (15). Control of Chr2 replication initiation might be linked to cell mass or some “timer” on Chr1 that, when replicated, signals initiation on Chr2. We used marker frequency analysis (MFA) to precisely analyze the replication pattern of the two chromosomes of a culture of wild-type V. cholerae El Tor N16961 strain (WT) grown under steady-state conditions. MFA provides an unprecedented resolution of the replication timing and the replication fork speed, can pinpoint the origin and the terminus of chromosome replication, and can detect chromosomal rearrangements (17). Indeed, the MFA plot of WT compared to the reference genome sequence (AE003852) registered a discrepancy in Chr1 organization (fig. S1). Rectification of the Chr1 reference sequence corrected the deviation. MFA of WT confirmed that both chromosomes are replicated bidirectionally, with each chromosome having one origin (ori1 and ori2) and one terminus (ter1 and ter2) of replication. The linearity and the slopes of the graphs for the four replichores indicate that replication speed is constant for both chromosomes (Fig. 1A, bottom). This analysis confirmed that Chr1 and Chr2 terminate replication concomitantly and that Chr2 is initiated when about two-thirds of Chr1 is replicated, as hypothesized.
Fig. 1. Chr1 and Chr2 replication coordination is promoted by the presence of a timer on Chr1 and not by the requirement to terminate their replication synchronously.
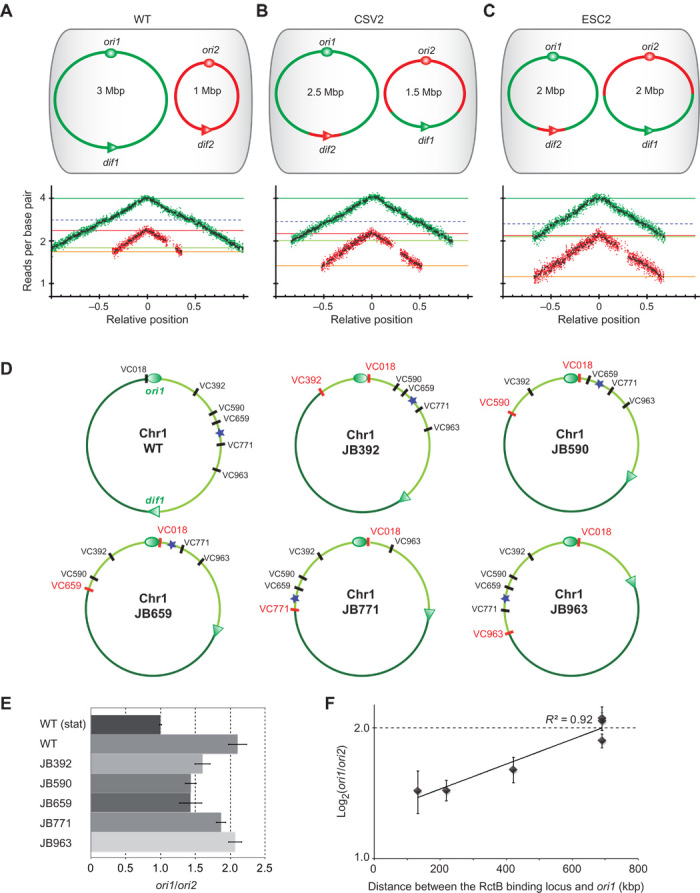
(A) Top: Genome structure of wild-type (WT) V. cholerae. Ovals indicate the origins of replication (ori1 and ori2) and triangles show dif sites (dif1 and dif2) on Chr1 (green) and Chr2 (red). Bottom: MFA of exponentially growing WT cultures using a corrected reference sequence of Chr1 (fig. S1). Log2 of number of reads starting at each base (normalized against reads from a stationary phase WT control) is plotted against their relative position on Chr1 and Chr2. Positions of ori1 and ori2 are set to 0 for a better visualization of the bidirectional replication. Any window containing repeated sequences is omitted; thus, the large gap observed in the right arm of Chr2 consists of filtered repeated sequences within the superintegron (28). Green (Chr1) and red (Chr2) dots indicate the average of 1000-bp windows; black dots indicate the average of 10,000-bp windows. Dark green, light green, red, and orange lines indicate ori1, ter1, ori2, and ter2 number of reads, respectively; dashed blue lines indicate the Chr1 RctB binding locus (12). The same color code is used for all MFA figures. (B and C) Genomic variants CSV2 (Chr1 = 2.5 Mbp and Chr2 = 1.5 Mbp) (B) and ESC2 (Chr1 = 2 Mbp, Chr2 = 2 Mbp) (C) are the same as in (A). The genetic exchanges made between Chr1 and Chr2 are shown in green and red. (D) Chr1 map of WT and genomic variants (JB392, JB590, JB659, JB771, and JB963) with large chromosomal inversions around a fixed locus (VC018) and other loci located at increasing distances from ori1 (VC392, VC590, VC659, VC771, and VC963), respectively. For each genomic mutant, the loci flanking the DNA inversion are shown in red. The left (dark green) and right (light green) replichores are separated by ori1 (oval) and dif1 (triangle). The position of the Chr1 RctB binding locus is indicated by a blue star. (E) Histogram representing quantitative PCR–measured ori1/ori2 ratios from relative gDNA quantification of exponentially fast-growing strains (gDNA from WT stationary culture was used for normalization). Bars display means (±SD) of at least three experiments. (F) Log2(ori1/ori2) plotted as a function of the distance between ori1 and the RctB binding locus displays a linear relationship (R2 = 0.92). Dots show means (±SD) of three experiments.
Chr2 replication initiation depends on the location of a timer region on Chr1
Chr2 replication initiation, after two-thirds of Chr1 has been replicated, could be linked to an unknown mechanism for coordination of replication termination of the two chromosomes, consistent with their relative sizes, 3 and 1 Mbp. To evaluate the impact of the chromosome sizes on their relative timing of replication, we generated two mutants with altered chromosome sizes using a dual site-specific recombination tool to transfer DNA from one chromosome to the other (18). In the CSV2 mutant, Chr1 and Chr2 sizes remained unbalanced at 2.5 and 1.5 Mb, respectively, whereas in the ESC2 strain, the two chromosomes are each at 2 Mb (Fig. 1, B and C). Increasing the size of Chr2 abolished synchronous termination in both ESC2 and CSV2 mutants, with the now larger Chr2 terminating replication after Chr1 (Fig. 1, B and C). MFA also revealed that Chr2 systematically initiates replication when a discrete position along one (or both) Chr1 replichore(s) is being replicated (Fig. 1, B and C). This finding suggested that a region located on Chr1 may trigger Chr2 replication initiation, and should be localized within the first two-thirds of either or both replichores and act as a “checkpoint” in replication of Chr2.
The relative timing of initiation of Chr1 and Chr2 can be followed by qPCR analysis of the ori1/ori2 ratio. We reasoned that a change in this ratio in isogenic mutants, where Chr1 size was unaltered but replichores were rearranged, would yield a signal for the region of interest. This was performed by inversion between a fixed intergenic locus (downstream of ORF VC018, that is, near ori1 on the left replichore) and other intergenic loci located at increasing distances from ori1 along the right replichore (Fig. 1D). Such inversions either caused no fitness cost (JB392) or were similarly affected (JB590, JB659, JB771, and JB963) compared to WT (fig. S2). We monitored the impact of each inversion on the ori1/ori2 ratio in exponentially growing cultures (Fig. 1E). In WT, the ori1/ori2 ratio is around 2, matching the observations of fast-growing V. cholerae (15). In mutants JB392, JB590, and JB659, ori1/ori2 ratios decreased, indicating that the region triggering Chr2 replication may be closer to ori1, causing earlier Chr2 replication initiation. In mutant JB771 and JB963, the ori1/ori2 ratio remains ~2, indicating the wild type–like timing of replication and that the locus triggering Chr2 initiation must be at the same distance from ori1 as in wild type. These results indicate that a locus located between VC659 and VC771 and its distance from ori1 play a role in the regulation of Chr2 replication. This region contains the Chr1 RctB binding locus, located in a noncoding region upstream of VC765 (12). Strikingly, the log2(ori1/ori2) ratio increases linearly with the distance between ori1 and the Chr1 RctB binding locus (Fig. 1F). This observation indicates that the timing of replication of the Chr1 RctB binding locus exerts a control on Chr2 replication initiation.
Replication of the Chr1 RctB binding locus (crtS) triggers Chr2 replication initiation
We tested the Chr1 RctB binding locus for its role in the coordination of the timing of replication between Chr1 and Chr2. Hereafter, we will refer to the Chr1 RctB binding locus as crtS. crtS was relocated to four intergenic loci on Chr1 at varying distances from ori1 (Fig. 2A). crtS-positional mutants display wild type–like fitness and phenotype (fig. S3). Therefore, all the determinants for crtS proper function appeared contained within its sequence. MFA showed that in crtSVC23 and crtSVC392, where crtS is closer to ori1, Chr2 initiates earlier than in WT (Fig. 2B). In crtSVC2238, where crtS is positioned at the same distance from ori1 but on the other replicore, Chr2 initiates roughly at the same time as in WT (Fig. 2B). In crtSVC963, with crtS farther from ori1, Chr2 initiates later than in WT (Fig. 2B). We calculated the ori1/ori2 and ori2/crtS ratios from the MFA (Fig. 2C). The log2 of ori1/ori2 ratio is linearly correlated with the ori1-crtS distance (R2 = 0.9988), suggesting that the timing of replication of crtS controls the timing of Chr2 replication initiation. The crtS/ori2 ratio remains constant (~0.8), indicating that there is a constant delay between crtS replication and Chr2 replication initiation.
Fig. 2. Timing of replication of the Chr1 RctB binding locus (crtS) controls the timing of initiation of Chr2.
