

Abstract
Humans are virtually identical in their genetic makeup, yet the small differences in our DNA give rise to tremendous phenotypic diversity across the human population. By contrast, the metagenome of the human microbiome—the total DNA content of microbes inhabiting our bodies—is quite a bit more variable, with only a third of its constituent genes found in a majority of healthy individuals. Understanding this variability in the “healthy microbiome” has thus been a major challenge in microbiome research, dating back at least to the 1960s, continuing through the Human Microbiome Project and beyond. Cataloguing the necessary and sufficient sets of microbiome features that support health, and the normal ranges of these features in healthy populations, is an essential first step to identifying and correcting microbial configurations that are implicated in disease. Toward this goal, several population-scale studies have documented the ranges and diversity of both taxonomic compositions and functional potentials normally observed in the microbiomes of healthy populations, along with possible driving factors such as geography, diet, and lifestyle. Here, we review several definitions of a ‘healthy microbiome’ that have emerged, the current understanding of the ranges of healthy microbial diversity, and gaps such as the characterization of molecular function and the development of ecological therapies to be addressed in the future.
Background
Humans have co-evolved with the trillions of microbes that inhabit our bodies and that create complex, body–habitat-specific, adaptive ecosystems that are finely attuned to relentlessly changing host physiology. Dysbioses in the microbiome have been associated with numerous diseases, including inflammatory bowel disease, multiple sclerosis, diabetes (types 1 and 2), allergies, asthma, autism, and cancer [1–5]. Like the concept of the pathogenicity of a single microbial taxon, dysbiosis of a microbial community can be difficult to define but could be considered as a perturbation that departs from an otherwise balanced ecology [1] to prolong, exacerbate, or induce a detrimental health effect. Thus, finding features that broadly distinguish healthy from unhealthy microbiomes will aid in the diagnosis of microbiome-related diseases and could potentially provide new means to prevent disease onset or to improve prognosis. Many potential features common to healthy microbiomes have been proposed, including prevalent organisms or molecular pathways [6] as well as norms of certain ecological properties, such as diversity or stability [7, 8]. Microbiomes regularly show a large degree of interpersonal diversity even in the absence of disease [7, 9]. This complicates the identification of simple microbial constituents or imbalances that either cause disease or reflect a diseased state. An understanding of the properties of a healthy microbiome, and the many different microbial ecologies that are encountered in the absence of overt disease, is therefore a necessary first step to identifying and correcting microbial configurations that are implicated in disease.
In this review, we use “healthy” to refer to the absence of any overt disease (as defined in [10], unless otherwise specified for particular studies). Most available data describe the gut microbiome and so many of the findings discussed here are from this area, though most principles apply to microbial habitats throughout the body. Early research into the ecology of the microbiome sought to identify a “core” set of microbial taxa universally present in healthy individuals who lack overt disease phenotypes, under the hypothesis that the absence of such microbes would indicate dysbiosis [11]; but studies of ecological diversity among healthy individuals revealed sufficient variation in the taxonomic composition of the microbiome to rapidly render such a hypothesis unlikely [11, 12]. Even shared taxa, from individual species to entire phyla, were found to vary in abundance by more than an order of magnitude among healthy individuals [7, 11]. Characterizing a “healthy” microbiome as an ideal set of specific microbes is therefore no longer a practical definition [2, 6].
An alternative hypothesis is that of a healthy “functional core”: a complement of metabolic and other molecular functions that are performed by the microbiome within a particular habitat but are not necessarily provided by the same organisms in different people [6]. Such a core might need to be present as genetic potential (that is, encoded within DNA metagenomes) much as the human genome must not encode serious deleterious mutations to be healthy or it may need to be expressed and well-regulated within an individual for him/her to remain healthy (that is, it must be encoded by RNA metatranscriptomes or present in the form of protein or small molecule products), or of course a combination thereof. The functional core must, of course, include at least the housekeeping functions necessary for individual microbial life, which must be present genomically and correctly expressed; interestingly, these properties may also include functions specific to microbes’ niches in the human ecosystem. Such functions may include processes that are not carried out by human cells and thus represent a potential basis for symbiotic host–microbial relationships. A healthy microbiome may be characterized further by its behavior over time [2, 8]; intuitively, a health-associated microbiome must have a degree of resilience to external (for example, dietary or pharmaceutical) or internal (for example, age- or stochastic-drift-related) changes. Even if a particular community structure provided all necessary core functions, without this resilience it could not guarantee these functions for long. Thus, the resistance of a microbiome to stress and perturbation and its ability to recover to a healthy functional profile afterwards are among the potential properties that characterize a healthy microbiome [2, 13].
Here, we review the current characterization of the healthy microbiome in terms of the normal microbial residents and their core functions, ecological properties, and temporal dynamics. We conclude by identifying key outstanding questions and research directions in this field and speculate on their solutions and impact. A combination of recent technological advances and activity within the field has driven a surge of interest in the human microbiome in health and disease (Table 1) and thus this review aims to summarize the variety of current perspectives on what may constitute a healthy microbiome.
Table 1.
Diversity of recent microbiome research, which has focused mainly on the gut
| Publications | ||
|---|---|---|
| Terms | All | 2011–2016 |
| Gut | colon | intestinal | 17,546 | 10,707 |
| 0ral | mouth | tongue | tooth | subgingival | supragingival | 4843 | 2089 |
| Urogenital | vaginal | penile | 1477 | 706 |
| Skin | cutaneous | 1372 | 754 |
| Airway | lung | 764 | 524 |
| Placenta | breast milk | 702 | 426 |
| Ocular | eye | 152 | 82 |
Number of results obtained by searching for “(microbiome | microbiota | microflora) (<Terms>)” on PubMed (retrieved 31 March 2016)
Our evolving understanding of the healthy microbiome
Early studies sought to identify the normal set of microbes that colonize healthy people, primarily in the gut, by culture and characterization of physiological properties. Such studies best highlight organisms that grow well in the lab environment, such as Escherichia coli. This bias led to the perception that E. coli is an abundant and prevalent member of the human gut microbiome [14]. The introduction of strictly anaerobic techniques in the 1970s allowed the recovery of more than 300 bacterial species from the gut alone [15]; furthermore, the counting of viable cells within standardized serial dilutions in selective media permitted quantification of these species. A summary of four large studies from this era [12] looking at stool samples from 141 Americans on different diets found that bacteria of the genus Bacteroides and anaerobic cocci were both prevalent and abundant, whereas the genus Clostridium was ubiquitous in lower abundance, though no single species (as then defined) was observed in all subjects. Other prevalent but lower-abundance bacteria included members of the genera Bifidobacterium, Eubacterium, Lactobacillus, and Streptococcus, as well as facultative anaerobes such as Escherichia.
It was already suspected at this time that a large number of human-associated microbial species remained undiscovered, with one study estimating the simultaneous presence of some 400 microbial species in a healthy colon [16, 17]. However, the fastidious requirements of some microbes and the labor-intensive nature of the work required to culture them presented a significant barrier to their discovery [12]. Further, not all microbes can be well-distinguished as species or strains by culturing on selective media alone; for example, the different high-abundance Bacteroides species are particularly difficult to disentangle [12, 17]. In addition, such studies of community composition were even more difficult to extend to non-bacterial microbes, such as viruses and fungi, and were even more impractical for studies of body habitats that are less microbially rich than the gut. New methods were required to study these aspects of the healthy microbiome.
