

Abstract
The CRISPR-Cas9 system in bacteria and archaea has recently been exploited for genome editing in various model organisms, including mice. The CRISPR-Cas9 reagents can be delivered directly into the mouse zygote to derive a mutant animal carrying targeted genetic modifications. The major components of the system include the guide RNA which provides target specificity, the Cas9 nuclease that creates the DNA double-strand break, and the donor oligonucleotide or plasmid carrying the intended mutation flanked by sequences homologous to the target site. Here we describe the general considerations and experimental protocols for creating genetically modified mice using the CRISPR-Cas9 system.
Keywords: CRISPR, genome editing, knockout, knock in, mouse model
INTRODUCTION
Mice are important model organisms to study human biology, diseases and therapeutics (Zambrowicz and Sands, 2003). Historically, using well-established gene targeting method, targeted mutagenesis in mice has been achieved, first in mouse embryonic stem cells and subsequently derivation of mice from the targeted ES clones (Capecchi, 2005). Recently, site-directed DNA endonucleases have been developed into powerful tools for genome editing in various model organisms (Hai et al., 2014; Hwang et al., 2013; Li et al., 2013; Niu et al., 2014), including mice (Wang et al., 2013; Yang et al., 2013). Compared with conventional gene targeting in ES cells, genome editing by site-directed DNA endonucleases is highly efficient and can be performed directly on the zygotes, circumventing the need of a germline competent embryonic stem cell line.
Site-directed nucleases include homing endonuclease, Zinc Finger Nuclease (ZFN), Transcription Activator-Like Effector Nuclease (TALEN) and recently, Clustered Regularly Interspaced Palindromic Repeat (CRISPR) and CRISPR associated endonuclease (Cas) (Doudna and Charpentier, 2014; Gaj et al., 2013; Hsu et al., 2014). Owing to its simplicity in design and superior efficiency, the CRISPR-Cas9 system has swiftly become the technology of choice for genome editing in both cell lines and animal model creation (Doudna and Charpentier, 2014; Hsu et al., 2014). For CRISPR-Cas9, the most widely used system is derived from Streptococcus Pyogenes (SP), although several orthologous systems from other strains have also been characterized (Doudna and Charpentier, 2014). This protocol is based on the SP CRISPR-Cas9 system.
The CRISPR-Cas9 system for genome editing involves several components, including the single guide RNA (sgRNA) that specifies target specificity and the Cas9 enzyme that creates the DNA double-strand break (DSB) (Figure 1). sgRNA is an artificial fusion of two naturally occurring RNA species, the CRISPR RNA (crRNA) and the trans-activating RNA (tracrRNA) (Cong et al., 2013; Jinek et al., 2012). The 20 nt (nucleotide) RNA sequence located at the 5′ end of the sgRNA specifies the target specificity by sequence complementarity to the protospacer sequence located in the genomic target site. In the SP CRISPR-Cas9 system, target specificity of the Cas9 protein is also dictated by the Protospacer Adjacent Motif (PAM), a trinucleotide sequence of “NGG”, directly adjacent to and continuous from the 3′ end of the protospacer sequence of the noncomplementary strand. In an effort to improve targeting efficiency (Chen et al., 2013), two modifications were made to the conventional sgRNA backbone, including “A-U flip” and “stem extension” (Figure 1). We routinely use this improved backbone in our CRISPR-Cas9 experiment.
Figure 1. The CRISPR-Cas9 System for Genome Editing.
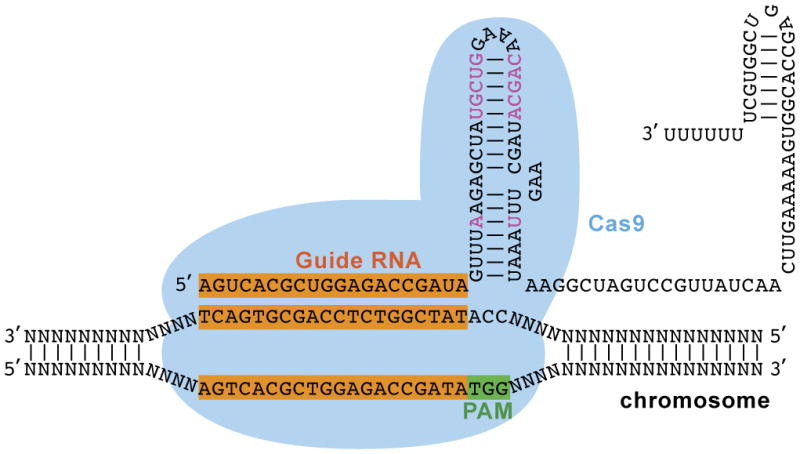
The CRISPR-Cas9 system has two components, the single guide RNA and the Cas9 nuclease. The guide RNA provides target specificity by Watson-Crick base pairing with the protospacer sequence located in the genome DNA target site, along with the “NGG” PAM sequence adjacent to and immediately 3′ from the protospacer sequence of the non complementary strand. Shown in black fond is the sgRNA in its conventional backbone (Cong et al., 2013; Jinek et al., 2012) and colored in pink the two modifications (“A-U flip” and “stem loop extension”) from the improved sgRNA backbone (Chen et al., 2013).
Various types of mutations can be created using the CRISPR-Cas9 system, depending on use of the endogenous DNA repair pathways (Doudna and Charpentier, 2014; Hsu et al., 2014; Wiles et al., 2015). When a DNA DSB is created, non homologous end joining (NHEJ) can come into play and repair the damage by ligating the two ends together, often with nucleotides inserted or deleted in between the two broken ends. This is the Indel (insertion/deletion) based knockout model. Alternatively, when a donor oligonucleotide or plasmid carrying the intended mutation flanked by sequences homologous to the target site is provided, homology directed repair (HDR) may be mobilized and incorporate the precise mutation into the target site. This is the HDR-based knock in model. The nature of the knock in mutation can be point mutation incorporation, tag or transgene insertion, or replacement of the murine sequence with its human ortholog. As is evident, if the goal is to disrupt or delete a genomic sequence, only sgRNA(s) and the Cas9 enzyme are needed, while if the goal is to create a precise genome modification, donor oligonucleotide or plasmid with homology arms flanking the intended mutation will need to be provided.
General Considerations for Generating a Mouse Model Using CRISPR-Cas9
The process for generating a mouse model can be arbitrarily divided into 5 steps, in sequence:
Step 1: Model Design (deliverable: in silicon design of the allele, design and validation of the genotyping strategies, and experimental plan)
Step 2: Reagent Synthesis (deliverable: Cas9 mRNA or protein, sgRNA, donor oligonucleotide or donor plasmid)
Step 3: Founder Generation (deliverable: founder mice carrying intended mutation)
Step 4: Germline Transmission (deliverable: F1 mice carrying intended mutation)
Step 5: Study Cohort Generation (deliverable: study cohort carrying intended mutation)
Before starting on bench work, one should always map out the entire process and derive the final edited allele in silico. Some important considerations are:
Region to Target
For a knockout model based on Indel mutation mediated by NHEJ, a common practice is to rely on frameshift mutation using one sgRNA or delete a segment of the genomic sequence using two sgRNAs flanking the region to be deleted. These are some of the important considerations:
Avoid targeting the “ATG” start site, if there is a downstream inframe “ATG” which may be used and produce a truncated protein.
Target a region that is relatively 5′ to the gene, downstream from the starting “ATG”, within an exon.
Target a 5′ exon commonly shared among the transcript variants, if the intent is to target all transcript variants associated with the gene of interest. Alternatively, target an exon used in a transcript variant, if the intent is to delete a specific transcript variant.
Target safely within an exon and preserve the splice donor and acceptor sites flanking a particular exon to avoid creation of an aberrant splicing product. Indel mutations created by CRISPR-Cas9 are often small, less than 20 nt in size, and as such, one should choose an exon that is large enough to accommodate the mutation while leaving the splice donor and acceptor sites unaffected.
Alternatively, sequences coding for a functionally important domain could be targeted, such as those mediating ligand binding, catalytic activity, or protein/protein interactions.
Another important consideration is sequence complexity in the area of interest. An unusually high GC content could post challenge for a polymerase chain reaction (PCR) for genotyping or long strings of “A” or “T” could prevent successful sequencing reaction reading through the region. It is essential to have a genotyping strategy designed and validated before finalizing a project plan, including the choice of a region to target or one may risk not being able to screen for the founder mice.
For a knock in model, the DSB is ideally placed as close to the mutation incorporation site as possible, therefore there will be very limited choices for the target site. For these projects, one must evaluate the region to be sure there is a valid CRISPR guide choice before embarking on a CRISPR project.
Guide Choices
For a model that relies on NHEJ to create an Indel allele, there may be multiple choices for a guide sequence. In this case, the primary consideration may be to minimize off target damage, while maximizing targeting efficiency. There are many sgRNA design programs that identify off-target sequence matches for a given guide sequence (Hsu et al., 2013; Wiles et al., 2015). For designing sgRNAs with high efficiency, in addition to the use of software programs that make an attempt to calculate “efficiency score” (Doench et al., 2014), it is a common practice to transfect the CRISPR guides into a cell line and evaluate their efficacy in vitro or to inject the guides and analyze targeting efficiency using the blastocysts or fetuses at embryonic day 10.5 or beyond. Also, for Indel models preference may be given to a guide sequence that has a better chance of producing a frameshift mutation based on analysis of microhomology directed repair (Bae et al., 2014).
