

Abstract
Electron impact excitation of ions from organics (EIEIO), also referred to as electron-induced dissociation, was applied to singly charged SM molecular species in the gas phase. Using ESI and a quadrupole TOF mass spectrometer equipped with an electron-ion reaction device, we found that SMs fragmented sufficiently to identify their lipid class, acyl group structure, and the location of double bond(s). Using this technique, nearly 200 SM molecular species were found in four natural lipid extracts: bovine milk, porcine brain, chicken egg yolk, and bovine heart. In addition to the most common backbone, d18:1, sphingosines with a range of carbon chain lengths, sphingadienes, and some sphinganine backbones were also detected. Modifications in natural SMs were also identified, including addition of iodine/methanol across a carbon-carbon double bond. This unparalleled new approach to SM analysis using EIEIO-MS shows promise as a unique and powerful tool for structural characterization.
Keywords: sphingolipids, lipidomics, lipids/chemistry, electron-induced dissociation
SMs comprise a class of lipids within the sphingolipid category that are found ubiquitously in the human body, but are enriched in brain lipids where they play a crucial role in insulating nerve cell axons. This class of lipids is also found in relatively high concentrations in cell membrane microdomains, termed lipid rafts, which are believed to be involved in diverse cell functions, such as cell trafficking, cell signaling, lipid-protein interactions, and apoptosis [for a general review of SM, see (1, 2)]. The use of ESI MS to fully characterize the diverse SM molecular species has proven to be challenging (3, 4). Structural elucidation of SM using collision-induced dissociation (CID), a commonly used fragmentation method for lipid analysis, is limited due the low-energy collisions (<100 eV) and generally only provides information regarding lipid class, carbon chain length, and the total number of double bonds that comprise the lipid backbone and the acyl group (3). Molecular species-specific information such as backbone type (sphinganine, sphingosine, and sphingadiene with different carbon chain lengths) and the structure of the amide-linked acyl chains (i.e., carbon chain length and the number and stereo-configuration of double bonds) are not provided in a typical CID MS/MS spectrum. The analysis of glycerophospholipids by CID also provides limited structural information and requires polarity switching to identify both the phospholipid class and the fatty acid composition (5–7). In order to fully characterize the structure of lipids, a different fragmentation technique is needed. Recently, Campbell and Baba (8) reported a “nearly” complete identification method of phosphatidylcholines (PCs) using electron-induced dissociation or electron impact excitation of ions from organics (EIEIO) (9) in a branched radio-frequency electron-ion reaction device (10) (referred to as ExD cell in this work). Using EIEIO to fragment glycerophospholipids, information regarding lipid class (or head group), acyl chain length, the number and location of double bonds, and the regioisomeric structure were obtained in a single experiment in the positive ion mode. Using an alternative fragmentation method, Deimler, Sander, and Jackson (4) recently reported a radical induced dissociation of PCs using bombardment of metastable helium atoms (MAD-MS). These reports on novel approaches to structural elucidation of glycerophospholipids suggest that characterization of other lipid categories, such as SM, may be improved by alternative fragmentation strategies to CID.
In this study, we applied EIEIO to SM structural characterization using a branched ion trap (10) installed in a quadrupole TOF mass spectrometer with a differential mobility spectrometry (DMS) cell (SelexIONTM Technology) installed between the ion source and the mass spectrometer (11–13). The DMS was used to isolate SM from other interfering lipid classes, namely PCs and triacylglycerols (TAGs), prior to analysis. This is particularly important because SMs and PCs share the same head group (i.e., phosphocholine) and are roughly in the same mass range. Using DMS to isolate the SM, we obtained nearly 200 EIEIO-based MS/MS spectra on individual SM molecular species in various biological lipid extracts without isobaric interference from other lipid classes or categories, which generated in-depth structural details, including the sphingolipid backbone length and number of double bonds, acyl chain structure, including carbon chain length, and the number and locations of double bond(s). Additionally, the fragments appeared in the product ion spectrum that enabled differentiation between sphinganines (SMs that have no carbon-carbon double bond in the sphingoid backbone), sphingosines (SMs that have one carbon-carbon double bond in the sphingoid backbone), and sphingadienes (SMs containing two carbon-carbon double bonds in the sphingoid backbone). Consequently, EIEIO yields “near-complete” characterization of SM molecular species.
EXPERIMENTS
Samples
All lipid standards and lipid mixtures were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) and are identified here using the guidelines of Liebisch et al. (14). The specific lipid molecular species, SM(d18:1,12:0) and PC[16:0,18:1(9Z)] were used for method validation. Commercial extracts of SM derived from porcine brain (brain SM), bovine milk (milk SM), and chicken egg (egg SM) were used to characterize SM from biological sources, and bovine heart extract (BHE) was used as a complex biological extract. The solvent for all working solutions was HPLC-grade dichloromethane:methanol (50/50, v/v) with 10 mM ammonium acetate. These solvents and ammonium acetate were purchased from Caledon Laboratory Chemicals (Georgetown, ON, Canada) and Sigma-Aldrich Canada Co. (Oakville, ON, Canada), respectively. Concentrations of working solution were 250 μg/ml for BHE; 100 μg/ml for brain SM, milk SM and egg SM; and 1 μg/ml for the standard SM and PC samples.