Culture-independent techniques such as DNA sequencing [18] and fluorescence in situ hybridization (FISH) [19] are now widespread and their democratization has allowed the DNA content of microbial samples to be interrogated directly [20]. Early studies using FISH targeting the 16S ribosomal RNA gene suggested that at least two-thirds of the gut bacteria in a western European cohort could be attributed to a set of six groups at approximately the species/genus level: two Bacteroides, two Clostridium, Streptococcus/Lactococcus, and Eubacterium rectale [19]. This has since proved to be optimistic and, even at the time, large variability was observed in the abundances of these groups between samples (standard deviations of ~60–80 % of their means) [19].
Some of the earliest efforts to sequence 16S rRNA genes directly from samples showed that 85–95 % of bacterial abundance corresponding to known species could be attributed to three bacterial groups related to Bacteroides, Clostridium cluster XIVa, and Clostridium cluster IV [21, 22]. 16S studies also showed a large diversity in the taxonomic composition both between healthy people and among closely linked biogeographical sites within a single person (such as mucosal and stool samples [23]). However, in all of these studies, the majority (75–80 %) of sequence clusters did not match any documented species at the time [21–23], explaining much of the underestimation of diversity in previous work.
The advent of massively parallel shotgun sequencing (high-throughput sequencing technologies) has substantially resolved the taxonomic composition of this microbial “dark matter” [24], although a striking percentage of functional diversity remains to be characterized (up to 50 % [25]) as does the composition of non-reference populations (discussed below). Initial findings echoed the large interpersonal differences, even between twins [26], but also implied the existence of a set of microbial genes that are common to all individuals [26, 27]. This helped seed the model that, like conserved housekeeping genes in individual organisms, a “core microbiome” can be defined at the functional rather than at the taxonomic level [26, 27].
Population-scale baseline cohorts
Large-scale projects have since been launched to characterize the diversity of microbial composition and its functional potential, building on the still-increasing throughput and cost-effectiveness of sequencing and other molecular assays. In 2010, the Metagenomes of the Human Intestinal Tract (MetaHIT) study reported gut metagenomes from stool samples from a cohort of 124 European adults (predominantly ‘healthy’), which at the time exceeded the sequencing volume of all previous microbiome studies by almost 200-fold [9]. In 2012, the Human Microbiome Project (HMP) reported the results of 16S profiling on 242 healthy adults from the United States and metagenomic sequencing on a subset of 139 individuals, with samples representing 18 body habitats distributed between five major body areas [7]. A large Chinese study on type 2 diabetes soon contributed an additional 145 gut metagenomes, approximately half of which were from non-diabetic controls [28]. Further, the MetaHIT consortium has since continued to publish new gut metagenomes from European adults [29–31]. Altogether, the number of population-scale healthy microbiomes surveyed in the gut and other body sites now exceeds 2000 individuals spanning multiple continents.
Typical components and diversity of the microbiome
Bacterial components of a healthy microbiome
The ecosystem of the colon has been the most intensively studied body habitat (Table 1) as it boasts a remarkable diversity between people and a microbial biomass (cell count) that eclipses that of other body sites by more than an order of magnitude [32]. In combination with the early rise of 16S rRNA gene sequencing and anaerobic culture techniques, these properties of the gut have led to a particularly strong focus in the literature on bacterial gut microbiome residents. Over 1000 gut bacterial species have now been characterized [15], providing a significant “parts list” of bacterial constituents. Interestingly, molecular phylogenetics has led to the reclassification of many of these species in the past 20 years. Of particular interest, species within Bacteroides, previously considered the most prevalent and abundant bacterial genus in the gut, have been reclassified into five genera: Alistipes, Prevotella, Paraprevotella, Parabacteroides, and Odoribacter, with additional culture-based and culture-independent molecular work ongoing [15]. An estimated 1000–1150 bacterial species were prevalent in the MetaHIT cohort’s gut microbiomes, of which each person carried ~160 species on average [9]. Healthy gut microbiomes as assessed by sequencing are consistently dominated by bacteria of two phyla—Bacteroidetes and Firmicutes [7, 9]—though even when considering this broad level of classification, individuals vary by more than an order of magnitude in their Firmicutes/Bacteroidetes ratios [7]. Prevalent bacteria in feces that have been identified through molecular techniques have broadened the lists above to include bacteria from at least eight families (Fig. 1a).
Fig. 1.
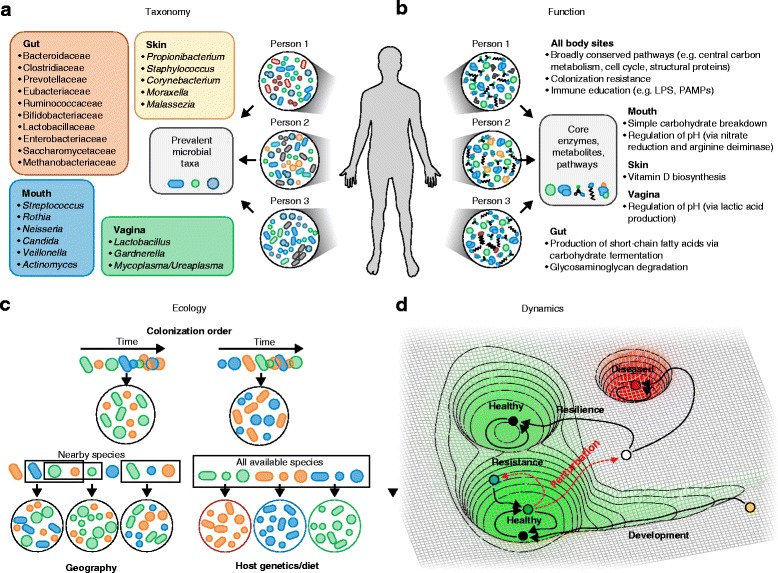
Possible definitions of a healthy microbiome: composition, function, dynamics, and ecology. a Early definitions of a “healthy” microbiome generally focused on sets of taxa that might be expected to be found prevalently in healthy people. While purely taxonomic cores of any type have remained elusive, even in relatively narrowly defined populations, each body-site habitat possesses strong phylogenetic enrichments. Typical genera (or families in the gut) in healthy populations at different sites are shown here [7, 9, 15, 33–35]. b Metagenomic measurements have allowed the functional potential of the microbiome at different sites to be assessed. These studies have yielded more consistently shared functional cores of body-wide and niche-specific pathways that are maintained in health [6, 7, 9, 98]. LPS lipopolysaccharide, PAMP pathogen-associated molecular pattern. c Ecological assembly patterns provide another possible definition of a healthy microbiome, because each host may draw from a “typical” meta-population of potential microbes through a mix of partially stochastic processes. These processes may include the order in which microbes colonize their respective human habitat (affected by geography and early exposures, for example), the prolonged availability of each microbe in the host’s local environment, and host selection (through diet or genetics, adapted from Fig. 1 of [101]). d The healthy microbiome can also be characterized in terms of its dynamics, depicted here in a simplified model as a conceptual energy landscape. The infant microbiome (yellow point) starts out in an unstable state and gradually descends towards one of potentially several healthy adult attractor states. Perturbations (dashed red arrows) can either be resisted (green point) or can move the microbiome out of the healthy state, after which a resilient microbiome will return to a healthy state (not necessarily the original healthy state) or fall into an unhealthy state (red)
Although less well-studied than the gut, many other body habitats within healthy individuals are occupied by microbial communities [7]. Community composition is more similar within than between habitats (for example, oral communities share greater similarity with oral communities in other people than with other habitats within the same person), although, in turn, inter-individual differences within habitats are much greater than intra-individual variability over time [7]. Oral sites harbor particularly diverse microbiomes [33], similar in complexity to the microbiome of the gut [7], and tend to be dominated by Streptococcus spp. [7]. Skin sites differ primarily with the local properties of the skin (dry versus moist versus sebaceous [34]) and are colonized primarily by Corynebacterium, Propionibacterium, and Staphylococcus [34]. The healthy vagina contains one of the most remarkably structured microbial ecosystems, with at least five reproducible community types, or “community state types”, each dominated by a single species of Lactobacillus (L. crispatus, L. iners, L. jensenii, or L. gasseri) or by a mixture of other microbes including Gardnerella [35, 36]. Significant determinants of a woman’s community state type include race/ethnicity [35, 37] and pregnancy [37], although even in this structured ecosystem within-subject longitudinal variation is substantial and, to date, has no fully explained causes.
