

Abstract
Sudden cardiac death (SCD) is the result of a change of cardiac activity from normal (typically sinus) rhythm to a rhythm that does not pump adequate blood to the brain. The most common rhythms leading to SCD are ventricular tachycardia (VT) or ventricular fibrillation (VF). These result from an accelerated ventricular pacemaker or ventricular reentrant waves. Despite significant efforts to develop accurate predictors for the risk of SCD, current methods for risk stratification still need to be improved. In this article we briefly review current approaches to risk stratification. Then we discuss the mathematical basis for dynamical transitions (called bifurcations) that may lead to VT and VF. One mechanism for transition to VT or VF involves a perturbation by a premature ventricular complex (PVC) during sinus rhythm. We describe the main mechanisms of PVCs (reentry, independent pacemakers and abnormal depolarizations). An emerging approach to risk stratification for SCD involves the development of individualized dynamical models of a patient based on measured anatomy and physiology. Careful analysis and modelling of dynamics of ventricular arrhythmia on an individual basis will be essential in order to improve risk stratification for SCD and to lay a foundation for personalized (precision) medicine in cardiology.
Abbreviations
- CI
coupling interval
- DAD
delayed afterdepolarization
- EAD
early afterdepolarization
- ECG
electrocardiogram
- ICD
implantable cardioverter defibrillator
- NIB
number of intervening beats (between two consecutive PVCs)
- NN interval
time between two sinus beats
- MRI
magnetic resonance imaging
- PVC
premature ventricular complex
- SCD
sudden cardiac death
- SDCI
standard deviation of the coupling interval
- SNIB
score of NIB
- VF
ventricular fibrillation
- VT
ventricular tachycardia
- VV interval
time between two consecutive PVCs
Introduction
Sudden cardiac death (SCD) refers to death within 2 h of onset of symptoms or during sleep due to a cardiac cause (Zipes & Wellens, 1998). Although SCD can occur due to a slow heart rhythm (bradycardia) caused by a stopping or blocking of the normal sinus pacemaker, more commonly it is due to a rapid heart rhythm (tachycardia), usually originating in the ventricles – ventricular tachycardia (VT) or ventricular fibrillation (VF). One major route to SCD is secondary to a heart attack (coronary infarct) in which a coronary artery is blocked. Although bradyarrhythmias and coronary infarcts are also causes of SCD, developing risk stratification for these occurrences lies outside the scope of this review. Rather, we consider SCD associated with a transition from a normal sinus rhythm to VT or VF. Since an implantable cardioverter defibrillator (ICD) may be an effective therapy for this class of arrhythmias (Moss et al. 2002; Bardy et al. 2005; Tung et al. 2008), a major clinical question involves risk stratification for SCD (Goldberger et al. 2008, 2011; Bastiaenen et al. 2012; Bilchick et al. 2012; Huikuri, 2015).
Recent reviews summarize studies on risk stratification for SCD (Wellens et al. 2014; Deyell et al. 2015). The paradigms for these studies are similar. First measure some set of parameters and dynamical features based on cardiac anatomy and physiology in some large subset of patients meeting a predefined clinical profile. Then assess the incidence of SCD over several years to see which factors show a strong correlation with the future incidence of SCD. These reviews underscore the conclusions that: (i) many different factors are associated with an increased risk for SCD; and (ii) current methods for risk stratification still need significant improvement in order to better define the roles for various existent and emerging medical and device therapies. One strategy to improve risk stratification for SCD is to carry out large observational studies, similar to those carried out previously (Huikuri, 2015). Although large observational studies play an important role in clinical decision making, we believe that alternative strategies will also be useful to improve risk stratification for SCD.
In this review, we address SCD from a basic science perspective. In SCD, the spatio‐temporal organization of cardiac activity changes typically from a pattern in which the normal sinus pacemaker is setting the rhythm of the entire heart, to a rhythm in which an accelerated ventricular pacemaker or ventricular reentrant waves set a rapid ventricular rhythm leading to VT or VF. Dynamical transitions such as these are called bifurcations and can be studied mathematically using techniques developed in the field of nonlinear dynamics (Glass et al. 1987; Krogh‐Madsen & Christini, 2012; Weiss et al. 2015).
In the next section, we briefly summarize the various physiological and electrocardiographic factors that have been the main focus of clinical investigations for risk stratification. After that we discuss generic mathematical properties of cardiac tissue and summarize conditions leading to blocking of excitation, alternans, and initiation of extra beats and pacemakers. These basic processes are implicated in the transition from sinus rhythm to the onset of arrhythmias. In the following section, we focus on mechanisms of premature ventricular complexes (PVCs) and discuss evidence that shows that not all PVCs are equal – some mechanisms confer a higher risk of SCD than others. The section after that discusses a new method for identifying mechanisms of PVCs based on their dynamic properties measured over 24 h. Such an analysis could provide a basis for better identifying mechanisms of PVCs and improving risk stratification for SCD. Then we discuss realistic and patient‐specific modelling of cardiac arrhythmias with a view towards improving the diagnosis and therapy for SCD. Finally, we identify directions for future research.
Prediction of sudden cardiac death
Clinical studies of risk stratification for SCD have investigated and identified a large number of physiological and anatomical characteristics that have been associated with an elevated risk of SCD (Wellens et al. 2014; Deyell et al. 2015). These different characteristics include anatomical substrate, autonomic function, genetic mutations, electrophysiological function and provocative testing.
Anatomical substrate
According to current guidelines, the main criterion for implantation of an ICD for primary prevention of SCD is a low left ventricular ejection fraction (Moss et al. 2002; Bardy et al. 2005; Epstein et al. 2008). Although this criterion has been verified in numerous clinical studies, a large fraction of patients who meet the criterion do not receive an ICD, a large fraction of patients who meet the criterion and do receive an ICD do not have triggering events, and a large number of people who do not meet the criterion do experience SCD (Goldberger et al. 2011; Albert & Stevenson, 2013). Indication of scarring in the ventricles is provided by fragmented QRS complexes on the electrocardiogram and this has also been used for risk stratification (Jain et al. 2014). Alternative approaches to assessing the anatomical substrate for arrhythmia involve imaging the heart using a variety of methods including echocardiography, magnetic resonance imaging and photon emission tomography (Vadakkumpadan et al. 2014). Prospective assessments of these modalities have not yet been carried out.
Autonomic function
The period of the cardiac cycle is continuously modulated by activity from the sympathetic and parasympathetic systems where greater sympathetic activity increases the heart rate and greater parasympathetic activity reduces the heart rate. Reduced heart rate variability is associated with an increased risk of SCD (Kleiger et al. 1987; Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology, 1996). The observation that blocking sympathetic activity using beta blockers reduces the incidence of SCD provides additional strong evidence of the importance of autonomic function in the pathophysiology of SCD (Shen & Zipes, 2014).
Patients with diseased hearts typically have increased heart rate and reduced fluctuations in the heart rate associated with increased sympathetic hyperactivity, decreased vagal activity, or both (La Rovere et al. 1998; Shusterman et al. 1998). Reduced heart rate variability (HRV) as measured by assessment of standard deviation of interbeat intervals (Goldberger et al. 2008), power spectral density of HRV (Huikuri et al. 2009), analysis of the complexity of HRV using entropy‐related measures (Wessel et al. 2000; Costa et al. 2002; Norris et al. 2008), and reduced values of scaling coefficients associated with fluctuations of heart rate (Au‐Yeung et al. 2015) show correlations with impaired health and increased risk for serious disease and/or SCD. Decreased entropy of rapid cardiac rhythm may also be useful to distinguish VT and VF from atrial fibrillation, thereby reducing the occurrence of inappropriate ICD shocks (DeMazumder et al. 2013).
Another measure associated with autonomic function is heart turbulence, measured by determining the transient heart rates following a PVC. Heart rate turbulence describes the short‐term fluctuations in sinus cycle length that follow a PVC (Barthel et al. 2003). After a PVC, the next sinus beat is typically blocked leading to a pause before the next sinus beat. This leads to an increase in blood pressure due to prolonged filling time and consequent slowing of the heart rate through operation of the baroreceptor reflex mediated by decreased sympathetic activity and increased parasympathetic activity. In patients who survived an acute myocardial infarction, indices reflecting increased sympathetic activity and reduced parasympathetic activity following a PVC were predictors of mortality (Barthel et al. 2003) and life‐threatening arrhythmias (Huikuri et al. 2010). In patients with congestive heart failure these indices were associated with a higher risk of SCD (Au‐Yeung et al. 2015). Although the measures of autonomic function are correlated with the risk for SCD, the correlations are not sufficiently strong to provide a basis for risk stratification.