DMS-ExD-TOF mass spectrometer
All experiments were conducted using a modified TripleTOF® 5600 system (SCIEX) that was equipped with both a DMS device and a branched radio-frequency ExD cell (Fig. 1) (10). The fundamental behavior of DMS and its application to lipid analysis are described elsewhere (11–13). Briefly, the DMS cell is mounted in the atmospheric pressure region between the ESI source (held at 5,000 V) and the spectrometer’s sampling orifice. The DMS operating parameters were optimized to isolate SM ions from potential isobaric interferences from ionized PCs and TAGs (see below). These parameters included a DMS cell temperature of 200°C, nitrogen (99.999%) as the curtain gas (transport gas for the DMS cell), and addition of isopropanol to the curtain gas at 1.5% (v/v) as a chemical modifier. For the DMS experiments, the separation voltage (SV) was held at an optimum value (3,900 V peak-to-peak), while the compensation voltage (COV) was scanned from −5 V to 20 V in 0.25 V increments. At each increment of COV, MS or EIEIO-MS spectra were recorded.
Fig. 1.

A: An ESI-quadrupole TOF mass spectrometer with an ExD cell and a DMS used in these EIEIO experiments. The TOF mass spectrometer is not shown in this figure. B in this figure represents the direction of the magnetic field to assist electrons traveling along the radio frequency potential minimum. B: MS spectrum of the BHE total lipid. C: DMS spectrum of lipid class separation by DMS. COV was scanned on constant SV. The sample is total lipid in BHE. We optimized the system as the yield of SMs was maximized. D: Separated SM spectrum.
The configuration of the ExD cell was reported previously (8, 10). To cool the ions in the device, helium buffer gas was introduced with a partial pressure in the ExD cell of ∼3 mTorr. This is the same pressure in Q2 due to the 5 mm diameter holes present in the IQ2B electrode that provide efficient gas conductance. The electron beam used in the EIEIO experiments was generated by a yttria-coated iridium disk cathode (ES-525; Kimball Physics, Wilton, NH). The cathode was operated at the space charge limit of the electron cloud in the ExD cell by a direct current power supply in constant voltage mode. The kinetic energy of the electron beam (KEe) was controlled by biasing the cathode negatively to the branched ion trap electrodes.
Simultaneous trapping mode (10) was mainly used in this work to enhance the production and detection of EIEIO fragment ions. In simultaneous trapping mode, ions were irradiated by the electron beam as ions were injected into the ExD cell (i.e., electron beam was applied when the IQ2A lens was set at open and the IQ2B was closed). All of the ions remaining in the ExD cell after the EIEIO reaction were subsequently injected into Q2 and beyond (i.e., the TOF pusher region) for mass analysis by opening the IQ2B lens for 1 ms. In contrast, trapping mode is a conventional scan function in which the electron beam irradiates the trapped precursor ions.
For some SM species, background subtraction of their EIEIO mass spectra was performed to augment detection of those species in the presence of low-level background chemical interferences (see below). To accomplish this, we first recorded an EIEIO mass spectrum without adding SM analyte ions to the ExD cell; as a result, only hydrocarbon-based background molecules in the ExD cell (presumed to arise from roughing pump oil contamination) were ionized. After this initial survey of the chemical background, SM analyte ions were admitted to the ExD cell and EIEIO was performed, resulting in a mixture of fragment ions derived from both the SM and the hydrocarbon chemical noise. However, background subtraction (removing the chemical noise-derived EIEIO peaks from the SM-derived mass spectrum) improved the analysis of the low-abundance SM species tremendously. This characterization of the chemical noise in the ExD cell was obtained approximately every 30 min during EIEIO experiments.
In order to perform CID, voltages on the two lens electrodes of the ExD cell (IQ2A and IQ2B) were set to allow precursor ions to be injected into the Q2 collision cell. Here, the electron beam was turned off and nitrogen gas was introduced into Q2 (and the ExD cell) as a collision gas. In order to apply kinetic energy to isolated ions, the upstream ion path before the IQ2A was biased 60–90 V to the ExD cell-Q2 assembly. The ExD cell was controlled by in-house software coded using LabView (National Instruments Co., Austin, TX).
RESULTS AND DISCUSSION
Like PCs, SMs yield rich EIEIO fragment ion spectra
In initial experiments, EIEIO (KEe = 10 eV) was applied to a synthetic standard, SM(d18:1,12:0) (Fig. 2A), and the resulting spectrum was rich enough in diagnostic fragment ions to provide near complete structural characterization for this SM molecular species. In the spectrum, the following specific regions of this EIEIO spectrum contained information on SM structural features: m/z 180–260, SM head group fragments and dual chain loss fragments; m/z 400∼500, SM backbone cleavage with losses of the acyl group and the head group present; and m/z >400, fragments from both carbon chains with a weaker, yet complete, fragment series for the carbon-carbon bond cleavages. In contrast, CID spectra acquired on the same instrument (Fig. 2B) contained fewer fragment ions and much less structural information, which only identified the head group (m/z 184) and the chain length of the sphingosine backbone (m/z 264) (3). As expected, CID did not produce fragments containing information on the backbone or the acyl group.
Fig. 2.

Comparison between dissociation product spectra by EIEIO (A) and CID (B). The sample was a synthesized standard SM, SM(d18:1,12:0). The EIEIO spectrum of a PC lipid [1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylcholine or PC(16:0/18:1)] is shown in (C) as a typical spectrum with the same head group as SM.