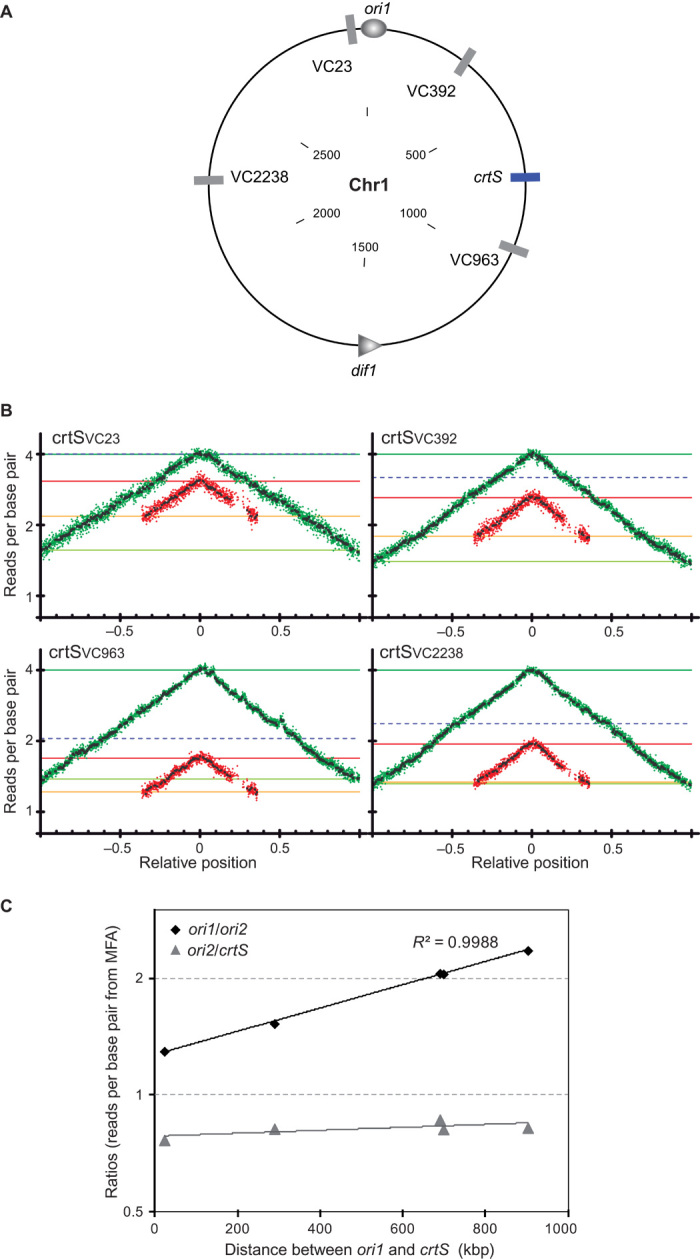
(A) Circular map of WT Chr1 showing the native location of crtS (blue bar) and the various loci where crtS was relocated (gray bars) with respect to ori1 (oval) and dif1 (triangle). The inside scale designates DNA size in kilobase pair. (B) MFA of relocated crtS mutants (crtSVC23, crtSVC392, crtSVC963, and crtSVC2238). The dashed blue lines indicate the number of reads of the loci where crtS has been relocated. (C) Log2(ori1/ori2) and log2(ori2/crtS) plotted as a function of the distance between crtS and ori1 in kilobase pair. The ratios were calculated as the ratios of the number of reads per base pair (from MFA) for each designated loci.
To confirm these observations, we tracked pairwise combinations of fluorescently labeled chromosomal positions using epifluorescence microscopy (19). We compared the distribution of ori1 with ori2 foci, VC783 (near crtS) with ori2 foci, and ter1 with ter2 foci in both WT and crtSVC23 strains (Fig. 3 and figs. S4 and S5). Images of exponentially growing cells were acquired, and cell length, along with the position of each tagged loci, was followed. WT cells ranged in size from 2 μm (newborn cells) to 4.5 μm (dividing cells), with two ori1 foci appearing in cells from 2.5 to 3 μm and two ori2 foci from 3 to 3.5 μm (Fig. 3, A, C, and E, and fig. S6, left). This observation indicates that ori1 is replicated and segregated before ori2, as previously observed (19). In crtSVC23, cells with two ori1 and two ori2 foci appeared in the same cell size range from 2.5 to 3 μm (Fig. 3, B, D, and F, and fig. S6, right), consistent with the MFA results showing that ori2 is replicated shortly after ori1 (Fig. 2B). In WT, duplication of VC783 foci (near crtS) occurs shortly before ori2 foci duplication (fig. S4, C and E), which is in accordance with MFA, showing a delay between crtS replication and ori2 replication (Fig. 1A). In crtSVC23, when crtS is no longer near VC783, ori2 foci duplicate before VC783 foci (fig. S4, D and F), consistent with MFA results (Fig. 2B). Duplications of the ter1 and ter2 foci in WT were only visible at the end of the cell cycle in dividing cells (fig. S5, A, C, and E, and fig. S6, left). Surprisingly, most crtSVC23 cells with two ter1 and two ter2 foci appeared in the same cell size range as WT (fig. S5, B, D, and F, and fig. S6, right). Because MFA studies showed that ter2 is replicated long before ter1 in crtSVC23 mutant (Fig. 2B), this result indicates that duplicated ter1 and ter2 remain colocalized at mid-cell until cell division occurs.
Fig. 3. Segregation of ori2 (but not ter2) occurs earlier when crtS is transposed near ori1.
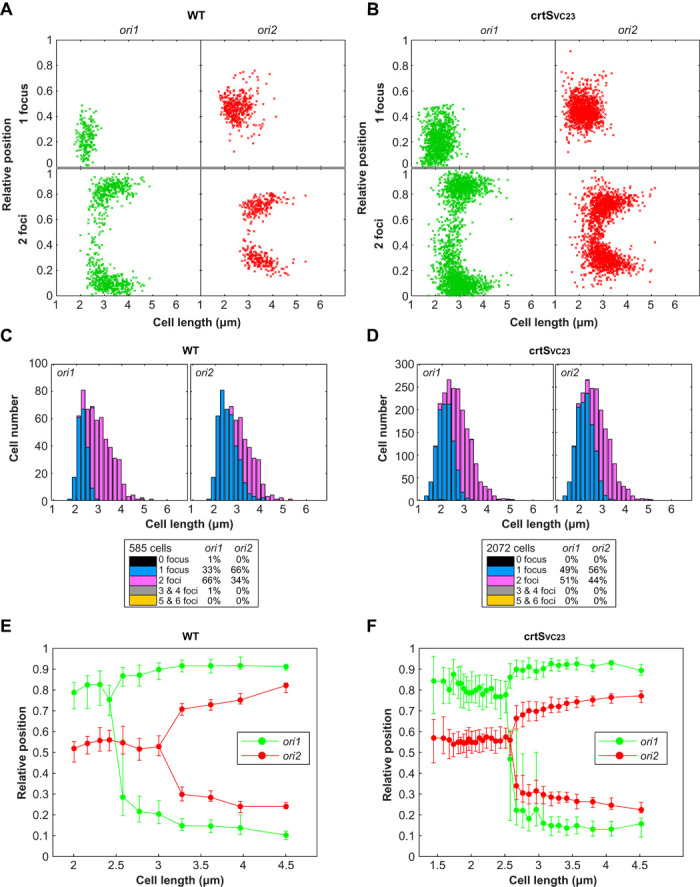
(A and B) Plot showing the position of ori1 (left panel) and ori2 (right panel) foci inside WT (A) and mutant crtSVC23 (B) cells. Foci are oriented longitudinally relative to the old pole of the cell as a function of cell length. The old pole of the cells was defined as the closest pole to an ori1 focus. The x axis represents the cell length (in micrometers). The y axis represents the relative position of the focus in bacterial cells, 0 being the old pole and 1 the new pole. Snapshot images of 585 WT and 2072 crtSVC23 mutant cells were analyzed. (C and D) Histograms displaying the amount of cells that exhibit zero, one, two, three, four, or five and six fluorescent foci according to cell size (in micrometers) in WT (C) and mutant crtSVC23 (D) cells. (E and F) By correlating the longitudinal position of ori1 and ori2 foci as a function of cell length, the segregation choreographies of the ori1 and ori2 were reconstituted throughout the cell cycle of WT (E) and mutant crtSVC23 (F) bacterial cells. Cells were classified according to their size and grouped by 30 to define each size interval. For most loci and cell length intervals, there were cells with either a single focus or two separated foci, the relative proportions of each type varying as a function of cell length. Only the position of the foci corresponding to the dominant cell type in each cell length interval was plotted. The median positions of the observed foci (filled circles), along with the 25th to 75th percentiles (error bars), were plotted for each cell size bin. The x axis represents the cell length (in micrometers). The y axis represents the relative position of the focus in bacterial cells (0, new pole and 1, old pole).