Several significant body habitats tend to have particularly low microbial biomass in healthy individuals and are thus more difficult to characterize. The lung, for example, is near-sterile in the absence of infection or chronic disease, leading to great interest in identifying its normal residents but also to substantial technical challenges in sampling and sequencing the site [38–40]. Likewise, breast milk [41] and the placenta [42] are of interest for the early establishment of both a healthy microbiome and the potential circulating blood [43] or tissue [44, 45] microbiomes for normal immune control of opportunists. There are considerable difficulties in acquiring metagenomes from such environments and thus most studies have relied on contamination-sensitive amplicon surveys [46] and relatively low-throughput single-cell techniques, such as FISH or microfluidics. Larger-scale carefully controlled studies are thus needed to establish the functionality of these challenging low-density microbial habitats.
Archaea, viruses, fungi, and other eukaryotes
The study of the healthy microbiome has been greatly enriched for bacteria [7, 9], with less attention given to other microbial domains. The human microbiome, though, spans the tree of life and thus includes archaea, viruses, and eukaryotes. A small number of archaeal genera have been identified in the healthy human microbiome, primarily in the gut. Species of the Methanobrevibacter genus are the most prevalent [47] in the gut, with their status as “healthy” members of other body sites’ communities remaining somewhat unclear [48]. Methanobrevibacter smithii in particular has been found to be well-adapted to the human gut, optimizing the digestion of dietary polysaccharides by other microbes [49] and adapting its gene expression in the presence of common gut bacteria such as Bacteroides thetaiotaomicron [49]. The human virome is particularly extensive and, while under-characterized, is recognized as an integral part of the healthy human ecosystem [50]. With the hypervariable nature of viruses, each person is expected to harbor a unique virome [51, 52], consisting primarily of bacteriophages [50] (an estimated 5 % of the gut bacterial gene complement codes for prophage proteins [9]). Phages also provide an additional means of horizontal gene transfer among otherwise distantly related bacteria [53]. As molecular-profiling techniques for archaea, viruses, and eukaryotes are still less well-developed than those for bacteria (even those using culture-independent approaches [47, 54]), information on the molecular functionality of these organisms within in situ communities remains limited.
Although the best-known eukaryotic microorganisms found in or on the human body (principally fungi and protists) are typically pathogens, it is important to remember that many such eukaryotes, in particular Candida, Malassezia, and Saccharomyces, are pervasive even in healthy populations [55–58]. Trans-kingdom interactions are responsible for at least part of the ecological and immune balance of the healthy microbiome; for example, there is apparent competition between bacteria and fungi across skin biochemical environments [59] or in Lactobacillus control of fungi in the gut [55] and vagina [60]. Although few examples exist, direct mutualistic relationships between humans and fungi have been found, of which the best-characterized involves the probiotic yeast Saccharomyces boulardii, originally isolated to combat cholera [61]. Some protozoa are even common inhabitants of healthy microbiomes [58, 62], albeit (like viruses) with even greater interpersonal variability than bacteria [58]. Further, the presence of some protozoa, such as the common Blastocystis, has been associated with reduced risk of gastrointestinal disease [63]. Finally, although multicellular eukaryotes such as helminths have generally been eliminated from gut microbiomes in Western cultures, they have been a component of the gut microbiome for a significant portion of our recent evolutionary history [64]. Given their potent immunomodulatory capabilities and interactions with the other inhabitants of the normal gut microbiome (such as Lactobacilli [65]), their elimination may have removed an important educator of our immune systems [64].
Geographical variation in the healthy microbiome
Studies contrasting the gut microbiomes from different countries have identified systematic differences in microbial composition, although it remains challenging to tease apart inter-batch technical effects from inter-population biology. Comparison between the largest cohorts from three continents—MetaHIT (European), HMP (American), and Chinese diabetes cohorts—found that the inter-country variation in taxonomic composition significantly exceeded inter-personal variation, which was not solely attributable to technical differences in experimental methodologies [29]. Nevertheless, smaller international studies have also identified geography as one of the major sources of large-scale variation in the microbiome, including between North and South America [66], Europe and Africa [67], Korea and Japan [68], and between rural and urban populations of Russia [69] and China [70]. Among possible drivers of this variation, diet has been suggested as an important contributor [67], along with other factors including geography, early-life exposures, and genetics [29, 71]. No one study has yet shown any of these factors to be causal in the large observed inter-population differences in healthy microbiomes [72].
Geographic differences at the strain level are also of interest, particularly as strain signatures exhibit greater temporal stability than do microbial abundance profiles [8, 73, 74]. Research in this area is preliminary but shows that strain differences are not particularly pronounced between countries or continents. Species such as Bacteroides coprocola and Prevotella copri show the greatest differences [73] and strain-level variants in antibiotic resistance genes spanning populations [75]. Strain-level microbial forensics on highly heritable species such as Helicobacter pylori have been remarkably insightful in tracing historical effects on the microbiome [76, 77] and culture-independent techniques should be leveraged for thorough large-scale population surveys in the future.
Microbiome establishment and early colonization
Factors that influence early-life microbiome dynamics are important precipitators of a healthy microbiome. Microbial introduction and persistence is a semi-stochastic process influenced by many elements (Fig. 1c), yielding a healthy adult-like configuration only after the first few years of life [66, 78–80]. Enrichment of the infant gut microbiome for symbionts such as Bacteroides, Parabacteroides, Clostridium, Lactobacillus, Bifidobacterium, and Faecalibacterium prausnitzii provides several determinants of a healthy microbiome. Once established, these are the main producers of short chain fatty acids (SCFAs), an important source of energy from non-digestible carbohydrates [81]. SCFAs are immunomodulatory [82], inhibit common pathogens, and are hypothesized to possess tumor-suppressive properties [83, 84]. The gut microbiome is an inextricable requirement for immune system education and the establishment of these beneficial genera early in life promotes immune tolerance and can consequently attenuate or abrogate autoimmune diseases [1, 85–89].