Optimization of the KEe utilized for EIEIO
The selection of the optimum level of KEe was essential for SM analysis using EIEIO, as we observed during our study on PCs (8). Besides overall EIEIO fragmentation efficiency, we sought to optimize signals to obtain the clearest chain fragment profiles with their characteristic radical and nonradical products. For the case of SMs, we chose SM(d18:1,18:0) from a brain extract as a model compound to optimize KEe, paying particular attention to the diagnostic fragmentation around the sphingosine backbone near the head group and the acyl chain. EIEIO spectra were recorded while ramping the KEe (Fig. 3), revealing each fragment ion’s KEe dependence. While the majority of fragments shared a common threshold of ∼5–6 eV, one exception was the acyl validation peak (head group loss and backbone cleavage between C2 and C3), which had a threshold of ∼9 eV. To obtain optimal signal intensity and information quality for the acyl validation fragments, there was a need to compromise between overall signal intensity and information quality at KEe values. For example, when KEe was set >10 eV, EIEIO spectra were unacceptable for identifying SMs because: 1) the acyl chain fragmentation simply became too extensive, making structural interpretation unduly complex, just like PC case (8); and 2) electron ionization (EI)-based chemical noise (from ionization of background contaminants) was drastically increased. In some cases, background chemical noise was still present in EIEIO spectra, but we established a protocol for such instances (see below). Thus, we set KEe = 10 eV for the SM analyses, thereby obtaining signals with good signal to noise ratio (S/N) for EIEIO spectra accumulated for periods ranging from a few seconds to several minutes (depending upon precursor SM signal intensity).
Fig. 3.

Signal intensity of each diagnostic peak on kinetic energy of electrons. Intensities of fragments were expanded vertically by the values shown in the figure. To obtain good S/N signal, the acyl validation peak (dashed line with round circle markers) and to avoid electron ionization (EI) of residual gas in the ExD cell, we have chosen electron energy of 10 eV.
EIEIO reaction speed for SM analysis
Figure 4A shows the fragmentation reaction speed at KEe of 10 eV, when electron beam intensity was at its space charge limit (i.e., reaction speed was not improved by increasing the heater current applied on the electron emitter). Conventional trapping mode was used in this measurement instead of the continuous trapping mode in order to define reaction time between electrons and ions. Isolated precursor ions were introduced into the ExD cell for 100 ms without electron beam. After ion loading, the IQ2A lens was closed before the electron beam was applied. The electron irradiation time, or reaction time, was the variable parameter of this measurement. The trapped ions were released from the ExD cell into the TOF mass analyzer by opening IQ2B after electron beam irradiation was terminated. Precursor ion intensity before electron irradiation, precursor ion intensity after electron irradiation, and the head group fragments are shown in Fig. 4A. Reaction of SMs was slower than was previously reported for PC (8), typically 1/5 in the same instruments. The mechanism that explains the reaction rate difference between SM and PC is not clear, but the interaction cross-section between the ions and electrons can be significantly different.
Fig. 4.

A: Reaction rate between singly charged SM and electron beam with kinetic energy of 10 eV in the conventional trapping mode. The sample was SM(d18:1,18:0) in brain SM. The reaction time was 440 ms when the electron beam was in space charge limit. B: Reaction time optimization in simultaneous trapping mode to obtain maximum yield of product ions. The consumed amount of precursor ions (or precursor used in the figure) was defined as the subtracted value of remained precursor intensity after reaction from the initial precursor intensity before reaction. The errors, mainly the fluctuation of the ion source, were large because of error propagation in the subtraction. The consumed precursor ions and obtained fragments were maximized at reaction time of 150 ms.
Rapid SM analysis can be achieved using simultaneous trapping mode, as has been shown for PCs (8) as well as for electron capture dissociation of peptides and proteins (10). Figure 4B shows precursor ion consumption and product yield in simultaneous trapping mode, which was the primary method used in this work. When irradiation time was increased, a maximum product yield was achieved at 150 ms.
EIEIO provides a generalized identification method for SMs
The EIEIO-based fragmentation pattern observed during the analysis of SM(d18:1,12:0) provides a good generic case for characterizing SMs, with the m/z values of the diagnostic peaks appearing in the EIEIO spectra shown in Table 1. The intense peak at m/z 184.074 indicates the presence of a phosphocholine head group in the lipid, which is often used to identify either PCs or SMs by traditional CID methods. In those workflows, SMs are only distinguished from PCs by the fact that SM precursor ions are of odd nominal m/z and even for PCs due to the nitrogen rule. However, this may not be the case if an SM exists in a modified form; alternatively, an SM could be present in low abundance within the isotope envelope of an interfering PC (e.g., the 13C isotope of a PC would be present as an odd m/z ion, possibly masking a monoisotopic form of SM). The latter issue can also be abrogated by the use of DMS separations (see below). Fortuitously, EIEIO provides additional SM diagnostic fragment ions at m/z 225.100 and 253.095 (Figs. 2A, 3) not displayed by PCs, which instead provide diagnostic fragments at m/z 224.104 and 226.083 (Fig. 2A, C). This results in product ion spectra that can be clearly attributed to SM, and enabled us to ascribe specific diagnostic fragments to SM molecular species in standards as well as complex biological lipid extracts without the need for chromatographic separation (12).
TABLE 1.