Genetic mutations
Advances in genetics and cardiac electrophysiology have led to the identification of a large number of different genetic mutations affecting ion channels that have been associated with increased incidence of cardiac arrhythmias and SCD. The best understood are mutations of potassium and sodium channels that lead to longer action potentials manifested by an increased duration of QT interval on the electrocardiogram (long QT syndrome) (Priori et al. 2003; Amin et al. 2013). The longer durations of action potentials may lead to PVCs as a consequence of early afterdepolarizations, and these may in turn induce a potentially lethal VT, torsade de pointes. However, since other factors including sympathetic activity and drugs also affect repolarization, it is not simple to assess the risk for SCD based solely on the presence of a mutation (Priori et al. 2003). Another group of mutations was discovered from genetic analysis of individuals that were identified initially by electrocardiographic abnormalities in a family that had several instances of cardiac arrest (Chen et al. 1998; Brugada et al. 2013). These mutations of the sodium channel, associated with the Brugada syndrome, lead to slow conduction predisposing to reentrant tachycardias (Brugada et al. 2013). Another class of mutations in receptors associated with calcium handling lead to calcium overload and VT induced by sympathetic stimulation (catecholaminergic polymorphic ventricular tachycardia) (Mohamed et al. 2007). Because there can be multiple sites of mutation in a single gene and there are other modulating factors of cardiac activity that are poorly understood, observation of mutations must be combined with electrophysiological data to assess risk.
Electrophysiological function
One of the earliest recognized risk factors for SCD is an increased numbers of PVCs (Kotler et al. 1973; Berkowitsch et al. 2004; Carrim & Khan, 2005). As is well known, the cardiac arrhythmia suppression trial demonstrated that reducing the incidence of PVCs with drugs led to an increased incidence of SCD (Echt et al. 1991). This study is partially responsible for a reduced emphasis on the role of PVCs in the assessment of risk and the genesis of SCD. However, a recent study, that reexamined ventricular ectopy in heart failure patients, showed increased risk with increased ectopy (Dukes et al. 2015) and concluded that there is a need for better understanding mechanisms of PVCs.
Another electrophysiological marker for increased risk for SCD is alternans of the T‐wave (Weiss et al. 2006; Verrier et al. 2011; Nieminen et al. 2014). From a dynamics perspective, T‐wave alternans is of particular interest since analysis of mathematical models of action potential duration have demonstrated the possibility for instabilities (bifurcations) leading to alternating action potential durations (Qu et al. 2010). T‐wave alternans on the electrocardiogram (ECG) could arise from this instability. However, it could also arise in other ways such as from 2:1 conduction in some parts of the heart and 1:1 conduction in other regions of the heart.
The stability of ventricular repolarization, as evaluated by the maximum slope of the relationship between the action potential duration and the diastolic interval (restitution curve), is a known factor for the onset of ventricular arrhythmias (Garfinkel et al. 2000). From the ECG, an estimated restitution curve is obtained from the relationship between the QT interval and the preceding TQ interval, to estimate the QT interval dynamics stability. In patients with myocardial infarction, the instability of QT interval dynamics is increased before VT (Chen et al. 2011), and a QT instability has predictive value for ventricular arrhythmia in patients with an implanted ICD (Chen et al. 2013). However, direct measurement of action potential restitution curves in patients failed to show a direct correlation between steep restitution curves and T‐wave alternans (Narayan et al. 2007). Since alternation of the action potential voltage can occur in the absence of alternans of the action potential duration (Bayer et al. 2010), an important question in basic science is to clarify the mechanisms of alternans in normal and heart failure patients and to clarify the role of calcium handling (Narayan et al. 2008; Wilson et al. 2009).
Electrophysiological testing
The development of clinical cardiac electrophysiology has been greatly advanced by the development of intracardiac catheters that can be used to record local activity, deliver electrical stimuli, and ablate cardiac tissue using radio‐frequency stimulation (Josephson, 2008). Provocative testing involves pacing the heart at a fixed rate and delivering a sequence of up to three premature stimuli from one or more sites in the ventricles in an effort to induce VT that would be substantially the same as a VT that would arise spontaneously (Roy et al. 1983; Buxton et al. 2000). Such procedures have provided a basis for testing antiarrhythmic drugs as well as for risk stratification for SCD. However, such procedures are invasive and impossible to implement on a routine basis. Although electrophysiological testing has the ability to predict patients at increased risk for SCD in patients with ischaemic heart disease and reduced ejection fraction, it has poor negative predictive power, and its predictive power for individuals in other populations at elevated risk for SCD is largely unknown (Thomas & Josephson, 2008). As discussed below, work is underway to develop patient‐specific models that can be used to assess the anatomical substrate for arrhythmias to help guide risk stratification and to assess targets for ablation in individuals (Trayanova, 2014).
Dynamics of wave propagation
Given the difficulties in developing risk stratification for SCD using standard clinical approaches, we think it is useful to discuss this question from a more basic theoretical perspective. Excitable media, such as the heart, can support propagation of waves of electrical activity. Simple and complex mathematical models reproduce and in some cases can be used to predict dynamic features associated with the onset of arrhythmias in cardiac tissues (Moe et al. 1977; Keener, 1981; Guevara et al. 1984; Glass et al. 1986; Courtemanche et al. 1989; Fenton & Karma, 1998; Chialvo et al. 1990; Lewis & Guevara, 1990; Vinet et al. 1990; Ito & Glass, 1992; Courtemanche et al. 1993; Starmer et al. 1993; Karma, 1994; Schulte‐Frohlinde et al. 2002). We discuss conduction block, alternans, and initiation of extra beats.
Conduction block
Following an excitation wave, cardiac tissue has a refractory period and a stimulus or excitation will not conduct if it is delivered too close to a previous stimulus. In addition, the velocity of propagation of cardiac excitation typically slows as the latency from a previous conducted beat decreases. As a consequence, as the pacing frequency of cardiac tissue increases, either from an external pacemaker or from an intrinsic source (the sinus pacemaker, an ectopic atrial or ventricular pacemaker, or a reentrant circuit) conduction may be blocked locally (Keener, 1981; Shrier et al. 1987; Courtemanche & Winfree, 1991; Karma, 1994). Such blocking of conduction plays a crucial role in atrio‐ventricular heart block, unidirectional block leading to reentry and termination of reentry as a consequence of anti‐tachycardia pacing, and transitions from monomorphic to polymorphic tachycardias. If there is blocking of cardiac excitation locally due to refractory tissue, there may be initiation of rotors or three‐dimensional reentrant waves (Krinsky, 1966; Courtemanche & Winfree, 1991; Karma, 1994; Fenton & Karma, 1998; Bub et al. 2002).
Alternans
During rapid pacing of cardiac tissue, in addtion to blocked beats, an instability may lead to alternation of electrophysiological properties (Chialvo et al. 1990; Lewis & Guevara, 1990; Vinet et al. 1990; Echebarria & Karma, 2002). The action potential duration decreases as the time from a preceding action potential decreases. If this decrease is steep enough, there will be an alternation of action potential duration (the technical term for this instability is a period‐doubling bifurcation). Since local alternation of action potential duration in ventricular tissue would lead to T‐wave alternans, this may provide insight into the reason why T‐wave alternans is associated with an increased risk of SCD (Rosenbaum et al. 1994; ten Tusscher & Panfilov, 2006; Weiss et al. 2006; Qu et al. 2010).
Initiation of extra beats
Cardiac tissue can also generate extra beats. Early afterdepolarizations and delayed afterdepolarizations may arise due to genetic abnormalities (Amin et al. 2013) or drug effects (Farkas & Nattel, 2010; Gonano et al. 2011). Theoretical models rely on detailed analyses of the underlying ionic mechanisms (Qu et al. 2013). It is also possible to have localized regions of cardiac tissue make a transition from an excitable cell to a pacemaker leading to an ectopic focus (Antzelevitch et al. 1983). If the localized pacemaker has a lower frequency than the sinus rhythm, then the pacemaker may be entrained in a one‐to‐one fashion to the sinus rhythm, and the localized pacemaker properties would not be evident. If, however, the pacemaker was in an anatomical region that had entrance block, the pacemaker might act as an ectopic focus generating a parasystolic rhythm (Antzelevitch et al. 1983), such as sometimes occurs in the right ventricular outflow tract. Although mathematical models for transtions from oscillatory to non‐oscillatory states are well developed (the transition can take place via a Hopf bifurcation) (Qu et al. 2013), theoretical models of physiological mechanisms that lead to such transtions, such as variation of catecholamine levels (Shen & Zipes, 2014), need to be developed better. Independent of specific mechanisms for ectopic pacemaker initiation, the consequence of ectopic pacemakers can be analysed theoretically (see the next section).