To further probe the influence of crtS doubling on the initiation of Chr2 replication, we analyzed two mutant strains, crtSWT/VC23 and crtSWT/VC2238, carrying two chromosomal copies of crtS. The crtSWT/VC23 mutant carries the native crtS site, as well as an extra copy near ori1 (VC23), so that one crtS is replicated before the other. The two crtS copies of the crtSWT/VC2238 mutant are located at equal distances from ori1 on each replichore; thus, the two crtS are replicated at the same time. Both mutants displayed a significant loss of fitness compared to WT, with crtSWT/VC2238 being the least affected (fig. S7). Both crtSWT/VC23 and crtSWT/VC2238 mutants displayed a wider cell size range, from 2 μm (newborn cells) to 5.5 μm (dividing cells) (Fig. 4 and fig. S8), indicating a flaw in the cell cycle control. Tracking of fluorescently labeled ori1 and ori2 loci reveals that most crtSWT/VC23 newborn cells exhibit two ori2 foci but only one ori1 focus (Fig. 4, A and B) compared to WT (Fig. 3A). Most crtSWT/VC23 dividing cells present two ori1 foci and four ori2 foci (Fig. 4, A to C), suggesting that whereas Chr1 goes from one to two copies, Chr2 goes from two to four copies per cell cycle. Similar results were obtained with the crtSWT/VC2238 mutant (fig. S8). In both mutants, daughter cells usually receive one copy of ori1 and two copies of ori2, whereas WT daughter cells normally receive a single copy of both ori1 and ori2 (Fig. 4, C to E). Sporadically, in both mutant strains, we observed that cell division occurs asymmetrically, leading to uneven chromosome partitioning. The proportion of cells with three ori2 foci in crtSWT/VC2238 (15%) is equivalent to the proportion of cells with one ori2 foci (14%) (Fig. 4E), suggesting that these cells could have arisen from the asymmetric cell division of four ori2 foci cells. However, crtSWT/VC23 cells display a larger fraction of cells containing three ori2 foci (30%) and a smaller fraction of cells with only one ori2 foci (7%) (Fig. 4D), meaning that uneven chromosome partitioning alone does not explain the existence of three ori2 foci cells. We speculate that in crtSWT/VC23, one of the two ori2 duplicates before the other. These differences between crtSWT/VC23 and crtSWT/VC2238 suggest that the difference in location and/or timing of replication of the extra crtS site alters the synchrony of initiation of multiple origins. These observations are puzzling because they do not fit with the E. coli paradigm where multiple origins initiate in synchrony (20). To explain that one ori2 is duplicated before the other in crtSWT/VC23, we hypothesized that the duplication of one crtS 3triggers the firing of only one ori2. This may require a direct contact between the replicated crtS and ori2.
Fig. 4. Two chromosomal copies of crtS doubles Chr2 copy number.
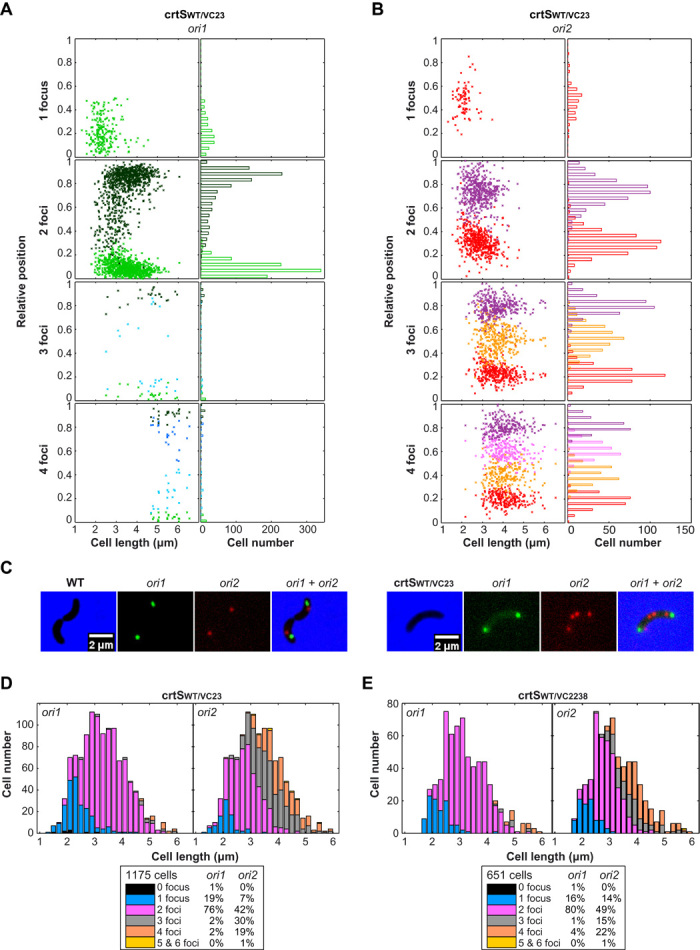
(A and B) Position of ori1 (A) and ori2 (B) foci inside crtSWT/VC23 cells. Foci are oriented longitudinally relative to the old pole of the cell as a function of cell length. The old pole of the cell was defined as the closest pole to an ori1 focus. On the left panel, the x axis represents the cell length (in micrometers). On the right panel, the x axis indicates cell number. The y axis represents the relative position of the focus in bacterial cells, 0 being the old pole and 1 the new pole. (C) Representative pictures of dividing cells (WT and crtSWT/VC23) observed by fluorescence microscopy. Cells were fluorescently labeled near ori1 and ori2. Merged pictures of ori1 (green) and ori2 (red) and phase-contrast (blue) micrographs show 2× more red spots than green spots in mutant crtSWT/VC23. (D and E) Amount of crtSWT/VC23 (D) and crtSWT/VC2238 (E) cells exhibiting zero, one, two, three, four, or five and six ori1 foci (left panel) and ori2 foci (right panel) according to cell size (in micrometers).
Chromosome conformation capture reveals a preferential contact between Chr1 and Chr2
To test for the possibility that Chr1-located crtS site regulation of Chr2 replication initiation involves physical contacts between the two chromosomes, we applied chromosome conformation capture [3C; (21)] to exponentially growing cultures in rapid (LB) and slow [minimal medium (MM)] growth conditions (Fig. 5A and fig. S9). As expected, the two chromosomes in the resulting contact map turned out as two well-individualized entities, each exhibiting a strong diagonal signal reflecting frequent contacts between adjacent loci (Fig. 5A) (22). These chromosomes presented local domains of increased contact frequencies, separated by barriers (fig. S10), similar to the chromatin interaction domains (CIDs) previously described in Bacillus subtilis (23, 24) and Caulobacter crescentus (25). The intrachromosomal contact maps of Chr1 and Chr2 differ with respect to the presence of a secondary perpendicular diagonal that reflects the bridging of left and right replichores by cohesin complexes in other bacteria (23–25). Whereas Chr1 did not present such a signal, Chr2 displayed a secondary diagonal revealing radically different folding of the two chromosomes (Fig. 5A). The conversion of contact maps into three-dimensional (3D) structures (fig. S9C and movies S1 and S2) (26) shows that whereas Chr1 adopts a largely open structure, Chr2 folds into a helicoidally shape with its two replichores tightly interlaced. It appears from this matrix that Chr2 folds into a structure similar to that of B. subtilis (23, 24) or C. crescentus (27), whereas Chr1 adopts a structure closer to the one observed for E. coli chromosome (22). The superintegron, a large gene capture and excision system localized on Chr2 (28), defines a clear CID surrounded by two highly transcribed genes (fig. S10). This observation suggests a specific topological structure, which can reflect the generally low transcription in this DNA element.
Fig. 5. Intra- and interchromosomal interactions in V. cholerae.
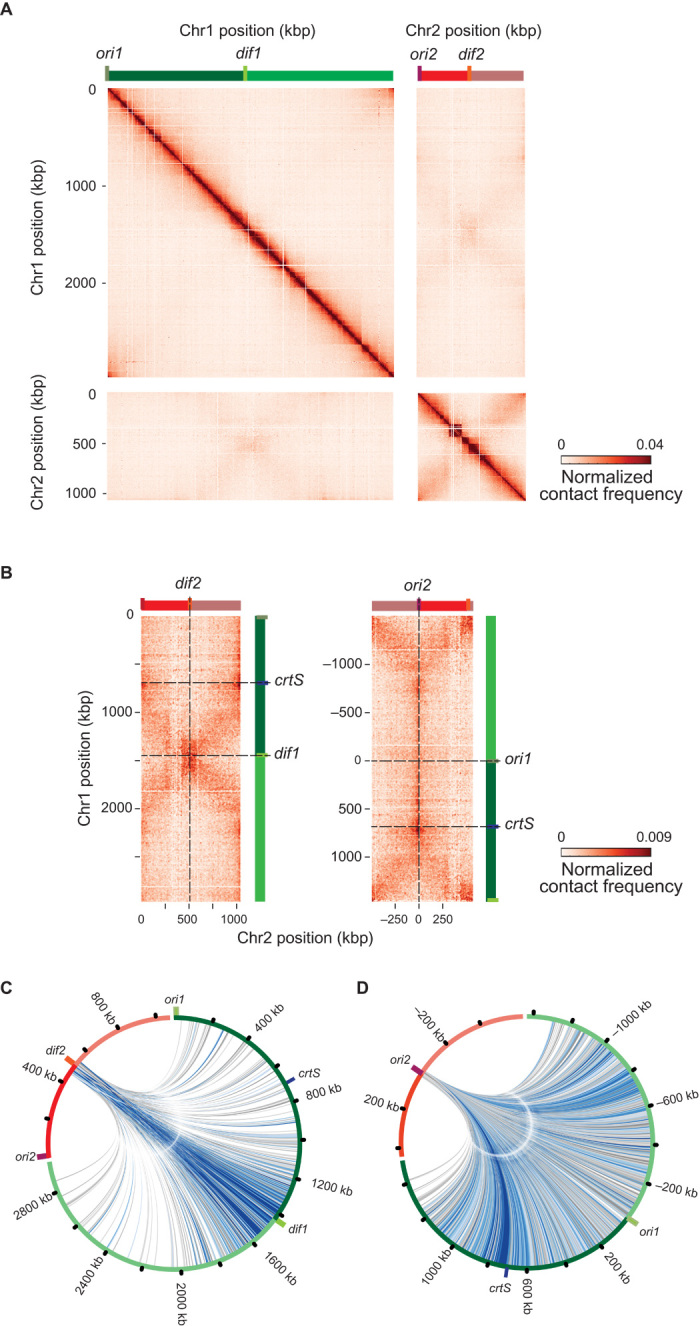
(A) Normalized and filtrated genomic contact map obtained from an asynchronous population of WT cells growing exponentially in LB. x and y axes represent genomic coordinates of each chromosome centered on dif sites (light green bar, dif1; orange bar, dif2). Origins of replication are shown as a dark green bar (ori1) and a dark red bar (ori2). Chr1 and Chr2 are represented by dark green (right arm) or light green (left arm) and dark red (right arm) or light red (left arm). The color scale reflects the frequency of contacts between two regions of the genome (arbitrary units), from white (rare contacts) to dark red (frequent contacts), and is conserved across all panels of all figures. (B) Interchromosomal contact map centered on dif (left panel) or ori sites (right panel). The crtS site is indicated as a blue bar. (C and D) Circos representation of interactions of 100 kbp (20 bin) around dif2 with Chr1 (C) and circos representation of interactions of 50 kbp (10 bin) around ori2 with Chr1 (D).