SM diagnostic in EIEIO MS
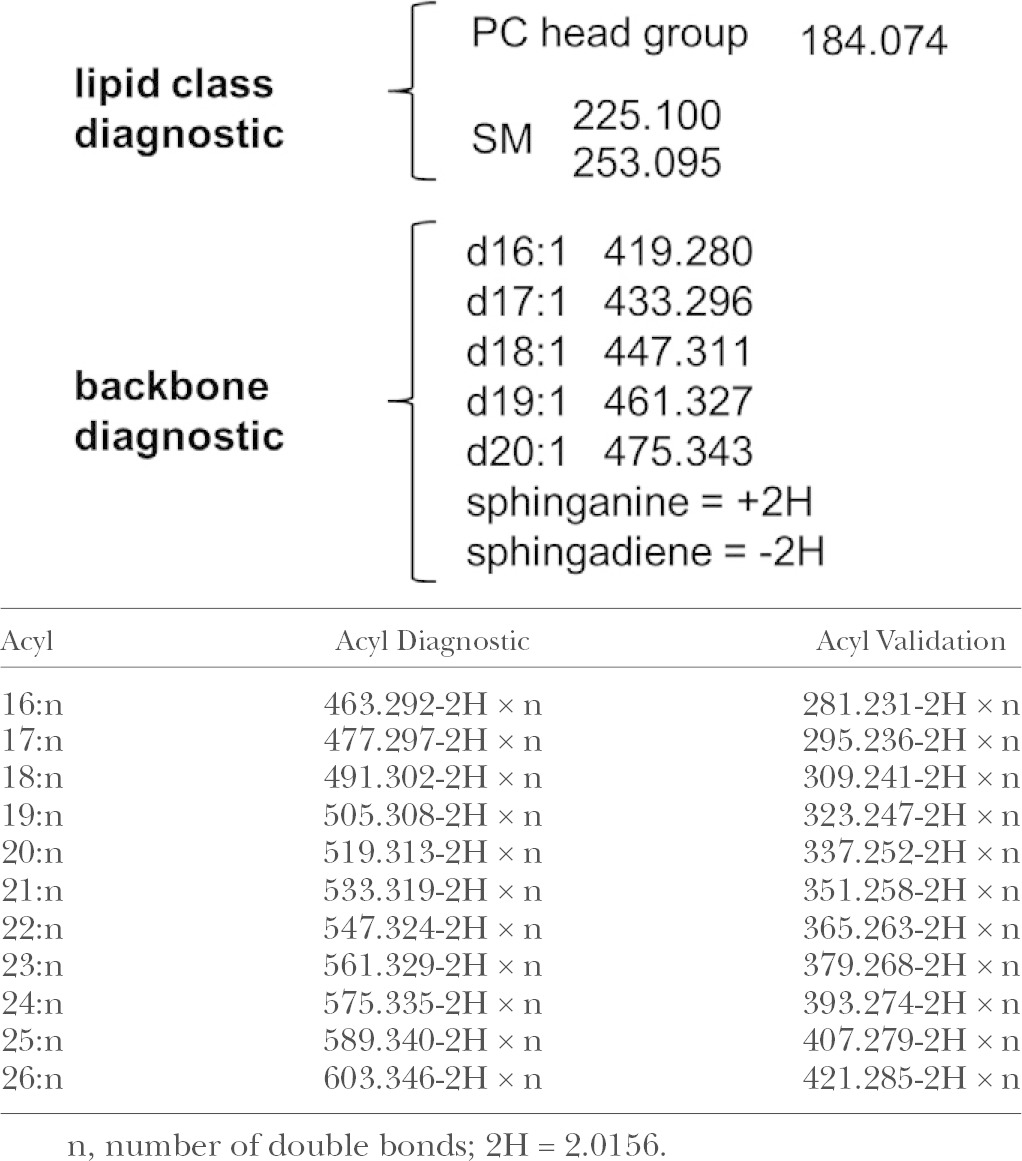
EIEIO of SMs also yielded diagnostic fragment ions resulting from bond cleavage around the branching point on the SM backbone (Fig. 3). These fragment ions (e.g., m/z 449.326-2H = m/z 447.311 in Fig. 2A) are unique to SM molecular species and are useful, especially in cases where less common SM forms are present, such as sphingosines with odd numbers of carbons, sphingadienes, and sphinganines. The acyl chain diagnostic peaks (Table 1) are easy to locate in EIEIO spectra, as they are often the most intense peaks located mid-range (m/z ∼200–500) in an EIEIO spectrum. For example, the fragment ion of m/z 407.264 in Fig. 2A is an example of such an acyl chain diagnostic peak. An additional acyl chain diagnostic peak (m/z 225.269 in Fig. 2A) was used for supplemental diagnostics to confirm the acyl chain. Here, we identified four peaks to identify the two chains in this SM molecular species, and these backbone and acyl chain fragments are consistent with a reconstructed SM precursor and its measured precursor m/z.