Dynamics of PVCs
The preceding section summarized several dynamic features of cardiac models. Transitions from sinus rhythm to VT often occur as a consequence of a PVC. Consequently, it is of interest to consider possible mechanisms of PVCs. In this section we briefly review this work from a perspective of improving methods for risk stratification. Although PVCs are commonly found in normal individuals and may be considered benign, there is also significant clinical data that show that frequent PVCs confer a risk for SCD (Kotler et al. 1973; Berkowitsch et al. 2004; Carrim & Khan, 2005; Dukes et al. 2015). The onset of ventricular tachyarrhythmia preceded by PVCs has been documented in different clinical contexts. In idiopathic VF patients, spontaneous VF episodes are initiated by PVCs with very short coupling intervals (Viskin et al. 1997; Haïssaguerre et al. 2008). Spontaneous VF initiated by PVCs with short–long–short sequences have been documented in 72% of VF episodes in patients with early repolarization and in 15% of VF episodes in Brugada syndrome patients (Nam et al. 2010). In long QT syndrome patients, the onset of polymorphic VT (torsade de pointes) is typically pause dependent: most episodes of polymorphic VT are preceded by either a single PVC or short–long–short sequences that arise due to PVCs in complex rhythms, such as ventricular bigeminy (Viskin et al. 1996). In other clinical contexts such as myocardial infarction, there is a less clear relationship between PVCs and the onset of VT or VF. An early study with 77 patients describes 492 episodes of VT that occurred during the first 48 h following the onset of myocardial infarction. PVCs were observed in 76% of cases during the 10 min preceding VT, but the incidence dropped to 40% during the minute prior to VT (Bluzhas & Lukshiene, 1985). The frequency of PVCs per minute had a large range (1 to 20) and the complexity of PVCs also varied, with groups of PVCs (24%), early PVCs (12%) and multifocal PVCs (30%). Since transition from VT to VF was observed in only 1% of cases, the frequency and complexity of PVCs were concluded to be of little predictive value for VF in patients with myocardial infarction. Regardless of the lack of a clear link between PVCs and the onset of VT or VF in myocardial infarction patients, the assessment of PVCs as precursor of VT or VF is still important for certain groups of myocardial infarction patients (Makikallio et al. 2005; Lerma et al. 2013). We now consider different mechanisms for the generation of PVCs.
Reentry
Reentry of ventricular beats can lead to PVCs. One scenario for this involves entrance of ventricular activity from a sinus beat into a ventricular scar resulting from an infarct via an isthmus of viable tissue (Josephson, 2008). If the conduction velocity is sufficiently slow, the exit from the scar tissue would occur after the refractory time of the sinus beat and lead to a PVC. The conduction time through the scar would be expected to be affected by a variety of factors including the sinus rate, prior conduction through the scar, drugs, and levels of catecholamines. Depending on these factors, it is possible that the PVC would lead to VT using the same pathway (Bogun et al. 2008). In that case one would expect that the morphology of the VT would be close to the morphology of the PVC (Pogwizd & Corr, 1987; Bogun et al. 2008; Chinushi et al. 2011). Conduction through the isthmus would be expected to show similar properties to conduction in excitable media including decremented conduction and various patterns of block (Callans et al. 1993). Theoretical models have addressed the dynamics of PVCs arising from reentry (Kinoshita et al. 1990; Schulte‐Frohlinde et al. 2002).
Parasystole
Ventricular parasystole results from the interaction between an ectopic ventricular pacemaker and the normal sinus rhythm. In the classic manifestation both rhythms are independent. Given different mechanisms for PVCs, this is the mechanism best understood. A PVC will be observed if the timing of the parasystolic beats falls outside the refractory period of the sinus beat; the ectopic beat will be blocked if it falls during the refractory period of the sinus beat; and there will be a fusion beat with different morphology if the ectopic beat falls simultaneously with the onset of the QRS complex of the ECG. Classically, parasystole is identified on the ECG from a triad of characteristics: intervals between PVCs are multiples of a common divisor, the coupling interval from the sinus beat to a PVC varies, and there are fusion beats. Mathematical analysis of parasystole revealed surprising properties for the sequence of integers that gives the number of sinus beats between two consecutive PVCs. In this sequence assuming that sinus frequency and the ectopic frequencies are fixed, there will in general be three integers that can be predicted based on the periods of the sinus and ectopic beats and the refractory time following the sinus beat (Glass et al. 1986). An important early paper by Moe and Jalife demonstrated that it is also possible for the parasystolic focus to be re‐set (or modulated) by the sinus beat (Moe et al. 1977). If only one sinus beat falls between two consecutive PVCs, plotting the interval between two consecutive PVCs as a function of the timing of the sinus beat in the PVC cycle generates a resetting curve. Modulated parasystole can lead to repetitive sequences such as bigeminy, in which there is one sinus beat between two consecutive PVCs, or trigeminy, in which there are two sinus beats between two consecutive PVCs (Antzelevitch et al. 1983; Courtemanche et al. 1989). An early paper that assessed malignancy of arrhythmias in individual patients concluded that although parasystole could lead to frequent ectopy, it did not confer an elevated risk of SCD (Kotler et al. 1973). Thus, identification of a parasystolic mechanism for PVCs based on dynamic features could play an important role in risk stratification. Because of the independent timing of sinus and parasystolic beats, in parasystole there will be occurrences of PVCs at different times in the sinus cycle and in particular on the T‐wave of the ECG. If one assumes that PVCs occurring during the T‐wave are proarrhythmogenic (Smirk, 1949), one would expect that parasystole would be a malignant rather than a benign arrhythmia. This apparent inconsistency indicates the possible importance of mechanism of PVCs, and bears further investigation.
Early afterdepolarizations and delayed afterdepolarizations
Early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs) are depolarizations following a cardiac action potential that occur earlier than would otherwise be expected. EADs occur during the action potential and DADs occur shortly after the action potential. In contrast to parasystolic PVCs, PVCs that arise as a consequence of EADs and DADs give rise to malignant arrhythmias. Although EADs and DADs are typically identified based on intracellular recordings, optical recordings from tissue culture provide a powerful new method to assess repolarization abnormalities on a tissue level. Several different mechanisms can lead to abnormal depolarizations; for a review see Roberts et al. (2012). They include genetic mutations of sodium channels leading to enhanced automaticity (Brugada syndrome; Brugada et al. 2013), genetic mutations of potassium channels leading to prolonged action potentials (long QT syndrome; Amin et al. 2013), drug effects leading to prolonged action potentials (acquired long QT syndrome; Farkas & Nattel, 2010), defects in calcium handling that may be exacerbated in conditions of catecholaminergic stimulation (catecholaminergic polymorphic ventricular tachycardia; Mohamed et al. 2007), drug effects from digitalis and other drugs leading to calcium overload and DADs (Gonano et al. 2011). Recent papers have developed mathematical models of the cardiac action potential and demonstrated bifurcations that can lead to the onset of arrhythmias induced by EADs (Chang et al. 2012; Karagueuzian et al. 2013; Qu et al. 2013).
Detailed ionic models have been useful in understanding the circumstances that can lead to PVCs. EADs that occur during phase 2 of the action potential (phase‐2 EADs) can markedly prolong the action potential duration but cannot generate propagating PVCs (Sato et al. 2009). An animal model of isolated ventricular rabbit myocytes exposed to H2O2 produced phase‐2 EADs due to slowed IKs activation (Qu et al. 2013). Phase‐3 EADs, produced for example with IKr blockade using dofetilide in isolated rabbit ventricular myocites (Guo et al. 2007), can prolong the action potential duration and also generate propagating PVCs (Qu et al. 2013). Phase‐3 EADs can also result from electrotonic coupling between regions with and without phase‐2 EADs in heterogeneous tissue (Maruyama et al. 2011). Finally, late phase‐3 EADs, which are similar to DADs, can arise from intact tissue (not from isolated myocytes) when the Ca2+ transient outlasts the action potential duration and Ca2+ inward currents depolarize the membrane voltage until it reaches the I Na threshold (Qu et al. 2013).
Heartprint
Since some mechanisms for PVCs are associated with lethal arrhythmias, distinguishing mechanisms of PVCs in individual patients may provide useful clinical information. Based on theoretical analysis of models of PVC generation, we have developed a visual method, called the heartprint, to characterize long‐term dynamics of the mechanism of PVCs. It was created as a visual and qualitative method to display statistical properties of intervals related to the PVCs: (i) the coupling interval (CI) between each PVC and the preceding sinus beat, (ii) the time interval between PVCs (VV interval), and (iii) the number of intervening sinus beats (NIBs) between two consecutive PVCs (Schulte‐Frohlinde et al. 2002). To explore the dependencies between these indexes and the underlying cardiac rhythm (represented by NN intervals, usually sinus rhythm), bivariate histograms are represented as grey scale plots where the ordinate is the NN interval, the abscissa axis is one of the heartprint indexes, and the incidence of VV intervals, NIB values and CI are indicated in the greyscale plots respectively, where the relative frequency of occurrence is indicated by the shading (darker shading representing more events) (Fig. 1). The example in Fig. 1 is derived from a Holter recording in the Sudden Cardiac Death Database in a patient who experienced an episode of sustained VT of unknown cause (Goldberger et al. 2000). The CI, observed in the ECG (Fig. 1 A), remained fixed throughout the 24 h recording (Fig. 1 B).