Strain Related Considerations
If the genome sequence of the intended strain is not available, a segment of the genomic DNA sequence from the strain of choice encompassing the target site should be retrieved by PCR and sequence verified before a guide and PAM sequence is chosen to avoid failure due to sequence divergence between the reference strain and the strain of choice. Also one must bear in mind that different strains of mice may behave differently to superovulation, microinjection and surgery (Byers et al., 2006; Yamauchi et al., 2007). As such, protocols may need further refinement when a new strain of mice is used.
Breeding Considerations
Founder mice from a CRISPR-Cas9 experiment often are mosaic, with each founder mouse carrying more than two alleles. Thus, it is a good idea to breed these founder mice first with the wild type mice of the chosen strain to transmit and segregate the alleles among the F1 mice. Under certain circumstances, a breeding scheme could be designed to accomplish two purposes, to transmit the allele and to recruit another mutant allele, in one round of breeding. For example, for a conditional knockout model, the founder mice may be bred with a Cre line to transmit the loxP allele to F1 mice and to recruit the Cre transgene. For a double knockout model, the founder mice may be bred with the second knockout model. In addition, in vitro fertilization may be considered to generate a large number of F1 mice, if male founder mice are available. Of particular notice is that the alleles that appear among the F1 mice must be carefully examined and the desired mutant allele carried forward for further breeding.
Here we describe the protocols that cover the entire process of generating mouse models using the CRISPR-Cas9 system, including synthesis of sgRNA, Cas9 mRNA and donor, preparing the injection mixture for use in microinjection or electroporation to deliver the CRISPR-Cas9 reagents into the mouse zygotes, and genotyping strategies used in a genome editing experiment.
Materials Commonly Share Among All Protocols
SeaKem LE Agarose (Cat. No. 50004, Lonza)
10X TAE buffer – 4 liter (Cat. No. 50841, Lonza)
I Kb Plus DNA Ladder (Cat. No. 10787-018, Life Technologies)
DNA Gel Loading Dye (6X) (Cat. No. R0611, Life Technologies)
RNase AWAY Decontamination Reagent (Cat. No. 10328-011, Life Technologies)
Submerged horizontal electrophoresis cells (various models, Bio-Rad)
Microwave oven
Phusion® High-Fidelity DNA Polymerase (Cat. No. M0530L, New England BioLabs)
PrimeSTAR GXL DNA Polymerase (Cat. No. R050B, Clontech)
QIAquick PCR Purification Kit (Cat. No. 28106, Qiagen)
Thermal-Lok 2-Position Dry Heat Bath (Cat. No: 2510-1102, USA Scientific)
Thermal Cycler with Dual 48/48 Fast Reaction Module (Cat. No.1851148, BioRad)
Thermal Cycler with 96-Well Fast Reaction Module (Cat. No., 1851196, BioRad)
BioDrop μLite spectrophotometer (Isogen Life Science)
Galaxy 20R centrifuge (VWR)
EC 105 Electrophoresis Power Supply (E-C Apparatus Corporation)
Manual Gel Documentation Systems (InGenius3, Syngene)
BASIC PROTOCOL 1: Microinjection Mixture Preparation
Components of the CRISPR-Cas9 system, including the sgRNA, Cas9 mRNA or protein, and donor DNA if it is a HDR knock in experiment, are brought together into a mixture and achieve the desired final concentrations and the mixture injected into the mouse zygotes. It is imperative that the preparation is free of particulate matters and most importantly, RNase contamination.
Materials
TE buffer: 10 mM Tris-HCl, 0.1 mM EDTA, pH 7.5, DNase and RNase free
Cas9 mRNA: 500 ng/μL
sgRNA: 500 ng/μL
donor oligonucleotide: 500 ng/μL
donor plasmid: 500 ng/μL
RNase-free PCR tubes (0.2 mL) (Cat. No. AM12225, Life Technologies)
RNase-free Microfuge Tubes (0.5 mL) (Catalog No. AM12300, Life Technologies)
RNase-free Microfuge Tubes (1.5 mL) (Cat. No. AM12400, Life Technologies)
Protocol steps
-
Add CRISPR-Cas9 components, including Cas9 mRNA, sgRNA and donor oligonucleotide or plasmid, into an RNase free vial, and bring up to the final volume with TE buffer.
Component Stock (ng/μL) Volume (μL) Final (ng/μL) Comments Cas9 mRNA 500 3 100 sgRNA 500 1.5 50 Oligonucleotide 500 (3) 100 Only needed for HDR model Plasmid 500 (0.6) 20 Only needed for HDR model TE variable Added to achieve the 15 μL total volume Total 15 The volumes in parenthesis indicate the amount of donor DNA needed, which is only relevant for making HDR knock-in models. Place the 0.2 mL tube into a 1.5 mL microfuge tube stuffed with a small piece of Kimwipe at the bottom to support the 0.2 mL tube.
Centrifuge for 15 minutes at 20,000 × g to separate supernatant from any possible particulate matters.
Transfer the supernatant to a new vial for use in microinjection: Carefully take 10 μL of the supernatant and split into 5 μL/vial, 2 vials, in 0.2 mL RNase free tube, pre labeled with project number.
Check for integrity of the preparation by taking absorbance at 230 nm, 260 nm and 280 nm on a spectrophotometer of the remaining 5 μL of the mixture to be sure all components are accounted for (indicative free of RNase contamination). Alternatively, perform non-denaturing agarose (Figure 2) or denaturing polyacrylamide gel electrophoresis (not shown) to be sure of integrity of the components.
Place on ice the 2 vials containing the supernatant portion of the preparation and bring to the microinjection laboratory.
You may want to take the preparation when microinjection has been completed and take absorbance as described above or perform agarose gel electrophoresis to be sure of integrity of the preparation throughout the day.
Figure 2.
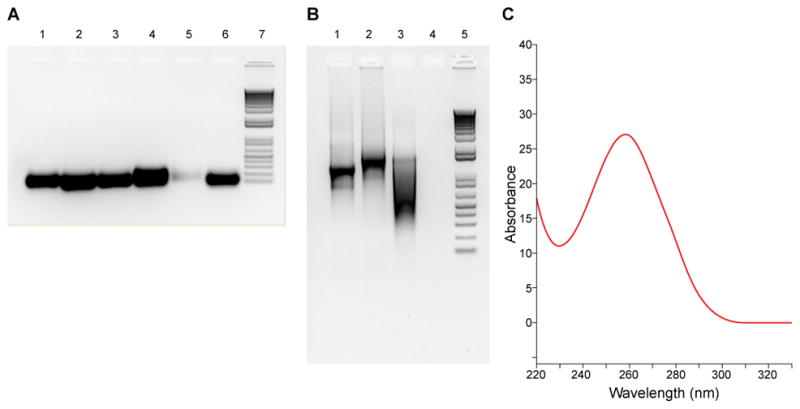
Quality control of the sgRNA and Cas9 mRNA preparations. (A) sgRNA seen on a nondenaturing agarose gel, stained with GelRed (Biotium). Lanes are samples 1 to 6, with sample 5 having a lower yield. Lane 7 is 1 Kb plus DNA ladder (Cat. No. 10787-018, Invitrogen). (B) Cas9 mRNA seen on a nondenaturing agarose gel, stained with GelRed. Lane 1: 2.5 μL withdrawn from the 96 μL in vitro transcription reaction; lane 2: 2.5 μL withdrawn from the 100 μL after poly adenylation was completed; Lane 3, 2.5 μL withdrawn from the 50 μL elution volume after purification with the MEGAclear kit. Lane 4: blank; Lane 5: 1 Kb plus DNA ladder. (C) Cas9 mRNA examined by spectrophotometry. The A260/A280 ratio is 2.3 and A260/A230 ratio 2.45 for this sample. The concentration is 1,125 ng/μL.
SUPPORT PROTOCOL 1: Guide Sequence Selection
For each target site, a guide sequence needs to be chosen and evaluated for targeting efficiency as well as possibility of off target damage, based on current understanding of the parameters that influence these outcomes. There are software programs that take into consideration of these parameters and can be used to assist with selection of a guide sequence. In addition, a guide can also be evaluated by transfection into a cell line or injection into the mouse zygotes and targeting efficiency analyzed (protocol not provided).
Materials
Internet access
Sequence management software (Vector NTI Geneious, ApE-A plasmid editor, etc.)
Protocol steps
Go to NCBI homepage (http://www.ncbi.nlm.nih.gov/) and from the dropdown menu, select “gene”, type the name of the gene and perform “search”.
From the table listing the gene choices that appears, select the gene from the species of interest (for example, Mus musculus).
From the NCBI page for the gene that appears, scroll down to “NCBI Reference Sequences (RefSeq)” and under “Genomic”, select “NC_0000XX” for C57BL/6J or “NT_XXXXXX” for other strains, such as NOD/ShiLtJ.
Click on “GenBank”.
From the GenBank nucleotide page that appears, click on the arrow under “Send” on the upper right hand corner to expand and see the drop down menu, and under “choose destination”, choose “file” and under “format”, choose “GenBank” and click “Create File”.
From the “Opening sequence.gb” window that appears, choose to open the file with VectorNTI format or other format of choice, if you do not have one set up as default already.
In Vector NTI, under “File”, choose “Save As” and save the genomic sequence of the gene of interest in “DNA/RNAs Database”.
Go to Ensembl genome browser (http://www.ensembl.org/index.html) and under “Search”, select “Mouse” and enter the name of the gene.
From the selections that appear, select the gene of interest.
In the table for the gene that appears, under the “Transcript ID” column, select the transcript of choice.
On the left hand column, under “Sequence”, click on “Exons”.