Overall, the chromosomes of V. cholerae present known features of chromosome organization in bacteria, as well as new ones. Indeed, the contact maps unveiled trans contacts between the two chromosomes at an unprecedented resolution. The two replichores of Chr2 present enrichment in contact with the bottom third of Chr1 replichore, as indicated by the cross-shaped trans contacts (Fig. 5B). These contacts initiate at dif sites and extend along the length of the chromosome up to ori2 for Chr2 and midway to Chr1. The spatial proximity of the terminus regions (ter), surrounding dif1 and dif2, is a striking driver of organization, with strong contacts between the ter regions, visible in both growth conditions (fig. S9A). The circos representation of the strongest interactions of 100 kbp surrounding dif2 (Fig. 5C) illustrates the enrichment of contacts between the ter regions. This is also visible in the 3D contact maps (fig. S9C and movies S1 and S2). These results are consistent with the imaging of the ter1 and ter2 showing a colocalization of the regions to the mid-cell at the end of the cell cycle (fig. S5E). The circos representation of the contacts made by a 50-kbp window centered on ori2 reveals preferential contacts with the right replichore of Chr1 (Fig. 5D). These contacts initiate at crtS and become stronger immediately after. More precisely, the contacts involve a region on the Chr2 left replichore adjacent to ori2, pointing toward a mechanical interplay that would drive this interaction.
crtS is crucial for Chr2 replication initiation at ori2
To assess the importance of crtS on Chr2 replication, we deleted a 150-bp sequence (coordinates 817950-818100), which closely corresponds to the chrI-4 sequence (coordinates 817947-818099) deleted by Baek and Chattoraj (12). Baek and Chattoraj (12) reported that chrI-4–deleted mutants showed minimal phenotypic changes and no growth defects, indicating that its action was probably modest. In contrast, our ΔcrtS mutants exhibited strong fitness defects and suffered marked physiological changes, with a large proportion of filamentous cells (Fig. 6A, top, and fig. S11). To corroborate the marked phenotype of ΔcrtS mutants with a problem in ori2 replication initiation, we deleted crtS in a V. cholerae strain where ori2 was replaced by a second ori1 locus and is thus no longer dependent on RctB (ICO1) (18). ICO1ΔcrtS mutants displayed no marked filamentous phenotypes and no additional growth defects compared to ICO1 (Fig. 6A, bottom, and fig. S11), confirming that the ΔcrtS phenotype is linked to a deficiency in replication initiation of Chr2 at ori2. By performing MFA on mutant ΔcrtS #7 (with two separate chromosomes), we could detect the location of ori2 by the overrepresentation of sequences (sharp peak), indicating that replication initiation still occurs at ori2. However, the MFA also highlighted an imbalance in the copy number of Chr1 and Chr2 with an ori1/ori2 ratio of 5.2 and a ter1/ter2 ratio of 2.9 (Fig. 6B). This imbalance was also observed by imaging fluorescently labeled Chr1 and Chr2 loci in a ΔcrtS mutant strain. Figure 6C shows a typical ΔcrtS filamentous cell with more VC783 foci than ori2 foci, whereas in WT, the two loci duplicate around the same time (fig. S4E), and most cells display the same number of VC783 and ori2 foci (fig. S4C). Time-lapse fluorescence microscopy of ΔcrtS cells shows that filaments grow and accumulate many Chr1 foci (movie S3). These filaments divide irregularly and occasionally give rise to a Chr2-less cell, which does not grow (movie S4).
Fig. 6. crtS is crucial for Chr2 replication initiation at ori2.
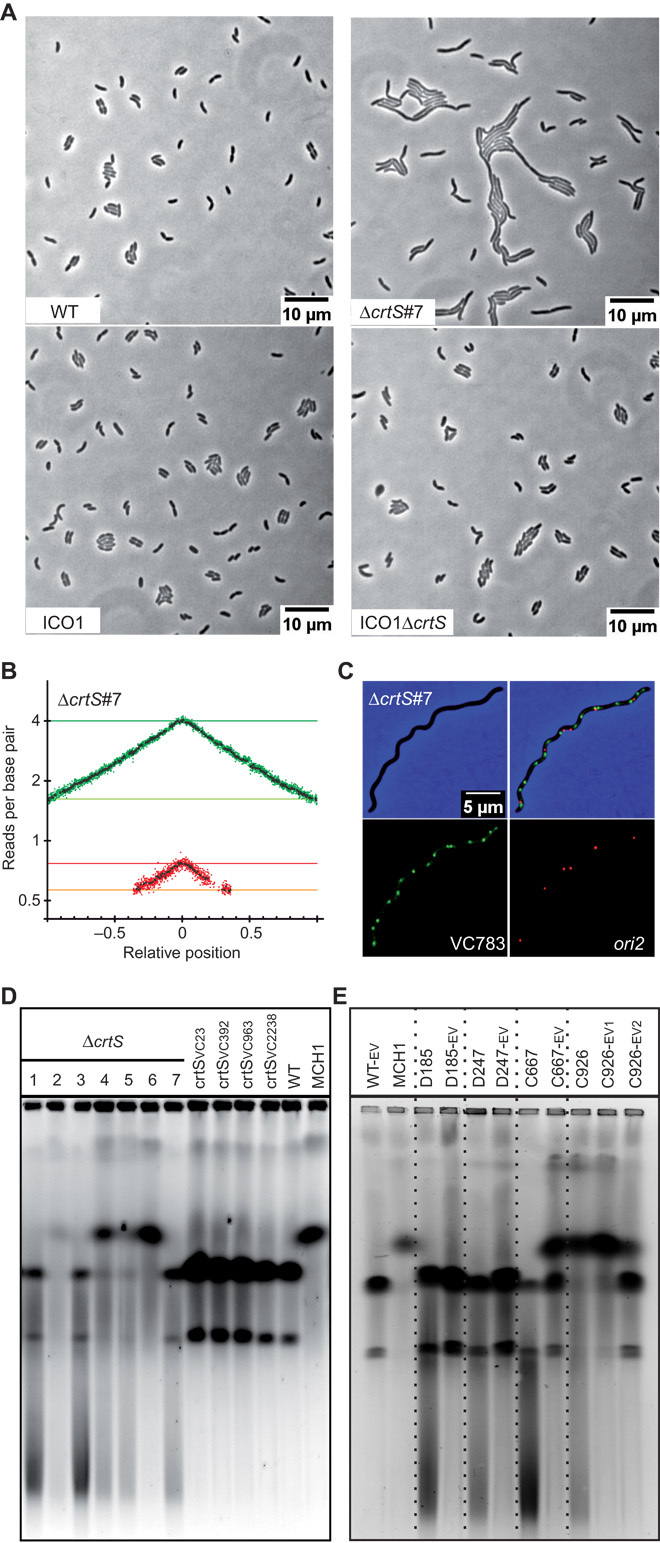
(A) Phenotype of crtS-deleted mutants in WT and ICO1. Representative pictures of phase-contrast microscopy of live cells growing on LB agar pads: WT, ΔcrtS #7 (with two separate chromosomes), ICO1, and ICO1ΔcrtS. (B) MFA of WTΔcrtS #7. (C) Representative picture of a filamentous ΔcrtS cell observed with fluorescence microscopy. Loci near crtS (VC783) and ori2 were fluorescently labeled. Merged pictures of VC783 (green) and ori2 (red) and phase-contrast (blue) micrographs show a higher number of green spots than red spots. (D) Ethidium bromide–stained pulsed-field gel electrophoresis (PFGE) of native gDNA. From left to right: Independent clones of crtS-deleted mutants (WTΔcrtS #1 to #7), mutants with relocated crtS (crtSVC23, crtSVC392, crtSVC963, and crtSVC2238), and WT (two chromosomes) and MCH1 (one synthetic fused chromosome), which are used as size reference (18). (E) Same as (D). From left to right: WT-EV (control), MCH1 (one synthetic fused chromosome), and independent clones of ΔcrtS mutants before (D185, D247, C667, and C926) and after a 200-generation evolution (-EV). For C926, samples were harvested after 100 (-EV1) and 200 generations (-EV2) to track the tendency of the fused chromosome to revert to two separate chromosomes.
