

Summary
Synaptic plasticity is a fundamental feature of the nervous system that allows adaptation to changing behavioral environments. Most studies of synaptic plasticity have examined the regulated trafficking of postsynaptic glutamate receptors that generates alterations in synaptic transmission. Whether and how changes in the presynaptic release machinery contribute to neuronal plasticity is less clear. The SNARE complex mediates neurotransmitter release in response to presynaptic Ca++ entry. Here we show that the SNARE fusion clamp Complexin undergoes activity-dependent phosphorylation that alters the basic properties of neurotransmission in Drosophila. Retrograde signaling following stimulation activates PKA-dependent phosphorylation of the Complexin C-terminus that selectively and transiently enhances spontaneous release. Enhanced spontaneous release is required for activity-dependent synaptic growth. These data indicate that SNARE-dependent fusion mechanisms can be regulated in an activity-dependent manner and highlight the key role of spontaneous neurotransmitter release as a mediator of functional and structural plasticity.
Keywords: Exocytosis, Synapse, Neurotransmitter Release, SNARE complex, minis, spontaneous release, retrograde signaling
Introduction
Synaptic transmission is initiated by the rapid fusion of docked synaptic vesicles via Ca++ influx in response to an action potential (AP). Synaptic vesicles can also fuse independent of APs, resulting in individual spontaneous quantal events termed minis (Fatt and Katz, 1952). Recent studies suggest spontaneous release may function independently as a regulated channel for neuronal signaling distinct from AP-driven release. Minis have been shown to activate specific biochemical signaling pathways that regulate structural and homeostatic plasticity (Andreae and Burrone, 2015; Choi et al., 2014; Lee et al., 2010; McKinney et al., 1999; Sutton et al., 2006). Moreover, minis modulate AP firing in certain neurons, suggesting spontaneous release can contribute to the integration of signals within neuronal ensembles (Carter and Regehr, 2002). Finally, accumulating evidence suggests that regulation of spontaneous versus evoked release can occur via distinct molecular interactions at the level of the fusion machinery (Cho et al., 2010; Kaeser-Woo et al., 2012; Kavalali, 2014; Martin et al., 2011).
Synaptic vesicle fusion is mediated by the regulated assembly of the SNARE complex to generate docked and primed synaptic vesicles (Sudhof and Rothman, 2009). Zippering of the SNAREs into a four-helix bundle provides the energy necessary to drive bilayer fusion (Weber et al., 1998). At the synapse, SNARE assembly and zippering is tightly controlled to achieve the precision and speed required for effective neurotransmission. One such regulator is Synaptotagmin 1 (Syt 1), a Ca++ sensor for AP-dependent synaptic vesicle fusion (Geppert et al., 1994; Littleton et al., 1993). In addition to Syt 1, the SNARE-binding protein Complexin (Cpx) has emerged as an important co-regulator for vesicle fusion (McMahon et al., 1995; Sudhof, 2013). In in vitro cell-cell fusion and lipid mixing assays, Cpx reduces membrane fusion, acting as a clamp to block premature exocytosis in the absence of Ca2+ (Giraudo et al., 2006; Malsam et al., 2012; Schaub et al., 2006). Genetic studies in Drosophila and C. elegans revealed a dramatic increase in minis in cpx null mutants (Hobson et al., 2011; Huntwork and Littleton, 2007; Jorquera et al., 2012; Martin et al., 2011). In addition to preventing spontaneous release, Cpx facilitates AP-dependent vesicle fusion (Hobson et al., 2011; Huntwork and Littleton, 2007; Martin et al., 2011; Xue et al., 2008). Structure-function studies indicate Cpx regulates minis and AP-dependent release via distinct molecular mechanisms (Cho et al., 2014; Cho et al., 2010; Hobson et al., 2011; Kaeser-Woo et al., 2012; Martin et al., 2011; Maximov et al., 2009). Binding of the Cpx central helix to the SNARE complex is required for both clamping and stimulating properties (Cho et al., 2010; Xue et al., 2010), while the C-terminus is required for clamping spontaneous fusion (Buhl et al., 2013; Cho et al., 2010; Kaeser-Woo et al., 2012; Wragg et al., 2013). Interestingly, the C-terminus of mammalian Cpxs I and II is phosphorylated in vivo, suggesting its function might be regulated through post-translational modifications (Shata et al., 2007).
The Drosophila NMJ undergoes acute forms of activity-dependent structural and functional plasticity, providing a model synapse to examine the role of the release machinery in synaptic plasticity (Ataman et al., 2008; Ballard et al., 2014; Berke et al., 2013; Frank et al., 2009; Goold and Davis, 2007; McCabe et al., 2003; Piccioli and Littleton, 2014). We previously identified a retrograde signaling pathway that drives synaptic plasticity at this glutamatergic synapse. High-frequency stimulation of motor neurons induces a long-lasting enhancement of spontaneous release, a process termed high-frequency-induced miniature release (HFMR). At embryonic NMJs, HFMR induction results in 100-fold increase in minis for several minutes before returning to baseline (Yoshihara et al., 2005). This process is dependent on the presynaptic cAMP-PKA kinase signaling pathway and postsynaptic Syt 4, a Ca++-sensitive regulator of postsynaptic vesicle fusion (Yoshihara et al., 2005). HFMR is also expressed at mature larval NMJs and is required for activity-dependent synaptic growth (Barber et al., 2009; Piccioli and Littleton, 2014; Yoshihara et al., 2005). Interestingly, cpx null mutants exhibit enhanced synaptic growth in addition to enhanced minis (Huntwork and Littleton, 2007). Indeed, minis have recently been found to be necessary and sufficient to drive developmental synaptic growth at Drosophila NMJs (Choi et al., 2014). These results suggest that HFMR and synaptic growth signals may be mechanistically linked through Syt 4-dependent retrograde signaling.
Here we demonstrate that minis are acutely regulated by activity-dependent phosphorylation of the C-terminus of the fusion clamp, Cpx. PKA-dependent phosphorylation of Cpx is required for synaptic structural plasticity and growth. Our findings suggest that Syt 4-dependent retrograde signaling regulates presynaptic PKA-dependent phosphorylation of Cpx to modulate its clamping properties, allowing short-term increases in spontaneous neurotransmitter release to control functional and structural plasticity.
Results
Synaptic growth correlates with spontaneous neurotransmitter release rate
We previously observed that cpxSH1 null mutants exhibit enhanced minis at 3rd instar larval NMJs. Increased spontaneous release was correlated with enhanced synaptic growth (Choi et al., 2014; Huntwork and Littleton, 2007), suggesting that minis may play a unique role in regulating the synaptic growth machinery at Drosophila synapses. To examine the role of minis in regulating synaptic growth, we used UAS-RNAi against Cpx to specifically remove the protein in motor neurons using the c164-GAL4 driver (Figure 1A). This approach allowed Cpx to be reduced in a motor neuron-specific manner, with minis enhanced to more physiological levels compared to the ~70–80 Hz range observed in the complete absence of Cpx. Electrophysiological recordings from 3rd instar larvae revealed enhanced mini frequency in Cpx knockdowns compared to controls (w1118; ; = 9.9 ± 1.5 Hz (n = 6) vs. w1118; = 2.1 ± 0.2 Hz (n = 5); p <0.01; Figures 1B and 1C). Similar to synaptic overgrowth observed in cpxSH1 nulls (see also Figure 2E, 2G, and 5G), Cpx knockdown larvae exhibited enhanced synaptic growth at muscle 6/7 of 3rd instar larvae (w1118; ; = 120.8 ± 5.5 boutons (n=13) vs. w1118; = 89.2 ± 3.8 boutons (n=18); p<0.0001; Figures 1D and 1E).
Figure 1.
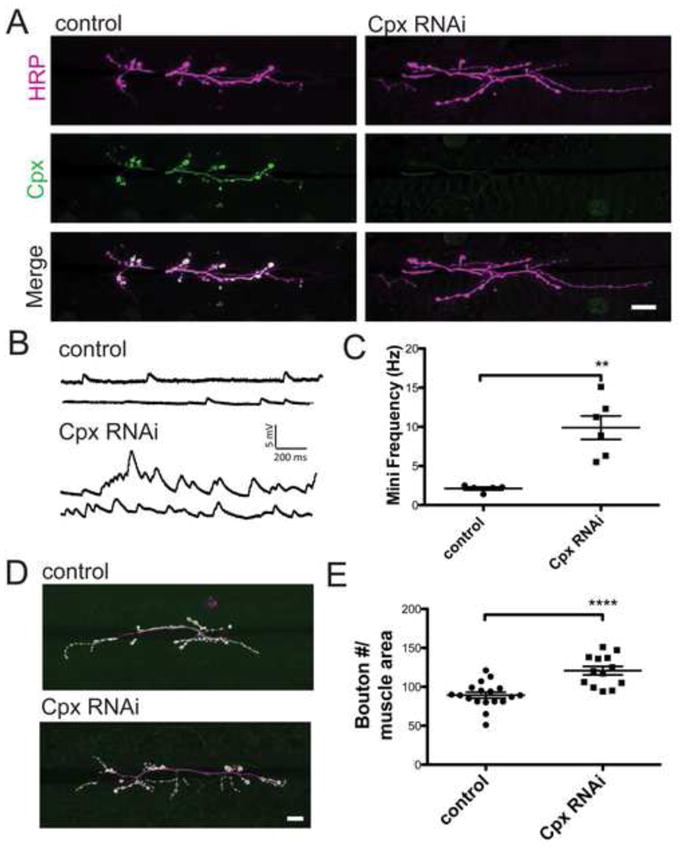
Cpx knockdown larvae exhibit increased spontaneous release and enhanced synaptic growth. (A) Expression of Cpx protein was reduced in Cpx RNAi knockdown lines (w1118; ; ) compared to control (w1118; ) using a motor neuron driver (c164-GAL4) to express Cpx-specific RNAi. Larvae were co-stained with anti-HRP and anti-Cpx specific antisera. Images were obtained from muscle 6/7 NMJs of 3rd instar larvae (B) Sample traces of spontaneous release from control and Cpx RNAi knockdown larvae. (C) Mean mini frequency (Hz ± SEM) recorded for each line. (D) Representative images of NMJs from control and Cpx RNAi knockdown larvae. Larvae were stained with anti-HRP (magenta) and anti-GluRIII (green) specific antisera to identify synaptic boutons. (E) Mean synaptic bouton number for control and Cpx RNAi knockdowns. Bouton number was normalized using muscle 6/7 area. Indicated comparisons were made using Student’s T-tests. Mean ± SEM is indicated. Scale bar = 20 μm.