This set of simple diagnostic rules was encoded for post-acquisition data analysis using LabView. Multiple isomeric SMs were often found in a single EIEIO spectrum [e.g., SM(d18:1,16:0) and SM(d19:0,15:1)], as they share identical precursor m/z values. However, while these isomers can share backbone and acyl diagnostic peaks, the presence of the other characteristic EIEIO fragment ions verified the presence of multiple isomers in those experiments. Data analysis also identified double bond locations from the pattern of the chain region using the same concept that was previously reported (8) (i.e., −2H mass shift at the double bond location and characteristic intensity profile with “V shape” in the EIEIO spectrum). While automatic identification of double bond position using these criteria was relatively facile for PCs, this was not always the case for SMs, where the classic “V shape” intensity profile was not always evident. Consequently, an assumption was made that an acyl chain with double bonds has one of ω-3 (or n-3), ω-6 (n-6), ω-7 (n-7), or ω-9 (n-9) form with two carbon-carbon single bond spacing in cases of polyunsaturated acyl chains. In cases of sphingadiene backbones, the second double bond position was easily identified at ω-4 (n-4) as the common location (15).
Optimal EIEIO characterization of SMs from natural extracts benefits from DMS
When applying EIEIO to characterize SMs in more complex lipid extracts, the challenge of isobaric interference can be addressed by using an orthogonal means of separation. In this study, we employed DMS to enhance the performance of EIEIO during this infusion-based lipidomics workflow, whereby DMS isolated SM from other potentially interfering lipid classes, namely PCs and TAGs, prior to EIEIO analysis. This scenario is particularly relevant when surveying the total lipid profile of BHE (Fig. 1B) using ESI. The resulting mass spectrum contains multiple lipid classes and categories, including phosphoethanolamine, PC, and TAG, with SM present at less than ∼1% to the total lipid signal (Fig. 1C)
Using optimized DMS voltages for selective transmission of SM (see below), the SM component of the BHE was easily identified (Fig. 1D). The optimization of the SM-specific DMS parameters was performed using the standard sample, SM(d18:1,12:0), as well as SMs from brain, egg yolk, and milk extracts. In addition to protonated SMs, we also observed sodiated SMs among the precursor ions (∼10%). The procedure for optimizing the DMS parameters is very straightforward. First, the SV was set to 3,900 V, where good separation of lipid classes had previously been demonstrated (12). Then, as COV was scanned from −5 V to 15 V (in 0.2 V increments, typically), full-scan TOF MS data were acquired for each step in COV. Then, the signal from each m/z value corresponding to a known SM species could be extracted as a function of COV, providing profiles where optimal DMS transmission of SM occurred. In this case, a COV set to 9 V provided good separation of SM from other lipid classes and categories, as demonstrated by the DMS-dependent MS spectra recorded only at COV = 9 V. These filtered signals provided MS/MS targets that were 1/10,000 the base peak intensity of the unfiltered (no DMS) full-scan mass spectra. With DMS engaged, we employed ion accumulation times of 1 s for a single TOF spectrum and 60–360 s for individual MS/MS spectra.
With DMS filtering engaged to transmit SM-based ions selectively, the study of SM profiles for a variety of biological lipid extracts was greatly enhanced. Figure 5 shows an MS spectrum obtained during the analysis of brain SM, with the identified SMs from the resulting EIEIO experiments listed in Table 2. The percentages reported in Table 2 represent each identified SM isomer’s relative contribution to the overall signal produced by that precursor m/z ion. The percent value following each identified SM represents the intensity ratio of the acyl diagnostic peaks in the same precursor m/z as an indicator of expressed ratio. Results of other SM sources are shown in the supplementary information (supplementary Figs. 1–3).
Fig. 5.

MS spectrum of porcine brain SM and identified SM. DMS was on to separate protonated SMs from sodiated SMs, which were ∼10% compared with protonated SMs.
TABLE 2.
Identified SMs in porcine brain SM shown in Fig. 5
| Precursor (m/z) | Precursor Intensity (%) | Identified SMs | |||
| 675.505 | 0.15 | SM(d18:1,14:0) [90%] | |||
| 689.52 | 0.03 | SM(d17:1,16:0) [73%] | SM(d18:1,15:0) [27%] | ||