PFGE revealed a high instability in the genome structure of ΔcrtS mutants. In many instances, the two chromosomes had spontaneously fused (Fig. 6D; ΔcrtS #2 and #4 to #6), and in all ΔcrtS mutants, a smear was clearly visible in the PFGE (Fig. 6D; ΔcrtS), indicating either genomic DNA (gDNA) degradation or a high level of genomic instability. crtS-positional mutants, however, did not display gDNA degradation/instability, thus evidencing the proper activity of crtS when transposed to ectopic chromosomal positions (Fig. 6D; crtSVC23, crtSVC392, crtSVC963, and crtSVC2238). The high instability observed in the genome of ΔcrtS mutants suggested a positive selection toward chromosome fusion. To test for this constraint, we grew four independent cultures of ΔcrtS mutants for 200 generations to investigate spontaneous fusion and other potential suppressors. PFGE analysis was performed on samples collected at the beginning and at the end of the experiment. At generation 0, three mutants (D185, D247, and C667) carried two chromosomes, whereas the fourth (C926) had already undergone a fusion. After 200 generations, both C926 and C667 displayed a mixed population of cells with fused and separate chromosomes, whereas D185 and D247 showed no sign of chromosome fusion (Fig. 6E). Evolved mutants significantly improved their fitness over 200 generations (fig. S12). Moreover, the DNA smear trademark of a ΔcrtS deletion was no longer detectable in any of the evolved mutants (D185-EV, D247-EV, C667-EV, and C926-EV2) (Fig. 6E). Microscopy and flow cytometry showed that D185-EV recovered a wild-type morphology with less than 1% of aberrantly sized cells (fig. S13). However, D247-EV, C667-EV, and C926-EV2 populations still displayed some filamentation. MFA of D247, before experimental evolution, is very similar to the MFA of ΔcrtS #7 and shows that no genome rearrangement had yet occurred (fig. S14). However, MFA of D185 shows that gene amplification (triplication) had occurred before experimental evolution and reverted in the evolved mutant D185-EV (fig. S14). Stress-induced gene amplification has been observed in E. coli and may confer a selective advantage to cells under stress (29). After 200 generations, MFA of D185-EV and of D247-EV reveals an up-regulation of ori2 initiation, presumably compensating for the lack of crtS activation, as shown by the increase in the Chr2/Chr1 copy number ratio compared to their parental strains (fig. S14). Such a remarkable rescue of phenotype of all ΔcrtS mutants over 200 generations suggested that compensatory mutations were required to restore, even partially, the growth defects. D185-EV whole-genome sequencing revealed a single mutation, leading to a F230S substitution in RctB, not present in D185. D247-EV genome sequence also revealed a single mutation (C to A transversion), 25 nucleotides upstream of the rctB start codon within the 29-mer RctB binding site, which is important for autorepression of RctB expression and ori2 initiation negative regulation via handcuffing with iterons (30). Further analysis of the ori2 regions of C667-EV and C926-EV2 revealed two new single mutations in RctB, which were absent from their parental strain (table S1). In all four mutants, ΔcrtS phenotype was partially rescued by rctB-related mutations, further supporting the direct connection between crtS and RctB.
DISCUSSION
Approximately 10% of bacterial species have genomes split over multiple chromosomes. The machineries controlling replication initiation of secondary chromosomes are always of plasmid origin (2). An important difference between plasmids and chromosomes is that the latter replicate once, and only once, per cell cycle. In V. cholerae, the two chromosomes satisfy this rule; furthermore, they are known to have a synchronous termination of replication. However, it remains unclear how this is coordinated. Here, we show that the timing of Chr2 replication initiation directly depends on the position of a short intergenic sequence (crtS) on Chr1. Our results provide strong evidence that the replication of crtS triggers initiation of Chr2 replication. Additionally, we reveal preferential contacts between Chr1 and Chr2, suggesting the existence of mechanistic functional interactions between these two chromosomes. This study unravels a novel checkpoint control mechanism allowing the replication coordination of multiple chromosomes in bacteria.
crtS is crucial for Chr2 replication
Here, deletion of crtS severely impairs growth and is associated with filamentation and DNA damage (Fig. 6A, top, and fig. S11). The large cell size heterogeneity in ΔcrtS mutants prevented statistically robust analysis based on fluorescence microscopy data. Nevertheless, filaments containing fewer Chr2 than Chr1 were consistent with MFA analysis showing Chr2 underrepresentation in ΔcrtS mutants (Fig. 6, B and C). In ΔcrtS filaments, we observed more VC783 foci than ori2 foci, suggesting that failure of proper regulation/activation of Chr2 replication is responsible for the observed filamentation. It was reported that deletion of the chrI-4 sequence (ΔchrI-4) led only to mild phenotypic changes (12). ΔchrI-4 mutants were also characterized by a delay in ori2 foci duplication (12). Why ΔcrtS differs from ΔchrI-4 phenotype previously characterized could be explained by some differences in the way these sequences were deleted (see the Supplementary Materials). We further show that crtS is directly involved in the regulation of replication initiation at ori2 through an RctB-associated mechanism because its deletion does not affect the physiology of the ICO1 mutant, in which Chr2 replication is no longer dependent on RctB at ori2 (Fig. 6A, bottom). Strains with crtS relocated to different positions along Chr1 did not show any of the ΔcrtS phenotypes, indicating that the 150-bp crtS DNA sequence is both necessary and sufficient to trigger Chr2 replication initiation, independently of its genetic context (Fig. 6D and fig. S3).
Replication of crtS orchestrates the replication of Chr1 and Chr2
The replication pattern of WT and mutant strains was investigated by MFA. WT MFA results show that the copy number of ori1 is higher than that of ori2, whereas the copy numbers of ter1 and ter2 are similar (Fig. 1A), in agreement with former hypotheses and results, suggesting a synchronized termination (15). Displacing the crtS locus to different positions on Chr1 modified Chr1 and Chr2 replication synchronization (Fig. 2, A and B). The ori1/ori2 ratio is perfectly correlated with the distance between ori1 and crtS (Fig. 2C), demonstrating that Chr2 replication initiation timing is dependent on this parameter, with crtS replication triggering Chr2 replication. According to MFA, there is a constant delay between crtS replication and the firing at ori2 (Fig. 2C), which roughly corresponds to the replication of 200 kbp, suggesting that the transfer of information to ori2 is not immediate. This delay may correspond to the time needed to activate RctB and the ori2 initiation system. Therefore, both the position of crtS and this delay account for the Chr1 and Chr2 termination synchrony. The crtS position may have been selected throughout evolution by the constraint imposed by this activation delay.
Transient genome remodeling suppresses crtS deletion
Spontaneous chromosome fusions are common suppressor events of crtS deletion, but these marked rearrangements are transient and can revert back to a two-chromosomal genomic configuration over time (Fig. 6, D and E). We previously showed that recombination-induced chromosome fusions occur spontaneously within wild-type populations of V. cholerae (31). These rearrangements are quickly reversed or outcompeted, probably as a result of slower growth linked to an increase in replication duration due to the larger chromosome size and/or metabolic cost due to gene dosage imbalance. Fused chromosomes can be stabilized upon depletion of Dam methylase, which is essential for Chr2 replication initiation (31). Here, we hypothesize that chromosome fusions are a transient and conveniently accessible suppressor step when confronted with crtS deletion, ensuring replication of Chr2 through piggybacking of the Chr1 replication machinery. The higher cost of this genome state would subsequently favor the acquisition and maintenance of compensatory mutations, permitting the restoration of the wild type–like two-chromosome structure. This was observed in mutant C926-EV2, which acquired a compensatory mutation in RctB (R195C) and resolved the chromosome fusion (Fig. 6E and table S1).
RctB-associated mutations compensate ΔcrtS by altering Chr2 initiation regulation
Our 200-generation evolution experiment results showed that the phenotype of ΔcrtS mutants was largely rescued by the selection of spontaneous mutations in the origin region of Chr2. Although the phenotypes of the evolved ΔcrtS mutants vary, they all involve an increase in fitness, a decrease in cell filamentation, and an absence of gDNA degradation compared to their parental strain (figs. S12 and S13). MFA of the evolved crtS mutants showed a higher Chr2/Chr1 copy number ratio, suggesting an up-regulation of ori2 initiation (fig. S14). This rapid acquisition of suppressor mutations can explain why our observations on ΔcrtS mutants differ from those of Baek and Chattoraj (12). Considering the rapid genome remodeling and acquisition of compensatory mutations in ori2 of ΔcrtS mutants, we speculate that the ΔchrI-4 mutant previously characterized (12) has acquired compensatory mutations to overcome the marked effect of crtS deletion. All ΔcrtS compensatory mutations were directly related to RctB (table S1), including three in the coding sequence (R195C, F230S, and L357I). RctB is a large, relatively poorly characterized protein that presents a 70-AA C-terminal region (451 to 521) involved in both iterons and 39-mer binding, as well as in dimerization (8). Previous rctB mutants were selected in the E. coli heterologous host for causing replication overinitiation of mini-Chr2 (9, 32–34). These “copy-up” mutants of RctB show reduced dimer binding to iterons (32), reduced dimerization (33), or reduced 39-mer binding (32) or fail to bind ATP (9), all consistently preventing RctB to repress replication. Most of these mutations were found outside the 70-AA C-terminal region (34), implying that important functions for ori2 replication control remains to be identified in RctB. One of our ΔcrtS suppressor mutants (R195C) was already documented as a copy-up mutant (34). An additional mutation was found in the promoter region of RctB within the 29-mer RctB binding site, possibly impairing binding. Up-regulation of Chr2 replication initiation could result from the suppression of RctB autorepression leading to an increase in RctB expression, or from the abolition of the 29-mer negative regulatory function by handcuffing with iterons (30). The mutations obtained in our study were selected directly in V. cholerae on Chr2, therefore taking into account the native physiological levels of all partners interacting to regulate Chr2 replication initiation. The experimental evolution screening of crtS mutants provides a promising approach to unravel new regulatory factors involved in V. cholerae Chr2 replication.
Chr1 and Chr2 termini cohesion before cell division
MFA shows that V. cholerae Chr1 and Chr2 finish replication at ter1 (around dif1) and ter2 (around dif2), respectively, in a nearly synchronous manner (Fig. 1A). In WT, both ter1 and ter2 are recruited early to mid-cell and remain together until the end of the cell cycle (fig. S5E), and 3C analysis shows strong interactions between the two ter regions (Fig. 5C). When Chr2 replication completes before Chr1 in crtSVC23 mutant (Fig. 2B), ter2 foci relocate earlier to mid-cell but remain at mid-cell until cell division, and segregate roughly at the same time as ter1 foci, like in WT (fig. S5, E and F, and fig. S6). These results suggest that ter1 and ter2 localization at mid-cell, where septum formation occurs at the end of the cell cycle, is important to coordinate their proper segregation before cell division.