Highlight the sequence for the exon of interest and copy the sequence. You may want to use a program (http://reverse-complement.com/cleanup.html) to clean up the sequence (get rid of spacing, etc.).
Copy the sequence and find this sequence in the gene in the VectorNTI file. Pay special attention to the region intended to be targeted, such as the region downstream from the 5′ ATG start codon, sequence around the 3′ stop codon, or any other region of interest.
Copy about 100 nt including the region to be targeted and paste into the window at CRISPR Design (http://crispr.mit.edu/).
Select the guide overlapping with the site intended for targeting and with the least sequence match elsewhere in the genome.
Alternatively, copy and paste sequence into the Benchling program (https://benchling.com/) and select guide(s) that has the best “efficiency score” and “specificity score”.
For Indel models, select guide(s) with best possibilities of creating a frameshift mutation based on microhomology directed repair analysis (http://www.rgenome.net/mich-calculator/).
You may want to create a composite sequence in VectorNTI format that is the DNA template for in vitro transcription, including the T7promoter sequence, the guide sequence and the rest of the sgRNA sequence. You can then copy and paste the guide sequence into this file every time when you want to synthesize a new guide. This file will provide you with the sequence for the forward primer, including T7 promoter, guide, and a part of the crRNA sequence that overlaps with sequence of the reverse primer, as provided in Table 1.
Table 1. Primers Used in the studies.
Oligonucleotides used in a PCR reaction to generate the DNA templates for in vitro transcription of the Cas9 mRNA and sgRNA
| Gene | Direction | Sequence (5′ to 3′) | Reference |
|---|---|---|---|
| Cas9 | F | TAATACGACTCACTATAGGGAGACCACCATGGACTATAAGGACCACGAC | (Cong et al., 2013) |
| R | GCGAGCTCTAGGAATTCTTAC | ||
| sgRNA, conventional backbone | F1 | gaaattaatacgactcactatag(N20)gttttagagctagaaatagc | (Cong et al., 2013) |
| R1 | aaaagcaccgactcggtgccactttttcaagttgataacggactagccttattttaacttgctatttctagctctaaaac | ||
| sgRNA improved backbone | F2 | taatacgactcactatag(N20)gtttaagagctatgctggaaac | (Chen et al., 2013) |
| R2 | aaaaaagcaccgactcggtgccactttttcaagttgataacggactagccttatttaaacttgctatgctgtttccagcatagctcttaaac |
Support PROTOCOL 2: Single Guide RNA Synthesis
sgRNA is an artificial fusion of two naturally occurring RNA species, the CRISPR RNA (crRNA) and the trans activating crRNA (tracrRNA) (Jinek et al., 2012) and about 124 to 130 nt in size, depending on the sgRNA backbone used (Figure 1). We routinely use the improved sgRNA backbone that has been shown to improve targeting efficiency in cell lines (Chen et al., 2013). A sgRNA can be transcribed from a linearized plasmid or a linear DNA template generated from a PCR reaction. In this protocol, we describe synthesis of the sgRNA from a DNA template generated from a PCR reaction and incorporated the T7 promoter from the forward primer sequence (Table 1).
Materials
sgRNA forward primer (Table 1, ordered as 4 nmole ultramer from IDT)
sgRNA reverse primer (Table 1, ordered as 4 nmole ultramer from IDT)
MEGAshortscript™ T7 Kit (Cat. No. AM1354, Life Technologies)
Protocol steps
Reconstitute the oligonucleotides to 10 μM (10 pmoles/μL) with nuclease free water
-
Set up the PCR reaction as indicated in the table. In this case, the sgRNA_forward and sgRNA_reverse primers are both the template and the primers:
Component 50 μL Final Concentration 1 Nuclease–free water 30.0 2 5X PrimeSTAR GXL buffer 10 1X 3 dNTP Mixture (2.5 mM each) 4 200 μM 4 10 μM sgRNA_forward 2.5 0.5 μM 5 10 μM sgRNA_reverse 2.5 0.5 μM 6 PrimeSTAR GXL DNA Polymerase 1.0 1.25 unit/50 μL reaction total 50 -
Perform PCR using the following cycling conditions:
Initial denaturation: 98°C, 30 seconds
Amplification: (98°C, 10 seconds; 55°C, 15 seconds; 68°C, 30 seconds), 35 cycles
Final extension: 68°C, 5 minutes
Hold: 4°C
Separate an aliquot of the PCR product on a 2% (weight/volume) agarose gel in TAE buffer to assess its yield and to verify that the product is unique and of the expected size. The size of the PCR product is 124 to 130 nt, depending on the choice of the sgRNA backbone sequence.
Purify the T7 promoter/sgRNA PCR product using the QIAQuick PCR purification kit according to manufacturer’s instructions. Reconstitute to 50 ng/μL or higher concentration.
Perform in vitro transcription to produce sgRNA using the MEGAshortscript T7 kit according to manufacturer’s instructions, using the entire 8 μL to accommodate DNA template (400 ng to 1000 ng).
Purify the sgRNA using the MEGAclear kit and elute with elution buffer according to the kit protocol. If the yield is found to be low, use an alternative protocol and purify with phenol: chloroform extraction/alcohol precipitation, or ammonium acetate precipitation as described in the kit manual.
Mix an aliquot of the sgRNA with the Gel Loading Buffer II provided in the MEGAshortscript T7 kit and separate on 2% nondenaturing agarose gel in 1× TAE solution. Stain the gel with GelRed (Cat. No. 41003, Biotium) to assess yield and quality (Figure 2A).
More conveniently, examine yield and quality by taking absorbance at 230 nm, 260 nm and 280 nm, similar to the example provided for a Cas9 mRNA preparation (Figure 2C), paying special attention to 260/280 ratio (a ratio of 2.0 or above is indicative of a clean RNA preparation, free of protein, phenol or other contaminants that absorb at or near 280 nm) and 260/230 ratio (expected 260/230 ratios are commonly in the range of 2.0–2.2, indicative of absence of contaminants that absorb at 230 nm, including EDTA and phenol).
Dilute the sgRNA to 500 ng/μL, aliquot into 5 μL per vial, and store at −80°C until use.
Support Protocol 3: Cas9 mRNA Synthesis
Cas9 mRNA is 4.3 Kb in size and could be synthesized in an in vitro transcription reaction. The T7 promoter is added to the forward primer (Table 1) and the DNA template produced from the pX330 plasmid in a PCR reaction using the forward and reverse primers specified in Table 1. The PCR template will carry the T7 promoter at the 5′ end followed by coding sequence for the SpCas9 gene that has been codon optimized to that from human. All reagents for mRNA synthesis and poly adenylation are included in the kit. Alternatively, Cas9 mRNA and protein can be purchased from various vendors.
Materials
Cas9 forward primer (Table 1, ordered from IDT)
Cas9 reverse primer (Table 1, ordered from IDT)
pX330 plasmid (plasmid #42230, Addgene)
mMESSAGE mMACHINE® T7 ULTRA Transcription Kit (Cat. No. AM1345, Life Technologies)
MEGAclear™ Kit (Cat. No. AM1908, Life Technologies)
Protocol steps
Reconstitute the oligonucleotides to 10 μM (10 pmoles/μL) with nuclease free water
-
Set up the PCR reaction as indicated in the table
Component 50 μL Final Concentration 1 Nuclease–free water 33 2 5X Phusion HF buffer 10 1X 3 10 mM dNTPs 1 200 μM 4 10 μM Cas9_forward 2.5 0.5 μM 5 10 μM Cas9_reverse 2.5 0.5 μM 6 pX330 (50 ng/μL) 0.5 <250 ng 7 Phusion DNA Polymerase 0.5 1.0 units/50 μL total 50 -
Perform PCR using the following cycling conditions:
Initial denaturation: 98°C, 30 seconds
Amplification: (98°C, 15 seconds; 55°C, 30 seconds; 72°C, 4 minutes), 35 cycles
Final extension: 72°C, 5 minutes
Hold: 4°C
Separate an aliquot of the PCR product (2 μL) on a 1% (weight/volume) agarose gel in TAE buffer to assess yield and to verify that the product is unique and of the expected size (4.3 Kb).
Purify the T7 promoter/Cas9 PCR product using the QIAQuick PCR purification kit according to manufacturer’s instructions. Reconstitute to 100 ng/μL or higher concentration. Alternatively, the PCR product can also be cloned into a plasmid and the plasmid linearized and used as the template for in vitro transcription.
Perform in vitro transcription to produce the Cas9 mRNA using the mMESSAGE mMACHINE® T7 ULTRA Transcription Kit according to manufacturer’s instructions, using the entire 6 μL to accommodate DNA template (500 ng to 1000 ng). Save 2.5 μL out of the 96 μL total as the “before pA addition” preparation for analysis by agarose gel electrophoresis later on.
Add poly (A) tail following manufacturer’s instructions and save 2.5 μL as the “after pA addition” preparation for analysis by agarose gel electrophoresis later on.
Purify the Cas9 mRNA using the MEGAclear kit and elute with 50 μL elution buffer according to the kit protocol. Alternatively, purify with phenol: chloroform extraction/isopropanol precipitation, or lithium chloride precipitation as described in the kit manual. Save an aliquot as the “after column” preparation for analysis by agarose gel electrophoresis.
Perform nondenaturing agarose gel electrophoresis of the 3 preparations (before pA, after pA and after column) on 1% agarose (weight/volume) gel in TAE buffer (Figure 2B). Make sure all setups have been treated with RNase Away and are RNase free. Alternatively, denaturing polyacrylamide gel electrophoresis could be used.