Figure 2.
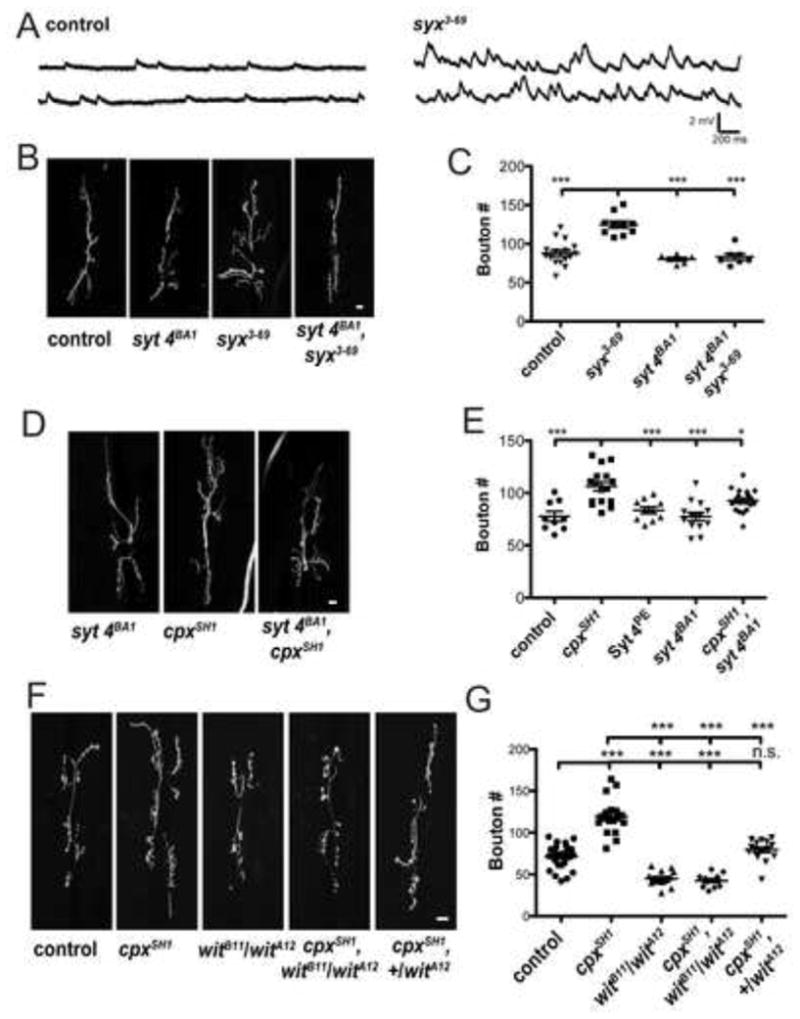
Synaptic growth associated with enhanced minis is suppressed by synaptotagmin 4 (syt 4BA1) and BMP receptor (wit) mutants. (A) Representative traces of spontaneous release recorded from syntaxin 1A mutant (w1118;; syx3-69) and control (w1118) larval muscle 6 NMJs. (B) Representative images of muscle 6/7 NMJs of control (w1118), syx3-69 mutant (w1118;; syx3-69), syt 4BA1 null (w1118;; syt 4BA1), and syx3-69, syt 4BA1 double mutant (w1118;; syx3-69, syt 4BA1). (C) Mean synaptic bouton number measured for indicated genotypes. (D) Representative images of muscle 6/7 NMJs of the indicated genotypes. (E) Mean synaptic bouton number measured for control (w1118;; CpxPE), cpxSH1 null (w1118;; cpxSH1), control Syt 4PE (w1118;; Syt 4PE), syt 4BA1 null (w1118;; syt 4BA1), and syx3-69, syt 4BA1 double mutant (w1118;; syt 4BA1, cpxSH1) (F) Representative images of muscle 6/7 NMJs from control (w1118;; CpxPE), cpxSH1 null, wit null (w1118;; ), wit, cpxSH1 double mutant (w1118;; ), and wit heterozygotes in cpx null background (w1118;; ). (G) Mean synaptic bouton number measured for the indicated genotypes. Indicated comparisons were made using ANOVA with post hoc Tukey analysis. Mean ± SEM is indicated. Scale bars = 10 μm.
Figure 5.
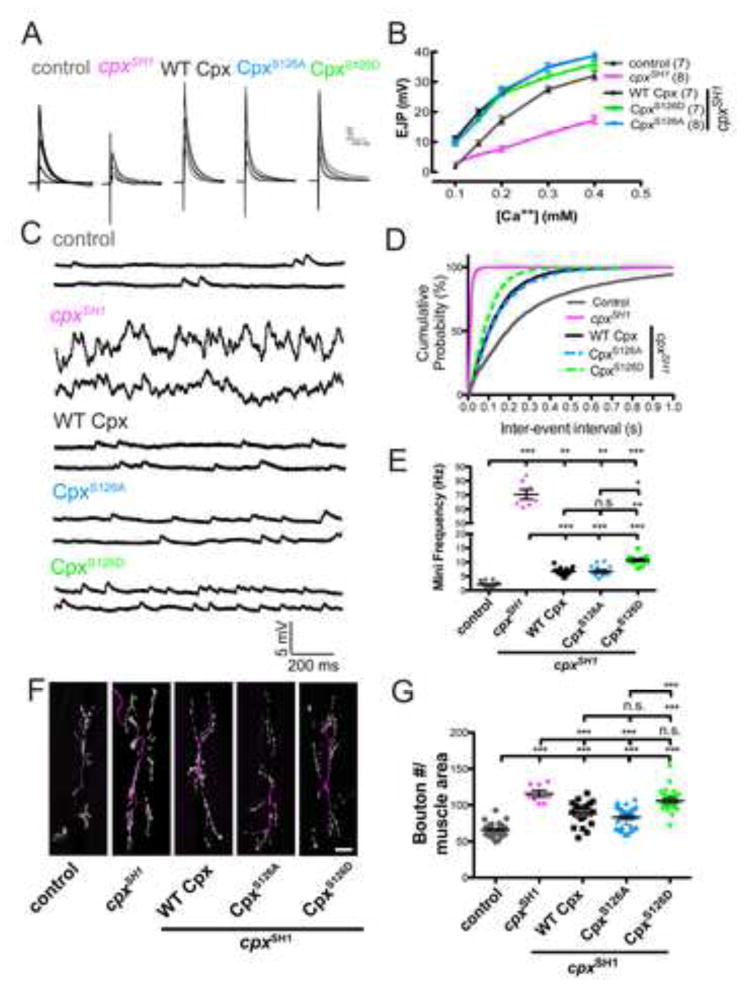
Mimicking phosphorylation of Cpx at S126 selectively modulates Cpx function as a fusion clamp and regulates synaptic growth. (A) Averaged traces of EJPs from control (w1118;; CpxPE), cpx null (w1118;; cpxSH1), WT Cpx rescues (C155;; WT Cpx, cpxSH1), and CpxS126A and CpxS126D mutant rescues (C155;; CpxS126A, cpxSH1 and C155;; CpxS126D, cpxSH1, respectively) recorded over a range of external Ca++ concentrations (0.1, 0.15, 0.2, 0.3, and 0.4 mM). Numbers in parentheses (n) represent individual muscle recordings at the indicated [Ca++]. (B) Summary of mean EJP amplitude (mV ± SEM) plotted at the indicated [Ca++]. (C) Representative traces of spontaneous release events from muscle 6 of control, cpxSH1 null, and WT Cpx, CpxS126A and CpxS126D rescue lines. (D) Cumulative probability plot of inter-event intervals (seconds) between mini events recorded indicated genotypes. (E) Summary of mean mini frequency (Hz ± SEM) for indicated genotypes. (F) Representative images of muscle 6/7 NMJs of control, cpxSH1 null, and WT Cpx, CpxS126A, CpxS126D rescue lines. Larvae were stained with anti-HRP (magenta) and anti GluRIII (green) specific antisera to identify synaptic boutons. Scale bar = 20 μm (G) Summary of mean synaptic bouton number normalized to muscle area for the indicated genotypes. Indicated comparisons were made using ANOVA with post hoc Tukey analysis. Mean ± SEM is indicated.