In bacteria, chromosome replication, segregation, and cell division are precisely orchestrated mechanisms. In E. coli, the Ter domain relocates to mid-cell as replication completes and becomes accessible to the divisome machinery for the final steps of cell division, including chromosome compaction, dimer resolution, and decatenation (35–38). MatP, a DNA binding protein, bridges distant matS sites within the Ter domain to organize it into a compact structure (39, 40) that interacts with the divisome to coordinate segregation and cell division (38). Chromosome dimer resolution at dif is mediated by the XerCD site-specific recombinases and the DNA translocase FtsK, which is anchored at the septum (36). In V. cholerae, MatP is known to play a role in Ter confinement of the two chromosomes at mid-cell after replication (41) and both Chr1 and Chr2 require the septal protein FtsK to resolve their dimers (42). Therefore, ter1 and ter2 cohesion at mid-cell may be important for their proper segregation, which may be coordinated by MatP or another mechanism that remains to be found.
Proposed mechanisms that mediate the signal between crtS replication and ori2 initiation
Our study shows that Chr2 initiation requires crtS replication (Fig. 2C), meaning that either DNA conformational changes caused by the passage of the replication fork across crtS, or the doubling in copy number of crtS after duplication triggers Chr2 replication. The binding activity of RctB to chrI-4 (crtS) was observed by ChIP-chip (12). It was reported that RctB binding could only be observed in vitro by DNase I footprinting when chrI-4 was carried by a supercoiled plasmid (12). Mutations of the protected bases abolished the enhancer activity of chrI-4, suggesting a direct interaction between chrI-4 and RctB (12). Baek and Chattoraj (12) suggested that chrI-4 could act as a DNA chaperone to remodel RctB to an active form with altered DNA binding activities. Indeed, by using a mini-Chr2 in E. coli, they show that addition of chrI-4 in trans abolishes the requirement for DnaK/J chaperones, which are normally important to promote RctB-dependent initiation at ori2 (12, 32). Initiator remodeling upon interaction with their cognate DNA binding sites has been observed for iteron-bearing plasmids (43), as well as for E. coli DnaA (44). RctB could form a bidentate protein simultaneously using two DNA binding domains to contact two DNA loci (for example, crtS and ori2). In mutants with two chromosomal copies of crtS, we observed that most of newborn cells have two ori2 and only one ori1 (Fig. 4 and fig. S8), suggesting that it is the duplication of the crtS sequence itself that triggers Chr2 replication initiation, and that the duplication of the single-copy crtS sequence in wild type is limiting initiation at ori2. Once crtS is duplicated, dimers of RctB could simultaneously contact two crtS sites, allowing a higher DNA binding affinity to iterons. In mutants with two chromosomal copies of crtS, a large proportion of cells divide with two ori1 and four ori2 foci, giving rise to newborn cells already having one ori1 and two ori2 foci (Fig. 4, C to E). Hence, passage of the replication fork across crtS might still be an option for triggering Chr2 replication if the signal had been sent in the mother cell. crtS contains two GATC sites that are substrates for Dam methylase. RctB binding is sensitive to the methylation state of iterons sites, which need to be fully methylated (45). The passage of the replication fork across crtS would generate transiently hemimethylated GATC sites that may affect RctB binding. However, mutation of the two GATC sites had no impact on crtS function. Passage of the replication fork also generates single-stranded DNA on the template for lagging-strand synthesis. RctB could recognize a DNA hairpin structure formed by the single-stranded state of crtS, thereby modifying its binding affinities for iterons and/or 39-mers. A better understanding of the genetic determinants enclosed within the crtS sequence will provide new clues to decipher the crtS-RctB–mediated mechanism that triggers Chr2 replication, and perhaps a better understanding of the mechanisms controlling the activation/deactivation of RctB.
A large fraction of crtSWT/VC23 cells display three ori2 foci, suggesting that one of the two ori2 duplicates before the other (Fig. 4D). These observations are different from E. coli, where multiple origins initiate in synchrony (20). To accommodate for a generation time shorter than the replication time in fast-growing cells, overlapping rounds of replication occur with multiple origins of replication. In E. coli, multiple origins fire synchronously, such that either 2, 4, or 8 (that is, 2n) origins of replication are present at the same time (20). In E. coli, several mechanisms are responsible for the coordinated initiation of multiple origins (DnaA titration, regulatory inactivation of DnaA, origin sequestration, and DnaA reactivation sequences) (46). All these mechanisms control the availability of the active form of DnaA for initiating replication from oriC. If the control of ori2 initiation by crtS was done only by controlling the availability of the RctB active form, we would expect a similar synchrony in the firing of multiple ori2, and this would be observed by cells containing only 2n ori2 foci (for example, 2 or 4). Because a large portion of crtSWT/VC23 cells have three ori2 foci, we hypothesize that the duplication of one crtS triggers the firing of only one ori2. This reinforces the hypothesis that firing could necessitate a contact between crtS and ori2. Fluorescence microscopy of WT fluorescently labeled near crtS (VC783) and ori2 loci shows that the two loci localize to the same region throughout the cell cycle (fig. S4E). The contacts between ori2 and Chr1 revealed by 3C may be caused by the simultaneous binding of RctB to ori2 and crtS. The most frequent contacts between ori2 and Chr1 occur downstream of crtS (Fig. 5D). A possible explanation is that, following the duplication of the crtS locus, the replication machineries of Chr1 and Chr2 are in the vicinity of each other until the end of replication of the two chromosomes. Nonreplicating cells (that is, stationary phase) lose the cross-shape contacts observed between Chr1 and Chr2 replichores during exponential growth (Fig. 5B, right), suggesting that replication is indeed responsible for the contacts of the two chromosomes along their chromosomal arms. Overall, the 3C analysis of the V. cholerae chromosomes points to a direct interplay between 3D organization and replication regulation. How trans topological contacts would drive a functional interaction between the two chromosomes remains unknown.
Concluding remarks
Bacteria can be subjected to sudden environmental changes and must adapt their replication rate to their growth rate. Fast-growing bacteria often initiate multiple overlapping rounds of DNA replication. Interrelated mechanisms control the availability of the active form of DnaA to regulate chromosome initiation, some of which are modulated by growth rate (46). V. cholerae is a fast-growing bacterium that encounters a broad spectrum of habitats (for example, aquatic environments and human beings). Here, we describe a system for the replication control of multiple chromosomes. We demonstrate that Chr2 surveys the replication of Chr1 via the crtS locus and only initiates replication when this site has been replicated. Repositioning crtS closer to or further from the ori1 causes Chr2 initiation to be initiated earlier or later, respectively, in the cell cycle. If the signal is not received (for example, ΔcrtS mutant), initiation of Chr2 will be impaired and the cell will filament, accumulating many copies of Chr1. We suggest this mechanism to be an example of a bacterial cell cycle checkpoint; coordination of passage from early cell cycle, with only Chr1 replicating, into the late part of the cell cycle, with both chromosomes replicating. The location of crtS on Chr1 dictates the timing of replication initiation at ori2 so that replication of the two chromosomes terminate simultaneously. This, in turn, ensures coordinated and faithful segregation of the chromosomes in line with cell division. The crtS-RctB–mediated checkpoint control of Chr2 initiation is a simple and flexible mechanism to ensure a cell cycle–specific time setting of Chr2 replication relative to Chr1 replication. This novel mechanism is an elegant and cost-effective way for secondary chromosomes to benefit from the already well-adapted replication regulatory system of the host main chromosome.
Replication of eukaryotic genomes is initiated from multiple origins located on each chromosome, enabling the complete genomic replication within the S phase of the cell cycle (47). Thus, bacteria with multiple replicons may share similarities with eukaryotes in the control of their genomic replication. However, it is observed in eukaryotes that (i) not all origins are activated within a single replication round and (ii) activated origins do not start replication simultaneously (48). The observed tight control of initiation at ori2 is different from the stochastic nature of initiation at eukaryotic origins (49). Nevertheless, we envisage that replication-dependent controls, such as the checkpoint described here, may contribute to the orchestration of the complex eukaryotic DNA replication.
MATERIALS AND METHODS
Experimental design
Our objective was to determine the regulatory pathway responsible for triggering Chr2 replication at a specific point of the cell cycle, so that the two chromosomes terminate replication at nearly the same time. To achieve this, we created several V. cholerae derivatives with altered chromosome organization and monitored their replication pattern to determine whether this synchronization was directly linked to the replication of a specific region in Chr1. We used genome engineering, MFA, microscopy, and chromosome conformation capture to identify the elements involved in this regulatory pathway.
Bacterial strains and plasmids
The bacterial strains and plasmids used in this study are listed in table S2. Large genome rearrangements were performed following the procedures described by Val et al. (18). Details are given in the Supplementary Materials.
Quantitative PCR
qPCR was performed on the gDNA of cells growing exponentially in LB at 37°C [OD450 (optical density at 450 nm), ~0.15]. Primers used to determine ori1/ori2 ratios are listed in table S3. Details are given in the Supplementary Materials.
Marker frequency analysis
MFA was performed on the gDNA of cells growing exponentially in LB (glucose) at 30°C (OD450, ~0.15). Libraries were sequenced using an Ion Proton sequencer (Life Technologies). MFA was performed essentially as described by Skovgaard et al. (17). Details are given in the Supplementary Materials.
Pulsed-field gel electrophoresis
PFGE was done following the procedure described by Val et al. (10). Details are given in the Supplementary Materials.
