

Abstract
Genetically targeted therapies for rare Mendelian conditions are improving patient outcomes. Here, we present the case of a 20-mo-old female suffering from a rapidly progressing neurological disorder. Although diagnosed initially with a possible autoimmune condition, analysis of the child's exome resulted in a diagnosis of Brown–Vialetto–Van Laere syndrome 2 (BVVLS2). This new diagnosis led to a change in the therapy plan from steroids and precautionary chemotherapy to high-dose riboflavin. Improvements were reported quickly, including in motor strength after 1 mo. In this case, the correct diagnosis and appropriate treatment would have been unlikely in the absence of exome sequencing and careful interpretation. This experience adds to a growing list of examples that emphasize the importance of early genome-wide diagnostics.
Keywords: drooling, gait imbalance, neck muscle weakness, neurodegeneration, seesaw nystagmus, upper motor neuron abnormality
INTRODUCTION
Approximately 90 patients have been reported with Brown–Vialetto–Van Laere syndrome (BVVLS), inclusive of those with mutations in SLC52A2 and SLC52A3 (Bosch et al. 2012). SLC52A3 mutations causing BVVLS1 disease are hypothesized to disrupt the riboflavin transporter (RFVT3) that is responsible for riboflavin uptake from food into serum (Haack et al. 2012). Consistent with that role, RFVT3 deficiency is often detected in patients through reduced riboflavin or flavin adenine dinucleotide (FAD) plasma levels. However, the gene disrupted among patient with BVVLS2, SLC52A2, encodes the transmembrane riboflavin transporter (RFVT2) hypothesized to be responsible for riboflavin uptake from the serum into the target cells (Haack et al. 2012). Consistent with this hypothesis, children with SLC52A2 mutations (BVVLS2 disease) have been described to have flavin adenine dinucleotide (FAD) plasma levels within the normal range (Haack et al. 2012) before riboflavin supplementation. For this reason, BVVLS2 will be consistently missed in current routine biochemical screening. Guided by the underlying disruption of riboflavin transporters, Foley et al. (2014) reported 16 patients with SLC52A2 mutations who were treated with high-dose riboflavin therapy. All patients tolerated the therapy well with no evidence of toxicity. Critical to patient care, these data also showed that the sooner the riboflavin supplementation is initiated in patients with riboflavin transporter deficiency, the better the prognosis (Foley et al. 2014).
There is increasing evidence that exome sequencing provides a substantial clinical benefit in a minority of patients with undiagnosed or unresolved genetic conditions. Here, we present a striking example in which the early exome sequencing of a child with a devastating undiagnosed condition resulted directly in a transformative change in the patient's management that would not have otherwise been achieved.
RESULTS
Clinical Presentation and Family History
At the age of 15 mo, with previously normal developmental milestones and no pertinent family medical history, a young girl was evaluated after an abrupt onset of vertical nystagmus (Human Phenotype Ontology term [Köhler et al. 2014]: HP:0010544). Contrast-enhanced cranial magnetic resonance imaging was normal.
At age 16 mo, an ophthalmic examination showed subtle pallor of both optic nerves. An electroretinogram showed 20%–50% reduction of amplitude of b-waves under both scotopic and photopic conditions—consistent with outer retina pathology involving both the rods and the cones (HP:0000548). At the same time, the child started showing unsteady gait (HP:0002317) and hand tremors (HP:0002378). Electroencephalogram, hearing, infectious biochemical profiles were normal, except for borderline elevation of acylcarnitine. Laboratory investigations for an autoimmune disorder were normal.
She was treated for 2 mo with biweekly pulse intravenous corticosteroids and immunoglobulin, with transient improvements in the nystagmus and gait abnormality but recurrence each time. By 17 mo, she began showing weakness of both upper extremities (HP:0003484), increased gait unsteadiness, a head tilt to the right side, and excessive drooling (HP:0002307). Physical examination showed no upper motor neuron signs in the lower extremities.
By 19 mo, she had shown no sustained response to steroid treatment. Given the rapidly progressing neurodegeneration and the continued possibility of an autoimmune disorder, treatment with cyclophosphamide—a chemotherapy agent—was considered as the next step. At the same time, an expedited genetic analysis was initiated given the possibility that a genetic diagnosis might change the treatment plan (approved by the Institutional Review Board of Duke University Medical Center).