Figure 1. Example of ECG trace (A) and heartprint (B) from a Holter recording of the Sudden Cardiac Death database (no. 47) with repetitive arrhythmia with fixed coupling interval (CI) .
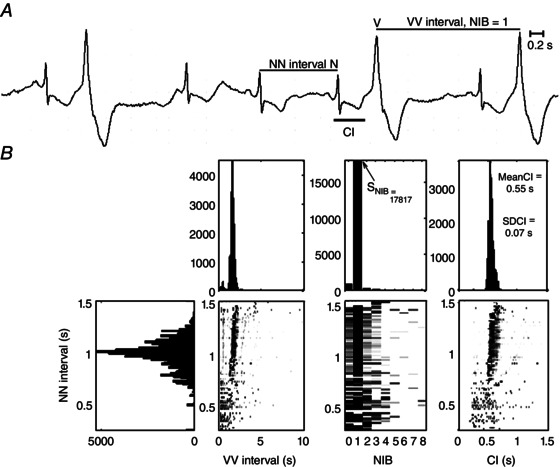
NIB, number of intervening sinus beats between two PVCs; SNIB, NIB index.
Based on the hearprint, new quantitative indices have been found which are associated with a higher risk of SCD, including the standard deviation of the CI (SDCI) and the NIB score (SNIB), which is the number of incidences (i.e. the height of the histogram in the heartprint) of the most prevalent values in the range 1–8 (Lerma et al. 2013). In survivors of acute myocardial infarction with depressed left ventricular function, repeating forms of PVCs (SNIB ≥ 83) was associated with a higher risk of fatal or near‐fatal sudden arrhythmias (primary endpoint of the study) with a hazard ratio of 3.5 (1.3–9.5). Although this is better than some other non‐invasive markers of risk for SCD based on the signal averaged ECG or heart rate turbulence (Huikuri et al. 2009), it requires further development and confirmation before it can be incorporated in new methods for risk stratification. A fixed CI (SDCI ≤ 80 ms) was associated with a higher risk of all‐cause mortality with a hazard ratio of 2.89 (1.13–7.38) and with cardiac death with a hazard ratio of 3.36 (1.00–11.22) (Lerma et al. 2013).
The heartprint was created as a visual tool to identify patterns in the interaction between the arrhythmia characteristics and the sinus interval, which could be associated to specific arrhythmogenic mechanisms (Schulte‐Frohlinde et al. 2002). Patterns with fixed CI may reflect either reentry or triggered activity, while patterns with broad CI are expected in parasystole. Using data from the Holter cardiac death database (Goldberger et al. 2000), a pattern of fixed CI and persistent ventricular bigeminy (as is shown in the example of Fig. 1) was found in 6 out of 15 recordings (Lerma et al. 2007). Since the recordings with such a pattern had also long QT interval and polymorphic VT (torsade de pointes), it is possible that EADs are the mechanism underlying the PVCs.
We conclude that it is important to understand the mechanisms of PVCs (Glass et al. 2011). Others have a similar perspective. A recent study found an increased risk associated with frequent PVCs (Dukes et al. 2015). A commentary on this paper concludes, ‘A patient‐specific physiological characterization is crucial to better understand the mechanistic links between [PVCs] and increased risk of adverse cardiovascular events’ (Santangeli & Marchlinski, 2015).
Realistic and patient‐specific diagnosis and modelling
In contrast to the simplified models discussed above (Dynamics of wave propagation), realistic models attempt to incorporate detailed features of cardiac tissue including multiple types of ion channels, anisotropy, extracellular medium, and three‐dimensional geometries including measured geometries in the individual. Of course, depending on the particular objectives, only a subset of these factors may be included in any particular model. The realistic models are playing an important role in understanding the effects of genetic mutations on ion channels, and the analysis of anti‐ and pro‐arrhythmic drugs on cardiac function (Fenton & Cherry, 2008).
Noble and colleagues pioneered the development of ionic models for cardiac cells. Detailed models now exist for a broad range of cell types and a broad range of species. Since the development of ionic models is based on voltage clamp experiments, development of these models is difficult due to many technical issues, including effects of tissue preparation on ionic properties, differences between homologous ion channels in different species, and variability between different cells of the same tissue type in the same species (Carusi et al. 2012; Britton et al. 2013; Sanchez et al. 2014).
The main observable of the models is the membrane voltage, and after fitting individual currents to available data, the model parameters are adjusted to fit the membrane voltage under some set of physiological conditions. One limitation of the approach is that the resulting models do not necessarily agree with other sorts of experimental data including dependence of cell and tissue properties on stimulation frequency (restitution properties) and the resetting properties of pacemaker cells. Indeed, different models of the same tissue often have important differences when exercised under various stimulation routines (Cherry & Fenton, 2007). A discussion of the problems involved in parameter estimation and possible approaches can be found in an article in this issue (Krogh‐Madsen et al. 2016).
In parallel with development of realistic ionic models, work is also underway on the development of whole‐heart models that incorporate three‐dimensional geometry of the heart based on magnetic resonance imaging (MRI). Although the potential for this has been recognized for a long time (Vigmond et al. 2001; Virag et al. 2002), there are many challenges given the complexity of the anatomy and physiology. However, significant progress has been made in simulation of both atrial and ventricular arrhythmias. Computational models can provide insight into determining potential loci for ablations (Hwang et al. 2014; Trayanova, 2014; McDowell et al. 2015). Whole‐heart modelling of the ventricles based on MRI imaging may also provide a novel approach to risk stratification. Trayanova and colleagues have pioneered this analysis with the study of the induction of VT in 13 patient‐specific models (Ashikaga et al. 2013; Trayanova et al. 2014). Inducibility of VT in the computer model correlates well with the risk of VT or VF in patients with implanted ICDs (Dickfeld et al. 2011; Ringenberg et al. 2014, 2015) and may play a potential role in risk stratification.
Challenges for risk stratification for SCD
Risk stratification for SCD poses a major problem for clinical medicine. Although ICDs offer the prospect of a life‐saving therapy for a subset of patients, they are highly invasive and expensive, and have associated risks due to infection and various types of malfunction including mis‐sensing and lead fracture. Since ICDs are not triggered in a large fraction of the patients who receive them, and many patients who experience SCD do not meet current criteria for implantation, there is a need for new approaches (Goldberger et al. 2008, 2011; Bastiaenen et al. 2012; Bilchick et al. 2012; Huikuri, 2015).
Our review of current approaches to risk stratification, theoretical modelling of cardiac dynamics, and dynamics of PVCs makes clear that the range of normal and pathological dynamics is large. Although simple algorithms and criteria for medical diagnoses and treatments have traditionally provided a basis for decision making in clinical medicine, recent work emphasizes that individual differences must be evaluated in decision making. Personalized or precision medicine, in which individual variability is taken into account for prevention, diagnosis and therapy of disease, has the potential to improve medical care (Collins & Varmus, 2015). Although precision medicine is most often discussed in the context of the influence of genetic differences on cancer biology, we believe that individualized analysis of cardiac dynamics will be an essential component of the application of personalized medicine to cardiology.
Some aspects of cardiac electrophysiology already adopt a personalized approach. Medical procedures involving cardiac ablation to treat arrhythmias involve mapping the intracardiac substrate using a variety of protocols to determine the arrhythmic loci and pathways in each individual. Such procedures are invasive, expensive and require specialized personnel. As mentioned earlier, significant progress is being made to assess the cardiac arrhythmia substrate using MRI combined with whole‐heart modelling to identify ablation targets and to do risk stratification for SCD (Trayanova, 2014). Since whole heart models simulate complex anatomy and physiology, these methods now require powerful computational facilities and high levels of technical expertise.
In a recent review, Goldberger et al. write, ‘Given the desirability of accurate risk stratification [for sudden cardiac death] and the long history of research in this area, it is important to understand why the field is not further advanced.’ (Goldberger et al. 2011).