To determine whether enhanced synaptic growth was unique to the cpx mutant and Cpx knockdowns that exhibit elevated mini frequencies, we examined synapses of an independent mutant that also exhibits elevated spontaneous release. syx3-69 mutants harbor a point mutation in the t-SNARE syntaxin 1A (Littleton et al., 1998) that gives rise to elevated mini rates (w1118;; syx3-69 = 14.2 ± 1.3 Hz (n=8)) similar to those observed in Cpx RNAi animals ((Bykhovskaia et al., 2013; Lagow et al., 2007), Figure 2A). Consistent with the cpxSH1 mutant and RNAi Cpx knockdown, synapses in syx3-69 mutants exhibited enhanced growth compared to control (w1118;; syx3-69 = 124.8 ± 4.4 boutons (n = 10) vs. control (w1118) = 87.8 ± 3.9 boutons (n = 16); p < 0.001; Figures 2B and 2C). Together, these results suggest that elevated rates of spontaneous release positively correlate with synaptic growth at Drosophila NMJs.
A Syt 4-mediated retrograde signaling pathway is required for mini-dependent synaptic growth
Retrograde signaling from postsynaptic compartments regulates both synaptic growth and homeostatic plasticity during larval development (Goold and Davis, 2007; Marques et al., 2002; McCabe et al., 2003; Paradis et al., 2001). We previously identified a retrograde signaling pathway that controls spontaneous release rates at Drosophila NMJs mediated by Syt 4, a Ca++-sensitive regulator of postsynaptic vesicle fusion. To examine if this pathway is required for the enhanced synaptic growth in animals that exhibit enhanced minis, we generated double mutants of syx3-69 with syt 4 null mutants (syt 4BA1). Introducing the syt 4BA1 mutant allele into the syx3-69 background suppressed synaptic growth (w1118;; syx3-69 = 124.8 ± 4.4 boutons (n=10) vs. w1118;; syx3-69, syt 4BA1 = 80.2 ± 2.3 boutons (n=6); p<0.001; Figures 2B and 2C). We next examined if Syt 4 is similarly required for synaptic growth in the cpxSH1 null background. cpxSH1 nulls exhibit greatly enhanced minis (w1118;; cpxSH1 = 70.4 ± 3.5 Hz (n=7)), as well as enhanced synaptic growth compared to control (w1118;; cpxSH1 = 106.3 ± 4.2 boutons (n=16) vs. w1118;; CpxPE (precise excision control for Cpx) = 77.8 ± 2.8 boutons (n=9); p<0.001; Figures 2D and 2E). Loss of Syt 4 alone did not show any elevation in synaptic growth compared to both cpxSH1 and syt 4BA1 controls (w1118;; syt 4BA1 = 77.5 ± 4.0 boutons (n = 13) vs. [w1118;; CpxPE = 77.8 ± 4.7 boutons (n = 9) and w1118;; Syt 4PE (precise excision control for Syt 4) = 83.6 ± 3.2 boutons (n = 10)]; p>0.05 for both comparisons; Figures 2D and 2E). Introducing the syt 4BA1 allele into the cpxSH1 background reduced synaptic growth compared to cpxSH1 allele alone (w1118;; syt 4BA1, cpxSH1 = 92.6 ± 2.6 boutons (n=19) vs. w1118;; cpxSH1 = 106.3 ± 4.2 boutons (n=16); p<0.05; Figures 2D and 2E). Introduction of the syt 4BA1 allele did not suppress enhanced minis in the cpxSH1 or syx3-69 backgrounds (see Table S2). These results indicate that synaptic overgrowth induced by elevated spontaneous release requires the Syt 4-dependent retrograde signaling pathway.
One well-characterized retrograde growth signal at the Drosophila NMJ is the bone morphogenic protein (BMP), Gbb (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003). Gbb is released from muscle and activates the presynaptic type-II BMP receptor wishful thinking (Wit). wit null mutants (witA12/witB11) exhibit reduced synaptic growth at larval NMJs, indicating the BMP retrograde signaling pathway is required for synapse development (Marques et al., 2002). In addition to its role in synapse development, BMP signaling is also important for acute activity-regulated forms of synaptic structural plasticity through modulation of presynaptic actin cytoskeletal dynamics (Piccioli and Littleton, 2014). Given BMP signaling contributes to both developmental synapse formation and acute structural plasticity, we examined if BMP signaling is required for the enhanced synaptic growth in the context of elevated minis. In the absence of the BMP receptor (witA12/witB11), synapses are undergrown compared to control (w1118;; = 45.1 ± 2.2 boutons (n=15) vs. w1118;; CpxPE = 71.1 ± 2.8 boutons (n=26); p<0.001; Figures 2F and 2G) consistent with previous studies (Aberle et al., 2002; Marques et al., 2002). Synaptic overgrowth in the cpxSH1 null background was strongly suppressed by the wit null mutant (w1118;; cpxSH1 = 119.2 ± 5.0 boutons (n=18) vs. w1118;; = 42.4 ± 2.2 boutons (n=12); p<0.001). Introduction of a single wit allele in the cpxSH1 background was also sufficient to suppress growth (w1118;;; cpxSH1 = 119.2 ± 5.0 boutons (n=18) vs. w1118;; = 79.4 ± 3.1 boutons (n=18); p<0.001), indicating a dose-dependent genetic interaction. Introducing wit alleles did not suppress elevated minis in the cpxSH1 null background (see Table S2). In summary, these results indicate that Syt 4 and BMP signaling pathways contribute to enhanced mini-dependent synaptic growth.
HFMR is transient and its expression is Syt 4- and PKA-dependent
High-frequency stimulation of motor neurons induces a long-lasting enhancement of spontaneous release (HFMR) that is dependent on postsynaptic Syt 4 and the presynaptic cAMP-PKA kinase pathway (Yoshihara et al., 2005). HFMR is also expressed at more mature 3rd instar larval NMJs in a Syt 4-dependent manner (Barber et al., 2009). To examine if Cpx regulation of spontaneous release might contribute to this process, we further characterized HFMR in 3rd instar larvae. In response to strong stimulation (1 second 100 Hz stimulation 4x, spaced 2 seconds apart), the frequency of mini events rapidly doubled compared to the baseline prior to stimulation (w1118;; CpxPE and w1118;; Syt 4PE genotypes = 1.9 ± 0.2 fold (n=6) 100 sec after induction; Figures 3A–3C). To determine the kinetics of this activity-dependent plasticity, we extended our recordings up to 8 minutes after stimulation. HFMR was transient, with spontaneous release rates returning to baseline within ~ 4 minutes following induction (Figure 3B). Consistent with prior work, syt 4BA1 null animals did not exhibit HFMR in response to stimulation within our 8-minute recording session, indicating HFMR expression is not simply delayed in the absence of Syt 4 (w1118;; CpxPE and w1118;; Syt 4PE genotypes = 1.6 ± 0.1 fold (n=7) vs. w1118;; syt 4BA1 = 1.1 ± 0.1 fold (n=7) at 100 sec after induction; p<0.001). Given the requirement for the presynaptic cAMP-PKA pathway in embryonic HFMR, we assayed whether PKA was required for activity-induced elevations in mini frequency at mature NMJs as well. Using a pharmacological approach to inhibit the cAMP-PKA pathway, we found that bath application of H89, a PKA inhibitor, strongly suppressed HFMR compared to controls (w1118;; CpxPE and w1118;; Syt 4PE genotypes = 1.6 ± 0.1 fold (n=7) vs. w1118;; Syt 4PE + H89 = 1.2 ± 0.1 fold (n=6) at 100 sec after induction; p<0.01). Together, these results suggest that HFMR expression at mature NMJs requires Syt 4 and PKA.
Figure 3.

Activity-dependent enhancement of minis requires Syt 4 and PKA activity. (A) Representative traces of minis before (top trace) and after (bottom trace) tetanic stimulation recorded from control (w1118;; CpxPE and w1118;; Syt 4PE) and syt 4BA1 nulls (w1118;; syt 4BA1). Representative traces of minis are also shown for control animals pre-treated with the PKA-specific inhibitor H89. (B) Mean mini frequency plotted as a function of time. Tetanic stimulation occurs at the 0 sec time point (Red Arrow). Mini frequency is normalized to average baseline mini frequency 60 seconds prior to stimulation for each preparation. (C) Average fold enhancement of mini frequency compared to baseline mini frequency 100 seconds after tetanic stimulation. Statistically significant differences in fold enhancement of minis were observed in control animals compared to syt 4BA1 nulls and H89 treated controls at each indicated time point starting at 100 seconds following HFMR stimulation up to 300 seconds, when minis started to return to pre-stimulus baseline mini frequency. (D) Pseudo-color activity heatmaps showing the distribution of spontaneous fusion rates for individual release sites at the same NMJ of muscle 4 prior (left panel) and after (right panel) tetanus stimulation. Each circle is a single region of interest (ROI) that represents the detection of a release event indicated by postsynaptic myrGCaMP6s fluorescence. Color-coding in the heatmaps indicate the number of mini events detected over a period of 2 minutes before and after HFMR stimulation. (E) Number of ROIs exhibiting indicated event frequency before (black) and after (magenta) HFMR stimulation (F) Representative images of Ca++ dynamics detected by presynaptic myrGCAMP6s expression before, during, and after stimulation. (G) Mean fluorescence change as a function of time during tetanus is shown for one representative NMJ stimulation trial. Red arrows indicate each 100 Hz stimulation event. Mean ± SEM is indicated. Indicated comparisons were made using ANOVA with post hoc Tukey analysis.