| 701.52 | 0.04 | SM(d18:2,16:0) [92%] | |||
| 703.535 | 2.68 | SM(d18:1,16:0) [79%] | SM(d16:1,18:0) [21%] | ||
| 717.55 | 0.29 | SM(d17:1,18:0) [46%] | SM(d18:1,17:0) [45%] | SM(d19:1, 16:0) [4%] | SM(d16:1,19:0) [3%] |
| 729.551 | 1.69 | SM(d18:2,18:0) [85%] | SM[d18:1,18:1(n-9)] [10%] | ||
| 731.539 | 40.77 | SM(d18:1,18:0) [94%] | |||
| 743.564 | 0.04 | SM(d19:2,18:0) [81%] | |||
| 759.589 | 6.56 | SM(d20:1,18:0) [52%] | SM(d18:1,20:0) [44%] | ||
| 771.595 | 0.03 | SM[d18:1,21:1(n-7)] [33%] | SM[d17:1,22:1(n-7)] [25%] | SM[d16:1,23:1(0-7)] [24%] | SM(d18:2,21:0) [11%] |
| 783.594 | 0.15 | SM[d18:1,22:2(n-6,-9)] [84%] | SM[d18:2,22:1(n-6)] [16%] | ||
| 785.604 | 4.82 | SM[d18:1,22:1(n-6)] [95%] | |||
| 787.616 | 5.45 | SM(d18:1,22:0) [83%] | SM[d18:0,22:1(n-6)] [11%] | ||
| 797.61 | 0.06 | SM(d18:1,23:2) [64%] | |||
| 799.622 | 1.52 | SM[d18:1,23:1(n-7)] [89%] | |||
| 801.635 | 1.26 | SM(d18:1,23:0) [83%] | SM(d18:0,23:1) [10%] | ||
| 809.607 | 0.06 | SM[d18:1,24:3(n-6,*,*)] [80%] | |||
| 811.622 | 1.55 | SM[d18:1,24:2(n-6,-9)] [82%] | SM[d18:2,24:1(n-9)] [10%] | SM[d18:2,24:1(n-6)] [4%] | |
| 813.605 | 29.99 | SM[d18:1,24:1(n-9)] [81%] | SM[d18:1,24:1(n-6)] [13%] | ||
| 825.636 | 0.11 | SM[d18:1,25:2(n-6,-9)] [75%] | |||
| 827.649 | 0.89 | SM(d18:1,25:1) [89%] | SM(d19:1,24:1) [11%] | (n-6) [17%], (n-9) [83%] | |
| 837.636 | 0.03 | SM[d18:1,26:3(n-6,*,*)] [51%] | SM(d18:1,26:3(n-9,*,*)) [38%] | ||
| 839.65 | 0.16 | SM[d18:1,26:2(n-6,-9)] [52%] | SM[d18:1,26:2(n-9,-12)] [17%] | SM[d20:2,24:1(n-9)] [9%] | SM[d20:2,24:1(n-6)] [6%] |
| 841.663 | 0.88 | SM(d18:1,26:1) [80%] | SM(d20:1,24:1) [14%] | (n-6) [31%], (n-9) [69%] | |
| 855.678 | 0.01 | SM(d20:1,25:1) [55%] | SM(d18:1,27:1) [32%] | SM(d19:1,26:1) [13%] | |
| 869.693 | 0.01 | SM(d18:1,28:1) [100%] | |||
| 871.69 | 0.03 | SM(d18:1,24:1) + 58.05 [100%] | |||
| 873.692 | 0.01 | SM(d18:2,24:1) + 58.05 [100%] | |||
| 887.471 | 0.01 | SM(d18:1,21:1) +101.11 [100%] | |||
| 914.744 | 0.01 | SM(d18:1,18:0) with CH3IO | SM(d18:1,24:1) + 101.11 | ||
| 943.523 | 0.04 | SM(d18:1,22:0) with CH3IO [100%] | |||
| 957.609 | 0.01 | SM(d18:1,23:0) with CH3IO [100%] | |||
| 971.551 | 0.19 | SM(d18:1,24:0) with CH3IO [100%] | |||
Precursor intensity [%] represents the amount of each precursor in the total lipids, which contains other impurities. Percent values following identified SMs show the relative intensities of acyl diagnostic peaks in the isobaric precursor ions. Total value in each precursor may not be 100% because of ignoring low intensity species. Underlined double bond locations in m/z 827 and 841 were not deconvoluted mathematically in each SM. Asterisks (*) represent unidentified double bond locations.
A typical spectrum of sphingadiene SM is shown in Fig. 6A, which is found in bovine brain SM. The backbone diagnostic peak was identified at m/z 445, which clearly indicates the backbone is sphingadiene. The second double bond location was identified at the n-4 site. Fig. 6B and Fig. 6C are the examples of SMs that had one double bond and two double bonds in the acyl groups, respectively.
Fig. 6.

Typical EIEIO spectra observed in porcine brain SM. A: The backbone is a sphingadiene, d18:2(n-4). B: The acyl chain has a carbon-carbon double bond at n-9. C: The acyl chain has two carbon-carbon double bonds at n-6 and n-9. [M-H+O2]•+ is a product of a radical-ion, neutral reaction from the hydrogen lost precursor species, which is often observed in ExD (16).
Backbone and acyl constituents
Abundant sphingosines and some sphingadienes were observed in all the extracts, but the sphinganine backbone was only observed in the bovine brain and milk SM (supplementary Fig. 4). The predominant molecular species were d18:1, which is conventionally recognized, i.e., ∼90% of sphingosine backbones were d18:1 in brain, egg, and heart, but only 43% were d18:1 and another 30% were d16:1 in milk. Chain lengths from 16 to 20, including odd number of carbons, were also detected in brain, milk, and BH. Sphingadiene existed at less than a few percent in brain, egg, and BH, but it was more significant in milk (10%). Although odd-chain fatty acids in mammalian systems are generally not considered, cows are ruminants and hence have extensive bacterial biomes in their digestive tract that can produce odd-chain fatty acids and be incorporated into the cow lipidome.
Acyl chains expressed in each SM are shown in supplementary Fig. 4. The identified profile of abundant acyl chains using this EIEIO method showed good agreement to the fatty acid distribution given by the vendor (17).