Examine the yield and quality of the Cas9 mRNA preparation by taking absorbance at wavelengths of 230 nm, 260 nm and 280 nm (Figure 2C). As described earlier for the sgRNA preparation, pay special attention to 260/280 and 260/230 ratios.
Dilute the Cas9 mRNA to 500 ng/μL in elution buffer.
Aliquot into 5 μL per vial and store at −80°C until use.
Support PROTOCOL 4: Donor Oligonucleotide Synthesis
An oligonucleotide up to 200 nt may be synthesized with a commercial vendor. A donor oligonucleotide is used when the intended mutation is relatively small and in addition to the intended mutation, the left and right homology arms of minimally 30 to 50 nt each could all be accommodated into the oligonucleotide. These include point mutation incorporation and tag insertion (3× FLAG, His, V5, loxP, etc.). The oligonucleotide is designed such that the mutation is centered with homology arms flanking each side. Please notice at 99.5% coupling efficiency, a preparation of a 200 nt oligonucleotide carries only 36.7% and a 124 nt 53.7% full length, wild type product. It may be desirable to use oligonucleotide of shorter lengths whenever possible and without compromising targeting efficiency or one may want to use PAGE or HPLC to enrich for the full length product.
Protocol steps
Design donor oligonucleotide with these guiding principles:
Total length is less than 200 nt, ideally around 125 nt.
The two homology arms flank the intended mutation, with minimally 40 to 50 nt allocated to each arm.
The intended mutation is positioned such that it disrupts (for tag or loxP insertion models) or replaces (for point mutation incorporation models) the PAM or the guide sequence, particularly the guide sequence proximal to the PAM sequence.
If such arrangement of the intended mutation is not possible, consider creating synonymous mutations such that the newly created HDR allele will not be a substrate for CRISPR-Cas9 cleavage. Choose a synonymous mutation that has a comparable codon usage level as that of the wild type codon or the synonymous mutation may work out as a knockdown or a knockout mutation and complicate interpretation of the phenotype associated with the intended mutation (Kimchi-Sarfaty et al., 2007). In addition, make sure that the silent mutation will not create a cryptic splice donor or acceptor sites that lead to production of aberrant splicing product.
However, it is possible to create mutant alleles free of synonymous mutations, if a larger number of mice could be generated to compensate for the possible lower efficiency of recovering the mutant allele, due to CRISPR-Cas9 cleaving the HDR allele.
After finalizing the design, order the oligonucleotide with a vendor, considering using PAGE or HPLC to enrich for the full length, wild type oligonucleotide. Store at 4°C until use.
Support Protocol 5: Donor Plasmid Assembly
When the intended mutation is transgene insertion or replacement of murine genome sequence with its human ortholog, a donor plasmid could be assembled by molecular cloning techniques carrying the exogenous gene flanked by homology arms of a few thousand nt. Different from a targeting vector intended for a conventional gene targeting experiment, the door vector created for an HDR experiment mediated by CRISPR-Cas9 does not require positive/negative selection cassettes. Also, the donor plasmid is used as a circular, supercoiled preparation to minimize random integration into the genome.
Materials
Restriction enzymes (New England Biolabs)
Shrimp alkaline phosphatase (Cat. No. M0371S, New England Biolabs)
T4 DNA ligase (Cat. No. M0202T, New England Biolabs)
LB broth, powder (Lennox) (Cat. No. 22700-025, Life Technologies)
NucleoBond Xtra Midi EF (Cat. No. 740420.50, Clontech)
Protocol steps
-
Design the donor plasmid with these guiding principles
Perform “repeat masker” analysis (http://www.repeatmasker.org/) to understand the sequence around the region to be mutated.
Position the homology arms around the region of interest to maximize inclusion of unique sequences and total combined length of the homology arms is between 5 to 15 Kb.
Take into consideration of the need for long range PCR, restriction fragment length polymorphism analysis and the 5′ and 3′ Southern blot strategies (Figure 5) and incorporate restriction sites, if necessary.
Assemble donor plasmid using molecular cloning techniques, including PCR, gene synthesis, restriction digestion/ligation, Gibson assembly, or recombineering, and clone all components, including the exogenous gene, the left homology arm and the right homology arm into a plasmid (pUC57, etc.).
Perform restriction fragment length polymorphism analysis to identify the correct clones and sequence the selected clones to be sure of the sequences, particularly around the junctions of the fragments.
Produce a midi prep of DNA, free of RNase contamination, using the Clontech NucleoBond Xtra Midi EF kit, according to manufacturer’s instructions.
Elute and adjust to 500 ng/μL.
Figure 5.

Genotyping Strategies. (A) Genotyping by conventional PCR. For Indel models, a pair of PCR primers encompassing the target region is used. For HDR models mediated by a donor oligonucleotide, the pair of primers should be positioned 5′ and 3′ to the region overlapping with the donor oligonucleotide. In addition, mutant specific primers can be designed to amplify only the mutant allele. (B) Genotyping by long range PCR. If a model is created mediated by a donor plasmid, the mice can be genotyped by a pair of primers with the upstream or downstream primer positioned outside of the homology arm and the second primer unique to the transgene sequence. (C) Genotyping by Southern blot. Southern blot can be used to assess successful integration of the transgene into the target genomic locus. In this example, Nco1 digestion releases an 11.5 Kb genomic DNA fragment from the Nanog locus from the wild type mice. When the mCherry transgene has been successfully inserted into the Nanog locus, it brings along an additional Nco1 site, generating a 5.6 Kb genome fragment in mutant mice that could be detected by the 3′ probe located outside of the 3′ homology arm. Southern blot with a probe from the mCherry gene confirms there is only one integration event per genome.
Basic Protocol 2: Microinjection to Deliver the CRISPR-Cas9 Reagents into Mouse Zygotes (Figures 3, 4, and Video 1)
Figure 3. Flow scheme of CRISPR-Cas9 mediated mouse model generation.
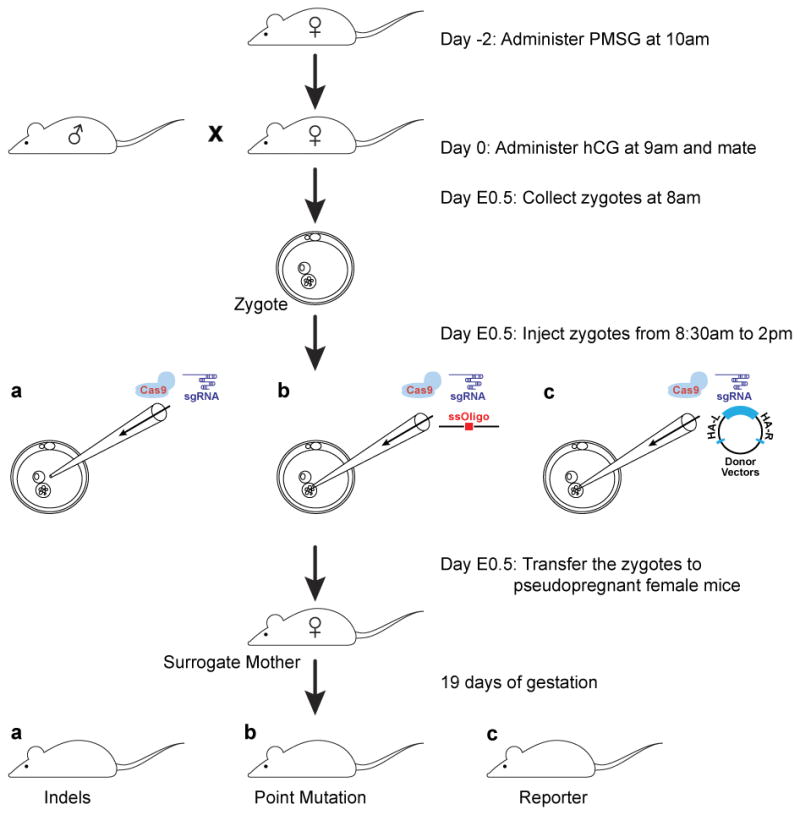
This is the flow scheme for the C57BJ/6J strain. Female donor mice are administered PMSG 2 days prior (5 IU), followed 47 hours later by hCG (5 IU), and mated with a stud male mouse immediately. Embryos are collected in the morning, injected with the CRISPR-Cas9 reagents and transferred the same day. Founder mice could be genotyped when they are 10 to 14 days old.
Figure 4. Microinjection of CRISPR-Cas9 reagents into the mouse zygote.
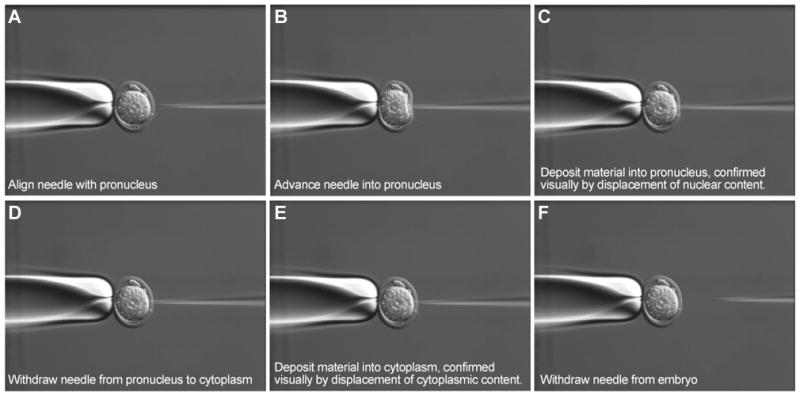
Panels “A” to “F” show in frames how to deposit the CRISPR-Cas9 reagents into both the pronucleus and the cytoplasm.