We next assayed how HFMR is expressed at individual release sites. Drosophila NMJs contains spatially segregated active zones that are directly apposed to distinct clusters of glutamate receptors in postsynaptic muscle. By measuring postsynaptic Ca++ influx from individual glutamate receptor clusters following release, single fusion events at individual active zones can be monitored (Melom et al., 2013; Peled and Isacoff, 2011). To measure Ca++ influx at postsynaptic sites, we employed a genetically encoded myristolated (membrane-tethered) variant of a Ca++-sensitive GFP (GCaMP6s). Postsynaptic expression of UAS-GCaMP6s using the muscle driver mef2-GAL4 allowed robust detection of postsynaptic responses from both evoked and spontaneous synaptic vesicle fusion events (Melom et al., 2013). Using this method, we quantified individual release events at NMJ synapses for 2 minutes before and 2 minutes after HFMR induction (Figures 3D and 3E). A large increase in spontaneous release over a broad population of individual release sites was detected following HFMR induction, including the recruitment of release sites that were previously inactive (Figures 3D, 3E, and 3F). Using this optical method to detect individual release events, we conclude that HFMR is mediated by a broad increase in spontaneous fusion probability over a large population of release sites at the NMJ.
Intracellular presynaptic Ca++ modulates mini frequency at certain synapses (Xu et al., 2009). To examine if a long-lasting increase in basal presynaptic Ca++ levels occurs after HFMR induction that contributes to enhanced minis beyond retrograde signaling, we monitored presynaptic intracellular Ca++ levels using a presynaptically expressed myristolated UAS-GCaMP6s. Intracellular presynaptic Ca++ levels rapidly increased during HMFR induction, peaking with each stimulation train (Figures 3F and 3G). Immediately after the HFMR induction protocol, GCaMP6s signal rapidly returned to background levels within a second, indicating prolonged elevated presynaptic Ca++ levels are not the cause of the long-lasting increase in mini release that occurs over minutes. The transient local increase in Ca++ in response to HFMR induction suggests that long-lasting synaptic modifications downstream of presynaptic Ca++ influx, such as PKA phosphorylation of substrates associated with the vesicle fusion machinery, are likely to mediate HFMR.
PKA phosphorylation of Cpx modulates its function as a fusion clamp
These data indicate retrograde signaling impinges on activation of presynaptic PKA to regulate HFMR. Additionally, the elevation of release rates over a broad population of active zones within minutes of stimulation suggest HFMR expression mechanisms might occur at the post-translational level at individual release sites. The downstream presynaptic targets of PKA that modulate HFMR are unknown, but may include components of the SNARE-complex fusion machinery (Sudhof and Rothman, 2009). Based on Cpx’s ability to directly regulate mini release frequency as a fusion clamp, we examined whether Cpx is a downstream target of the Syt 4 retrograde signaling pathway, and if its synaptic function as a fusion clamp is regulated by PKA phosphorylation in response to stimulation that leads to HFMR expression at NMJs (Figure 4A).
Figure 4.

Cpx is a substrate for PKA phosphorylation. (A) Proposed retrograde Syt 4-dependent signaling pathway that is activated by strong stimulation. Postsynaptic Ca++ elevation following stimulation drives release of a retrograde signal that activates the presynaptic PKA pathway. PKA is hypothesized to phosphorylate Cpx, altering its clamping properties. In addition, PKA may phosphorylate additional targets to facilitate structural growth. (B) Structure of Drosophila (Dm) Cpx and the location of the predicted PKA phosphorylation site (S126) in the C-terminus. The N-terminus and C-terminus flank the indicated accessory and central helix domains. The alignment of C-terminal regions of Dm Cpx, mouse (Mm) Cpx I, and C. elegans (Ce) Cpx is shown below. Predicted amphipathic helix domains are highlighted in yellow, and predicted phosphorylation sites indicated in bold. S115 of Mm Cpx I is phosphorylated by protein kinase CK2 (Shata et al., 2007). T119 of Mm Cpx I is a predicted substrate for PKC, and T129 of Ce Cpx is a predicted substrate for AKT (Wong et al., 2007). (C) Helical wheel diagrams of the amphipathic region for Dm Cpx, Mm Cpx I, and Ce Cpx. Hydrophilic resides are indicated in magenta, hydrophobic residues in green, and nonpolar and uncharged residues in grey. Predicted phosphorylation sites are indicated by *. (D) CpxS126A exhibited greatly decreased [32P] incorporation compared to WT Cpx (CpxS126A 32P incorporation was reduced to 35.2 ± 10.6 % relative band intensity of WT Cpx 32P incorporation (n=4)). Recombinant control Cpx (WT) and phosphoincompetent CpxS126A were incubated with [32P]ATP and recombinant active PKA. Phosphorylation was visualized by autoradiography after proteins were separated by SDS-PAGE (bottom panel). Gels were also stained with Coomassie blue to assay protein loading (top panel).
Acute and local regulation of Cpx function as a vesicle clamp at the post-translational level is likely to involve the C-terminus of Cpx (Figure 4B). We previously identified two transcripts that arise from the cpx locus that result in the expression of Cpx isoforms with distinct C-terminal domains – one containing a CAAX preynylation motif (termed 7A) and one lacking this lipid tethering domain (7B) (Buhl et al., 2013; Cho et al., 2010). The C-terminus of Cpx is required to inhibit spontaneous vesicle fusion, likely via its ability to interact with lipid membranes (Cho et al., 2010; Iyer et al., 2013; Kaeser-Woo et al., 2012; Snead et al., 2014; Wragg et al., 2013; Xue et al., 2009). Although Cpx 7B does not contain a CAAX motif, it contains a predicted amphipathic helix that likely mediates protein/lipid association important for Cpx function (Wragg et al., 2013). Interestingly, S126 in the C-terminus of Cpx 7B is predicted to be a PKA phosphorylation site (http://mendel.imp.ac.at/pkaPS/) based on the consensus sequence R-R-X-S/T-Φ (Figure 4B; (Ubersax and Ferrell, 2007)). This phosphorylation site flanks the hydrophobic face of the predicted amphipathic region near then end of the Cpx C-terminus (Figures 4B and 4C). The amphipathic region is conserved in other Cpx orthologs, and mammalian and C. elegans phosphorylation sites that similarly flank the respective hydrophobic face of the amphipathic region have either been identified or predicted (see Figures 4B and 4C; (Shata et al., 2007)). Phosphorylation of Cpx 7B (hereafter referred to as Cpx) at this site could disrupt C-terminal membrane interactions occurring along the hydrophobic face of the amphipathic helix. To assay if Cpx is indeed a substrate for PKA phosphorylation, recombinant Cpx was purified and incubated with PKA and [32P]ATP. In the presence of PKA, Cpx readily incorporated 32P, indicating Cpx is a substrate for PKA in vitro (Figure 4D). Mutation of the predicted PKA phosphorylation site S126 to a phosphoincompetent alanine residue (CpxS126A) exhibited greatly reduced PKA-dependent phosphorylation compared to WT Cpx (32P incorporation into CpxS126A is reduced to 35.2 ± 10.6 % of WT Cpx 32P incorporation (n=4)), suggesting PKA phosphorylates Cpx primarily at the predicted PKA phosphorylation residue S126.
To examine how Cpx phosphorylation at S126 might alter its in vivo function, we generated transgenic animals that neuronally express WT Cpx or Cpx with S126 substitutions in the cpxSH1 null background. We generated serine→alanine (CpxS126A) and serine→aspartic acid (CpxS126D) substitutions to mimic phosphoincompetent and phosphomimetic Cpx states, respectively. We first assayed the ability of the CpxS126 mutants to rescue basal electrophysiological properties compared to WT Cpx rescues by recording from 3rd instar NMJs and measuring minis and evoked responses. cpxSH1 null larvae exhibited reduced evoked responses across all Ca++ concentrations compared to neuronally expressed WT Cpx rescues (w1118;; cpxSH1 = 7.7 ± 1.0 mV (n=8) vs. C155;; WT Cpx, cpxSH1 = 26.6 ± 1.1 mV (n=7) at [Ca++] = 0.2 mM; p<0.001; Figures 5A and 5B). Both neuronally expressed CpxS126A and CpxS126D point mutants rescued the cpxSH1 evoked release phenotype, with responses comparable to WT Cpx ([C155;; CpxS126A, cpxSH1 = 27.1 ± 1.3 mV (n=8) and C155;; CpxS126D, cpxSH1 = 25.8 ± 0.8 (n=7)] vs. C155;; WT Cpx, cpxSH1 = 26.6 ± 1.1 mV (n=7) at [Ca++] = 0.2 mM; p>0.05). These results indicate C-terminal modification at S126 does not alter evoked release properties regulated by Cpx.
We next examined the ability of phosphoincompetent and phosphomimetic Cpx to rescue the cpxSH1 null mini phenotype. cpxSH1 null larvae exhibited an elevated rate of minis that are significantly reduced by neuronal expression of WT Cpx (w1118;; cpxSH1 = 70.4 ± 3.4 Hz (n=7) vs. C155;; WT Cpx, cpxSH1 = 6.6 ± 0.4 Hz (n=15); p<0.001; Figures 5C and 5D). Phosphoincompetent CpxS126A rescue lines exhibited mini frequencies similar to WT Cpx rescues (C155;; CpxS126A, cpxSH1 = 6.7 ± 0.5 Hz (n=14) vs. C155;; WT Cpx, cpxSH1 = 6.6 ± 0.4 Hz (n=15); p>0.05), indicating the basal clamping function of CpxS126A is intact. In contrast, CpxS126D rescue animals exhibited a significant elevation in mini frequency compared to WT Cpx and CpxS126A rescues (C155;; CpxS126D, cpxSH1 = 10.7 ± 0.5 Hz (n=13) vs. [C155;; WT Cpx, cpxSH1 = 6.6 ± 0.4 Hz (n=15); p<0.001 and C155;; CpxS126A, cpxSH1 = 6.7 ± 0.5 Hz (n=14); p<0.01]). These results suggest that PKA phosphorylation of CpxS126 may selectively regulate Cpx’s ability to act as a vesicle fusion clamp, leaving its function in evoked release unperturbed.