Modified SMs in brain SM
The brain SM contained a great variety of SM molecular species derived from the numerous combinations of different sphingoid backbones and acyl groups, but this sample also showed existence of SM molecular species containing unique chemical modifications. The most prevalent irregular SM molecules found in these studies had precursor ion masses of m/z 914.775, 943.523, 957.609, and 971.551. A significant differentiating characteristic of these peaks, compared with the majority of SM molecular species, is their precursor masses that are 0.1 amu smaller than standard SM molecular species. These molecules are clearly SM, because all lipid class diagnostic peaks for SM are clearly observed, but the acyl group and backbone masses are inconsistent with the results in Table 1. The −0.1 amu difference suggests that the molecules have an unusually heavy atom in their structures. In their EIEIO and CID spectra, an abundant neutral loss of m/z 126.903+H was observed. This mass is exactly the same as that of an iodine atom. The EIEIO spectra showed further consecutive neutral losses from the iodine loss: those corresponding to losses of CH3 and O. Detailed analysis of the chain fragments shows that an iodine atom and a methoxy group are attached to double bonds present in unmodified SM (Fig. 7A–C). The iodine modification was found at the methyl terminal side and the methoxy group was at the head group side in all four precursor cases. Interestingly, the double bond in the sphingadiene backbone (n-4) was modified in the case of m/z 914.775.
Fig. 7.

Iodine-containing SM observed in brain SM. Normal SM[d18:1,24:1(n-9)] (A) and iodine-containing SM (C). The carbon-carbon double bond region is expanded in (B). White arrows indicate backbone fragments from the normal SM. Black arrows indicate acyl group fragments. The 2H difference started at the n-9 position indicates the double bond location in the normal SM (D). In the case of the modified SM, fragment series of CH, O, and CH3 were appeared. This indicates the modified SM has the structure shown in E.
The source of iodinated SM is unclear. It is certainly possible that the iodine and methoxy group adducts are artifactually generated. Indeed, iodine is traditionally used to detect double bonds in lipid analysis (18). To evaluate the structural characteristics of SM that had reacted with adventitious iodine, SM extracts were exposed to iodine and methanol vapor. However, from this process, we did not find any evidence of SM molecular species containing iodine or methoxy group adducts, despite the fact the SM mixture was replete with SM molecular species with acyl chains containing carbon-carbon double bonds. Instead, replacement of a hydrogen by an iodine was observed as the main process (supplementary Fig. 5). Dual iodine adduct reaction at a double bond, which is often used for estimation of double bond number (known as iodine number), was also observed, but it was 1/100 weaker than the hydrogen replacement by an iodine atom. From these observations, we can conclude that the iodinated SM found in bovine brain extracts is not formed via a typical iodine addition across a double bond. In the context of a biological reaction mechanism, it is interesting that this iodine-methoxy type of modification was observed only in brain, which is known to have relatively higher levels of iodine, rather than in other tissues (with the exception of the thyroid) (19). Further studies that are outside the scope of this report are needed to identify the mechanism of the iodine-methoxy adduction. However, it should be noted that this new methodology has the analytical power to analyze such unknown modifications.
The potential of high-throughput lipid structural characterization using DMS-EIEIO-TOF MS
In this study, EIEIO spectra were accumulated for a long duration (2–5 min) to obtain high quality spectra to survey EIEIO characteristics on SMs with no concern about ion statistics and S/N. For high throughput analysis, such as LC-MS measurements, much shorter accumulation should be applied. To identify the diagnostic peaks, the peak intensities should at least be above the detection limits given by noise fluctuation. Theoretical minimum accumulation time was calculated using the stored multiple EIEIO spectra accumulated every second in BHE. Minimum accumulation time can be defined as the time when intensities of all diagnostic peaks become higher than the detection limits, which is a triple of the standard deviation of the EI noise at the same m/z of each diagnostic peak (supplementary Fig. 1). Though the intensity of SMs in BHE was not strong because SMs were minor components in the total lipids, the accumulation for a typical LC time scale (a few seconds) was sufficient to identify many SM molecular species. A longer accumulation than LC time scale will be required for lower concentrated species, typically lower than 0.1 μM, but infused measurements will also be promising, like this work, because DMS separates SMs from other lipid classes efficiently without the need for chromatographic separation.
CONCLUSION
The combination of DMS and EIEIO MS is a promising tool for SM analysis. Lipid class, backbone structure, and acyl chain characterizations are clearly identified using fragmentation information in a single EIEIO spectrum. This method also revealed the power to identify unknown modifications in SM molecular species. While previous studies employing atmospheric-pressure chemical ionization (APCI) have shown promise in characterizing SMs (20, 21), their findings have been limited to identifying intact species (Brutto level identification) and inferring FA composition from limited information derived from MS/MS spectra. As noted previously, APCI must be employed, as ESI-MS/MS can provide little to no structural information for SMs, only confirmation of the phosphocholine head group at m/z 184. In addition, these studies must rely upon separation by LC for sample simplification (due to in-source fragmentation from APCI), requiring additional analysis time and resources. In comparison, the DMS-EIEIO workflow for characterizing SMs requires no chromatographic separation and provides much greater in-depth structural analyses of these biologically valuable lipid biomarkers.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. James Hager (SCIEX) for helpful discussions.
Footnotes
Abbreviations:
- APCI
- atmospheric-pressure chemical ionization
- BHE
- bovine heart extract
- CID
- collision-induced dissociation
- COV
- compensation voltage
- DMS
- differential mobility spectrometry
- EI
- electron ionization
- EIEIO
- electron impact excitation of ions from organics
- ExD cell
- electron-ion reaction device
- KEe
- kinetic energy of the electron beam
- PC
- phosphatidylcholine
- S/N
- signal to noise ratio
- SV
- separation voltage
- TAG
- triacylglycerol
The online version of this article (available at http://www.jlr.org) contains a supplement.