We believe that it is possible to do a better job for risk stratification for SCD. However, there is a need for new strategies employing richer data subjected to mathematical analysis and modelling. Until recently, most research has been carried out by clinicians based on data collected over a limited time frame. Since cardiac arrhythmias often have significant fluctuations during the day and tachycardias have paroxysmal onsets, determining the physiological changes that lead to the arrhythmia onset and mechanism presents a problem in data collection as well as a problem in analysis. However, the increasing development of wearable devices, cloud computing, and big data offers the prospect of carrying out analysis of electrocardiographic data with a perspective of developing a better understanding of the mechanisms of arrhythmias and the factors that lead to their onset. We believe that it will be useful to examine fluctuating dynamics of arrhythmias over many hours or days, develop quantitative methods to identify distinguishing features, and carry out simulations of dynamics to compare with the data. Since the changing physiological conditions during the day may lead to bifurcations in the dynamics, it will be necessary for people with mathematical expertise to collaborate with cardiac electrophysiologists to develop appropriate theoretical models. Although this is still at a research stage, it provides the potential for understanding mechanisms of arrhythmias in individuals. We believe that this is a crucial step for the development of better methods for risk stratification for SCD.
Additional information
Competing interests
The authors hold a patent in the subject matter.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
L.G. thanks the Heart and Stroke Foundation of Canada for support of this research (grant number HSFCG‐15‐0009251).
Biographies
Claudia Lerma received a PhD in Biomedical Sciences from the Universidad Nacional Autónoma de México and was a Postdoctoral Fellow at the Center for Nonlinear Dynamics in McGill University. She is a Medical Sciences Researcher at the Instituto Nacional de Cardiología Ignacio Chávez, México. Her research interests include nonlinear dynamics of cardiovascular control and non‐invasive methods for prediction of sudden cardiac death.

Leon Glass obtained a PhD in Chemistry from the University of Chicago and was a Postdoctoral Fellow in the University of Edinburgh, University of Chicago and University of Rochester. He is currently a Professor of Physiology and the Isadore Rosenfeld Chair in Cardiology at McGill University, Montreal, Quebec, Canada. His research interests include nonlinear dynamics applied to the physiology of visual perception, cardiac arrhythmias and genetic networks.
This review was presented at the symposium Cardiac arrhythmias: challenges for diagnosis and treatment, which took place at McGill University, Montreal, QC, Canada, 6–7 November 2014.
References
- Albert CM & Stevenson WG (2013). Implantable cardioverter‐defibrillators for primary prevention of sudden cardiac death: too little and too late? Circulation 128, 1721–1723. [DOI] [PubMed] [Google Scholar]
- Amin AS, Pinto YM & Wilde AA (2013). Long QT syndrome: beyond the causal mutation. J Physiol 591, 4125–4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Bernstein MJ, Feldman HN & Moe GK (1983). Parasystole, reentry, and tachycardia: a canine preparation of cardiac arrhythmias occurring across inexcitable segments of tissue. Circulation 68, 1101–1115. [DOI] [PubMed] [Google Scholar]
- Ashikaga H, Arevalo H, Vadakkumpadan F, Blake RC III, Bayer JD, Nazarian S, Muz ZM, Tandri H, Berger RD, Calkins H, Herzka DA, Trayanova NA & Halperin HR (2013). Feasibility of image‐based simulation to estimate ablation target in human ventricular arrhythmia. Heart Rhythm 10, 1109–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Au‐Yeung WM, Reinhall P, Poole JE, Anderson J, Johnson G, Fletcher RD, Moore HJ, Mark DB, Lee KL & Bardy GH (2015). SCD‐HeFT: Use of RR interval statistics for long‐term risk stratification for arrhythmic sudden cardiac death. Heart Rhythm 12, 2058–2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, McNulty SE, Clapp‐Channing N, Davidson‐Ray LD, Fraulo ES, Fishbein DP, Luceri RM & Ip JH (2005). Amiodarone or an implantable cardioverter‐defibrillator for congestive heart failure. N Engl J Med 352, 225–237. [DOI] [PubMed] [Google Scholar]
- Barthel P, Schneider R, Bauer A, Ulm K, Schmitt C, Schomig A & Schmidt G (2003). Risk stratification after acute myocardial infarction by heart rate turbulence. Circulation 108, 1221–1226. [DOI] [PubMed] [Google Scholar]
- Bastiaenen R, Batchvarov V & Gallagher MM (2012). Ventricular automaticity as a predictor of sudden death in ischaemic heart disease. Europace 14, 795–803. [DOI] [PubMed] [Google Scholar]
- Bayer JD, Narayan SM, Lalani GG & Trayanova NA (2010). Rate‐dependent action potential alternans in human heart failure implicates abnormal intracellular calcium handling. Heart Rhythm 7, 1093–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitsch A, Zareba W, Neumann T, Erdogan A, Nitt SM, Moss AJ & Pitschner HF (2004). Risk stratification using heart rate turbulence and ventricular arrhythmia in MADIT II: usefulness and limitations of a 10‐minute Holter recording. Ann Noninvasive Electrocardiol 9, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilchick KC, Stukenborg GJ, Kamath S & Cheng A (2012). Prediction of mortality in clinical practice for Medicare patients undergoing defibrillator implantation for primary prevention of sudden cardiac death. J Am Coll Cardiol 60, 1647–1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluzhas J & Lukshiene D (1985). Ventricular tachycardia in myocardial infarction: relation to heart rate and premature ventricular contractions. Eur Heart J 6, 745–750. [DOI] [PubMed] [Google Scholar]
- Bogun F, Crawford T, Chalfoun N, Kuhne M, Sarrazin JF, Wells D, Good E, Jongnarangsin K, Oral H, Chugh A, Pelosi F & Morady F (2008). Relationship of frequent postinfarction premature ventricular complexes to the reentry circuit of scar‐related ventricular tachycardia. Heart Rhythm 5, 367–374. [DOI] [PubMed] [Google Scholar]
- Britton OJ, Bueno‐Orovio A, Van AK, Lu HR, Towart R, Gallacher DJ & Rodriguez B (2013). Experimentally calibrated population of models predicts and explains intersubject variability in cardiac cellular electrophysiology. Proc Natl Acad Sci USA 110, E2098–E2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brugada P, Brugada J & Roy D (2013). Brugada syndrome 1992–2012: 20 years of scientific excitement, and more. Eur Heart J 34, 3610–3615. [DOI] [PubMed] [Google Scholar]
- Bub G, Shrier A & Glass L (2002). Spiral wave generation in heterogeneous excitable media. Phys Rev Lett 88, 058101. [DOI] [PubMed] [Google Scholar]
- Buxton AE, Lee KL, DiCarlo L, Gold MR, Greer GS, Prystowsky EN, O'Toole MF, Tang A, Fisher JD, Coromilas J, Talajic M & Hafley G (2000). Electrophysiologic testing to identify patients with coronary artery disease who are at risk for sudden death. Multicenter Unsustained Tachycardia Trial Investigators. N Engl J Med 342, 1937–1945. [DOI] [PubMed] [Google Scholar]
- Callans DJ, Hook BG & Josephson ME (1993). Comparison of resetting and entrainment of uniform sustained ventricular tachycardia. Further insights into the characteristics of the excitable gap. Circulation 87, 1229–1238. [DOI] [PubMed] [Google Scholar]
- Carrim ZI & Khan AA (2005). Mean frequency of premature ventricular complexes as predictor of malignant ventricular arrhythmias. Mt Sinai J Med 72, 374–380. [PubMed] [Google Scholar]
- Carusi A, Burrage K & Rodriguez B (2012). Bridging experiments, models and simulations: an integrative approach to validation in computational cardiac electrophysiology. Am J Physiol Heart Circ Physiol 303, H144–H155. [DOI] [PubMed] [Google Scholar]