We next assayed if WT Cpx and CpxS126 mutant rescues altered synaptic growth. cpxSH1 nulls exhibit enhanced synaptic growth that correlate with enhanced synaptic minis (w1118;; cpxSH1 = 115.8 ± 4.3 (n=8) boutons vs. control (w1118;; CpxPE = 65.6 ± 1.8 boutons (n=28); p<0.001; Figures 5F and 5G). WT Cpx and both CpxS126A rescue lines show a mild elevation of mini frequency due to the lack of a complete rescue of the null phenotype (Figures 5D and 5E). Consistent with a positive correlation of minis and synaptic growth, WT Cpx and CpxS126A mutant rescues showed modest enhancements in synaptic growth compared to control (w1118;; CpxPE = 65.6 ± 1.8 boutons (n=28) vs. C155;; WT Cpx, cpxSH1 = 88.7 ± 3.0 boutons (n=24) and C155;; CpxS126A, cpxSH1 = 83.1 ± 2.9 boutons (n=27); p<0.001 for both comparisons; Figures 5F and 5G). Strikingly, the elevated mini frequency in the CpxS126D rescue animals compared to WT Cpx and CpxS126A rescues (Figures 5D) was correlated with significantly enhanced synaptic growth (C155;; CpxS126D, cpxSH1 = 105.8 ± 3.2 boutons (n=25) vs. [C155;; WT Cpx, cpxSH1 = 88.8 ± 3.0 boutons (n=24) and C155;; CpxS126A, cpxSH1 = 83.1 ± 2.9 boutons (n=27)]; p<0.001 for both comparisons). These data demonstrate that phosphomimetic CpxS126D displays elevated mini release and increased synaptic growth, suggesting acute regulation of Cpx function via PKA phosphorylation may regulate synaptic growth and plasticity.
HFMR expression requires PKA and Cpx phosphorylation site S126
Having established that the PKA phosphorylation site in Cpx (S126) can regulate the clamping properties of the protein, we examined if CpxS126 is required for HFMR expression. We first determined if WT Cpx rescues were capable of expressing HFMR. HFMR induction in larval NMJs neuronally expressing WT Cpx in a cpxSH1 null background resulted in HFMR that was blocked by the PKA inhibitor, H89 (C155;; WT Cpx, cpxSH1 = 1.6 ± 0.1 fold (n=7) vs. C155;; WT Cpx, cpxSH1 + H89 = 1.0 ± 0.1 (n=6) fold at 100 sec after induction; p<0.001; Figures 6A–6C). To examine the requirement of S126 for HFMR expression, we neuronally expressed CpxS126 mutants in the cpxSH1 null background. Compared to WT Cpx rescues, CpxS126A did not exhibit significant HFMR (C155;; WT Cpx, cpxSH1 = 1.6 ± 0.1 fold (n=7) vs. C155;; CpxS126A, cpxSH1 = 1.1 ± 0.1 fold (n=9) 100 sec after induction; p <0.001), demonstrating that S126 is necessary for HFMR expression. The phosphomimeitc CpxS126D exhibited elevated mini frequency under basal conditions (see Figure 5D and 5E). If activity-dependent phosphorylation of CpxS126 is necessary for HFMR expression, we hypothesized that the phosphomimetic CpxS126D would occlude HFMR. Examination of HFMR in CpxS126D rescues revealed that CpxS126D animals failed to express HFMR compared to WT Cpx recues (C155;; WT Cpx, cpxSH1 = 1.6 ± 0.1 fold (n=7) vs. C155;; CpxS126D, cpxSH1 = 1.0 ± 0.1 fold (n=7) 100 sec after induction; p <0.001), demonstrating that mimicking phosphorylation of Cpx at S126 is sufficient to elevate spontaneous release rate without stimulation and to occlude HFMR expression. Together, these results suggest that dynamic phosphorylation of Cpx at residue S126 is necessary for HFMR expression.
Figure 6.

Cpx PKA phosphorylation site S126 is required for HFMR expression. (A) Representative traces of minis before (top) and after (bottom) tetanic stimulation recorded from cpxSH1 rescue lines neuronally expressing WT Cpx rescues (C155;; WT Cpx, cpxSH1) with or without PKA inhibitor H89, and CpxS126A and CpxS126D mutant rescues (C155;; CpxS126A, cpxSH1 and C155;; CpxS126D, cpxSH1, respectively). (B) Mean mini frequency plotted as a function of time. Tetanic stimulation occurs at the 0 sec time point (Red Arrow). Mini frequency is normalized to average baseline mini frequency prior to stimulation for each preparation. (C) Average fold enhancement of mini frequency compared to baseline 100 seconds after tetanic stimulation. Indicated comparisons were made using ANOVA with post hoc Tukey analysis. Mean ± SEM is indicated.
PKA phosphorylation of Cpx S126 is required for activity-dependent structural plasticity
Our results suggest PKA phosphorylation of Cpx regulates its function as a vesicle fusion clamp to express HFMR. The cAMP-PKA pathway also regulates synaptic morphology at the Drosophila NMJ. cAMP metabolism mutants, dunce (dnc) and rutabaga (rut), exhibit synaptic growth alterations in addition to the behavioral learning defects by which they were originally identified (Davis et al., 1996; Guan et al., 2011; Zhong et al., 1992; Zhong and Wu, 1991). We previously observed that expression of a constitutively active PKA (CA-PKA) results in synaptic NMJ overgrowth (Yoshihara et al., 2005). 3rd instar larvae that express presynaptic CA-PKA exhibit enhanced synaptic growth compared to control (C155; = 111.6 ± 3.6 boutons (n=33) vs. control (C155) = 64.3 ± 1.8 boutons (n=30); p<0.001 Figures 7A & 7B). To examine the contribution of CpxS126 to PKA-dependent synaptic growth, we presynaptically co-expressed CA-PKA in the cpxSH1 null background with either WT Cpx or the phosphoincompetent CpxS126A. Co-expression of CA-PKA with WT Cpx results in synaptic overgrowth similar to CA-PKA expression alone (C155; ; = 115.6 ± 5.8 boutons (n=19) compared to control (C155) = 64.3 ± 1.8 boutons (n=30); p<0.001). However, co-expression of CA-PKA with PKA phosphoincompetent CpxS126A suppressed synaptic overgrowth compared to WT Cpx rescues (C155; ; = 85.4 ± 3.1 boutons (n=24) vs. C155; ; = 115.6 ± 5.8 boutons (n=19); p<0.001). We next examined synaptic growth when co-expressing CA-PKA and phosphomimetic CpxS126D. We found that co-expressing CA-PKA with phosphomimetic CpxS126D exhibited synaptic growth similar to WT Cpx rescues (C155; ; = 106.3 ± 3.5 boutons (n=18) vs. C155; ; = 115.6 ± 5.8 boutons (n=19); p>0.05). Interestingly, CA-PKA expressed in the cpxSH1 null background did not exhibit significant synaptic growth beyond CA-PKA alone or WT Cpx rescues (C155; ; cpxSH1 = 122.8 ± 5.0 boutons (n=12) vs. [C155; = 111.6 ± 3.6 boutons (n=33) and C155; ; = 115.6 ± 5.8 boutons (n=19)]; p>0.5 for both comparisons). This result suggests a shared signaling pathway between PKA and Cpx in regulating synaptic growth rather than parallel pathways. Overall, these results demonstrate that S126 is required for PKA-dependent synaptic growth and suggest Cpx is a downstream target of the PKA signaling pathway that regulates morphological growth and structural plasticity of synaptic terminals.
Figure 7.

Cpx PKA phosphorylation site S126 is required for PKA and activity-dependent synaptic growth. (A) Representative images of muscle 6/7 NMJs from control (C155 elav-GAL4), and lines neuronally overexpressing constitutively active PKA alone (C155; ) or in the cpxSH1 null (C155; ; cpxSH1), WT Cpx rescue (C155; ; ), CpxS126A rescue (C155; ; ), and CpxS126D rescue (C155; ; ) backgrounds. (B) Summary of mean synaptic bouton number normalized to muscle area for the indicated genetic backgrounds. Indicated comparisons were made using ANOVA with post hoc Tukey analysis. (C) Representative images of control (w1118;; CpxPE) and WT Cpx rescues (C155;; WT Cpx, cpxSH1), CpxS126A rescues (C155;; CpxS126A, cpxSH1), and CpxS126D rescues (C155;; CpxS126D, cpxSH1) reared at 25° or 29°. (D) Summary of % change in synaptic bouton number normalized to muscle area for the indicated genetic backgrounds and rearing temperatures. Synaptic bouton number was normalized to bouton number of larvae reared at 25° for each respective genotype to reflect % change. Indicated comparisons were made using Student’s T-test. Scale bars = 20 μm. Mean ± SEM is indicated in all figures.