REFERENCES
- 1.Slotte J. P. 2013. Biological functions of sphingomyelins. Prog. Lipid Res. 52: 424–437. [Erratum. 2013. Prog. Lipid Res. 52: 681.]. [DOI] [PubMed] [Google Scholar]
- 2.Chakraborty M., and Jiang X. C.. 2013. Sphingomyelin and its role in cellular signaling. Adv. Exp. Med. Biol. 991: 1–14. [DOI] [PubMed] [Google Scholar]
- 3.Shaner R. L., Allegood J. C., Park H., Wang E., Kelly S., Haynes C. A., Sullards M. C., and Merrill A. H. Jr. 2009. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 50: 1692–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deimler R. E., Sander M., and Jackson G. P.. 2015. Radical-induced fragmentation of phospholipid cations using metastable atom-activated dissociation mass spectrometry (MAD-MS). Int. J. Mass Spectrom. 390: 178–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stutzman J. R., Blanksby S. J., and McLuckey S. A.. 2013. Gas-phase transformation of phosphatidylcholine cations to structurally informative anions via ion/ion chemistry. Anal. Chem. 85: 3752–3757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekroos K., Ejsing C. S., Bahr T., Karas M., Simons K., and Shevchenko A.. 2003. Charting molecular composition of phosphatidylcholines by fatty acid scanning and ion trap MS3 fragmentation. J. Lipid Res. 44: 2181–2192. [DOI] [PubMed] [Google Scholar]
- 7.Pham H. T., Maccarone A. T., Thomas M. C., Campbell J. L., Mitchell T. W., and Blanksby S. J.. 2014. Structural characterization of glycerophospholipids by combinations of ozone- and collision-induced dissociation mass spectrometry: the next step towards “top-down” lipidomics. Analyst. 139: 204–214. [DOI] [PubMed] [Google Scholar]
- 8.Campbell J. L., and Baba T.. 2015. Near-complete structural characterization of phosphatidylcholines using electron impact excitation of ions from organics. Anal. Chem. 87: 5837–5845. [DOI] [PubMed] [Google Scholar]
- 9.Cody R. B., and Freiser B. S.. 1979. Electron impact excitation of ions from organics: an alternative to collision induced dissociation. Anal. Chem. 51: 547–551. [Google Scholar]
- 10.Baba T., Campbell J. L., Le Blanc J. C. Y., Hager J. W., and Thomson B. A.. 2015. Electron capture dissociation in a branched radio-frequency ion trap. Anal. Chem. 87: 785–792. [DOI] [PubMed] [Google Scholar]
- 11.Schneider B. B., Covey T. R., Coy S. L., Krylov E. V., and Nazarov E. G.. 2010. Planar differential mobility spectrometer as a pre-filter for atmospheric pressure ionization mass spectrometry. Int. J. Mass Spectrom. 298: 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker P. R. S., Armando A. M., Campbell J. L., Quehenberger O., and Dennis E. A.. 2014. Three-dimensional enhanced lipidomics analysis combining UPLC, differential ion mobility spectrometry, and mass spectrometric separation strategies. J. Lipid Res. 55: 2432–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lintonen T. P., Baker P. R., Suoniemi M., Ubhi B. K., Koistinen K. M., Duchoslav E., Campbell J. L., and Ekroos K.. 2014. Differential mobility spectrometry-driven shotgun lipidomics. Anal. Chem. 86: 9662–9669. [DOI] [PubMed] [Google Scholar]
- 14.Liebisch G., Vizcaíno J. A., Köfeler H., Trötzmüller M., Griffiths W. J., Schmitz G., Spener F., and Wakelam M. J. O.. 2013. Shorthand notation for lipid structures derived from mass spectrometry. J. Lipid Res. 54: 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Barenholz Y., and Thompson T. E.. 1980. Sphingomyelins in bilayers and biological membranes. Biochim. Biophys. Acta. 604: 129–158. [DOI] [PubMed] [Google Scholar]
- 16.Baba T., and Campbell J. L.. 2015. Capturing polyradical protein cations after an electron capture event: evidence for their stable distonic structures in the gas phase. J. Am. Soc. Mass Spectrom. 26: 1695–1701. [DOI] [PubMed] [Google Scholar]
- 17.Avanti Polar Lipids, Inc. Fatty Acid Distribution in Sphingolipids/Sphingomyelin/Natural. Accessed January 29, 2016, at http://www.avantilipids.com.
- 18.Williams J. N. Jr., Anderson C. E., and Jasik A. D.. 1962. A sensitive and specific method for plasmalogens and other enol ethers. J. Lipid Res. 3: 378–381. [Google Scholar]
- 19.Delange F. 2000. The role of iodine in brain development. Proc. Nutr. Soc. 59: 75–79. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson A. Å., Michélsen P., and Odham G.. 1998. Molecular species of sphingomyelin: determination by high-performance liquid chromatography/mass spectrometry with atmospheric pressure chemical ionization. J. Mass Spectrom. 33: 1192–1198. [DOI] [PubMed] [Google Scholar]
- 21.Byrdwell W. C., and Perry R. H.. 2007. Liquid chromatography with dual parallel mass spectrometry and 31P nuclear magnetic resonance spectroscopy for analysis of sphingomyelin and dihydrosphingomyelin II. Bovine milk sphingolipids. J. Chromatogr. A. 1146: 164–185. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.