- Chang MG, Chang CY, de Lange E, Xu L, O'Rourke B, Karagueuzian HS, Tung L, Marban E, Garfinkel A, Weiss JN, Qu Z & Abraham MR (2012). Dynamics of early afterdepolarization‐mediated triggered activity in cardiac monolayers. Biophys J 102, 2706–2714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz‐Lopez R, Wang Z, Antzelevitch C, O'Brien RE, Schulze‐Bahr E, Keating MT, Towbin JA & Wang Q (1998). Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature 392, 293–296. [DOI] [PubMed] [Google Scholar]
- Chen X, Hu Y, Fetics BJ, Berger RD & Trayanova NA (2011). Unstable QT interval dynamics precedes ventricular tachycardia onset in patients with acute myocardial infarction: a novel approach to detect instability in QT interval dynamics from clinical ECG. Circ Arrhythm Electrophysiol 4, 858–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Tereshchenko LG, Berger RD & Trayanova NA (2013). Arrhythmia risk stratification based on QT interval instability: an intracardiac electrocardiogram study. Heart Rhythm 10, 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry EM & Fenton FH (2007). A tale of two dogs: analyzing two models of canine ventricular electrophysiology. Am J Physiol Heart Circ Physiol 292, H43–H55. [DOI] [PubMed] [Google Scholar]
- Chialvo DR, Gilmour RF Jr & Jalife J (1990). Low dimensional chaos in cardiac tissue. Nature 343, 653–657. [DOI] [PubMed] [Google Scholar]
- Chinushi M, Furushima H, Hosaka Y, Izumi D & Aizawa Y (2011). Ventricular fibrillation and ventricular tachycardia triggered by late‐coupled ventricular extrasystoles in a Brugada syndrome patient. Pacing Clin Electrophysiol 34, e1–e5. [DOI] [PubMed] [Google Scholar]
- Collins FS & Varmus H (2015). A new initiative on precision medicine. N Engl J Med 372, 793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa M, Goldberger AL & Peng CK (2002). Multiscale entropy analysis of complex physiologic time series. Phys Rev Lett 89, 068102. [DOI] [PubMed] [Google Scholar]
- Courtemanche M, Glass L & Keener JP (1993). Instabilities of a propagating pulse in a ring of excitable media. Phys Rev Lett 70, 2182. [DOI] [PubMed] [Google Scholar]
- Courtemanche M, Glass L, Rosengarten MD & Goldberger AL (1989). Beyond pure parasystole: promises and problems in modeling complex arrhythmias. Am J Physiol Heart Circ Physiol 257, H693–H706. [DOI] [PubMed] [Google Scholar]
- Courtemanche M & Winfree AT (1991). Re‐entrant rotating waves in a Beeler–Reuter based model of two‐dimensional cardiac electrical activity. Int J Bifurcation Chaos 1, 431–444. [Google Scholar]
- DeMazumder D, Lake DE, Cheng A, Moss TJ, Guallar E, Weiss RG, Jones SR, Tomaselli GF & Moorman JR (2013). Dynamic analysis of cardiac rhythms for discriminating atrial fibrillation from lethal ventricular arrhythmias. Circ Arrhythm Electrophysiol 6, 555–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyell MW, Krahn AD & Goldberger JJ (2015). Sudden cardiac death risk stratification. Circ Res 116, 1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickfeld T, Tian J, Ahmad G, Jimenez A, Turgeman A, Kuk R, Peters M, Saliaris A, Saba M, Shorofsky S & Jeudy J (2011). MRI‐Guided ventricular tachycardia ablation: integration of late gadolinium‐enhanced 3D scar in patients with implantable cardioverter‐defibrillators. Circ Arrhythm Electrophysiol 4, 172–184. [DOI] [PubMed] [Google Scholar]
- Dukes JW, Dewland TA, Vittinghoff E, Mandyam MC, Heckbert SR, Siscovick DS, Stein PK, Psaty BM, Sotoodehnia N, Gottdiener JS & Marcus GM (2015). Ventricular ectopy as a predictor of heart failure and death. J Am Coll Cardiol 66, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echebarria B & Karma A (2002). Instability and spatiotemporal dynamics of alternans in paced cardiac tissue. Phys Rev Lett 88, 208101. [DOI] [PubMed] [Google Scholar]
- Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias‐Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL et al (1991). Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med 324, 781–788. [DOI] [PubMed] [Google Scholar]
- Epstein AE, DiMarco JP, Ellenbogen KA, Estes NA III, Freedman RA, Gettes LS, Gillinov AM, Gregoratos G, Hammill SC, Hayes DL, Hlatky MA, Newby LK, Page RL, Schoenfeld MH, Silka MJ, Stevenson LW, Sweeney MO, Smith SC Jr, Jacobs AK, Adams CD, Anderson JL, Buller CE, Creager MA, Ettinger SM, Faxon DP, Halperin JL, Hiratzka LF, Hunt SA, Krumholz HM, Kushner FG, Lytle BW, Nishimura RA, Ornato JP, Page RL, Riegel B, Tarkington LG & Yancy CW (2008). ACC/AHA/HRS 2008 Guidelines for Device‐Based Therapy of Cardiac Rhythm Abnormalities: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the ACC/AHA/NASPE 2002 Guideline Update for Implantation of Cardiac Pacemakers and Antiarrhythmia Devices) developed in collaboration with the American Association for Thoracic Surgery and Society of Thoracic Surgeons. J Am Coll Cardiol 51, e1–e62. [DOI] [PubMed] [Google Scholar]
- Farkas AS & Nattel S (2010). Minimizing repolarization‐related proarrhythmic risk in drug development and clinical practice. Drugs 70, 573–603. [DOI] [PubMed] [Google Scholar]
- Fenton FH & Cherry EM (2008). Models of cardiac cell. Scholarpedia 3, 1868. [Google Scholar]
- Fenton FH & Karma A (1998). Vortex dynamics in three‐dimensional continuous myocardium with fiber rotation: filament instability and fibrillation. Chaos 8, 20–47. [DOI] [PubMed] [Google Scholar]
- Garfinkel A, Kim YH, Voroshilovsky O, Qu Z, Kil JR, Lee MH, Karagueuzian HS, Weiss JN & Chen PS (2000). Preventing ventricular fibrillation by flattening cardiac restitution. Proc Natl Acad Sci USA 97, 6061–6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass L, Goldberger AL & Belair J (1986). Dynamics of pure parasystole. Am J Physiol Heart Circ Physiol 251, H841–H847. [DOI] [PubMed] [Google Scholar]
- Glass L, Guevara MR & Shrier A (1987). Universal bifurcations and the classification of cardiac arrhythmias. Ann N Y Acad Sci 504, 168–178. [DOI] [PubMed] [Google Scholar]
- Glass L, Lerma C & Shrier A (2011). New methods for the analysis of heartbeat behavior in risk stratification. Front Physiol 2, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger AL, Amaral LA, Glass L, Hausdorff JM, Ivanov PC, Mark RG, Mietus JE, Moody GB, Peng CK & Stanley HE (2000). PhysioBank, PhysioToolkit, and PhysioNet: components of a new research resource for complex physiologic signals. Circulation 101, E215–E220. [DOI] [PubMed] [Google Scholar]
- Goldberger JJ, Buxton AE, Cain M, Costantini O, Exner DV, Knight BP, Lloyd‐Jones D, Kadish AH, Lee B, Moss A, Myerburg R, Olgin J, Passman R, Rosenbaum D, Stevenson W, Zareba W & Zipes DP (2011). Risk stratification for arrhythmic sudden cardiac death: identifying the roadblocks. Circulation 123, 2423–2430. [DOI] [PubMed] [Google Scholar]
- Goldberger JJ, Cain ME, Hohnloser SH, Kadish AH, Knight BP, Lauer MS, Maron BJ, Page RL, Siscovick D, Stevenson WG & Zipes DP (2008). American Heart Association/American College of Cardiology Foundation/Heart Rhythm Society scientific statement on noninvasive risk stratification techniques for identifying patients at risk for sudden cardiac death: a scientific statement from the American Heart Association Council on Clinical Cardiology Committee on Electrocardiography and Arrhythmias and Council on Epidemiology and Prevention. Circulation 118, 1497–1518. [PubMed] [Google Scholar]
- Gonano LA, Sepulveda M, Rico Y, Kaetzel M, Valverde CA, Dedman J, Mattiazzi A & Vila PM (2011). Calcium‐calmodulin kinase II mediates digitalis‐induced arrhythmias. Circ Arrhythm Electrophysiol 4, 947–957. [DOI] [PubMed] [Google Scholar]
- Guevara MR, Ward G, Shrier A & Glass L (1984). Electrical alternans and period doubling bifurcations. IEEE Comput Cardiol 562, 167–170. [Google Scholar]
- Guo D, Zhao X, Wu Y, Liu T, Kowey PR & Yan GX (2007). L‐type calcium current reactivation contributes to arrhythmogenesis associated with action potential triangulation. J Cardiovasc Electrophysiol 18, 196–203. [DOI] [PubMed] [Google Scholar]
- Haïssaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, Pasquie JL, Nogami A, Babuty D, Yli‐Mayry S, De Chillou C, Scanu P, Mabo P, Matsuo S, Probst V, Le Scouarnec S, Defaye P, Schlaepfer J, Rostock T, Lacroix D, Lamaison D, Lavergne T, Aizawa Y, Englund A, Anselme F, O'Neill M, Hocini M, Lim KT, Knecht S, Veenhuyzen GD, Bordachar P, Chauvin M, Jais P, Coureau G, Chene G, Klein GJ & Clementy J (2008). Sudden cardiac arrest associated with early repolarization. N Engl J Med 358, 2016–2023. [DOI] [PubMed] [Google Scholar]