Synaptic expansion at the NMJ is regulated by the overall level of motor neuron activity. Genetic manipulations that enhance or decrease neuronal activity at the NMJ alter synapse formation at Drosophila NMJs (Budnik et al., 1990; Schuster et al., 1996). In addition to genetic manipulations that modulate activity, larvae reared under variable environmental temperatures modulate locomotor activity (Sigrist et al., 2003; Zhong and Wu, 2004). At elevated temperatures, enhanced larval locomotion is correlated with enhanced synaptic NMJ growth, suggesting locomotor experience and NMJ growth are directly coupled (Sigrist et al., 2003). Interestingly, enhancement of synaptic growth at elevated temperatures is suppressed in adenylyl cyclase mutants, indicating that increased neuronal activity and synaptic growth induced environmentally requires PKA signaling (Zhong and Wu, 2004). To determine if Cpx phosphorylation contributes to this form of activity-dependent structural plasticity, we examined temperature induced synaptic growth in WT Cpx and CpxS126 mutant rescue animals. Similar to previously published results, rearing control larvae at 29°C resulted in significant increase in synaptic growth compared to animals reared at 25°C (w1118;; CpxPE at 29°C = 123.8 boutons (n=36) vs. w1118;; CpxPE at 25°C = 100.0 ± 2.4 boutons (n=35); p<0.0001; Figures 7C and 7D). Rescue larvae expressing presynaptic WT Cpx in the cpxSH1 null background exhibited elevated synaptic growth at 29°C compared to growth at 25°C, similar to controls (C155;; WT Cpx, cpxSH1 at 29°C = 129.2 boutons (n=29) vs. C155;; WT Cpx, cpxSH1 at 25°C = 100.0 ± 2.7 boutons (n=36); p<0.0001). We next examined if phosphorylation of S126 might regulate this form of structural plasticity. Unlike control and WT Cpx rescues, phosphoincompetent CpxS126A rescues failed to exhibit enhanced synaptic growth at elevated temperatures (C155;; CpxS126A, cpxSH1 at 29°C = 93.9 ± 33.1 (n=36) boutons vs. C155;; CpxS126A, cpxSH1 at 25°C = 100.0 ± 2.4 boutons (n=35); p>0.05). Similar to earlier experiments in which HFMR expression was occluded in CpxS126D rescues, phosphomimetic CpxS126D rescues did not express temperature induced synaptic growth (C155;; CpxS126D, cpxSH1 at 29°C = 100.7 ± 6.0 (n=17) vs. C155;; CpxS126D, cpxSH1 at 25°C = 100.0 ± 3.8 boutons (n=20); p>0.05). These results indicate that the PKA phosphorylation site at S126 in Cpx is required for expression of temperature-induced structural plasticity at Drosophila NMJs. Taken together, these data suggest that a PKA-dependent retrograde signaling pathway mediated by postsynaptic Syt 4 converges on Cpx to regulate its function as a synaptic vesicle fusion clamp, and that this regulation is required for activity-dependent functional and structural plasticity that occurs downstream of enhanced spontaneous release.
Discussion
These findings indicate that the SNARE-binding protein Cpx is a key PKA target that regulates spontaneous fusion rates and presynaptic plasticity at Drosophila NMJs. We demonstrate that Cpx’s function can be modified to regulate activity-dependent functional and structural plasticity. In vivo experiments using Cpx phosphomimetic rescues demonstrate that Cpx phosphorylation at residue S126 selectively alters its ability to act as a synaptic vesicle fusion clamp. In addition, S126 is critical for the expression of HFMR, an activity-dependent form of acute functional plasticity that modulates mini frequency at Drosophila synapses. Our data indicate a Syt 4-dependent retrograde signaling pathway converges on Cpx to regulate its synaptic function (Figure 4A). Additionally, we find that elevated spontaneous fusion rates correlate with enhanced synaptic growth. This pathway requires Syt 4 retrograde signaling to enhance spontaneous release and to trigger synaptic growth. Moreover, the Cpx S126 PKA phosphorylation site is required for activity-dependent synaptic growth, suggesting acute regulation of minis via Cpx phosphorylation is likely to contribute to structural synaptic plasticity. Together, these data identify a novel mechanism of acute synaptic plasticity that impinges directly on the presynaptic release machinery to regulate spontaneous release rates and synaptic maturation.
How does acute phosphorylation of S126 alter Cpx’s function? The Cpx C-terminus associates with lipid membranes through a prenylation domain (CAAX motif) and/or the presence of an amphipathic helix (Cho et al., 2010; Iyer et al., 2013; Kaeser-Woo et al., 2012; Snead et al., 2014; Wragg et al., 2013; Xue et al., 2009). The Drosophila Cpx isoform used in our study (Cpx 7B) lacks a CAAX-motif, but contains a C-terminal amphipathic helix flanked by the S126 phosphorylation site. S126 phosphorylation does not alter synaptic targeting of Cpx (Figures S1A and S1B) or its ability to associate with SNARE complexes in vitro (Figure S2, S3A, S3B, and Table S3). As such, phosphorylation may instead alter interactions of the amphipathic helix region with lipid membranes and/or alter Cpx interactions with other proteins that modulate synaptic release. Given the well-established role of the Cpx C-terminus in regulating membrane binding and synaptic vesicle tethering of Cpx, phosphorylation at this site would be predicted to alter the subcellular localization of the protein and its accessibility to the SNARE complex (Buhl et al., 2013; Snead et al., 2014; Wragg et al., 2013). However, we did not detect large differences between WT Cpx and CpxS126D in liposome binding (Figure S4). This assay is unlikely to reveal subtle changes in lipid interactions by Cpx, as we found that C-terminal deletions (CTD) maintained its ability to bind membranes. The ability of the CTD versions of Drosophila Cpx to associate with liposomes is unlike that observed with C. elegans Cpx (Wragg et al., 2013), and indicate domains outside of Drosophila Cpx’s C-terminus contribute to lipid membrane association as well, potentially masking effects from S126 phosphorylation that might occur in vivo. Alternatively, phosphorylation of the Cpx C-terminus could alter its association with other SNARE complex modulators such as Syt 1 (Jorquera et al., 2012; Tang et al., 2006).
Our data indicate that enhanced minis regulate synaptic growth through several previously identified NMJ maturation pathways (Figure 4A). We find that the Wit signaling pathway is required for synaptic growth in the background of enhanced minis. The wit gene encodes a presynaptic type II BMP receptor that receives retrograde, transsynaptic BMP signals from postsynaptic muscles (Marques et al., 2002; McCabe et al., 2003; Piccioli and Littleton, 2014). Consistent with our data, other studies have demonstrated that downstream signaling components of the BMP pathway are necessary and sufficient for mini-dependent synaptic growth at the Drosophila NMJ (Choi et al., 2014). Additionally, we found that the postsynaptic Ca++ sensor, Syt 4, is required for enhancing spontaneous release and increasing synaptic growth (Barber et al., 2009; Yoshihara et al., 2005). Our data does not currently distinguish the interdependence of the BMP and Syt 4 retrograde signaling pathways, and other retrograde signaling pathways might contribute to mini-dependent synaptic growth as well (Ballard et al., 2014). Recently, several retrograde pathways have been identified that regulate functional homeostatic plasticity at the Drosophila NMJ (Davis and Muller, 2015). Future work will be required to fully define the retrograde signaling pathways necessary to mediate mini-dependent synaptic growth.
How might elevated spontaneous release through Cpx phosphorylation regulate synaptic growth? We hypothesize that the switch in synaptic vesicle release mode to a constitutive fusion pathway that occurs over several minutes following stimulation serves as a synaptic tagging mechanism. By continuing to activate postsynaptic glutamate receptors in the absence of incoming action potentials, the elevation in mini frequency would serve to enhance postsynaptic calcium levels by prolonging glutamate receptor stimulation (as observed in Fig. 3D). This would prolong retrograde signaling that initiates downstream cascades to directly alter cytoskeletal architecture required for synaptic bouton budding (Piccioli and Littleton, 2014). Given that elevated rates of spontaneous fusion still occur in cpx and syx3-69 in the absence of Syt 4 and BMP signaling, yet synaptic overgrowth is suppressed in these conditions, it is unlikely that spontaneous fusion itself directly drives synaptic growth. Rather, the transient enhancement in spontaneous release may serve to prolong postsynaptic calcium signals that engage distinct effectors for structural remodeling that fail to be activated in the absence of elevated spontaneous release. Results from mammalian studies indicate spontaneous release can uniquely regulate postsynaptic protein translation and activate distinct populations of NMDA receptors (Atasoy et al., 2008; Sutton et al., 2006) compared to evoked release, so it is possible that spontaneous fusion may engage unique postsynaptic effectors at Drosophila NMJs as well.
In the last few decades, intense research efforts have elucidated several molecular mechanisms of classic Hebbian forms of synaptic plasticity that include long-term potentiation (LTP) and long-term depression (LTD), alterations in synaptic function that lasts last minutes to hours (Mayford et al., 2012). The most widely studied expression mechanism for these forms of synaptic plasticity involve modulation of postsynaptic AMPA-type glutamate receptor (AMPAR) function and membrane trafficking (Huganir and Nicoll, 2013). In contrast, molecular mechanisms of short-term synaptic plasticity remain poorly understood. Several forms of short-term plasticity have been described, such as post-tetanic potentiation (PTP), which involves stimulation-dependent increases in synaptic strength, including changes in mini frequency (Habets and Borst, 2005; Korogod et al., 2005; Lou et al., 2008). Short-term plasticity expression is likely to impinge on the alterations to the presynaptic release machinery downstream of activated effector molecules (Zucker and Regehr, 2002). For example, Munc 18, a presynaptic protein involved in the priming step of vesicle exocytosis via its ability to associate with members of the SNARE machinery, is dynamically regulated by Ca++-dependent protein kinase C (PKC), and its regulation is required to express PTP at the Calyx of Held (Genc et al., 2014). Here we demonstrate that the presynaptic vesicle fusion machinery can also be directly modified to alter spontaneous neurotransmission via activity-dependent modification of Cpx function by PKA. Protein kinase CK2 and PKC phosphorylation sites within the C-terminus of mammalian and C. elegans Cpx have been identified (see Figure 4B and 4C; (Shata et al., 2007)). Therefore, activity-dependent regulation of Cpx function via C-terminal phosphorylation may be an evolutionarily conserved mechanism to regulate synaptic plasticity. Moreover, Cpx may lie downstream of multiple effector pathways to modulate various forms of short-term plasticity, including PTP, in a synapse-specific manner. Interestingly, Cpx is expressed both pre- and postsynaptically in mammalian hippocampal neurons and is required to express LTP via regulation of AMPAR delivery to the synapse, suggesting Cpx-mediated synaptic plasticity expression mechanisms may also occur postsynaptically (Ahmad et al., 2012).