For an Indel model mediated by NHEJ, the microinjection mixture, containing sgRNA(s) and Cas9 mRNA, is injected into the cytoplasm of the zygote. For a knock in model mediated by HDR, we inject into both the pronuclei and the cytoplasm. The embryos are transferred the same day into pseudopregnant female mice and carried to term. A latent period of a few hours is expected during which the Cas9 mRNA will be translated into the protein nuclease. Also it is possible that the CRISPR-Cas9 reagents may continue to be available, while the embryos divide and the chromosomes duplicate. This may explain the observation that founder mice from a CRISPR-Cas9 experiment are often mosaic, carrying a variety of alleles in each founder.
Materials
Pregnant Mare Serum Gonadotrophin (PMSG) (Cat. No., HOR-272, Prospec, East Brunswick, NJ)
Human chorionic gonadotrophin (hCG) (Cat. No., HOR-250, Prospec, East Brunswick, NJ)
Donor female mice (various strains, the Jackson Laboratory)
Stud male mice (various strains, the Jackson Laboratory)
Microdissecting forceps
Pseudopregnant female mice (CBYB6F1/J, 9–11 weeks)
M2 media (Cat. No. M7167-100ML, Sigma Aldrich)
hyaluronidase (Cat. No. H3506, Sigma-Aldrich)
K-RVCL-50 media (COOK Medical)
mineral oil (Cat. No. M8410, Sigma-Aldrich)
MINC Benchtop Incubator (Cook Medical)
Zeiss AxioObserver.D1 Microscope (Zeiss)
TransferMan NK2 micromanipulator (Eppendorf)
Narashige IM-5A Pneumatic Injector (Tritech Research, Inc.)
Thin Wall Glass Capillaries (Cat. No. TW100F-4, WPI)
P-97 Micropipette Puller (Sutter Instrument Company)
Cryogenic tube with round bottom, 1.8 mL (Cat. No. 363401, Fisher Scientific)
Protocol steps
Each donor female mouse (age determined by strain) receives 5 IU (IP, intraperitoneal) of PMSG followed 47 hours later by 5 IU (IP) of hCG.
Immediately post administration of hCG, each female mouse is mated 1:1 with a stud male mouse.
22 hours later, female mice are examined for the presence of a copulation plug in the vaginal tract.
Female mice displaying a copulation plug are euthanized and the oviducts excised and placed into M2 media.
The oviducts are transferred into M2 media containing hyaluronidase (0.3 mg/mL).
The oocyte clutch is released by puncturing the ampulla with a pair of microdissecting forceps and allowed to incubate in the M2 media containing hyaluronidase until the cumulus cells fall off.
Zygotes are transferred to a dish containing fresh M2 media and graded for fertilization and viability.
The graded zygotes are passed through two washes of fresh M2 media and then placed in microdrops of K-RVCL-50 media that have been equilibrated under mineral oil for 24 hours in a COOK MINC benchtop incubator (37°C, 5% CO2/5% O2/Nitrogen).
Zygotes are removed from culture and placed onto a slide containing 150 μL of fresh M2 media.
Microinjection is performed on a Zeiss AxioObserver.D1 using Eppendorf NK2 micromanipulators in conjunction with Narashige IM-5A injectors. Standard zygote microinjection procedure is followed (Hogan et al., 1986), with special care made to deposit the CRISPR-Cas9 reagents into both the pronucleus and the cytoplasm of the zygote (Figure 4). Needles for microinjection are pulled fresh daily using WPI TW100F-4 thin wall glass capillaries.
Fill the microinjection needle with the CRISPR-Cas9 reagents by placing the back end of the needle into the vial containing the reagents and allow it to fill via capillary action to the tip. When there is visual confirmation that the tip of the needle has filled, remove the needle from the vial and load into the injector (Narashige IM-5A).
To prevent the media on the slide from diluting the reagents inside the needle through the opening at the tip, back pressure is established in the needle before it is moved to the slide. On the slide, the needle is opened further (chipped) by gently flicking it against the holding pipette. Once the needle is opened, pressure is applied to establish flow of the reagents out of the tip of the needle. It is critical that positive pressure be maintained at all times to prevent influx of the media and diluting of the reagents.
To generate NHEJ-mediated knockout models, orientate the zygote so that the needle can enter without piercing the pronucleus. Move the needle along the x-axis to penetrate the zona and the oolemma layers of the zygote, and deposit reagents into cytoplasm. Visually confirm that reagents are being deposited by observing the displacement of the cytoplasm at the tip of the needle. If a bleb forms, the oolemma has not been pierced and the injection is not successful.
To generate HDR-mediated knock in models, orient the zygote to allow clear access to the pronucleus to be injected (Figure 4A). Move the needle along the x-axis to penetrate through the zona and cytoplasm of the zygote, enter and deposit reagents into the pronucleus (Figure 4B, C, D). The pronucleus will swell as reagents are deposited and once that has been confirmed, retract the needle into the cytoplasm and pause briefly to also deposit reagents there (Figure 4E, F). Displacement of the cytoplasm indicates a successful injection (Video 1).
Injected zygotes are removed from the slide, rinsed through three 30 μL drops of equilibrated K-RVCL-50, and then placed into a separate 30 μL microdrop of equilibrated K-RVCL-50..
Zygotes are removed from culture and placed into a 1.8 mL screw-cap cryogenic tube (Cat. No. 363401, Fisher Scientific) preloaded with 900 μL of pre-warmed M2 media for transport to the surgical suite.
The zygotes are removed from the tube and placed into culture (K-RVCL-50 under oil, COOK MINC benchtop incubator, 37°C, 5% CO2/5% O2/Nitrogen).
At the time of transfer, the zygotes are removed from culture and placed into pre-warmed M2 media and transferred via the oviduct into day 0.5 pseudopregnant CBYB6F1/J females (9–11weeks) following standard methodologies (Hogan et al., 1986).
Basic Protocol 3: Genotyping by Conventional PCR and Sequencing of PCR Product (Figures 5 & 6)
Figure 6.

Genotyping CRISPR-Cas9 Mice by Sequencing of PCR Product. (A) NHEJ mutation: Sequencing data from a founder mouse is provided, alongside with sequencing data from a wild type littermate. PAM sequence is boxed in red and guide sequence green. The wild type mouse exhibits clean wild type reference sequence. The founder mouse showed overlapping of 3 sequencing traces starting from the third nt counting from the PAM sequence. Sequence analysis using the “TIDE” program reveals the existence and relative abundance of 3 alleles in the founder mouse, including alleles with 1 nt inserted (52.9%), 4 nt deleted (31.7%) and no change in length (12.7%), when compared to the wild type sequence. (B) Point mutation incorporation. The founder mouse carries several alleles and the correctly targeted allele carrying the intended EcoRV to EcoR1 conversion is confirmed by cloning of the PCR product and sequencing of individual clones. Boxed in red is the PAM sequence and green guide sequence. (C) 3X FLAG Tag insertion. Successful insertion of the 3× FLAG tag was confirmed by sequencing of the cloned PCR product. Boxed in red is the PAM, green guide and blue the 3× FLAG tag sequences. The 3× FLAG insertion site is indicated by a red arrow.
To genotype NHEJ or HDR models mediated by CRISPR-Cas9 using a donor oligonucleotide, conventional PCR and sequencing of the PCR product may suffice. The PCR primers should be positioned to encompass the area to be targeted and should produce a product 500 to 600 nt in size to allow good quality sequencing data for genotype analysis (Figure 5A). If possible, for HDR models, mutant specific PCR primer should be designed to enable identification of founder mice carrying the intended mutation, among other mutant alleles (Figure 5A). This could be very helpful, considering that founder mice generated with the CRISPR-Cas9 nuclease are often mosaic and mutant allele may only be one of those existing in a particular founder mouse.
A tail snip or ear punch sample may be taken when the mice are 14 to 17 days old, processed by the HotSHOT method (Truett et al., 2000) and potential founder mice screened. As discussed earlier, founder mice from a CRISPR-Cas9 experiment are often mosaic, with the founder mouse carrying more than two alleles. Allele composition in a particular founder mouse could be analyzed using the “TIDE” software program that decomposes the alleles (Brinkman et al., 2014) (Figure 6A) or PCR products encompassing the target site cloned and individual clones sequenced.
Materials
96-Well Thermal Cycling Plate (Catalog Number 89049-178, VWR)
MicroAmp Clear Adhesive Film (Catalog Number 4306311, Life Technologies)
VIAFLO II electronic pipettes, 12 channels (12.5 μL, Part No. 4621)
VIAFLO II electronic pipettes, 12 channels (125 μL, Part No. 4622)
VIAFLO II electronic pipettes, 12 channels (1250 μL, Part No. 4624)
PrimeSTAR GXL DNA Polymerase (R050B, Clontech)
Alkaline lysis solution (25 mM NaOH, 0.2 mM EDTA, pH 12)
Neutralization solution (40 mM Tris-HCl, pH 5.0)
HighPrep PCR (AC-60050, MAGBIO, Gaithersburg, MD)
96R Magnet Plate (A001219, Alpaqua, Beverly, MA)
Tris-HCl, pH 8.0
TE buffer (Tris-HCl 10 mM, EDTA 0.1 mM, pH 8.0)
70% ethanol
Protocol steps
Place 2 mm tail tip (or ear punch) sample into a well in a 96-well thermal cycling plate and seal the plate with a plate cover. Tail samples can then be transported from animal facility to a molecular biology laboratory.