Genomic Analysis
Interpretation of the patient's whole-exome sequence identified a compound heterozygous genotype in the solute carrier family 52 (riboflavin transporter), member 2 gene (SLC52A2). Loss-of-function mutations in SLC52A2 are associated with the autosomal recessive disorder Brown–Vialetto–Van Laere Syndrome 2 (BVVLS2), a progressive neurodegenerative disease that is a consequence of severe riboflavin deficiency. BVVLS2 often results in respiratory failure and is associated with early mortality (Johnson et al. 2012; Foley et al. 2014).
The patient inherited a rare missense mutation (0.015% ESP6503 minor allele frequency) from her heterozygous father NM_001253815.1:c.1016T>C p.(Leu339Pro) and a rare (0.008% ESP6503 minor allele frequency) midprotein nonsense mutation from her heterozygous mother NM_001253815.1:c.808C>T p.(Gln270*) (Table 1). The paternal missense variant (L339P) has been reported as pathogenic in multiple patients (Johnson et al. 2012; Foley et al. 2014), with in vitro assays showing abolishment of 3H-Riboflavin uptake for the mutant protein (Foley et al. 2014). The maternal nonsense variant has not been previously linked to BVVLS2. This Q270* nonsense variant, however, would be predicted to abolish protein function. Consistent with this expectation we showed that the nonsense variant results in a truncated protein that appears subject to degradation (Fig. 1). In summary, the patient has a compound heterozygous genotype made of two loss-of-function mutations in SLC52A2. Both variants were confirmed to be present in the patient by a CLIA-certified laboratory. Consistent with expectation of loss of function in a gene intolerant to functional variation (Petrovski et al. 2013), this genotype means that the patient in fact has Brown–Vialetto–Van Laere Syndrome 2, an autosomal recessive condition caused by loss-of-function mutations in both copies of SLC52A2.
Table 1.
SLC52A2 (NM_001253815.1) variants
| Chr:Position GRCh37(hg19) | HGVS cDNA | HGVS protein | Predicted effect | dbSNP ID | Genotype | Parent of origin | Observed effect | EVS MAF (%) |
|---|---|---|---|---|---|---|---|---|
| 8:145583960 | c.808C>T | p.(Gln270*) | Stop gain | rs375088539 | Heterozygous | Mother | Degradation (Fig. 1) | 0.008 |
| 8:145584264 | c.1016T>C | p.(Leu339Pro) | Missense | rs148234606 | Heterozygous | Father | Abolished riboflavin uptake (Foley et al. 2014) | 0.015 |
Details about the two SLC52A2 loss-of-function variants identified in our patient. Chromosome and position coordinates based on GRCh37(hg19). Corresponding DNA and protein references are provided. EVS minor allele frequency (MAF) is based on up to 6503 samples. Both variants have been independently validated by a CLIA-certified laboratory.
Figure 1.
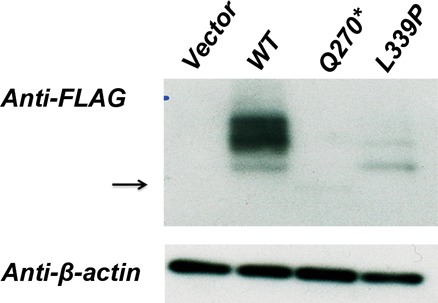
Western blot analysis using crude membrane fractions of HEK293 expressing the empty vector, WT-SLC52A2-Flag (WT), Q270*-SLC52A2-Flag (Q270X), and L339P-SLC52A2-Flag(L339P) variants. The blot was probed using anti-Flag and β-actin antibodies. The arrow highlights the location of the faint band of the truncated protein due to Q270* mutation at ∼30 kDa. The L339P mutation results in decreased protein expression. The functional effect of the L339P missense has been previously described (Foley et al. 2014).
Immediate Treatment Outcomes
In retrospect after the genetic diagnosis, the patient's presentation is considered a good match for the rare BVVLS2 diagnosis. Because of the urgent need to withdraw the scheduled precautionary chemotherapy for a possible autoimmune condition, and to introduce riboflavin, the whole-exome sequencing results and the correct diagnosis were communicated to the parents within 72 h of the genome interpretation. The same day, a 10 mg/kg/d dose of riboflavin was initiated (Fig. 2) and titrated up to 70 mg/kg/d over 2 mo. The child has tolerated the riboflavin therapy well. The parents reported a decrease in drooling within a week, followed by improved head tilt, an improved sense of well-being, and a renewed interest in play (Fig. 3). Two months after initiating therapy, the gait was noticed to be steadier and forearm strength had improved (power 3/5). Immediately upon initiating riboflavin therapy no new symptoms or further decline in any pretreatment symptoms were observed.