Fluorescence microscopy
All Chr1 loci were labeled with a parSpMT1 site, and Chr2 loci were labeled with a lacO array. The genes encoding for yGFP-Δ30ParBpMT1 and LacI-mCherry protein fusions were inserted in the lacZ gene using plasmid pAD19 (table S2) (19). Cultures for microscopy were grown in minimal fructose medium to limit replication rounds to once per cell cycle. Microscopy observations and data analysis were performed following procedures and using MATLAB scripts already described by David et al. (19). Details are given in the Supplementary Materials.
Chromosome conformation capture
3C libraries were built, sequenced, and analyzed as previously described (23). For all the data, we generated matrices with bins of 5 kbp using a four-base cutter (Hpa II). Both matrices (LB and MM) showed a strong correlation together using Spearman’s rank correlation coefficient (ρ = 0.488, P < 10 to 64). Experimental details are given in the Supplementary Materials.
Supplementary Material
Acknowledgments
We thank S. Aguilar Pierlé and A. Soler Bistué for useful discussion and I. Vallet-Gely and F.-X. Barre for sharing MATLAB functions. Funding: This research was funded by the Institut Pasteur, INSERM, and CNRS. This work was supported by a grant from the French National Research Agency (ANR-10-BLAN-131301). R.K. was funded by the European Research Council (FP7/2007-2013; ERC grant agreement 260822). M.M. was funded by the Association pour la Recherche sur le Cancer fellowship (20100600373). F.d.L.M. and M.J.B. were supported by the Pasteur-Paris University Program. Author contributions: Conceived and designed the experiments: M.-E.V., M.M., R.K., O.S., and D.M. Performed the experiments: M.-E.V., M.M., S.P.K., F.d.L.M., M.J.B., C.P., and H.K. Analyzed the data: M.-E.V., M.M., R.K., O.S., and D.M. Wrote the paper: M.-E.V., S.P.K., R.K., O.S., and D.M. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Sequence reads used for MFA and 3C experiments have been submitted to NCBI Sequence Read Archive under accession number SRP070634.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/4/e1501914/DC1
Materials and Methods
fig. S1. Chromosomal inversion detected around ori1 when WT sequences are mapped against the National Center for Biotechnology Information (NCBI) reference genome AE003852.
fig. S2. Large DNA inversions either caused no fitness cost (JB392) or were similarly affected (JB590, JB659, JB771, and JB963).
fig. S3. Complementation of ΔcrtS filamentous phenotype by addition of an ectopic chromosomal copy of crtS.
fig. S4. Duplication and segregation of VC783 and ori2 foci in WT and mutant crtSVC23 throughout the cell cycle.
fig. S5. Duplication and segregation of ter1 and ter2 foci in WT and mutant crtSVC23 throughout the cell cycle.
fig. S6. ori2 foci duplicate earlier when crtS is located near ori1.
fig. S7. The addition of an extra copy of crtS affects the growth of V. cholerae.
fig. S8. Doubling in ori2 copy number in mutant with two chromosomal copies of crtS.
fig. S9. Comparison of global chromosome organization of V. cholerae in different growth conditions.
fig. S10. Comparison of directional index analysis at a 100-kbp scale with transcription and GC content for fast-growing cells.
fig. S11. ΔcrtS mutants have a fitness defect, whereas ICO1ΔcrtS shows no additional growth defect.
fig. S12. Fitness improvement of ΔcrtS mutants by the acquisition of compensatory mutations.
fig. S13. Loss of filamentation phenotype of ΔcrtS mutants by the acquisition of compensatory mutations.
fig. S14. MFA of crtS mutants before and after acquisition of compensatory mutations.
fig. S15. The effect of MFA normalizations.
table S1. Compensatory mutations obtained after evolution of ΔcrtS mutants.
table S2. List of plasmids and bacterial strains.
table S3. Primers used in qPCR.
movie S1. 3D representations of the contact map from fig. S9 (exponential growth of the WT in LB).
movie S2. 3D representations of the contact map from fig. S9 (exponential growth of the WT in MM).
movie S3. Time-lapse fluorescence microscopy of ΔcrtS filamentous cells, tagged at VC783 (Chr1) and near ori2 (Chr2), growing on an M9 MM agar pad supplemented with fructose and thiamine.
movie S4. Time-lapse fluorescence microscopy of ΔcrtS filamentous cells, tagged at VC783 (Chr1) and near ori2 (Chr2), growing on an M9 MM agar pad supplemented with fructose and thiamine.
REFERENCES AND NOTES
- 1.C. Mackenzie, S. Kaplan, M. Choudhary, Microbial Evolution: Gene Establishment, Survival, and Exchange, R. V. Miller, M. J. Day, Eds. (ASM Press, Washington, DC, 2004). [Google Scholar]
- 2.Harrison P. W., Lower R. P. J., Kim N. K. D., Young J. P. W., Introducing the bacterial ‘chromid’: Not a chromosome, not a plasmid. Trends Microbiol. 18, 141–148 2010). [DOI] [PubMed] [Google Scholar]
- 3.Katayama T., Ozaki S., Keyamura K., Fujimitsu K., Regulation of the replication cycle: Conserved and diverse regulatory systems for DnaA and oriC. Nat. Rev. Microbiol. 8, 163–170 (2010). [DOI] [PubMed] [Google Scholar]
- 4.del Solar G., Espinosa M., Plasmid copy number control: An ever-growing story. Mol. Microbiol. 37, 492–500 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Okada K., Iida T., Kita-Tsukamoto K., Honda T., Vibrios commonly possess two chromosomes. J. Bacteriol. 187, 752–727 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heidelberg J. F., Eisen J. A., Nelson W. C., Clayton R. A., Gwinn M. L., Dodson R. J., Haft D. H., Hickey E. K., Peterson J. D., Umayam L., Gill S. R., Nelson K. E., Read T. D., Tettelin H., Richardson D., Ermolaeva M. D., Vamathevan J., Bass S., Qin H., Dragoi I., Sellers P., McDonald L., Utterback T., Fleishmann R. D., Nierman W. C., White O., Salzberg S. L., Smith H. O., Colwell R. R., Mekalanos J. J., Venter J. C., Fraser C. M., DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406, 477–483 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duigou S., Knudsen K. G., Skovgaard O., Egan E. S., Løbner-Olesen A., Waldor M. K., Independent control of replication initiation of the two Vibrio cholerae chromosomes by DnaA and RctB. J. Bacteriol. 188, 6419–6424 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jha J. K., Ghirlando R., Chattoraj D. K., Initiator protein dimerization plays a key role in replication control of Vibrio cholerae chromosome 2. Nucleic Acids Res. 42, 10538–10549 2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duigou S., Yamaichi Y., Waldor M. K., ATP negatively regulates the initiator protein of Vibrio cholerae chromosome II replication. Proc. Natl. Acad. Sci. U.S.A. 105, 10577–10582 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Val M.-E., Soler-Bistué A., Bland M. J., Mazel D., Management of multipartite genomes: The Vibrio cholerae model. Curr. Opin. Microbiol. 22, 120–126 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Venkova-Canova T., Chattoraj D. K., Transition from a plasmid to a chromosomal mode of replication entails additional regulators. Proc. Natl. Acad. Sci. U.S.A. 108, 6199–6204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baek J. H., Chattoraj D. K., Chromosome I controls chromosome II replication in Vibrio cholerae. PLOS Genet. 10, e1004184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nordström K., Dasgupta S., Copy-number control of the Escherichia coli chromosome: A plasmidologist’s view. EMBO Rep. 7, 484–489 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Egan E. S., Løbner-Olesen A., Waldor M. K., Synchronous replication initiation of the two Vibrio cholerae chromosomes. Curr. Biol. 14, R501–R502 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Rasmussen T., Jensen R. B., Skovgaard O., The two chromosomes of Vibrio cholerae are initiated at different time points in the cell cycle. EMBO J. 26, 3124–3131 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rocha E. P., The organization of the bacterial genome. Annu. Rev. Genet. 42, 211–233 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Skovgaard O., Bak M., Løbner-Olesen A., Tommerup N., Genome-wide detection of chromosomal rearrangements, indels, and mutations in circular chromosomes by short read sequencing. Genome Res. 21, 1388–1393 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Val M.-E., Skovgaard O., Ducos-Galand M., Bland M. J., Mazel D., Genome engineering in Vibrio cholerae: A feasible approach to address biological issues. PLOS Genet. 8, e1002472 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.David A., Demarre G., Muresan L., Paly E., Barre F.-X., Possoz C., The two cis-acting sites, parS1 and oriC1, contribute to the longitudinal organisation of Vibrio cholerae chromosome I. PLOS Genet. 10, e1004448 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skarstad K., Boye E., Steen H. B., Timing of initiation of chromosome replication in individual Escherichia coli cells. EMBO J. 5, 1711–1717 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dekker J., Rippe K., Dekker M., Kleckner N., Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Marbouty M., Cournac A., Flot J.-F., Marie-Nelly H., Mozziconacci J., Koszul R., Metagenomic chromosome conformation capture (meta3C) unveils the diversity of chromosome organization in microorganisms. ELife 3, e03318 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marbouty M., Le Gall A., Cattoni D. I., Cournac A., Koh A., Fiche J.-B., Mozziconacci J., Murray H., Koszul R., Nollmann M., Condensin- and replication-mediated bacterial chromosome folding and origin condensation revealed by Hi-C and super-resolution imaging. Mol. Cell 59, 588–602 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Wang X., Le T. B. K., Lajoie B. R., Dekker J., Laub M. T., Rudner D. Z., Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev. 29, 1661–1675 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Le T. B. K., Imakaev M. V., Mirny L. A., Laub M. T., High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342, 731–734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lesne A., Riposo J., Roger P., Cournac A., Mozziconacci J., 3D genome reconstruction from chromosomal contacts. Nat. Methods 11, 1141–1143 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Umbarger M. A., Toro E., Wright M. A., Porreca G. J., Baù D., Hong S.