Centrifuge briefly to collect the samples to the bottom of the wells and peel off the cover.
Add 50 μL of the alkaline lysis solution (25 mM NaOH, 0.2 mM EDTA, pH 12) into each well, using a multichannel pipette.
Cover the plate with MicroAmp Clear Adhesive Film.
Centrifuge briefly to make sure samples are submerged in lysis solution and there is no air bubble trapped in the solution.
Boil in a PCR machine with heated lid, 98°C for 30 minutes, and cool down to room temperature at the end of the incubation period.
Centrifuge briefly to collect all content and then peel off the cover.
Add 50 μL of the neutralization solution (40 mM Tris-HCl, pH 5.0) and mix well by pipetting up and down a few times and apply a new cover.
Centrifuge briefly to collect content in the well.
Use 1 μL of the supernatant in a 25 μL PCR reaction, avoid touching the bottom of the well and picking up hair and the tissue chunks that had not been dissolved.
Store the remaining tissue lysate at 4°C for a few months or for longer term storage, at −20°C.
-
Set up PCR reaction as recommended by the manufacturer:
Components 25 μL Reaction (μL) Final Concentration 1 5× PrimeSTAR GXL Buffer 5.0 1X 2 dNTP Mixture (2.5 mM each) 2.0 200 μM each 3 Forward Primer (10 pmoles/μL) 0.5 0.2 pmoles/μL 4 Reverse Primer (10 pmoles/μL) 0.5 0.2 pmoles/μL 5 Template (crude lysate) 1.0 6 PrimeSTAR GXL DNA Polymerase 0.5 0.625 U/50 μL 7 Sterile Distilled Water 15.5 Total 25.0 Cycle 30 times at 98°C for 10 seconds; 55°C for 15 seconds; 68°C for 1 minutes/Kb
Add 5 μL of the 6X DNA loading dye to the 25 μL PCR reaction, mix well by pipetting up and down several times, spin briefly in a centrifuge to collect content, and separate 5 μL on 1% (weight/volume) agarose gel to assess yield and uniqueness of the PCR product. If multiple PCR products are observed on the gel, you may want to increase the annealing temperature or optimize other parameters until a clean and robust PCR product is produced.
Mix the HighPrep PCR reagent (magnetic beads) well and add 45 μL of the reagent to the 25 μL PCR reaction in the 96 well Sample Plate.
Mix well by pipetting up and down 6 to 8 times, centrifuge briefly and incubate at room temperature for 5 minutes (PCR product should now bind to the magnetic beads).
Place the Sample Plate on the Magnetic Separation Plate for 2 to 3 minutes until the solution clears (beads will pull to the side of the well).
Remove and discard the supernatant by pipetting (be careful not to disturb the beads).
Wash minimally twice by adding 100 μL of 70% ethanol to each well, incubate for 30 seconds at room temperature, and remove and discard the supernatant by pipetting.
Air dry the samples for about 5 minutes until all traces of ethanol have evaporated.
Remove the Sample Plate from the Magnetic Separation Plate and add 40 μL of elution buffer (water, Tris-HCl, pH 8.0 or TE buffer).
Mix thoroughly by pipetting up and down a few times, incubate for about 2 minutes, and place back on the Magnetic Separation Plate to clear the content.
Use 5 μL of PCR product and 1 μL of the sequencing primer (5 pmoles/μL) for a sequencing reaction.
Download sequencing data and use any of the sequence analysis software (Sequencher, Vector NTI, etc.) for analysis, comparing to sequencing data from a wild type mouse (Figure 6A).
As discussed earlier, founder mice from a CRISPR-Cas9 experiment are often mosaic. The number and nature of the alleles each founder carries can be determined by a software program (tide.nki.nl) (Figure 6A) or by cloning of the PCR products and sequencing of individual clones.
Mice carrying successful point mutation incorporation and tag insertion can also be genotyped by conventional PCR and sequencing of the PCR product (Figure 6B & C).
Support Protocol 6: Long Range PCR Analysis
When a donor plasmid is used in a CRISPR-Cas9 experiment, it is necessary to differentiate the HDR allele from random integration of the donor plasmid into the genome. This could be accomplished by long range PCR or Southern blot analysis. Long range PCR experiment could be designed such that one primer is located in the genomic region beyond the region overlapping with the homology arms of the donor plasmid and the second primer unique to the intended mutation (Figure 5B). In this arrangement, a successful PCR product will only be produced when the intended mutation has been incorporated by HDR, as compared to having been integrated randomly into the genome.
Materials
Gentra Puregene Mouse Tail Kit (Cat. No. 158267, Qiagen)
PrimeSTAR GXL DNA Polymerase (R050B, Clontech)
QIAquick PCR Purification Kit (Cat. No. 28106, Qiagen)
70% ethanol
10 mM Tris-HCl pH 8.0
Steps
Extract genomic DNA from mouse biopsy samples using the Gentra Puregene Mouse Tail Kit, following manufacturer’s instructions.
Perform long range PCR as in “Conventional PCR Analysis”, with extension time adjusted to allow amplification of longer PCR product (with each additional 1 Kb of sequences, add 1 minute to the extension time).
Purify PCR product using the QIAquick PCR purification kit, following manufacturer’s instructions (adequate for recovery of PCR products 100 nt to 10,000 nt in size).
Depending on the genotyping strategies designed for the model, perform restriction fragment length polymorphism (RFLP) analysis, or sequence the PCR product to determine whether intended mutation has been incorporated.
Founder mice from a CRISPR-Cas9 experiment are often mosaic. It may be necessary to clone the product products and sequence individual clones to determine whether the intended mutation has been incorporated.
Support protocol 7: Southern blot analysis (Southern, 2006)
Southern blot is a well-established protocol in molecular biology. The major advantage over long range PCR analysis is that it provides unequivocal data regarding integrity of the targeted allele. If the targeting event had occurred as predicted, a well thought out Southern blot design would produce a banding pattern that support the notion that the targeted allele is correct and does not involve aberrant recombinant product at the intended site. Southern blot analysis should always be performed for HDR allele mediated by CRISPR-Cas9 with a donor plasmid.
Here are some basic considerations (see Figure 5C for an example):
Construct DNA sequences for the wild type and the targeted alleles in silico.
-
Perform restriction analysis to identify a restriction site that would satisfy these criteria:
It is not a substrate for CpG methylation, or if it is, the cognate restriction enzyme is not sensitive to CpG methylation. Some examples are BamH1 (GGATCC), BglII (AGATCT), HindIII (AAGCTT), KpnI (GGTACC), Mfe1 (CAATTG), Nco1 (CCATGG), Spe1 (ACTAGT), and Xba1 (TCTAGA).
In the wild type allele, a pair of restriction sites should exist, with one restriction site positioned outside of the homology arm, extending into the genomic locus. The second restriction site is best positioned also outside of the other homology arm. Use two different restriction sites, if a pair of the same restriction sites is not available.
For the targeted allele, a convenient design is for the exogenous gene to bring in this same restriction site that, combining with the restriction site outside of the homology arm, produces a restriction fragment that differs in size from that of the wild type allele. Alternatively, if the inserted transgene is large enough, the use of the same two naturally occurring restriction sites outside of the homology arms will produce a mutant fragment that is larger than the wild type sequence and the two bands can be resolved on the blot.
The fragment encompassed by the two restriction sites should be less than 20 Kb in size to allow efficient transfer of the DNA fragment from the agarose gel to the membrane during the blotting process.
There should be enough sequence in between the end of the homology arm and the restriction site outside of the homology arm to allow its use as the probe for Southern blot. A typical probe for a Southern blot is from 500 nt to 800 nt in size and unique in sequence. Uniqueness of this sequence could be verified using a software program, such as RepeatMasker analysis (http://www.repeatmasker.org/).
The DNA fragment released by this restriction enzyme between the wild type allele and mutant allele would differ in size such that they will be separated during agarose gel electrophoresis and subsequently, differentiated on the blot. If the two bands are close in size, they may be separated by prolonged electrophoresis.
Follow standard procedures for Southern blot.
Southern blot analysis performed for founder mice will identify the targeted allele if this allele is a major allele in the founder mice. However, when the targeted allele exists as a minor allele, Southern blot may not be adequate to discern this allele on the blot. In this case, long range PCR product could be produced, cloned and individual clones sequenced to identity this allele. In addition, a mutant specific PCR strategy could be designed to enable identification of the mutant allele among other alleles.
Basic Protocol 4: CRISPR-Cas9 Delivery by Electroporation (Figure 7 and Video 2)
Figure 7.