Figure 2.

Timeline of events starting from the day of phlebotomy (D0) to the day that the SLC52A2 sequencing results were communicated to the family (D24).
Figure 3.

Head tilt, pre- and posttreatment with riboflavin supplementation. (A) Three weeks pretreatment; (B) day 2 of treatment; (C) 3 wk posttreatment; (D) 4 wk posttreatment.
The patient's sister, a healthy appearing newborn female, had cord blood sent for genotyping of the familial SLC52A2 mutations. As a precautionary measure, she was initiated on 10 mg/kg/d of riboflavin while awaiting the results. The genotyping results indicated that the newborn sister was not a carrier for either SLC52A2 loss-of-function allele and was appropriately weaned off the precautionary riboflavin supplementation.
DISCUSSION
The carrier frequency of SLC52A2 pathogenic mutations suggests approximately 1 in 4.3 million children will be affected globally (Exome Aggregation Consortium; exac.broadinstitute.org/). The population rate is consistent with the clinically cited rate of autosomal recessive BVVLS2 being less than 1 in a million. Interestingly, there have been fewer reported diagnoses in the United States than the 73 the carrier frequency would suggest. Such differences between the expected and reported numbers of cases could in part stem from early and aggressive, yet inaccurate, treatment compromising recognition of clinical features that might have enabled an eventually correct diagnosis. Consistent with this, in BVVLS2, riboflavin or FAD biochemical plasma screening generally return normal values, making BVVLS2 challenging to diagnose early. This expected rate of BVVLS2 also suggests that there could be patients in the United States that are being mismanaged because of the failure to correctly diagnose their condition as BVVLS. We emphasize that sequencing and careful interpretation could result in a correct genetic diagnosis for such patients and ultimately facilitate changes in patient management.
To our knowledge, this is the first and earliest diagnosis of an isolated case of BVVLS2 through exome sequencing in the absence of clinical ascertainment for BVVLS, and it has resulted in the treatment of a patient at one of the youngest ages yet reported. This case adds to a rapidly growing number of examples in which exome sequencing provides a diagnosis that would not otherwise have been obtained and an immediate clinical benefit in the form of a genomic guided treatment (Choi et al. 2009; Bainbridge et al. 2011; Worthey et al. 2011; Bearden et al. 2014; Canna et al. 2014; Romberg et al. 2014). Our case also emphasizes that a timely genetic diagnosis using exome sequencing has avoided alternative toxic treatments and a patient management plan that would have been unlikely to help our patient's genetic condition. Finally, the expedited exome sequencing allowed us to provide prophylactic treatment to the at-risk newborn sibling while awaiting the results of her genotyping test. Such precautionary newborn screening highlights the possibility of implementing safe and effective presymptomatic treatment from birth.
It remains unclear what proportion of patients with serious genetic diseases would derive such clear clinical benefit from exome sequencing. Nevertheless, the accumulating evidence strongly suggests that this happens with sufficient regularity to warrant systematic application of exome sequencing, and eventually genome sequencing, in all unresolved childhood diseases where a genetic cause is suspected. Moreover, in a number of cases, including BVVLS2, the earlier that treatment is initiated, the better the prognosis. This creates a strong clinical imperative for applying exome sequencing as early as possible.
MATERIALS AND METHODS
Sequencing and Analysis
DNA was extracted from maternal, paternal, and proband blood samples and was exome sequenced on the HiSeq2500 using KAPA Biosystem's library preparation kit followed by whole-exome capture using Nimblegen SeqCap EZ V3.0rapid. Paired-end 2 × 100 read lengths were used for the exome capture sequencing. After processing the raw reads using a pipeline based on the GATK best practice protocol, the resulting alignments indicated that 96.2% of the 33,266,994 consensus coding sequence CCDS (release 14) sites had ≥10-fold coverage in all three family members. Individual-level coverage data indicate that all three family members had 100% of SLC52A2 covered with at least 10-fold coverage (Table 2). The sequence data were analyzed using our established trio sequencing protocols (Zhu et al. 2015) that identify qualifying variants forming genotypes not observed in the parents or in an external database of 6503 controls of convenience provided by the Exome Sequencing Project (ESP6500SI) (NHLBI GO Exome Sequencing Project; evs.gs.washington.edu).