- Huikuri HV (2015). Where to go in risk stratification for sudden cardiac death: Are P values enough? Heart Rhythm 12, 2067–2068. [DOI] [PubMed] [Google Scholar]
- Huikuri HV, Exner DV, Kavanagh KM, Aggarwal SG, Mitchell LB, Messier MD, Becker D, Sheldon RS & Bloch‐Thomsen PE (2010). Attenuated recovery of heart rate turbulence early after myocardial infarction identifies patients at high risk for fatal or near‐fatal arrhythmic events. Heart Rhythm 7, 229–235. [DOI] [PubMed] [Google Scholar]
- Huikuri HV, Raatikainen MJ, Moerch‐Joergensen R, Hartikainen J, Virtanen V, Boland J, Anttonen O, Hoest N, Boersma LV, Platou ES, Messier MD & Bloch‐Thomsen PE (2009). Prediction of fatal or near‐fatal cardiac arrhythmia events in patients with depressed left ventricular function after an acute myocardial infarction. Eur Heart J 30, 689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang M, Kwon SS, Wi J, Park M, Lee HS, Park JS, Lee YS, Shim EB & Pak HN (2014). Virtual ablation for atrial fibrillation in personalized in‐silico three‐dimensional left atrial modeling: comparison with clinical catheter ablation. Prog Biophys Mol Biol 116, 40–47. [DOI] [PubMed] [Google Scholar]
- Ito H & Glass L (1992). Theory of reentrant excitation in a ring of cardiac tissue. Physica D 56, 84–106. [Google Scholar]
- Jain R, Singh R, Yamini S & Das MK (2014). Fragmented ECG as a risk marker in cardiovascular diseases. Curr Cardiol Rev 10, 277–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson ME (2008). Clinical Cardiac Electrophysiology: Techniques and Interpretations, 4th edn Lippincott Williams and Wilkins, Philadelphia. [Google Scholar]
- Karagueuzian HS, Stepanyan H & Mandel WJ (2013). Bifurcation theory and cardiac arrhythmias. Am J Cardiovasc Dis 3, 1–16. [PMC free article] [PubMed] [Google Scholar]
- Karma A (1994). Electrical alternans and spiral wave breakup in cardiac tissue. Chaos 4, 461–472. [DOI] [PubMed] [Google Scholar]
- Keener JP (1981). On cardiac arrhythmias: AV conduction block. J Math Biol 12, 215–225. [Google Scholar]
- Kinoshita S, Konishi G & Kinoshita Y (1990). Mechanism of ventricular extrasystoles with fixed coupling. A theoretical model derived from the concept of longitudinal dissociation in the reentrant pathway of extrasystoles. J Electrocardiol 23, 249–254. [DOI] [PubMed] [Google Scholar]
- Kleiger RE, Miller JP, Bigger JT Jr & Moss AJ (1987). Decreased heart rate variability and its association with increased mortality after acute myocardial infarction. Am J Cardiol 59, 256–262. [DOI] [PubMed] [Google Scholar]
- Kotler MN, Tabatznik B, Mower MM & Tominaga S (1973). Prognostic significance of ventricular ectopic beats with respect to sudden death in the late postinfarction period. Circulation 47, 959–966. [DOI] [PubMed] [Google Scholar]
- Krinsky VI (1966). Excitation propagation in heterogeneous medium (modes similar to cardiac fibrillation). Biofizika 11, 676–683. [PubMed] [Google Scholar]
- Krogh‐Madsen T & Christini DJ (2012). Nonlinear dynamics in cardiology. Annu Rev Biomed Eng 14, 179–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh‐Madsen T, Sobie EA & Christini DJ (2016). Improving myocyte model fidelity and utility via dynamic electrophysiology protocols and optimization algorithms. J Physiol 594, 2525–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Rovere MT, Bigger JT, Marcus FI, Mortara A & Schwartz PJ (1998). Baroreflex sensitivity and heart‐rate variability in prediction of total cardiac mortality after myocardial infarction. ATRAMI (Autonomic Tone and Reflexes After Myocardial Infarction) Investigators. Lancet 351, 478–484. [DOI] [PubMed] [Google Scholar]
- Lerma C, Gorelick A, Ghanem RN, Glass L & Huikuri HV (2013). Patterns of ectopy leading to increased risk of fatal or near‐fatal cardiac arrhythmia in patients with depressed left ventricular function after an acute myocardial infarction. Europace 15, 1304–1312. [DOI] [PubMed] [Google Scholar]
- Lerma C, Lee CF, Glass L & Goldberger AL (2007). The rule of bigeminy revisited: analysis in sudden cardiac death syndrome. J Electrocardiol 40, 78–88. [DOI] [PubMed] [Google Scholar]
- Lewis TJ & Guevara MR (1990). Chaotic dynamics in an ionic model of the propagated cardiac action potential. J Theor Biol 146, 407–432. [DOI] [PubMed] [Google Scholar]
- Makikallio TH, Barthel P, Schneider R, Bauer A, Tapanainen JM, Tulppo MP, Schmidt G & Huikuri HV (2005). Prediction of sudden cardiac death after acute myocardial infarction: role of Holter monitoring in the modern treatment era. Eur Heart J 26, 762–769. [DOI] [PubMed] [Google Scholar]
- Maruyama M, Lin SF, Xie Y, Chua SK, Joung B, Han S, Shinohara T, Shen MJ, Qu Z, Weiss JN & Chen PS (2011). Genesis of phase 3 early afterdepolarizations and triggered activity in acquired long‐QT syndrome. Circ Arrhythm Electrophysiol 4, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDowell KS, Zahid S, Vadakkumpadan F, Blauer J, MacLeod RS & Trayanova NA (2015). Virtual electrophysiological study of atrial fibrillation in fibrotic remodeling. PLoS One 10, e0117110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moe GK, Jalife J, Mueller WJ & Moe B (1977). A mathematical model of parasystole and its application to clinical arrhythmias. Circulation 56, 968–979. [DOI] [PubMed] [Google Scholar]
- Mohamed U, Napolitano C & Priori SG (2007). Molecular and electrophysiological bases of catecholaminergic polymorphic ventricular tachycardia. J Cardiovasc Electrophysiol 18, 791–797. [DOI] [PubMed] [Google Scholar]
- Moss AJ, Zareba W, Hall WJ, Klein H, Wilber DJ, Cannom DS, Daubert JP, Higgins SL, Brown MW & Andrews ML (2002). Prophylactic implantation of a defibrillator in patients with myocardial infarction and reduced ejection fraction. N Engl J Med 346, 877–883. [DOI] [PubMed] [Google Scholar]
- Nam GB, Ko KH, Kim J, Park KM, Rhee KS, Choi KJ, Kim YH & Antzelevitch C (2010). Mode of onset of ventricular fibrillation in patients with early repolarization pattern vs. Brugada syndrome. Eur Heart J 31, 330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan SM, Bayer JD, Lalani G & Trayanova NA (2008). Action potential dynamics explain arrhythmic vulnerability in human heart failure: a clinical and modeling study implicating abnormal calcium handling. J Am Coll Cardiol 52, 1782–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan SM, Franz MR, Lalani G, Kim J & Sastry A (2007). T‐wave alternans, restitution of human action potential duration, and outcome. J Am Coll Cardiol 50, 2385–2392. [DOI] [PubMed] [Google Scholar]
- Nieminen T, Scirica BM, Pegler JR, Tavares C, Pagotto VP, Kanas AF, Sobrado MF, Nearing BD, Umez‐Eronini AA, Morrow DA, Belardinelli L & Verrier RL (2014). Relation of T‐wave alternans to mortality and nonsustained ventricular tachycardia in patients with non‐ST‐segment elevation acute coronary syndrome from the MERLIN‐TIMI 36 trial of ranolazine versus placebo. Am J Cardiol 114, 17–23. [DOI] [PubMed] [Google Scholar]
- Norris PR, Stein PK & Morris JA Jr (2008). Reduced heart rate multiscale entropy predicts death in critical illness: a study of physiologic complexity in 285 trauma patients. J Crit Care 23, 399–405. [DOI] [PubMed] [Google Scholar]
- Pogwizd SM & Corr PB (1987). Reentrant and nonreentrant mechanisms contribute to arrhythmogenesis during early myocardial ischemia: results using three‐dimensional mapping. Circ Res 61, 352–371. [DOI] [PubMed] [Google Scholar]
- Priori SG, Schwartz PJ, Napolitano C, Bloise R, Ronchetti E, Grillo M, Vicentini A, Spazzolini C, Nastoli J, Bottelli G, Folli R & Cappelletti D (2003). Risk stratification in the long‐QT syndrome. N Engl J Med 348, 1866–1874. [DOI] [PubMed] [Google Scholar]