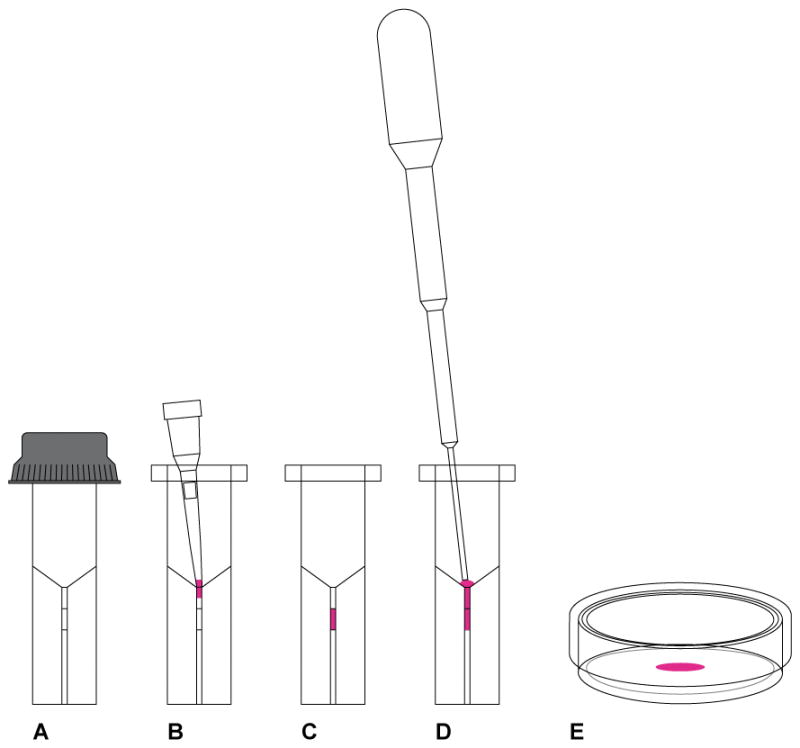
CRISPR-Cas9 delivery by electroporation. (A) We use the ECM 830 Square Wave Electroporation System from Harvard Apparatus and the electroporation cuvette from the same vendor (Cat. No. 45-0124) with 1 mm gap between the electrodes. The minimum volume is 20 μL and maximum 90 μL. (B) The 20 μL of embryos suspended in the CRISPR-Cas9 reagents is loaded into a 20 μL pipette tip and ready to be released into the chamber of the cuvette. (C) The 20 μL of embryos suspended in the CRISPR-Cas9 reagents is released into the chamber and spread evenly at the bottom of the chamber by gentle tapping. The cuvette is then transferred to the electroporator and pulses delivered. (D) Use the sterile plastic transfer pipette that comes with the cuvette to pick up 100 μL of prewarmed and pre equilibrated KSOMaa Evolve/BSA (1 mg/mL) media and release the content into the bottom of the cuvette. (E) Pipet a few times to release the embryos that may have adhered to the wall of the chamber and transfer to a 35 mm Petri dish and get the embryos ready for transfer into pseudopregnant female mice.
Although CRISPR-Cas9 reagents have traditionally been injected directly into the zygotes, several groups, including ours, showed that efficient genome editing can be achieved by using electroporation to deliver the reagents (Hashimoto and Takemoto, 2015; Kaneko et al., 2014; Qin et al., 2015).
Materials and Equipment
ECM830 Square Wave Electroporation System (UX-02894-18, Harvard Apparatus)
Electroporation Cuvette Plus, 1 mm Gap (Part Number 45-0124, Harvard Apparatus)
EmbryoMax M2 Medium (MR-015-D, EMD Millipore)
Acidic Tyrode’s Solution (T1788, Sigma-Aldrich)
Opti-MEM media (P/N 31985, Gibco)
KSOMaa Evolve (ZEKS-050, Zenith Biotech)
Bovine Serum Albumin (A2153, Sigma-Aldrich, use 1 mg/mL)
20 uL TipOne beveled sterile filter tip (Item No. 1120-1810, USA Scientific)
35 × 10 mm Petri dish (Catalogue No. 351008, Corning)
Steps (Figure 7 and Video 2)
Prepare the CRISPR-Cas9 reagents at 2× concentration (Cas9 mRNA, 1,200 ng/μL, sgRNA, 600 ng/μL, donor oligonucleotide, 2,000 ng/μL, reconstituted in TE buffer, pH 7.5), in a volume of 10 μL, in a DNase and RNase free vial, prepared fresh on the day of experiment.
Harvest the embryos as described earlier in Basic Protocol 2.
Remove the embryos from K-RVCL media and wash in pre-warmed M2 media (EmbryoMax, Millipore).
In groups of 50 to 150, depending on skill level of personnel handling the embryos, place the embryos in the Acidic Tyrode’s solution (T1788, Sigma-Aldrich) for 10 seconds.
Remove the embryos and wash through three 100 μL drops of pre-warmed M2 media.
Pick up the embryos with a pipette and immediately before electroporation, place into a 10 μL drop of Opti-MEM media (P/N 31985, Gibco) that has been pre-warmed and equilibrated.
Add the 10 μL of CRISPR-Cas9 reagents, constituting of the Cas9 mRNA, sgRNA, and donor oligonucleotide (if relevant) reconstituted in TE buffer (pH 7.5), to the embryos in Opti-MEM media and pipet gently up and down a few times to disperse the embryos into CRISPR-Cas9 reagents.
Pick up the 20 μL of embryos suspended in CRISPR-Cas9 reagents with a 20 μL pipet tip and deposit the content into the chamber of the 1 mm electroporation cuvette (P/N 45-0124, Harvard Apparatus), making sure the pipet tip is securely positioned between electrodes of the cuvette (Figure 7B).
Cap the cuvette and tap gently to collect all content to the bottom of the chamber, eliminating any bubbles and spreading the content evenly on the bottom of the chamber (Figure 7C).
Electroporate using the ECM830 Square Wave Electroporation System (BTX Harvard Apparatus) with the condition of 30 V, 1 ms pulse duration and two pulses separated by 100 ms pulse interval.
Following delivery of the pulses, with the sterile plastic transfer pipette that comes with the cuvette, pick up 100 μL of prewarmed and pre equilibrated KSOMaa Evolve/BSA (1 mg/mL) media and release into the bottom of the cuvette (Figure 7D).
Remove the embryos from the cuvette and place into a 35 × 10 mm Petri dish (Figure 7E).
Rinse the cuvette with additional 100 μL of KOSMaa Evolve/BSA (1 mg/uL) and add to the embryos in 35 × 10 mm Petri dish.
Transfer the embryos into CByB6F1/J pseudo pregnant female mice following standard embryo transfer protocol.
REAGENTS AND SOLUTIONS
-
Alkaline Lysis solution (25 mM NaOH, 0.2 mM disodium EDTA, pH 12)
Stock Concentration 1000 mL Total Volume Final Concentration 10 N NaOH 2.5 mL 25 mM NaOH 0.5 M disodium EDTA 0.4 mL 0.2 mM disodium EDTA water 997.1 Neutralization solution (40 mM Tris-HCl, pH 5.0)
Dissolve 6.3 g of Tris HCL in 1000 mL of water
COMMENTARY
Background Information
Although mouse models carrying targeted mutations have been created in the last 30 years using the gene targeting technology (Capecchi, 2005), they are limited to those strains that have available a germline competent ES cell line and in addition, prone to technical challenges inherent to the technology. The nuclease technologies, including ZFN, TALEN and CRISPR-Cas9, have recently been invented and found to be highly efficient such that they can be injected directly into the mouse zygotes and mutant mice derived among a limited number of mice born. We first reported the successful generation of mutant mice using a Piezo drill based injection method to deliver the CRISPR-Cas9 reagents into the mouse zygotes (Wang et al., 2013; Yang et al., 2013). In this protocol, we focus on pronuclear microinjection, which is a more widely used method, and present electroporation as an alternative effective method to deliver the CRISPR-Cas9 system.
Critical Parameters
Quality of the RNA Preparations
Central to the CRISPR-Cas9 protocols is the use of Cas9 mRNA and sgRNA, which, although straightforward to synthesize, are prone to degradation by RNase which is ubiquitous in a laboratory environment. Special care must be exercised to minimize the opportunities introducing the RNase into the process while handling the samples during synthesis and injection. All counter tops, glasswares and plastic wares must be cleansed with solution formulated to eliminate RNase activities (for example, RNase Away™ from Life Technologies) and tips and vials certified to be RNase free. RNA samples should be analyzed by spectrophotometry or agarose gel electrophoresis to assess integrity of the samples (Figure 2).
Guide Efficiency
Although parameters impacting efficiency of the CRISPR-Cas9 system are starting to be understood (Doench et al., 2014), it remains a challenge to predict in vivo efficacy of a given guide. It is imperative to use more than one guide per experiment if possible, to maximize the chance for a successful experiment. In addition, guides could be screened by transfecting into a cell line or injecting into mouse embryos to assess targeting efficiency. For a knock in experiment, depending on the site to be mutated, there may be a limited number of guide choices for the spCas9. In this case, one may want to look into the Cas9 with altered PAM specificities (Kleinstiver et al., 2015) which may provide additional genome coverage and flexibility.
Inbred Strains of Mice
Inbred strains of mice may respond differently to superovulation. To work with a particular inbred strain of mice, the protocol for superovulation may need to be adjusted accordingly, For example, in response to the same hormone regimen (5.0 IU PMSG and 5.0 IU of hCG), while the 129S1/SvlmJ produced 39.5 normal oocytes per female donor, the C57BL/6J 25 oocytes and the A/J only 5.4 (Byers et al., 2006). In addition, zygotes from inbred strains of mice may respond differently to the trauma of microinjection (Yamauchi et al., 2007) and electroporation. When dealing with unconventional or difficult inbred strains of mice, it may be a good idea to incorporate control experiments whenever possible to monitor the entire process and to identify the cause of any problem that may be encountered.
Genotyping
Although some founders may be homozygous or heterozygous for the intended mutation and can be readily identified, others may carry the intended mutation, along with other mutant alleles. As such, it may take effort to uncover the mutant allele. This could be accomplished by mutant specific PCR strategies and cloning of the PCR products and sequencing of the individual clones.