Table 2.
Sequencing coverage
| Sample | Percentage of reads aligned | Average read coverage | Number of CCDS r14 sites with ≥10-fold coverage | Percentage of CCDS r14 with ≥10-fold coverage | Percentage of CCDS r14 with ≥20-fold coverage | Percentage of SLC52A2 sites (1354 bases) with ≥10-fold coverage |
|---|---|---|---|---|---|---|
| Proband | 99.51 | 63.91 | 32,282,510 | 97.04% | 92.43% | 100 |
| Mother | 99.47 | 74.84 | 32,491,343 | 97.67% | 94.64% | 100 |
| Father | 99.62 | 93.58 | 32,658,451 | 98.17% | 95.93% | 100 |
Sequencing coverage information for each of the three exome-sequenced family members. The 33.3 Mbp of consensus coding sequence (CCDS release 14) is used as the reference for protein-coding sequence coverage—this includes a 2-bp extension at both ends of exons to include putative canonical splice variants.
Western Blot
The human cDNA clone of SLC52A2 was ordered from PlasmID (Harvard Medical School, clone ID: HsCD00372773). The cDNA sequence was amplified using PrimeSTAR GXL DNA Polymerase (Takara, ID: R050A) and primers (Table 3) that introduced a HindIII restriction site 3′ of the sequence and a BamH1 site 5′ of the sequence. The linear cDNA and the p3XFlag-CMV 7.1 vector (Sigma-Aldrich, ID: E7533) were then digested with HindIII and BamH1 and ligated together using T4-ligase (New England Biolabs, ID: M0202S). The wild-type construct, WT-SLC52A2-Flag, was used to create Q270*-SLC52A2-Flag and L339P-SLC52A2-Flag by site-directed mutagenesis introducing the C>T (Q270*) and T>C (L339P) mutations (Table 3). Each construct was sequenced by Sanger to verify mutation and full cDNA sequence. HEK293 cells were transfected in triplicate using each of the listed constructs, and lysates were collected 48 h after transfection using RIPA buffer (Sigma-Aldrich, ID: R0278) and 1× protease inhibitor (Thermo Scientific, ID:88266). Protein concentrations were determined using the Pierce BCA protein assay kit (Thermo Scientific, ID: 23227). Of note, 2.5 μg of lysate was subjected to SDS-PAGE gel and transferred to a polyvinylidene difluoride membrane (Millipore, ID: ISEQ15150). The membrane was incubated with anti-Flag antibody (1:2000; Sigma-Aldrich, ID: F3165) or anti-β-actin (1:5000; Santa Cruz, ID: sc-47778). Proteins were visualized with the ECL Plus Western Blotting Detection System (GE Healthcare, ID: RPN2232).
Table 3.
Primer sequences used to create constructs for immunoblotting
| Primer name | Primer sequence |
|---|---|
| SLC52A2-HindIII-F | ttAAGCTTGCAGCACCCACGCCCGCCCG |
| SLC52A2-BamH1 | gtGGATCCTCAGGAGTCACAGGGGTCTG |
| SLC52A2-MutaF-Q270* | TAGCTTCTATCAGCCCGCAG |
| SLC52A2-MutaR-Q270* | ATAGGCCTTAGGGTCTGGAC |
| SLC52A2-MutaF-L339P | CGGGCGGCCTCTCTCTGCT |
| SLC52A2-MutaR-L339P | GCCCTGCCAAGGACCTGCA |
ADDITIONAL INFORMATION
Ethics Statement
The Institutional Review Board of Duke University Medical Center approved this research protocol (Pro00032301). Written informed consent was received from both parents.
Data Deposition and Access
Our patient consent does not permit patient sequence data to be uploaded to a data repository. The NM_001253815.1:p.(Leu339Pro) missense variant can be accessed through dbSNP rs148234606, and ClinVar SCV000221313. The NM_001253815.1:p.(Gln270*) nonsense variant can be accessed through dbSNP rs375088539, and ClinVar SCV000218457.