Delivery mode can affect early-life establishment of microbiota such that Caesarean section is associated with enrichment for opportunists, including Haemophilus spp., Enterobacter cancerogenus/E. hormaechei, Veillonella dispar/V. parvula [78], and Staphylococcus [80]. These microbes continue to persist at least throughout the first year of life [78] and possibly contribute to infant infection burden. Diet also represents a strong selective pressure on the microbiome [71, 90] and breast-feeding (as the first diet) favors certain microbial clades from among the initial microbiota that may have assembled at random. For example, human milk oligosaccharides (HMO) can be used as the sole carbon source by only a handful of Bifidobacterium and Bacteroides species [91] and, more so, bovine milk oligosaccharides (BMO) were recently shown to promote growth and metabolism in a microbiota-dependent manner in animal infant models [92]. While this model may not translate directly to human infants because of the unique structural diversity, complexity, and high concentration of HMO [93, 94], it lends further support to the inference that the long-term benefits of breast-feeding [95] are mediated, in part, by the microbiome.
Hallmarks of health
Functional core
While large interpersonal differences are observed in the taxonomic composition of the microbiome at all sites, the abundance of metabolic pathways is considerably more consistent across people for a given site [7, 9, 26, 27]. Further, while the composition of the microbiome changes drastically over the first years of life, this functional profile is established early on and remains stable thereafter, at least in the gut [72]. This suggests that one definition of a “core” healthy microbiome might include specific microbial gene family combinations, metabolic modules, and regulatory pathways that together promote a stable host-associated ecology [96, 97]. This core includes functions from at least three groups: first, and most simply, the housekeeping functions necessary for all microbial life, such as transcription and translation, energy production, and structural components [6, 7, 9]. Second, this core includes processes that are specific to human-associated microbiomes across body-site habitats, such as adhesion to host cell surfaces and the production of compounds implicated in host–microbe interaction (including essential vitamins, such as vitamin K, and immunostimulatory compounds) [6, 7]. Finally, different body habitats each have their own specialized core functions [98]. For example, in the gut, core functions include glycosaminoglycan biodegradation, the production of several short-chain fatty acids, enrichment for specific lipopolysaccharides, and the production of vitamins and essential amino acids [6, 9, 98, 99] (Fig. 1b). Which of these functions tend to be enriched in a given population can be affected by long-term selective pressures such as diet [67]. A necessary condition for a healthy microbiome is therefore the presence of an assemblage of microbial species that can carry out specific sets of biomolecular functions in each of the niche-specific biochemical environments across the body.
Healthy community ecology
If microbial communities assemble on the basis of their coverage of a core set of functions while selecting from a large meta-population of potential colonizers, they are likely to be ecologically diverse [100–102], both in terms of richness (number of taxa present) and evenness (abundance of many microbial constituents). High diversity has been generally associated with health [11] and temporal stability [103]. The latter could, for example, be the result of the increased functional redundancy that comes with a more diverse set of microbes, even if the functional potential of the assembly is minimally achievable with fewer taxa. Conversely, a relative lack of diversity is apparent in the gut microbiome in diseases ranging from obesity [26] to inflammatory bowel disease [104] and types 1 [72] and 2 [28] diabetes; and in the skin microbiome in atopic dermatitis [105] and psoriasis [106]. Antibiotics also cause a drastic reduction in the diversity of the microbiome with highly variable recovery dynamics [107], potentially weakening the community’s ability to exclude pathogens. This may clear the way for infection by pathobionts—normal microbial community members that become detrimental under perturbation, such as Candida albicans [57]. The principle that high diversity is “healthy” does not hold for all body sites, however, as diversity in the vaginal microbiome can be associated with bacterial vaginosis [108], cervical intraepithelial neoplasia [109] (an abnormal growth on the cervix), pre-term birth [36], and inflammation [110].
Given the typical observation of increased microbiome diversity in health, it has been hypothesized [111] that developed countries’ consistently reduced gut microbial diversities may account for higher chronic disease rates relative to those seen in developing countries and primitive societies [66, 112, 113], termed the “disappearing microbiome hypothesis” [111]. This loss of diversity may be linked to a high-fat, high-refined-sugar, and low-fiber diet [114]. Humanized mice on such a diet exhibit a depletion in microbial diversity [114] and though this is recoverable by returning to a high-fiber diet within a generation, it becomes fixed after four generations [114]. If this result generalizes to human populations, it increases the urgency of developing rationally targeted microbiome maintenance or therapeutic methods, so as to steer less health-promoting microbiomes towards more natural assemblages. The disappearing microbiome hypothesis in some ways represents an evolution of the “hygiene” or “old friends” hypotheses [115], all of which suggest that while modern North American or European cohorts may represent “healthy” microbiomes, their relationship to what is evolutionarily “normal” may be more complex.
Resistance, resilience, and stability
Other hallmarks of health from the microbial ecology perspective are the ability to resist perturbation (which might result from the entry of a pathogen, alteration of diet, or medication) and to return to a healthy state afterwards. These properties have been termed resistance and resilience, respectively [2]. For example, after an antibiotic treatment, healthy gut communities generally recover to their previous state after a few weeks to months [116]. A recent definition of microbial health thus explicitly comprises not a single static state but rather a dynamic equilibrium [2]. In this view, a healthy microbiome corresponds to an attractor of an underlying dynamic system (Fig. 1d), in a similar manner to cell fate in a metazoan [117]. Attractors capture both resistance and resilience, in that the system will resist a departure from an attractor, and unless a fluctuation (which might be due to external perturbation or internal stochasticity) is sufficiently large, it will tend to return to the steady state area [117]. The most visible examples in the human microbiome may be transitions between community state types in the healthy vagina; although their specific health implications are not yet enumerated, not all community state types have the same degree of stability [36]. The gut microbiome is also in flux, gaining and losing species over time, with different taxa having different stabilities and with some consistently remaining in the gut for many years [8]. The mechanisms by which specific taxa persist are not yet well-delineated, but it is interesting to speculate whether such mechanisms might relate to the driving principles behind the assembly of the microbiome. If specific communities do assemble primarily to fill a suite of habitat-suited functional niches [6], then species that provide key metabolic, signaling, immunomodulatory, or other roles in a particular assembly may be more temporally stable than those in the functional periphery. Coupling dynamics with the taxonomic diversity and immense molecular functional potential of the microbiome is thus a reminder of the human microbiome’s complexity and, as a result, the difficulty of defining even the apparently simple concept of microbial health.
Outlook
The era of population-scale whole-microbiome epidemiology has only recently begun, with the HMP [7, 118] and MetaHIT [9, 29] among the first large cohorts to include broad reference data in health, and several more cohorts soon to come. Data to date have been dominated by cross-sectional, amplicon-based studies of Western populations, all of which are efficient and accessible but which do not yet paint a consistent, comprehensive picture of the global, dynamic, healthy microbiome. Large-scale epidemiology in other areas of human health, such as nutrition and lifestyle, has built a solid foundation for prospective, long-running cohorts, painstaking analyses, and carefully validated measurement instruments [119–121], all of which represent particularly promising avenues of exploration for the microbiome. Nesting longitudinal microbiome studies in existing cohorts has the advantage of utilizing long-term collected lifestyle, dietary, medical, and phenotype information, as well as integration with banked biospecimens. An example of an unconventional large-scale study, notable for its infrastructure and outreach, is the American Gut project: a crowd-funded source of microbiome reference data paired with subject-provided environmental metadata. Prospective studies with detailed molecular data, while more expensive and logistically challenging, will also be necessary to facilitate predictive models and to establish the causality of dysbioses. The ongoing “HMP2” or Integrative Human Microbiome Project (iHMP) [122] includes three such longitudinal studies, which are providing multi-omic data for health and chronic disease, along with protocols and computational tools as a foundation for future work.