- Qu Z, Xie LH, Olcese R, Karagueuzian HS, Chen PS, Garfinkel A & Weiss JN (2013). Early afterdepolarizations in cardiac myocytes: beyond reduced repolarization reserve. Cardiovasc Res 99, 6–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Xie Y, Garfinkel A & Weiss JN (2010). T‐wave alternans and arrhythmogenesis in cardiac diseases. Front Physiol 1, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringenberg J, Deo M, Filgueiras‐Rama D, Pizarro G, Ibanez B, Peinado R, Merino JL, Berenfeld O & Devabhaktuni V (2014). Effects of fibrosis morphology on reentrant ventricular tachycardia inducibility and simulation fidelity in patient‐derived models. Clin Med Insights Cardiol 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringenberg J, Deo M, Filgueiras‐Rama D, Pizarro G, Ibanez B, Peinado R, Trayanova N, Miller M, Merino JL, Berenfeld O & Devabhaktuni V (2015). Corrigendum to Effects of fibrosis morphology on reentrant ventricular tachycardia inducibility and simulation fidelity in patient‐derived models. Clin Med Insights Cardiol 8(Suppl 1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts BN, Yang PC, Behrens SB, Moreno JD & Clancy CE (2012). Computational approaches to understand cardiac electrophysiology and arrhythmias. Am J Physiol Heart Circ Physiol 303, H766–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum DS, Jackson LE, Smith JM, Garan H, Ruskin JN & Cohen RJ (1994). Electrical alternans and vulnerability to ventricular arrhythmias. N Engl J Med 330, 235–241. [DOI] [PubMed] [Google Scholar]
- Roy D, Waxman HL, Kienzle MG, Buxton AE, Marchlinski FE & Josephson ME (1983). Clinical characteristics and long‐term follow‐up in 119 survivors of cardiac arrest: relation to inducibility at electrophysiologic testing. Am J Cardiol 52, 969–974. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Bueno‐Orovio A, Wettwer E, Loose S, Simon J, Ravens U, Pueyo E & Rodriguez B (2014). Inter‐subject variability in human atrial action potential in sinus rhythm versus chronic atrial fibrillation. PLoS One 9, e105897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangeli P & Marchlinski FE (2015). Ventricular ectopy as a modifiable risk factor for heart failure and death: “deja vu all over again” may be a good thing. J Am Coll Cardiol 66, 110–112. [DOI] [PubMed] [Google Scholar]
- Sato D, Xie LH, Sovari AA, Tran DX, Morita N, Xie F, Karagueuzian H, Garfinkel A, Weiss JN & Qu Z (2009). Synchronization of chaotic early afterdepolarizations in the genesis of cardiac arrhythmias. Proc Natl Acad Sci USA 106, 2983–2988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte‐Frohlinde V, Ashkenazy Y, Goldberger AL, Ivanov PC, Costa M, Morley‐Davies A, Stanley HE & Glass L (2002). Complex patterns of abnormal heartbeats. Phys Rev E Stat Nonlin Soft Matter Phys 66, 031901. [DOI] [PubMed] [Google Scholar]
- Shen MJ & Zipes DP (2014). Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res 114, 1004–1021. [DOI] [PubMed] [Google Scholar]
- Shrier A, Dubarsky H, Rosengarten M, Guevara MR, Nattel S & Glass L (1987). Prediction of complex atrioventricular conduction rhythms in humans with use of the atrioventricular nodal recovery curve. Circulation 76, 1196–1205. [DOI] [PubMed] [Google Scholar]
- Shusterman V, Aysin B, Gottipaty V, Weiss R, Brode S, Schwartzman D & Anderson KP (1998). Autonomic nervous system activity and the spontaneous initiation of ventricular tachycardia. ESVEM Investigators. Electrophysiologic Study Versus Electrocardiographic Monitoring Trial. J Am Coll Cardiol 32, 1891–1899. [DOI] [PubMed] [Google Scholar]
- Smirk FH (1949). R waves interrupting T waves. Br Heart J 11, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starmer CF, Biktashev VN, Romashko DN, Stepanov MR, Makarova ON & Krinsky VI (1993). Vulnerability in an excitable medium: analytical and numerical studies of initiating unidirectional propagation. Biophys J 65, 1775–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Task Force of the European Society of Cardiology and the North American Society of Pacing and Electrophysiology (1996). Heart rate variability: standards of measurement, physiological interpretation and clinical use. Circulation 93, 1043–1065. [PubMed] [Google Scholar]
- ten Tusscher KH & Panfilov AV (2006). Alternans and spiral breakup in a human ventricular tissue model. Am J Physiol Heart Circ Physiol 291, H1088–H1100. [DOI] [PubMed] [Google Scholar]
- Thomas KE & Josephson ME (2008). The role of electrophysiology study in risk stratification of sudden cardiac death. Prog Cardiovasc Dis 51, 97–105. [DOI] [PubMed] [Google Scholar]
- Trayanova NA (2014). Mathematical approaches to understanding and imaging atrial fibrillation: significance for mechanisms and management. Circ Res 114, 1516–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trayanova NA, Boyle PM, Arevalo HJ & Zahid S (2014). Exploring susceptibility to atrial and ventricular arrhythmias resulting from remodeling of the passive electrical properties in the heart: a simulation approach. Front Physiol 5, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tung R, Zimetbaum P & Josephson ME (2008). A critical appraisal of implantable cardioverter‐defibrillator therapy for the prevention of sudden cardiac death. J Am Coll Cardiol 52, 1111–1121. [DOI] [PubMed] [Google Scholar]
- Vadakkumpadan F, Trayanova N & Wu KC (2014). Image‐based left ventricular shape analysis for sudden cardiac death risk stratification. Heart Rhythm 11, 1693–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verrier RL, Klingenheben T, Malik M, El‐Sherif N, Exner DV, Hohnloser SH, Ikeda T, Martinez JP, Narayan SM, Nieminen T & Rosenbaum DS (2011). Microvolt T‐wave alternans physiological basis, methods of measurement, and clinical utility–consensus guideline by International Society for Holter and Noninvasive Electrocardiology. J Am Coll Cardiol 58, 1309–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigmond EJ, Ruckdeschel R & Trayanova N (2001). Reentry in a morphologically realistic atrial model. J Cardiovasc Electrophysiol 12, 1046–1054. [DOI] [PubMed] [Google Scholar]
- Vinet A, Chialvo DR & Jalife J (1990). Irregular dynamics of excitation in biologic and mathematical models of cardiac cells. Ann N Y Acad Sci 601, 281–298. [DOI] [PubMed] [Google Scholar]
- Virag N, Jacquement V, Henriquez CS, Zozor S, Blanc O, Vesin JM, Pruvot E & Kappenberg L (2002). Study of atrial arrhythmias in a computer model based on magnetic resonance images of human atria. Chaos 12, 754–763. [DOI] [PubMed] [Google Scholar]
- Viskin S, Alla SR, Barron HV, Heller K, Saxon L, Kitzis I, Hare GF, Wong MJ, Lesh MD & Scheinman MM (1996). Mode of onset of torsade de pointes in congenital long QT syndrome. J Am Coll Cardiol 28, 1262–1268. [DOI] [PubMed] [Google Scholar]
- Viskin S, Lesh MD, Eldar M, Fish R, Setbon I, Laniado S & Belhassen B (1997). Mode of onset of malignant ventricular arrhythmias in idiopathic ventricular fibrillation. J Cardiovasc Electrophysiol 8, 1115–1120. [DOI] [PubMed] [Google Scholar]
- Weiss JN, Garfinkel A, Karagueuzian HS, Nguyen TP, Olcese R, Chen PS & Qu Z (2015). Perspective: a dynamics‐based classification of ventricular arrhythmias. J Mol Cell Cardiol 82, 136–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JN, Karma A, Shiferaw Y, Chen PS, Garfinkel A & Qu Z (2006). From pulsus to pulseless: the saga of cardiac alternans. Circ Res 98, 1244–1253. [DOI] [PubMed] [Google Scholar]
- Wellens HJ, Schwartz PJ, Lindemans FW, Buxton AE, Goldberger JJ, Hohnloser SH, Huikuri HV, Kaab S, La Rovere MT, Malik M, Myerburg RJ, Simoons ML, Swedberg K, Tijssen J, Voors AA & Wilde AA (2014). Risk stratification for sudden cardiac death: current status and challenges for the future. Eur Heart J 35, 1642–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wessel N, Ziehmann C, Kurths J, Meyerfeldt U, Schirdewan A & Voss A (2000). Short‐term forecasting of life‐threatening cardiac arrhythmias based on symbolic dynamics and finite‐time growth rates. Phys Rev E Stat Phys Plasmas Fluids Relat Interdiscip Topics 61, 733–739. [DOI] [PubMed] [Google Scholar]
- Wilson LD, Jeyaraj D, Wan X, Hoeker GS, Said TH, Gittinger M, Laurita KR & Rosenbaum DS (2009). Heart failure enhances susceptibility to arrhythmogenic cardiac alternans. Heart Rhythm 6, 251–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipes DP & Wellens HJ (1998). Sudden cardiac death. Circulation 98, 2334–2351. [DOI] [PubMed] [Google Scholar]