Acknowledgments
We thank the family for their enthusiasm to share their experience with the larger community. We thank members of the Institute for Genomic Medicine, formerly Center for Human Genome Variation, (Joshua Bridgers, Kenneth Cronin [former], Sally Gewalt, Jonathan Keebler [former], Zhong Ren, and Quanli Wang) for their ongoing commitment that enables this work. We would like to thank Angelica Struve and Tom Maniatis from the Maniatis Laboratory for their help and advice. We thank Dr. A. Reghan Foley of the Neuromuscular and Neurogenetic Disorders of Childhood Section, National Institute of Neurological Disorders and Stroke, National Institutes of Health, for her recommendations on the riboflavin dosing regimen and clinical surveillance while on this regimen, based on findings from studying an international cohort of BVVLS2 patients. We also thank the NHLBI GO Exome Sequencing Project and its ongoing studies that produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010). S.P. is a National Health and Medical Research Council (NHMRC) CJ Martin Fellow. The authors wish to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about.
Author Contributions
Patient recruitment and phenotyping: V.S., K.S., R.C., L.C., R.K., M.A.E-D., Y-H.J., and M.A.M. Sequence data analysis and interpretation: S.P. and D.B.G. Functional evaluation of variant: K.M.M., R.S.D., B.K., and S.P. Writing of manuscript: S.P., V.S., and D.B.G. All authors contributed to reviewing the final draft.
Competing Interest Statement
The authors have declared no competing interest.
REFERENCES
- Bainbridge MN, Wiszniewski W, Murdock DR, Friedman J, Gonzaga-Jauregui C, Newsham I, Reid JG, Fink JK, Morgan MB, Gingras MC, et al. 2011. Whole-genome sequencing for optimized patient management. Sci Transl Med 3: 87re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden D, Strong A, Ehnot J, DiGiovine M, Dlugos D, Goldberg EM. 2014. Targeted treatment of migrating partial seizures of infancy with quinidine. Ann Neurol 76: 457–461. [DOI] [PubMed] [Google Scholar]
- Bosch AM, Stroek K, Abeling NG, Waterham HR, Ijlst L, Wanders RJ. 2012. The Brown–Vialetto–Van Laere and Fazio Londe syndrome revisited: natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis 7: 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, DiMattia MA, Zaal KJ, Sanchez GA, Kim H, et al. 2014. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 46: 1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, Liu T, Tikhonova IR, Zumbo P, Nayir A, Bakkaloglu A, Ozen S, Sanjad S, et al. 2009. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci 106: 19096–19101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley AR, Menezes MP, Pandraud A, Gonzalez MA, Al-Odaib A, Abrams AJ, Sugano K, Yonezawa A, Manzur AY, Burns J, et al. 2014. Treatable childhood neuronopathy caused by mutations in riboflavin transporter RFVT2. Brain 137: 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack TB, Makowski C, Yao Y, Graf E, Hempel M, Wieland T, Tauer U, Ahting U, Mayr JA, Freisinger P, et al. 2012. Impaired riboflavin transport due to missense mutations in SLC52A2 causes Brown-Vialetto-Van Laere syndrome. J Inherit Metab Dis 35: 943–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JO, Gibbs JR, Megarbane A, Urtizberea JA, Hernandez DG, Foley AR, Arepalli S, Pandraud A, Simón-Sánchez J, Clayton P, et al. 2012. Exome sequencing reveals riboflavin transporter mutations as a cause of motor neuron disease. Brain 135: 2875–2882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler S, Doelken SC, Mungall CJ, Bauer S, Firth HV, Bailleul-Forestier I, Black GC, Brown DL, Brudno M, Campbell J, et al. 2014. The Human Phenotype Ontology project: linking molecular biology and disease through phenotype data. Nucleic Acids Res 42: D966–D974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. 2013. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet 9: e1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, Overton J, Meffre E, Khokha MK, Huttner AJ, et al. 2014. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 46: 1135–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, Serpe JM, Dasu T, Tschannen MR, Veith RL, et al. 2011. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med 13: 255–262. [DOI] [PubMed] [Google Scholar]
- Zhu X, Petrovski S, Xie P, Ruzzo EK, Lu YF, McSweeney KM, Ben-Zeev B, Nissenkorn A, Anikster Y, Oz-Levi D, et al. 2015. Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet Med 10.1038/gim.2014.191. [DOI] [PMC free article] [PubMed] [Google Scholar]