-H., Fero M. J., Zhu L. J., Marti-Renom M. A., McAdams H. H., Shapiro L., Dekker J., Church G. M., The three-dimensional architecture of a bacterial genome and its alteration by genetic perturbation. Mol. Cell 44, 252–264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazel D., Dychinco B., Webb V. A., Davies J., A distinctive class of integron in the Vibrio cholerae genome. Science 280, 605–608 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Slack A., Thornton P. C., Magner D. B., Rosenberg S. M., Hastings P. J., On the mechanism of gene amplification induced under stress in Escherichia coli. PLOS Genet. 2, e48 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Venkova-Canova T., Saha A., Chattoraj D. K., A 29-mer site regulates transcription of the initiator gene as well as function of the replication origin of Vibrio cholerae chromosome II. Plasmid 67, 102–110 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Val M.-E., Kennedy S. P., Soler-Bistué A. J., Barbe V., Bouchier C., Ducos-Galand M., Skovgaard O., Mazel D., Fuse or die: How to survive the loss of Dam in Vibrio cholerae. Mol. Microbiol. 91, 665–678 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Jha J. K., Demarre G., Venkova-Canova T., Chattoraj D. K., Replication regulation of Vibrio cholerae chromosome II involves initiator binding to the origin both as monomer and as dimer. Nucleic Acids Res. 40, 6026–6038 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koch B., Ma X., Løbner-Olesen A., rctB mutations that increase copy number of Vibrio cholerae oriCII in Escherichia coli. Plasmid 68, 159–169 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Yamaichi Y., Gerding M. A., Davis B. M., Waldor M. K., Regulatory cross-talk links Vibrio cholerae chromosome II replication and segregation. PLOS Genet. 7, e1002189 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stouf M., Meile J.-C., Cornet F., FtsK actively segregates sister chromosomes in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 110, 11157–11162 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kennedy S. P., Chevalier F., Barre F.-X., Delayed activation of Xer recombination at dif by FtsK during septum assembly in Escherichia coli. Mol. Microbiol. 68, 1018–1028 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Espeli O., Lee C., Marians K. J., A physical and functional interaction between Escherichia coli FtsK and topoisomerase IV. J. Biol. Chem. 278, 44639–44644 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Espéli O., Borne R., Dupaigne P., Thiel A., Gigant E., Mercier R., Boccard F., A MatP-divisome interaction coordinates chromosome segregation with cell division in E. coli. EMBO J. 31, 3198–3211 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mercier R., Petit M.-A., Schbath S., Robin S., El Karoui M., Boccard F., Espéli O., The MatP/matS site-specific system organizes the terminus region of the E. coli chromosome into a macrodomain. Cell 135, 475–485 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Dupaigne P., Tonthat N. K., Espéli O., Whitfill T., Boccard F., Schumacher M. A., Molecular basis for a protein-mediated DNA-bridging mechanism that functions in condensation of the E. coli chromosome. Mol. Cell 48, 560–571 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Demarre G., Galli E., Muresan L., Paly E., David A., Possoz C., Barre F.-X., Differential management of the replication terminus regions of the two Vibrio cholerae chromosomes during cell division. PLOS Genet. 10, e1004557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Val M.-E., Kennedy S. P., El Karoui M., Bonné L., Chevalier F., Barre F.-X., FtsK-dependent dimer resolution on multiple chromosomes in the pathogen Vibrio cholerae. PLOS Genet. 4, e1000201 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Díaz-López T., Lages-Gonzalo M., Serrano-López A., Alfonso C., Rivas G., Díaz-Orejas R., Giraldo R., Structural changes in RepA, a plasmid replication initiator, upon binding to origin DNA. J. Biol. Chem. 278, 18606–18616 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Fujimitsu K., Senriuchi T., Katayama T., Specific genomic sequences of E. coli promote replicational initiation by directly reactivating ADP-DnaA. Genes Dev. 23, 1221–1233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Demarre G., Chattoraj D. K., DNA adenine methylation is required to replicate both Vibrio cholerae chromosomes once per cell cycle. PLOS Genet. 6, e1000939 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Leonard A. C., Grimwade J. E., Regulation of DnaA assembly and activity: Taking directions from the genome. Annu. Rev. Microbiol. 65, 19–35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhind N., Gilbert D. M., DNA replication timing. Cold Spring Harb. Perspect. Biol. 5, a010132 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Agier N., Romano O. M., Touzain F., Cosentino Lagomarsino M., Fischer G., The spatiotemporal program of replication in the genome of Lachancea kluyveri. Genome Biol. Evol. 5, 370–388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hawkins M., Retkute R., Müller C. A., Saner N., Tanaka T. U., de Moura A. P. S., Nieduszynski C. A., High-resolution replication profiles define the stochastic nature of genome replication initiation and termination. Cell Rep. 5, 1132–1141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Le Roux F., Binesse J., Saulnier D., Mazel D., Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl. Environ. Microbiol. 73, 777–784 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marvig R. L., Blokesch M., Natural transformation of Vibrio cholerae as a tool - Optimizing the procedure. BMC Microbiol. 10, 155 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Soler-Bistué A., Mondotte J. A., Bland M. J., Val M.-E., Saleh M.-C., Mazel D., Genomic location of the major ribosomal protein gene locus determines Vibrio cholerae global growth and infectivity. PLOS Genet. 11, e1005156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bremer H., Churchward G., An examination of the Cooper-Helmstetter theory of DNA replication in bacteria and its underlying assumptions. J. Theor. Biol. 69, 645–654 (1977). [DOI] [PubMed] [Google Scholar]
- 54.Cooper S., Helmstetter C. E., Chromosome replication and the division cycle of Escherichia coliB/r. J. Mol. Biol. 31, 519–540 (1968). [DOI] [PubMed] [Google Scholar]
- 55.Sliusarenko O., Heinritz J., Emonet T., Jacobs-Wagner C., High-throughput, subpixel precision analysis of bacterial morphogenesis and intracellular spatio-temporal dynamics. Mol. Microbiol. 80, 612–627 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Imakaev M., Fudenberg G., McCord R. P., Naumova N., Goloborodko A., Lajoie B. R., Dekker J., Mirny L. A., Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat. Methods 9, 999–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cournac A., Marie-Nelly H., Marbouty M., Koszul R., Mozziconacci J., Normalization of a chromosomal contact map. BMC Genomics 13, 436 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Papenfort K., Förstner K. U., Cong J.-P., Sharma C. M., Bassler B. L., Differential RNA-seq of Vibrio cholerae identifies the VqmR small RNA as a regulator of biofilm formation. Proc. Natl. Acad. Sci. U.S.A. 112, E766–E775 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krzywinski M., Schein J., Birol İ., Connors J., Gascoyne R., Horsman D., Jones S. J., Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Feng L., Reeves P. R., Lan R., Ren Y., Gao C., Zhou Z., Ren Y., Cheng J., Wang W., Wang J., Qian W., Li D., Wang L., A recalibrated molecular clock and independent origins for the cholera pandemic clones. PLOS One 3, e4053 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lan R., Reeves P. R., Recombination between rRNA operons created most of the ribotype variation observed in the seventh pandemic clone of Vibrio cholerae. Microbiology 144, 1213–1221 (1998). [DOI] [PubMed] [Google Scholar]
- 63.Dixon J. R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J. S., Ren B., Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Meibom K. L., Blokesch M., Dolganov N. A., Wu C.-Y., Schoolnik G. K., Chitin induces natural competence in Vibrio cholerae. Science 310, 1824–1827 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/4/e1501914/DC1
Materials and Methods
fig. S1. Chromosomal inversion detected around ori1 when WT sequences are mapped against the National Center for Biotechnology Information (NCBI) reference genome AE003852.
fig. S2. Large DNA inversions either caused no fitness cost (JB392) or were similarly affected (JB590, JB659, JB771, and JB963).
fig. S3. Complementation of ΔcrtS filamentous phenotype by addition of an ectopic chromosomal copy of crtS.
fig. S4. Duplication and segregation of VC783 and ori2 foci in WT and mutant crtSVC23 throughout the cell cycle.
fig. S5. Duplication and segregation of ter1 and ter2 foci in WT and mutant crtSVC23 throughout the cell cycle.
fig. S6. ori2 foci duplicate earlier when crtS is located near ori1.
fig. S7. The addition of an extra copy of crtS affects the growth of V. cholerae.
fig. S8. Doubling in ori2 copy number in mutant with two chromosomal copies of crtS.
fig. S9. Comparison of global chromosome organization of V. cholerae in different growth conditions.
fig. S10. Comparison of directional index analysis at a 100-kbp scale with transcription and GC content for fast-growing cells.
fig. S11. ΔcrtS mutants have a fitness defect, whereas ICO1ΔcrtS shows no additional growth defect.
fig. S12. Fitness improvement of ΔcrtS mutants by the acquisition of compensatory mutations.
fig. S13. Loss of filamentation phenotype of ΔcrtS mutants by the acquisition of compensatory mutations.
fig. S14. MFA of crtS mutants before and after acquisition of compensatory mutations.
fig. S15. The effect of MFA normalizations.
table S1. Compensatory mutations obtained after evolution of ΔcrtS mutants.
table S2. List of plasmids and bacterial strains.
table S3. Primers used in qPCR.
movie S1. 3D representations of the contact map from fig. S9 (exponential growth of the WT in LB).
movie S2. 3D representations of the contact map from fig. S9 (exponential growth of the WT in MM).
movie S3. Time-lapse fluorescence microscopy of ΔcrtS filamentous cells, tagged at VC783 (Chr1) and near ori2 (Chr2), growing on an M9 MM agar pad supplemented with fructose and thiamine.
movie S4. Time-lapse fluorescence microscopy of ΔcrtS filamentous cells, tagged at VC783 (Chr1) and near ori2 (Chr2), growing on an M9 MM agar pad supplemented with fructose and thiamine.