While many current studies of the microbiome focus on disease, a better understanding of the healthy microbiome will itself help to develop new microbial community diagnostics and therapeutics [123]. To the degree that universal features of the healthy microbiome can be defined, their absence may be predictive of disease onset generally, much like the presence of features specific to any one condition’s dysbiosis (especially useful if it occurs prior to disease onset). Alternatively, personalized medicine and longitudinal monitoring may serve the same purpose with respect to departure from an individual’s own “healthy” state [1, 104]. Therapeutically, as targeted interventions are developed to manipulate the microbiome, the treatment of a dysbiosis need not return to the healthy state from which an individual departed (due to a perturbation such as antibiotic treatment or the invasion of a pathogen), but perhaps only to a healthy state (Fig. 1d). Likewise, even if a microbial dysbiosis proves to be responsive rather than causal in any given disease state, the return to a “healthy” state may still provide therapeutic benefit [73, 101, 124].
One of the biggest outstanding gaps in understanding the basic biology of the “healthy” microbiome is perhaps at the level of annotating its molecular function: up to 50 % of microbial gene families encountered in the human microbiome remain functionally uncharacterized, even in well-studied environments such as the gut [9, 25, 29]. This is to a degree true in individual microbial isolate genomes as well, where even the well-studied E. coli K12 contains some 18 % of gene products with no reported function [125], with appreciably more at the E. coli species pangenome level [126]. It is likely, for example, that some of these genes are responsible for microbe–microbe or host–microbe interactions and thus will only be expressed or characterizable in community settings. Population-scale studies of the microbiome can themselves be used to mitigate this situation partially, in that microbial gene families that are prevalent and abundant but not yet well-understood can be prioritized for characterization. Likewise microbial communities provide a new source of guilt-by-association information that can be used computationally to generate predictions of gene function [127, 128]. Nevertheless, returning to the field’s microbiological roots may ultimately prove most important in this area: the best biochemical characterizations still derive from culture-based physiology, microbial metabolism, co-culture and interactions, and controlled laboratory environments coupled with high-throughput molecular assays [15, 129, 130].
Studies of the microbiome, both in health and in disease, must continue to integrate population-scale epidemiology with narrow but deep clinical studies in the setting of personalized medicine. In both cases, studies of the body-wide microbiome can be seen as an extension of microbial techniques already used for infectious disease surveillance [131]: rather than waiting to monitor a pathogen’s outbreak in a population or its persistence within an individual, our complete microbial community could be monitored for health maintenance or departures into disease. This is equally true in the integration of microbiome activity with host immune, transcriptional, epigenetic, and clinical state: precision microbial community medicine must rely on host–microbiome interactions as a key component. This will help to identify potential pathogens rapidly [132] and will make it possible to determine the “right” interventions to restore health after dysbiosis, ranging from dietary or lifestyle changes through probiotics into microbially targeted pharmaceuticals [133]. A better understanding of the healthy microbiome must thus approach it as one aspect of deeply monitored personalized health (e.g., [121]) and must integrate population-scale assessment of the microbial community with a well-characterized molecular understanding and analyses of how beneficial community states are maintained body-wide and life-long.
Acknowledgements
We thank Eric Franzosa for providing helpful feedback regarding the content of this review.
Abbreviations
- FISH
fluorescence in situ hybridization
- HMO
human milk oligosaccharides
- HMP
Human Microbiome Project
- MetaHIT
Metagenomes of the Human Intestinal Tract
- SCFA
short chain fatty acid
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CH designed the review. All authors drafted the manuscript. All authors read and approved the final manuscript.
References
- 1.Petersen C, Round JL. Defining dysbiosis and its influence on host immunity and disease. Cell Microbiol. 2014;16:1024–1033. doi: 10.1111/cmi.12308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Backhed F, Fraser CM, Ringel Y, Sanders ME, Sartor RB, Sherman PM, et al. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe. 2012;12:611–622. doi: 10.1016/j.chom.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 3.Trompette A, Gollwitzer ES, Yadava K, Sichelstiel AK, Sprenger N, Ngom-Bru C, et al. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat Med. 2014;20:159–166. doi: 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- 4.Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell. 2013;155:1451–1463. doi: 10.1016/j.cell.2013.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garrett WS. Cancer and the microbiota. Science. 2015;348:80–86. doi: 10.1126/science.aaa4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shafquat A, Joice R, Simmons SL, Huttenhower C. Functional and phylogenetic assembly of microbial communities in the human microbiome. Trends Microbiol. 2014;22:261–266. doi: 10.1016/j.tim.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.The Human Microbiome Project Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, et al. The long-term stability of the human gut microbiota. Science. 2013;341:1237439. doi: 10.1126/science.1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aagaard K, Petrosino J, Keitel W, Watson M, Katancik J, Garcia N, et al. The Human Microbiome Project strategy for comprehensive sampling of the human microbiome and why it matters. FASEB J. 2013;27:1012–1022. doi: 10.1096/fj.12-220806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449:804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Finegold SM, Sutter VL, Mathison GE. Normal indigenous intestinal flora. In: Hentges DJ, editor. Human intestinal microflora in health and disease. New York: Academic Press, Inc.; 1983. pp. 3–31. [Google Scholar]
- 13.Bodelier PL. Toward understanding, managing, and protecting microbial ecosystems. Front Microbiol. 2011;2:80. doi: 10.3389/fmicb.2011.00080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rajilic-Stojanovic M, de Vos WM. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol Rev. 2014;38:996–1047. doi: 10.1111/1574-6976.12075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore WE, Holdeman LV. Human fecal flora: the normal flora of 20 Japanese-Hawaiians. Appl Microbiol. 1974;27:961–979. doi: 10.1128/am.27.5.961-979.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brown JP. Role of gut bacterial flora in nutrition and health: a review of recent advances in bacteriological techniques, metabolism, and factors affecting flora composition. CRC Crit Rev Food Sci Nutr. 1977;8:229–336. doi: 10.1080/10408397709527224. [DOI] [PubMed] [Google Scholar]