In summary, these results indicate minis serve as an independent and regulated neuronal signaling pathway that contributes to activity-dependent synaptic growth. We previously found Cpx’s function as a facilitator and clamp for synaptic vesicle fusion is genetically separable, demonstrating distinct molecular mechanisms regulate evoked and spontaneous release (Cho et al., 2014; Cho et al., 2010). Evoked and spontaneous release are also separable beyond Cpx regulation, as other studies have demonstrated that minis can utilize distinct components of the SNARE machinery, distinct vesicle pools, and distinct individual synaptic release sites (Kavalali, 2014; Melom et al., 2013). These findings suggest diverse regulatory mechanisms for spontaneous release that might be selectively modulated at specific synapses.
Experimental Procedures
Drosophila genetics and molecular cloning
Drosophila melanogaster were cultured on standard medium at 25°C. QuickChange (Stratagene, La Jolla, CA) was used for site directed mutagenesis on the existing cloned Cpx (Isoform 7B) (Buhl et al., 2013; Huntwork and Littleton, 2007) to generate specified Cpx mutants. PCR products were subcloned into a modified pValum construct, allowing use of the Gal4/UAS expression system (Brand and Perrimon, 1993). These constructs were injected into a yv; attP 3rd chromosome docking strains by Genetic Services Inc. (Cambridge, MA). Homozygous third chromosome UAS lines and as well as mutant alleles, syt 4BA1 and wit, were then recombined into the cpxSH1 null mutant background. The C155 elav-GAL4 driver was used for neuronal expression of transgenes unless otherwise indicated. Cpx RNAi knockdown line (transformant 21477) was obtained from the Vienna Drosophila Resource Center (Dietzl et al., 2007).
Immunostaining
Immunostaining was performed on wandering 3rd instar larvae reared at 25°C, unless otherwise indicated, as previously described (Huntwork and Littleton, 2007). See Supplemental Information for more information.
In vitro kinase assay
In vitro kinase assays were performed using the purified recombinant Cpx protein and the catalytic subunit of PKA (New England Biolabs, Ipswich, MA), according to manufacturer supplied instructions. Briefly, approximately 10 mg of purified GST-fusion protein (WT Cpx and CpxS126A) was used per reaction and incubated with 2500 units of recombinant kinase and [32P]ATP (Perkin Elmer, Waltham, MA). Reaction products were separated by SDS-PAGE. Gels were stained with Bio-Safe Coomassie Blue (Bio-Rad, Hercules, CA), dried, and exposed to autoradiography film at room temperature. Mean integrated density of each band (mean intensity/pixel multiplied by the area of the band) was quantified using ImageJ (Rasband, 1997–2009), and relative density of mutant Cpx was calculated by normalizing to WT Cpx band intensity.
Electrophysiological analysis
Electrophysiological analysis of wandering 3rd instar larva was performed in Drosophila HL3.1 saline containing (in mM): 70 NaCl, 5 KCl, 4 MgCl2, 10 NaHCO3, 5 Trehalose, 115 sucrose, 5 HEPES-NaOH, pH 7.2, and indicated concentrations of Ca++. Evoked EJPs were recorded intracellularly from muscle fiber 6 of segments A3 and A4 using an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA). Data acquisition and analysis was performed using Clampfit 9.0 software (Axon Instruments, Foster City, CA) and Mini Analysis 6.0.3 (Synaptosoft, Inc, Fort Lee, NJ) as previously described (Cho et al., 2010). Nerve stimulation at indicated frequencies in each experiment was performed using a programmable stimulator (Master-8; AMPI, Jerusalem, Israel). PKA inhibitor H89 ([25 μM in HL3.1) was bath applied to the dissected larvae and incubated for 10 min prior to stimulation (Tocris Bioscience). Mini frequency (number of individual mini events/second) was calculated from recordings of at least 2 minutes. HFMR was induced as previously described (Barber et al., 2009; Yoshihara et al., 2005). Briefly, HFMR was induced by 4 trains of 100 Hz stimuli spaced 2 seconds apart in 0.3 mM extracellular Ca++. Mini events 1 minute before and up to ~10 min after HFMR induction were continuously collected. Mini frequency at indicated time points were calculated in 20-second bins. Fold enhancement was calculated by normalizing to the baseline mini frequency recorded prior to HFMR induction. Each n value represents a single muscle recording, with data generated from at least three individual larvae of each genotype.
Ca++ imaging
Spontaneous release events were detected as previously described (Melom et al., 2013). Briefly, wandering 3rd instar larvae expressing postsynaptic UAS-myrGCaMP6s using the mef2-GAL4 driver were dissected in Ca++-free HL3.1. The motor neurons were cut just below the ventral nerve cord, washed several times with HL3.1 containing 1.5 mM Ca++, and allowed to rest for 5 min. Images were acquired with a PerkinElmer Ultraview Vox spinning disc confocal microscope equipped with a Hamamatsu C9100–13 ImagEM EM CCD at 8–35 Hz with a 40× 0.8 NA water-immersion objective (Carl Zeiss). A single confocal plane of muscle 4 NMJ in segment A3 was continuously imaged for 2 minutes before and after HFMR stimulation. Presynaptic Ca++ influx during HFMR stimulation was monitored in larvae expressing presynaptic UAS-myrGCAMP6s driven by the pan neuronal C155 elav-GAL4 driver.
Statistical Analysis
All error bars are standard error of the mean (SEM). Statistical significance was determined using one-way ANOVA with post hoc Tukey analysis for multiple comparisons and Student’s T-test when only two groups were compared (n.s. = p>0.05, * = p<0.05, ** = p<0.01, *** = p<0.001, ****=p<0.0001).
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grants NS064750 (to R.W.C.), NS40296 and MH104536 (to J.T.L.), and the JPB Foundation.
Abbreviations
- NMJ
neuromuscular junction
- EJP
excitatory junctional potential
- Cpx
complexin
- SNARE
soluble N-ethylmaleimide-sensitive fusion attachment protein receptor
- SEM
standard error of the mean
- WT
wildtype
- HRP
horse radish peroxidase
Footnotes
Author Contributions
R.W.C. performed and analyzed the electrophysiology, confocal imaging, biochemistry, and genetics. L.B. performed and analyzed confocal imaging, and designed experiments. D.V. performed the in vitro kinase experiment. Y.A. performed and analyzed the GCaMP6s experiments. A.T. and R.W.C. performed and analyzed the Cpx/SNARE pull down experiments. F.L. performed ITC experiments. J.T.L supervised the project. R.W.C. and J.T.L. designed experiments and wrote the paper.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Ahmad M, Polepalli JS, Goswami D, Yang X, Kaeser-Woo YJ, Sudhof TC, Malenka RC. Postsynaptic complexin controls AMPA receptor exocytosis during LTP. Neuron. 2012;73:260–267. doi: 10.1016/j.neuron.2011.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreae LC, Burrone J. Spontaneous Neurotransmitter Release Shapes Dendritic Arbors via Long-Range Activation of NMDA Receptors. Cell reports. 2015 doi: 10.1016/j.celrep.2015.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataman B, Ashley J, Gorczyca M, Ramachandran P, Fouquet W, Sigrist SJ, Budnik V. Rapid activity-dependent modifications in synaptic structure and function require bidirectional Wnt signaling. Neuron. 2008;57:705–718. doi: 10.1016/j.neuron.2008.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atasoy D, Ertunc M, Moulder KL, Blackwell J, Chung C, Su J, Kavalali ET. Spontaneous and evoked glutamate release activates two populations of NMDA receptors with limited overlap. J Neurosci. 2008;28:10151–10166. doi: 10.1523/JNEUROSCI.2432-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard SL, Miller DL, Ganetzky B. Retrograde neurotrophin signaling through Tollo regulates synaptic growth in Drosophila. J Cell Biol. 2014;204:1157–1172. doi: 10.1083/jcb.201308115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber CF, Jorquera RA, Melom JE, Littleton JT. Postsynaptic regulation of synaptic plasticity by synaptotagmin 4 requires both C2 domains. J Cell Biol. 2009;187:295–310. doi: 10.1083/jcb.200903098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke B, Wittnam J, McNeill E, Van Vactor DL, Keshishian H. Retrograde BMP signaling at the synapse: a permissive signal for synapse maturation and activity-dependent plasticity. J Neurosci. 2013;33:17937–17950. doi: 10.1523/JNEUROSCI.6075-11.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Budnik V, Zhong Y, Wu CF. Morphological plasticity of motor axons in Drosophila mutants with altered excitability. J Neurosci. 1990;10:3754–3768. doi: 10.1523/JNEUROSCI.10-11-03754.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhl LK, Jorquera RA, Akbergenova Y, Huntwork-Rodriguez S, Volfson D, Littleton JT. Differential regulation of evoked and spontaneous neurotransmitter release by C-terminal modifications of complexin. Mol Cell Neurosci. 2013;52:161–172. doi: 10.1016/j.mcn.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaia M, Jagota A, Gonzalez A, Vasin A, Littleton JT. Interaction of the complexin accessory helix with the C-terminus of the SNARE complex: molecular-dynamics model of the fusion clamp. Biophys J. 2013;105:679–690. doi: 10.1016/j.bpj.2013.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nature neuroscience. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Cho RW, Kummel D, Li F, Baguley SW, Coleman J, Rothman JE, Littleton JT. Genetic analysis of the Complexin trans-clamping model for cross-linking SNARE complexes in vivo. Proc Natl Acad Sci U S A. 2014;111:10317–10322. doi: 10.1073/pnas.1409311111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Song Y, Littleton JT. Comparative analysis of Drosophila and mammalian complexins as fusion clamps and facilitators of neurotransmitter release. Mol Cell Neurosci. 2010;45:389–397. doi: 10.1016/j.mcn.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BJ, Imlach WL, Jiao W, Wolfram V, Wu Y, Grbic M, Cela C, Baines RA, Nitabach MN, McCabe BD. Miniature neurotransmission regulates Drosophila synaptic structural maturation. Neuron. 2014;82:618–634. doi: 10.1016/j.neuron.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis GW, Muller M. Homeostatic control of presynaptic neurotransmitter release. Annual review of physiology. 2015;77:251–270. doi: 10.1146/annurev-physiol-021014-071740. [DOI] [PubMed] [Google Scholar]