Troubleshooting
| Problem | Possible Cause | Solution |
|---|---|---|
| Cas9 mRNA degraded | RNase might have been introduced into the system. | One must be mindful that RNase is ubiquitous in a laboratory environment and exercise vigilance by decontaminating countertop and all glassware and plastic wares that are being used to support Cas9 mRNA synthesis and use RNase free tips and vials. Also avoid talking while working with RNA samples. |
| No or low recovery of the sgRNA from column purification | The MEGAclear Kit (Part Number AM1908, Life Technologies) is a glass filter-based system for purification of single stranded RNA transcript and suitable for RNA transcripts greater than 100 nt. The sgRNA in a CRISPR/Cas9 experiment is often 120 nt in size and may not be adequately retained in the column. | Consider using phenol:chloroform extraction and alcohol precipitation to purify the sgRNA preparation instead. Or use alcohol precipitation directly. |
| Microinjection mixture clogging the needle | This may be caused by particulate matter carryovers from column purification or a high concentration of the donor DNA. | It can be resolved by centrifuging the microinjection mixture at 20,000 × g for 15 minutes and using the supernatant for microinjection. |
| No or low live birth for manipulated embryos | Embryos compromised in the process | Whenever available, transfer unmanipulated embryos as a control to determine whether media, reagents or environmental conditions (excessive noise, construction, etc.) may be the problem. |
| No targeting | The guide may not have worked | Choose another guide or carry more than one guide in parallel if possible to maximize the chance of successful delivery of the model at the end of the process. Also it may help to evaluate the guides in vitro in a cell line or injecting into mouse embryos and screen the guides before choosing a guide for a full-scale experiment. |
| Poor recovery of the embryos from the electroporation cuvette | The embryos may have adhered to the sides of the cuvette | Be sure to use the plastic transfer pipette that comes with the cuvette to transfer embryos. Also, the embryos can be recovered by an additional round of rinsing of the cuvette. |
Anticipated Results
NHEJ Knockout Models: We have been successful delivering founder mice carrying NHEJ mutations for the more than 60 models that we have screened, if we use minimally two guides per model. The average number of mice screened per guide is 15 and average founder efficiency 33% among 1,291 mice that we have genotyped (Table 2). It is understood that this efficiency rate may reflect the biology of the CRISPR-Cas9 system and in addition, our learning curve working with the CRISP-Cas9 system, as these are among the first of our CRISPR-Cas9 experience.
HDR Knock in Models Mediated by a Donor Oligonucleotide or a Donor Plasmid: We successfully delivered founder mice for 37 models out of the 42 models that we have genotyped. Founder efficiency differs widely among the models and strains but could be over 50%. Also mice carrying homozygous HDR alleles have been identified among the founder mice from certain projects.
Table 2.
NHEJ Founder Efficiency Among the Major Inbred Strains of Mice
| Strain | Projects | Guides Worked | Total Guides | Founder Mice | Total Mice | Founder Efficiency (%) |
|---|---|---|---|---|---|---|
| C57BL/6J | 6 | 9 | 10 | 94 | 214 | 44 |
| C57BL/6NJ | 2 | 3 | 3 | 8 | 74 | 11 |
| NOD/ShiLtJ | 22 | 38 | 44 | 200 | 643 | 31 |
| NSG™ | 10 | 15 | 18 | 91 | 291 | 31 |
| NRG | 1 | 2 | 2 | 16 | 29 | 55 |
| DBA/2J | 5 | 3 | 5 | 3 | 22 | 14 |
| BXSB/MpJ | 1 | 1 | 1 | 17 | 18 | 94 |
| Total | 47 | 71 | 83 | 429 | 1291 | 33 |
Time Considerations
NHEJ Knockout and HDR Knock in Models Mediated by a Donor Oligonucleotide: It is possible to derive founder mice within 2 months from concept to delivery of the founder mice, including 1–2 weeks to get reagents ready (donor oligonucleotide and sgRNA), 3 weeks for mice to be born, 3 weeks for mice to age and be genotyped.
HDR Knock in Models Mediated by a Donor Plasmid: Timeline depends on successful synthesis or assembly of a donor plasmid. It is possible to derive founder mice within 2 months, when all reagents (Cas9 mRNA, sgRNA and a donor plasmid) are available.
Supplementary Material
Video1 Microinjection of the CRISPR-Cas9 reagents into both the pronucleus and the cytoplasm.
Video 2 Electroporation of the CRISPR-Cas9 reagents into the mouse zygotes
Acknowledgments
We would like to thank the Molecular Biology, Cell Biology, and Microinjection laboratories of the Jackson Laboratory for their contribution exploring the CRISPR-Cas9 technology and Jesse Hammer for the excellent artwork. Research reported in this publication was partially supported by the National Cancer Institute under award number P30CA034196. H.W. is supported by National Natural Science Foundation of China (31471215), Strategic Priority Research Program of the Chinese Academy of Sciences (XDA01010409), and the State 863 project 2015AA020307.
Footnotes
INTERNET RESOURCES (optional)
To download genomic sequence for a gene, go to: http://www.ncbi.nlm.nih.gov/
For exon/intron structure and sequence analysis, go to: http://www.ensembl.org/index.html
For post translational processing and protein functional domains, go to: http://www.uniprot.org/
For off target sequence match analysis go to: http://crispr.mit.edu/
For on target and off target efficiency scores, go to : https://benchling.com/
For micro-homology mediated repair, go to: http://www.rgenome.net/mich-calculator/)
For repeatmasking, go to: http://www.repeatmasker.org/
For sequence divergence among mouse strains, go to: https://www.sanger.ac.uk/sanger/Mouse_SnpViewer/rel-1303
References
- Bae S, Kweon J, Kim HS, Kim JS. Microhomology-based choice of Cas9 nuclease target sites. Nature methods. 2014;11:705–706. doi: 10.1038/nmeth.3015. [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, van Steensel B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014;42:e168. doi: 10.1093/nar/gku936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers SL, Payson SJ, Taft RA. Performance of ten inbred mouse strains following assisted reproductive technologies (ARTs) Theriogenology. 2006;65:1716–1726. doi: 10.1016/j.theriogenology.2005.09.016. [DOI] [PubMed] [Google Scholar]
- Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet. 2005;6:507–512. doi: 10.1038/nrg1619. [DOI] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, Huang B. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nat Biotechnol. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends in biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hai T, Teng F, Guo R, Li W, Zhou Q. One-step generation of knockout pigs by zygote injection of CRISPR/Cas system. Cell research. 2014;24:372–375. doi: 10.1038/cr.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Takemoto T. Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing. Scientific reports. 2015;5:11315. doi: 10.1038/srep11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Costantini F, Lacy E. Manipulating the mouse embryo : a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, N.Y: 1986. [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, Cradick TJ, Marraffini LA, Bao G, Zhang F. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, Reyon D, Maeder ML, Tsai SQ, Sander JD, Peterson RT, Yeh JR, Joung JK. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Sakuma T, Yamamoto T, Mashimo T. Simple knockout by electroporation of engineered endonucleases into intact rat embryos. Scientific reports. 2014;4:6382. doi: 10.1038/srep06382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimchi-Sarfaty C, Oh JM, Kim IW, Sauna ZE, Calcagno AM, Ambudkar SV, Gottesman MM. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;315:525–528. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- Kleinstiver BP, Prew MS, Tsai SQ, Topkar VV, Nguyen NT, Zheng Z, Gonzales AP, Li Z, Peterson RT, Yeh JJ, Aryee MJ, Joung JK. Engineered CRISPR-Cas9 nucleases with altered PAM specificities. Nature. 2015 doi: 10.1038/nature14592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol. 2013;31:684–686. doi: 10.1038/nbt.2652. [DOI] [PubMed] [Google Scholar]
- Niu Y, Shen B, Cui Y, Chen Y, Wang J, Wang L, Kang Y, Zhao X, Si W, Li W, Xiang AP, Zhou J, Guo X, Bi Y, Si C, Hu B, Dong G, Wang H, Zhou Z, Li T, Tan T, Pu X, Wang F, Ji S, Zhou Q, Huang X, Ji W, Sha J. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell. 2014;156:836–843. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- Qin W, Dion SL, Kutny PM, Zhang Y, Cheng A, Jillette NL, Malhotra A, Geurts AM, Chen YG, Wang H. Efficient CRISPR/Cas9-Mediated Genome Editing in Mice by Zygote Electroporation of Nuclease. Genetics. 2015 doi: 10.1534/genetics.115.176594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern E. Southern blotting. Nature protocols. 2006;1:518–525. doi: 10.1038/nprot.2006.73. [DOI] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, Warman ML. Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT) BioTechniques. 2000;29:52, 54. doi: 10.2144/00291bm09. [DOI] [PubMed] [Google Scholar]
- Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. Onestep generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153:910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiles MV, Qin W, Cheng AW, Wang H. CRISPR-Cas9-mediated genome editing and guide RNA design. Mammalian genome : official journal of the International Mammalian Genome Society. 2015 doi: 10.1007/s00335-015-9565-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y, Doe B, Ajduk A, Ward MA. Genomic DNA damage in mouse transgenesis. Biology of reproduction. 2007;77:803–812. doi: 10.1095/biolreprod.107.063040. [DOI] [PubMed] [Google Scholar]
- Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154:1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrowicz BP, Sands AT. Knockouts model the 100 best-selling drugs--will they model the next 100? Nature reviews Drug discovery. 2003;2:38–51. doi: 10.1038/nrd987. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video1 Microinjection of the CRISPR-Cas9 reagents into both the pronucleus and the cytoplasm.
Video 2 Electroporation of the CRISPR-Cas9 reagents into the mouse zygotes