- 18.Lane DJ, Pace B, Olsen GJ, Stahl DA, Sogin ML, Pace NR. Rapid determination of 16S ribosomal RNA sequences for phylogenetic analyses. Proc Natl Acad Sci U S A. 1985;82:6955–6959. doi: 10.1073/pnas.82.20.6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franks AH, Harmsen HJ, Raangs GC, Jansen GJ, Schut F, Welling GW. Variations of bacterial populations in human feces measured by fluorescent in situ hybridization with group-specific 16S rRNA-targeted oligonucleotide probes. Appl Environ Microbiol. 1998;64:3336–3345. doi: 10.1128/aem.64.9.3336-3345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hugenholtz P. Exploring prokaryotic diversity in the genomic era. Genome Biol. 2002;3:Reviews0003. doi: 10.1186/gb-2002-3-2-reviews0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suau A, Bonnet R, Sutren M, Godon JJ, Gibson GR, Collins MD, Dore J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl Environ Microbiol. 1999;65:4799–4807. doi: 10.1128/aem.65.11.4799-4807.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol. 2002;39:33–9. doi: 10.1111/j.1574-6941.2002.tb00904.x. [DOI] [PubMed] [Google Scholar]
- 23.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tringe SG, von Mering C, Kobayashi A, Salamov AA, Chen K, Chang HW, et al. Comparative metagenomics of microbial communities. Science. 2005;308:554–557. doi: 10.1126/science.1107851. [DOI] [PubMed] [Google Scholar]
- 25.Joice R, Yasuda K, Shafquat A, Morgan XC, Huttenhower C. Determining microbial products and identifying molecular targets in the human microbiome. Cell Metab. 2014;20:731–741. doi: 10.1016/j.cmet.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457:480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60. doi: 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- 29.Li J, Jia H, Cai X, Zhong H, Feng Q, Sunagawa S, et al. An integrated catalog of reference genes in the human gut microbiome. Nat Biotechnol. 2014;32:834–841. doi: 10.1038/nbt.2942. [DOI] [PubMed] [Google Scholar]
- 30.Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, et al. Richness of human gut microbiome correlates with metabolic markers. Nature. 2013;500:541–546. doi: 10.1038/nature12506. [DOI] [PubMed] [Google Scholar]
- 31.Erickson AR, Cantarel BL, Lamendella R, Darzi Y, Mongodin EF, Pan C, et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS One. 2012;7 doi: 10.1371/journal.pone.0049138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. bioRxiv. 2016. http://dx.doi.org/10.1101/036103. [DOI] [PMC free article] [PubMed]
- 33.Wade WG. The oral microbiome in health and disease. Pharmacol Res. 2013;69:137–143. doi: 10.1016/j.phrs.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 34.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, et al. Topographical and temporal diversity of the human skin microbiome. Science. 2009;324:1190–1192. doi: 10.1126/science.1171700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci U S A. 2011;108(1):4680–4687. doi: 10.1073/pnas.1002611107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci U S A. 2015;112:11060–11065. doi: 10.1073/pnas.1502875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fettweis JM, Brooks JP, Serrano MG, Sheth NU, Girerd PH, Edwards DJ, et al. Differences in vaginal microbiome in African American women versus women of European ancestry. Microbiology. 2014;160:2272–2282. doi: 10.1099/mic.0.081034-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Charlson ES, Bittinger K, Haas AR, Fitzgerald AS, Frank I, Yadav A, et al. Topographical continuity of bacterial populations in the healthy human respiratory tract. Am J Respir Crit Care Med. 2011;184:957–963. doi: 10.1164/rccm.201104-0655OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris A, Beck JM, Schloss PD, Campbell TB, Crothers K, Curtis JL, et al. Comparison of the respiratory microbiome in healthy nonsmokers and smokers. Am J Respir Crit Care Med. 2013;187:1067–1075. doi: 10.1164/rccm.201210-1913OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hilty M, Burke C, Pedro H, Cardenas P, Bush A, Bossley C, et al. Disordered microbial communities in asthmatic airways. PLoS One. 2010;5 doi: 10.1371/journal.pone.0008578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hunt KM, Foster JA, Forney LJ, Schutte UM, Beck DL, Abdo Z, et al. Characterization of the diversity and temporal stability of bacterial communities in human milk. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kliman HJ. Comment on “the placenta harbors a unique microbiome”. Sci Transl Med. 2014;6:254le254. doi: 10.1126/scitranslmed.3009864. [DOI] [PubMed] [Google Scholar]
- 43.Paisse S, Valle C, Servant F, Courtney M, Burcelin R, Amar J, Lelouvier B. Comprehensive description of blood microbiome from healthy donors assessed by 16S targeted metagenomic sequencing. Transfusion. 2016 doi: 10.1111/trf.13477. [DOI] [PubMed] [Google Scholar]
- 44.Burcelin R, Serino M, Chabo C, Garidou L, Pomie C, Courtney M, et al. Metagenome and metabolism. The tissue microbiota hypothesis. Diabetes Obesity Metab. 2013;15(3):61–70. doi: 10.1111/dom.12157. [DOI] [PubMed] [Google Scholar]
- 45.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun. 2013;4:1431. doi: 10.1038/ncomms2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Salter SJ, Cox MJ, Turek EM, Calus ST, Cookson WO, Moffatt MF, et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014;12:87. doi: 10.1186/s12915-014-0087-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Horz HP. Archaeal lineages within the human microbiome: absent, rare or elusive? Life (Basel) 2015;5:1333–1345. doi: 10.3390/life5021333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matarazzo F, Ribeiro AC, Feres M, Faveri M, Mayer MP. Diversity and quantitative analysis of Archaea in aggressive periodontitis and periodontally healthy subjects. J Clin Periodontol. 2011;38:621–627. doi: 10.1111/j.1600-051X.2011.01734.x. [DOI] [PubMed] [Google Scholar]
- 49.Samuel BS, Hansen EE, Manchester JK, Coutinho PM, Henrissat B, Fulton R, et al. Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc Natl Acad Sci U S A. 2007;104:10643–10648. doi: 10.1073/pnas.0704189104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Virgin HW. The virome in mammalian physiology and disease. Cell. 2014;157:142–150. doi: 10.1016/j.cell.2014.02.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Minot S, Bryson A, Chehoud C, Wu GD, Lewis JD, Bushman FD. Rapid evolution of the human gut virome. Proc Natl Acad Sci U S A. 2013;110:12450–12455. doi: 10.1073/pnas.1300833110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reyes A, Haynes M, Hanson N, Angly FE, Heath AC, Rohwer F, Gordon JI. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466:334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang J, Gao Y, Zhao F. Phage-bacteria interaction network in human oral microbiome. Environ Microbiol. 2015 doi: 10.1111/1462-2920.12923. [DOI] [PubMed] [Google Scholar]
- 54.Norman JM, Handley SA, Virgin HW. Kingdom-agnostic metagenomics and the importance of complete characterization of enteric microbial communities. Gastroenterology. 2014;146:1459–1469. doi: 10.1053/j.gastro.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huffnagle GB, Noverr MC. The emerging world of the fungal microbiome. Trends Microbiol. 2013;21:334–341. doi: 10.1016/j.tim.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lukes J, Stensvold CR, Jirku-Pomajbikova K, Wegener PL. Are human intestinal eukaryotes beneficial or commensals? PLoS Pathog. 2015;11 doi: 10.1371/journal.ppat.1005039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Underhill DM, Iliev ID. The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol. 2014;14:405–416. doi: 10.1038/nri3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parfrey LW, Walters WA, Lauber CL, Clemente JC, Berg-Lyons D, Teiling C, et al. Communities of microbial eukaryotes in the mammalian gut within the context of environmental eukaryotic diversity. Front Microbiol. 2014;5:298. doi: 10.3389/fmicb.2014.00298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Findley K, Oh J, Yang J, Conlan S, Deming C, Meyer JA, et al. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013;498:367–370. doi: 10.1038/nature12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rizzo A, Losacco A, Carratelli CR. Lactobacillus crispatus modulates epithelial cell defense against Candida albicans through Toll-like receptors 2 and 4, interleukin 8 and human beta-defensins 2 and 3. Immunol Lett. 2013;156:102–109. doi: 10.1016/j.imlet.2013.08.013. [DOI] [PubMed] [Google Scholar]