- Davis GW, Schuster CM, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. III. CREB is necessary for presynaptic functional plasticity. Neuron. 1996;17:669–679. doi: 10.1016/s0896-6273(00)80199-3. [DOI] [PubMed] [Google Scholar]
- Dietzl G, Chen D, Schnorrer F, Su KC, Barinova Y, Fellner M, Gasser B, Kinsey K, Oppel S, Scheiblauer S, et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature. 2007;448:151–156. doi: 10.1038/nature05954. [DOI] [PubMed] [Google Scholar]
- Fatt P, Katz B. Spontaneous subthreshold activity at motor nerve endings. J Physiol. 1952;117:109–128. [PMC free article] [PubMed] [Google Scholar]
- Frank CA, Pielage J, Davis GW. A presynaptic homeostatic signaling system composed of the Eph receptor, ephexin, Cdc42, and CaV2.1 calcium channels. Neuron. 2009;61:556–569. doi: 10.1016/j.neuron.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genc O, Kochubey O, Toonen RF, Verhage M, Schneggenburger R. Munc18–1 is a dynamically regulated PKC target during short-term enhancement of transmitter release. eLife. 2014;3:e01715. doi: 10.7554/eLife.01715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Sudhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- Goold CP, Davis GW. The BMP ligand Gbb gates the expression of synaptic homeostasis independent of synaptic growth control. Neuron. 2007;56:109–123. doi: 10.1016/j.neuron.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan Z, Buhl LK, Quinn WG, Littleton JT. Altered gene regulation and synaptic morphology in Drosophila learning and memory mutants. Learning & memory. 2011;18:191–206. doi: 10.1101/lm.2027111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habets RL, Borst JG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobson RJ, Liu Q, Watanabe S, Jorgensen EM. Complexin maintains vesicles in the primed state in C. elegans. Curr Biol. 2011;21:106–113. doi: 10.1016/j.cub.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- Iyer J, Wahlmark CJ, Kuser-Ahnert GA, Kawasaki F. Molecular mechanisms of COMPLEXIN fusion clamp function in synaptic exocytosis revealed in a new Drosophila mutant. Mol Cell Neurosci. 2013;56:244–254. doi: 10.1016/j.mcn.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J Neurosci. 2012;32:18234–18245. doi: 10.1523/JNEUROSCI.3212-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Yang X, Sudhof TC. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci. 2012;32:2877–2885. doi: 10.1523/JNEUROSCI.3360-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavalali ET. The mechanisms and functions of spontaneous neurotransmitter release. Nature reviews Neuroscience. 2014;16:5–16. doi: 10.1038/nrn3875. [DOI] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci. 2005;25:5127–5137. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagow RD, Bao H, Cohen EN, Daniels RW, Zuzek A, Williams WH, Macleod GT, Sutton RB, Zhang B. Modification of a hydrophobic layer by a point mutation in syntaxin 1A regulates the rate of synaptic vesicle fusion. PLoS Biol. 2007;5:e72. doi: 10.1371/journal.pbio.0050072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MC, Yasuda R, Ehlers MD. Metaplasticity at single glutamatergic synapses. Neuron. 2010;66:859–870. doi: 10.1016/j.neuron.2010.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell. 1993;74:1125–1134. doi: 10.1016/0092-8674(93)90733-7. [DOI] [PubMed] [Google Scholar]
- Lou X, Korogod N, Brose N, Schneggenburger R. Phorbol esters modulate spontaneous and Ca2+-evoked transmitter release via acting on both Munc13 and protein kinase C. J Neurosci. 2008;28:8257–8267. doi: 10.1523/JNEUROSCI.0550-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Parisotto D, Bharat TA, Scheutzow A, Krause JM, Briggs JA, Sollner TH. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 2012;31:3270–3281. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O’Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol. 2011;21:97–105. doi: 10.1016/j.cub.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayford M, Siegelbaum SA, Kandel ER. Synapses and memory storage. Cold Spring Harbor perspectives in biology. 2012;4 doi: 10.1101/cshperspect.a005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O’Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Durr R, Gahwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nature neuroscience. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Missler M, Li C, Sudhof TC. Complexins: cytosolic proteins that regulate SNAP receptor function. Cell. 1995;83:111–119. doi: 10.1016/0092-8674(95)90239-2. [DOI] [PubMed] [Google Scholar]
- Melom JE, Akbergenova Y, Gavornik JP, Littleton JT. Spontaneous and evoked release are independently regulated at individual active zones. J Neurosci. 2013;33:17253–17263. doi: 10.1523/JNEUROSCI.3334-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron. 2001;30:737–749. doi: 10.1016/s0896-6273(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Peled ES, Isacoff EY. Optical quantal analysis of synaptic transmission in wild-type and rab3-mutant Drosophila motor axons. Nat Neurosci. 2011;14:519–526. doi: 10.1038/nn.2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccioli ZD, Littleton JT. Retrograde BMP signaling modulates rapid activity-dependent synaptic growth via presynaptic LIM kinase regulation of cofilin. J Neurosci. 2014;34:4371–4381. doi: 10.1523/JNEUROSCI.4943-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband WS. Image J. Bethesda, MD: U. S. National Institutes of Health; 1997–2009. [Google Scholar]
- Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat Struct Mol Biol. 2006;13:748–750. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. II. Fasciclin II controls presynaptic structural plasticity. Neuron. 1996;17:655–667. doi: 10.1016/s0896-6273(00)80198-1. [DOI] [PubMed] [Google Scholar]
- Shata A, Saisu H, Odani S, Abe T. Phosphorylated synaphin/complexin found in the brain exhibits enhanced SNARE complex binding. Biochem Biophys Res Commun. 2007;354:808–813. doi: 10.1016/j.bbrc.2007.01.064. [DOI] [PubMed] [Google Scholar]
- Sigrist SJ, Reiff DF, Thiel PR, Steinert JR, Schuster CM. Experience-dependent strengthening of Drosophila neuromuscular junctions. J Neurosci. 2003;23:6546–6556. doi: 10.1523/JNEUROSCI.23-16-06546.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead D, Wragg RT, Dittman JS, Eliezer D. Membrane curvature sensing by the C-terminal domain of complexin. Nature communications. 2014;5:4955. doi: 10.1038/ncomms5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC. Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron. 2013;80:675–690. doi: 10.1016/j.neuron.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Ubersax JA, Ferrell JE., Jr Mechanisms of specificity in protein phosphorylation. Nat Rev Mol Cell Biol. 2007;8:530–541. doi: 10.1038/nrm2203. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, Rothman JE. SNAREpins: minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Wong YH, Lee TY, Liang HK, Huang CM, Wang TY, Yang YH, Chu CH, Huang HD, Ko MT, Hwang JK. KinasePhos 2.0: a web server for identifying protein kinase-specific phosphorylation sites based on sequences and coupling patterns. Nucleic Acids Res. 2007;35:W588–594. doi: 10.1093/nar/gkm322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS. Synaptic vesicles position complexin to block spontaneous fusion. Neuron. 2013;77:323–334. doi: 10.1016/j.neuron.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Sudhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Craig TK, Xu J, Chao HT, Rizo J, Rosenmund C. Binding of the complexin N terminus to the SNARE complex potentiates synaptic-vesicle fusogenicity. Nat Struct Mol Biol. 2010;17:568–575. doi: 10.1038/nsmb.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol. 2008;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Adolfsen B, Galle KT, Littleton JT. Retrograde signaling by Syt 4 induces presynaptic release and synapse-specific growth. Science. 2005;310:858–863. doi: 10.1126/science.1117541. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Budnik V, Wu CF. Synaptic plasticity in Drosophila memory and hyperexcitable mutants: role of cAMP cascade. J Neurosci. 1992;12:644–651. doi: 10.1523/JNEUROSCI.12-02-00644.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Y, Wu CF. Altered synaptic plasticity in Drosophila memory mutants with a defective cyclic AMP cascade. Science. 1991;251:198–201. doi: 10.1126/science.1670967. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Wu CF. Neuronal activity and adenylyl cyclase in environment-dependent plasticity of axonal outgrowth in Drosophila. J Neurosci. 2004;24:1439–1445. doi: 10.1523/JNEUROSCI.0740-02.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annual review of physiology. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.