- 61.Hatoum R, Labrie S, Fliss I. Antimicrobial and probiotic properties of yeasts: from fundamental to novel applications. Front Microbiol. 2012;3:421. doi: 10.3389/fmicb.2012.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Scanlan PD, Stensvold CR, Rajilic-Stojanovic M, Heilig HG, De Vos WM, O’Toole PW, Cotter PD. The microbial eukaryote Blastocystis is a prevalent and diverse member of the healthy human gut microbiota. FEMS Microbiol Ecol. 2014;90:326–330. doi: 10.1111/1574-6941.12396. [DOI] [PubMed] [Google Scholar]
- 63.Rossen NG, Bart A, Verhaar N, van Nood E, Kootte R, de Groot PF, et al. Low prevalence of Blastocystis sp. in active ulcerative colitis patients. Eur J Clin Microbiol Infect Dis. 2015;34:1039–1044. doi: 10.1007/s10096-015-2312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Loke P, Lim YA. Helminths and the microbiota: parts of the hygiene hypothesis. Parasite Immunol. 2015;37:314–323. doi: 10.1111/pim.12193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reynolds LA, Smith KA, Filbey KJ, Harcus Y, Hewitson JP, Redpath SA, et al. Commensal–pathogen interactions in the intestinal tract: lactobacilli promote infection with, and are promoted by, helminth parasites. Gut Microbes. 2014;5:522–532. doi: 10.4161/gmic.32155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486:222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.De Filippo C, Cavalieri D, Di Paola M, Ramazzotti M, Poullet JB, Massart S, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A. 2010;107:14691–14696. doi: 10.1073/pnas.1005963107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Takeshita T, Matsuo K, Furuta M, Shibata Y, Fukami K, Shimazaki Y, et al. Distinct composition of the oral indigenous microbiota in South Korean and Japanese adults. Sci Rep. 2014;4:6990. doi: 10.1038/srep06990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tyakht AV, Kostryukova ES, Popenko AS, Belenikin MS, Pavlenko AV, Larin AK, et al. Human gut microbiota community structures in urban and rural populations in Russia. Nat Commun. 2013;4:2469. doi: 10.1038/ncomms3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Guo Z, Xue Z, Sun Z, Zhang M, Wang L, et al. A phylo-functional core of gut microbiota in healthy young Chinese cohorts across lifestyles, geography and ethnicities. ISME J. 2015;9:1979–1990. doi: 10.1038/ismej.2015.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Carmody RN, Gerber GK, Luevano JM, Jr, Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015;17:72–84. doi: 10.1016/j.chom.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kostic AD, Gevers D, Siljander H, Vatanen T, Hyotylainen T, Hamalainen AM, et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe. 2015;17:260–273. doi: 10.1016/j.chom.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schloissnig S, Arumugam M, Sunagawa S, Mitreva M, Tap J, Zhu A, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493:45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Franzosa EA, Huang K, Meadow JF, Gevers D, Lemon KP, Bohannan BJ, Huttenhower C. Identifying personal microbiomes using metagenomic codes. Proc Natl Acad Sci U S A. 2015;112:E2930–E2938. doi: 10.1073/pnas.1423854112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun. 2013;4:2151. doi: 10.1038/ncomms3151. [DOI] [PubMed] [Google Scholar]
- 76.Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, et al. Traces of human migrations in Helicobacter pylori populations. Science. 2003;299:1582–1585. doi: 10.1126/science.1080857. [DOI] [PubMed] [Google Scholar]
- 77.Dominguez-Bello MG, Blaser MJ. The human microbiota as a marker for migrations of individuals and populations. Annu Rev Anthropol. 2011;40:451–474. doi: 10.1146/annurev-anthro-081309-145711. [DOI] [Google Scholar]
- 78.Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17:852. doi: 10.1016/j.chom.2015.05.012. [DOI] [PubMed] [Google Scholar]
- 79.Mackie RI, Sghir A, Gaskins HR. Developmental microbial ecology of the neonatal gastrointestinal tract. Am J Clin Nutr. 1999;69:1035s–1045s. doi: 10.1093/ajcn/69.5.1035s. [DOI] [PubMed] [Google Scholar]
- 80.Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, Knight R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc Natl Acad Sci U S A. 2010;107:11971–11975. doi: 10.1073/pnas.1002601107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Byrne CS, Chambers ES, Morrison DJ, Frost G. The role of short chain fatty acids in appetite regulation and energy homeostasis. Int J Obes (Lond) 2015;39:1331–1338. doi: 10.1038/ijo.2015.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341:569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tang Y, Chen Y, Jiang H, Robbins GT, Nie D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int J Cancer. 2011;128:847–856. doi: 10.1002/ijc.25638. [DOI] [PubMed] [Google Scholar]
- 84.Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci U S A. 2014;111:2247–2252. doi: 10.1073/pnas.1322269111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Round JL, Lee SM, Li J, Tran G, Jabri B, Chatila TA, Mazmanian SK. The Toll-like receptor 2 pathway establishes colonization by a commensal of the human microbiota. Science. 2011;332:974–977. doi: 10.1126/science.1206095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Geuking MB, Cahenzli J, Lawson MA, Ng DC, Slack E, Hapfelmeier S, et al. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity. 2011;34:794–806. doi: 10.1016/j.immuni.2011.03.021. [DOI] [PubMed] [Google Scholar]
- 87.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ochoa-Reparaz J, Mielcarz DW, Wang Y, Begum-Haque S, Dasgupta S, Kasper DL, Kasper LH. A polysaccharide from the human commensal Bacteroides fragilis protects against CNS demyelinating disease. Mucosal Immunol. 2010;3:487–495. doi: 10.1038/mi.2010.29. [DOI] [PubMed] [Google Scholar]
- 89.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500:232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 90.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zivkovic AM, German JB, Lebrilla CB, Mills DA. Human milk glycobiome and its impact on the infant gastrointestinal microbiota. Proc Natl Acad Sci U S A. 2011;108(1):4653–4658. doi: 10.1073/pnas.1000083107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Charbonneau MR, O’Donnell D, Blanton LV, Totten SM, Davis JC, Barratt MJ, et al. Sialylated milk oligosaccharides promote microbiota-dependent growth in models of infant undernutrition. Cell. 2016;164:859–871. doi: 10.1016/j.cell.2016.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zivkovic AM, Barile D. Bovine milk as a source of functional oligosaccharides for improving human health. Adv Nutr. 2011;2:284–289. doi: 10.3945/an.111.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ninonuevo MR, Bode L. Infant formula oligosaccharides opening the gates (for speculation): commentary on the article by Barrat et al. on page 34. Pediatr Res. 2008;64:8–10. doi: 10.1203/PDR.0b013e3181752c2f. [DOI] [PubMed] [Google Scholar]
- 95.Stuebe A. The risks of not breastfeeding for mothers and infants. Rev Obstet Gynecol. 2009;2:222–231. [PMC free article] [PubMed] [Google Scholar]
- 96.Xu Z, Malmer D, Langille MG, Way SF, Knight R. Which is more important for classifying microbial communities: who’s there or what they can do? ISME J. 2014;8:2357–2359. doi: 10.1038/ismej.2014.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martiny JB, Jones SE, Lennon JT, Martiny AC. Microbiomes in light of traits: a phylogenetic perspective. Science. 2015;350:aac9323. doi: 10.1126/science.aac9323. [DOI] [PubMed] [Google Scholar]
- 98.Abubucker S, Segata N, Goll J, Schubert AM, Izard J, Cantarel BL, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLoS Comput Biol. 2012;8 doi: 10.1371/journal.pcbi.1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Flint HJ, Scott KP, Louis P, Duncan SH. The role of the gut microbiota in nutrition and health. Nat Rev Gastroenterol Hepatol. 2012;9:577–589. doi: 10.1038/nrgastro.2012.156. [DOI] [PubMed] [Google Scholar]
- 100.Lemon KP, Armitage GC, Relman DA, Fischbach MA. Microbiota-targeted therapies: an ecological perspective. Sci Transl Med. 2012;4:137rv135. doi: 10.1126/scitranslmed.3004183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336:1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Burke C, Steinberg P, Rusch D, Kjelleberg S, Thomas T. Bacterial community assembly based on functional genes rather than species. Proc Natl Acad Sci U S A. 2011;108:14288–14293. doi: 10.1073/pnas.1101591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Flores GE, Caporaso JG, Henley JB, Rideout JR, Domogala D, Chase J, et al. Temporal variability is a personalized feature of the human microbiome. Genome Biol. 2014;15:531. doi: 10.1186/s13059-014-0531-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Huttenhower C, Kostic AD, Xavier RJ. Inflammatory bowel disease as a model for translating the microbiome. Immunity. 2014;40:843–854. doi: 10.1016/j.immuni.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res. 2012;22:850–859. doi: 10.1101/gr.131029.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Statnikov A, Alekseyenko AV, Li Z, Henaff M, Perez-Perez GI, Blaser MJ, Aliferis CF. Microbiomic signatures of psoriasis: feasibility and methodology comparison. Sci Rep. 2013;3:2620. doi: 10.1038/srep02620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fredricks DN, Fiedler TL, Marrazzo JM. Molecular identification of bacteria associated with bacterial vaginosis. New Engl J Med. 2005;353:1899–1911. doi: 10.1056/NEJMoa043802. [DOI] [PubMed] [Google Scholar]
- 109.Mitra A, MacIntyre DA, Lee YS, Smith A, Marchesi JR, Lehne B, et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci Rep. 2015;5:16865. doi: 10.1038/srep16865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Anahtar MN, Byrne EH, Doherty KE, Bowman BA, Yamamoto HS, Soumillon M, et al. Cervicovaginal bacteria are a major modulator of host inflammatory responses in the female genital tract. Immunity. 2015;42:965–976. doi: 10.1016/j.immuni.2015.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Blaser MJ, Falkow S. What are the consequences of the disappearing human microbiota? Nat Rev Microbiol. 2009;7:887–894. doi: 10.1038/nrmicro2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Schnorr SL, Candela M, Rampelli S, Centanni M, Consolandi C, Basaglia G, et al. Gut microbiome of the Hadza hunter-gatherers. Nat Commun. 2014;5:3654. doi: 10.1038/ncomms4654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Clemente JC, Pehrsson EC, Blaser MJ, Sandhu K, Gao Z, Wang B, et al. The microbiome of uncontacted Amerindians. Sci Adv. 2015;1(3):e1500183. doi: 10.1126/sciadv.1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sonnenburg ED, Smits SA, Tikhonov M, Higginbottom SK, Wingreen NS, Sonnenburg JL. Diet-induced extinctions in the gut microbiota compound over generations. Nature. 2016;529:212–215. doi: 10.1038/nature16504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Strachan DP. Hay fever, hygiene, and household size. BMJ. 1989;299:1259–1260. doi: 10.1136/bmj.299.6710.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11) doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Furusawa C, Kaneko K. A dynamical-systems view of stem cell biology. Science. 2012;338:215–217. doi: 10.1126/science.1224311. [DOI] [PubMed] [Google Scholar]
- 118.The Human Microbiome Project Consortium A framework for human microbiome research. Nature. 2012;486:215–221. doi: 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, et al. Conducting a microbiome study. Cell. 2014;158:250–262. doi: 10.1016/j.cell.2014.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Higgins MW. The Framingham Heart Study: review of epidemiological design and data, limitations and prospects. Prog Clin Biol Res. 1984;147:51–64. [PubMed] [Google Scholar]
- 121.Zeevi D, Korem T, Zmora N, Israeli D, Rothschild D, Weinberger A, et al. Personalized nutrition by prediction of glycemic responses. Cell. 2015;163:1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 122.The Integrative Human Microbiome Project Research Network Consortium The Integrative Human Microbiome Project: dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe. 2014;16:276–289. doi: 10.1016/j.chom.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bornigen D, Morgan XC, Franzosa EA, Ren B, Xavier RJ, Garrett WS, Huttenhower C. Functional profiling of the gut microbiome in disease-associated inflammation. Genome Med. 2013;5:65. doi: 10.1186/gm469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Jenq RR, Ubeda C, Taur Y, Menezes CC, Khanin R, Dudakov JA, et al. Regulation of intestinal inflammation by microbiota following allogeneic bone marrow transplantation. J Exp Med. 2012;209:903–911. doi: 10.1084/jem.20112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Keseler IM, Mackie A, Peralta-Gil M, Santos-Zavaleta A, Gama-Castro S, Bonavides-Martinez C, et al. EcoCyc: fusing model organism databases with systems biology. Nucleic Acids Res. 2013;41(Database issue):D605–D612. doi: 10.1093/nar/gks1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rasko DA, Rosovitz MJ, Myers GS, Mongodin EF, Fricke WF, Gajer P, et al. The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J Bacteriol. 2008;190:6881–6893. doi: 10.1128/JB.00619-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Haft DH. Using comparative genomics to drive new discoveries in microbiology. Curr Opin Microbiol. 2015;23:189–196. doi: 10.1016/j.mib.2014.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein–protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41(Database issue):D808–15. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Anton BP, Chang YC, Brown P, Choi HP, Faller LL, Guleria J, et al. The COMBREX project: design, methodology, and initial results. PLoS Biol. 2013;11 doi: 10.1371/journal.pbio.1001638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Santiago-Rodriguez TM, Ly M, Daigneault MC, Brown IH, McDonald JA, Bonilla N, et al. Chemostat culture systems support diverse bacteriophage communities from human feces. Microbiome. 2015;3:58. doi: 10.1186/s40168-015-0124-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koser CU, Ellington MJ, Cartwright EJ, Gillespie SH, Brown NM, Farrington M, et al. Routine use of microbial whole genome sequencing in diagnostic and public health microbiology. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Boyd SD. Diagnostic applications of high-throughput DNA sequencing. Annu Rev Pathol. 2013;8:381–410. doi: 10.1146/annurev-pathol-020712-164026. [DOI] [PubMed] [Google Scholar]
- 133.Dutton RJ, Turnbaugh PJ. Taking a metagenomic view of human nutrition. Curr Opin Clin Nutr Metab Care. 2012;15:448–454. doi: 10.1097/MCO.0b013e3283561133. [DOI] [PubMed] [Google Scholar]