

SUMMARY
Induced regulatory T (iTreg) and T helper 17 (Th17) cells promote mucosal homeostasis. We used a T cell transfer model of colitis to compare the capacity of iTreg and Th17 cells to develop in situ following the transfer of naïve CD4+ CD45RBhi T cells into Rag1−/− C57BL/6 or BALB/c mice, the prototypical Th1/M1- and Th2/M2-prone strains. We found that the frequency and number of Foxp3+ iTreg cells and Th17 cells were significantly reduced in C57BL/6 mice compared to the BALB/c strain. C57BL/6 mice with colitis were also resistant to nTreg cell immunotherapy. Pre-treatment of C57BL/6 Rag1−/− mice with IL-4 plus IL-13, or with M2a but not M1 macrophages, dramatically increased the generation of iTreg and Th17 cells. Importantly M2a transfers, either as a pretreatment or in mice with established colitis, allowed successful immunotherapy with nTreg cells. M2a macrophages also reduced the generation of pathogenic ex-iTreg cells, suggesting that they stabilize the expression of Foxp3. Thus polarized M2a macrophages drive a directionally concordant expansion of the iTreg -Th17 cell axis and can be exploited as a therapeutic adjuvant in cell-transfer immunotherapy to reestablish mucosal tolerance.
INTRODUCTION
Mucosal tolerance involves maintaining a delicate balance between pro-inflammatory and anti-inflammatory immune responses in a complex and dynamic antigenic environment. This essential regulatory process requires considerable adaptability in the regulatory compartment, derived in part by the scalable production of iTreg cells that act to complement the more stable thymically drived nTreg cell lineage (1–3). Both iTreg and nTreg cells express the transcription factor forkhead boxP3 (Foxp3) and have similar gene expression profiles, suggesting that they utilize similar suppressive mechanisms and share a functional reciprocity (1). This has been demonstrated for Treg-produced IL-10 in a model of colitis, where either Treg cell type is sufficient for adequate delivery of this anti-inflammatory cytokine (4). In contrast to shared suppressive mechanisms, the TCR repertoires of iTreg and nTreg cells are distinct, a finding that is predicted by the developmental origins of the two cell types and is critical for Treg cell control (1). Together, these and other data are consistent with the generally held view that the nTreg TCR repertoires are biased toward self-recognition, while the iTreg TCR repertoires are focused on foreign antigens (5–7). Global Treg cell deficiency is associated with multi-organ autoimmune disease, as shown in thymectomyzed neonatal mice and in Foxp3-deficient mice and humans (8).
In several experimental models of autoimmune disease, iTreg cells are generated in vivo (9). Environmental factors, such as agonists for the aryl hydrocarbon receptor and the retinoic acid receptor, enhance their development (10–12). For example, polyclonal and TCR transgenic CD4+ T cells express Foxp3 during homeostatic expansion or after chronic exposure to intravenous antigen (13, 14). Similarly, oral administration of antigen acutely generates iTreg cells that promote antigen-specific tolerance in the gut (15). During the experimental colitis that follows the transfer of CD45RBhi naïve T cells into Rag1−/− hosts, iTreg cells are generated in situ (3). Similarly, iTreg cells are also generated when rescuing Foxp3 deficient mice with nTreg and Tconv cells. In rescued Foxp3− mice, selective iTreg cell depletion results in increased host T cell activation, increased host pro-inflammatory cytokine production and increased development of inflammatory tissue infiltrates (1). The collective data illustrates that mucosal tolerance depends upon the mechanisms that control iTreg cell development and stabilize iTreg cell function. Indeed, iTreg cells are unstable in models of GVHD, where IL-6 is abundant and decreases Foxp3 induction (16). Epigenetic studies of iTreg cells have confirmed increased methylation in conserved non-coding region 2 of the Foxp3 promoter, indicating unstable Foxp3 expression(1, 17).
TGF-β1 is a pleiotropic cytokine produced by many other cell types that is required for the generation of iTreg and Th17 cells. TGF-β1 has anti-inflammatory properties that counteract pro-inflammatory factors produced during inflammatory bowel disease (IBD) and other chronic autoimmune diseases. The pivotal role of TGF-β1 in the suppression of inflammation is highlighted in studies where the Tgfb1 or the Tgfbr2 genes were deleted, and the mice developed multi-organ inflammation that included the gut, similar to Foxp3- deficient mice (18–20). TGF-β1 can also act as a proinflammatory cytokine when coupled with other cytokine signals, as illustrated by requirement for TGF-β1 in the differentiation of Th17 cells (21). As a population, cells expressing IL-17 family members are associated with autoimmune diseases, although a proportion of Th17 cells are non-pathogenic. For example TGF-β1 and IL-6 promote the development of non-pathogenic Th17 cells that do not cause experimental autoimmune encephalomyelitis. However, IL-23 in combination with TGF-β1 and IL-6 promote pathogenic Th17 cells that also produce IFN-γ and are associated with severe EAE (22). Thus the role of TGF-βR signaling in counterbalancing iTreg or Th17 cell development is context-dependent.
In vivo, the critical cells necessary for the generation of iTreg and Th17 cells are unknown. Recently, cells of the innate immune system have emerged as important sources of support for a regulatory environment. For example, intestinal macrophages that produce IL-10 have been shown to support nTreg homeostasis in the gut (23, 24). Like T cells, macrophages play both pro-inflammatory and anti-inflammatory roles and are polarized depending on the cytokine environment. M1 macrophages produce TNF-α, IL-6, IL-12 and IL-23 and are inflammatory while M2a macrophages produce TGF-β1 and IL-10 and dampen inflammatory responses (25, 26). Thus polarized macrophages have the potential to influence the development of both effector and regulatory T cell responses.
Here, we used a T cell transfer model of colitis to determine the impact of polarized macrophages on the development and stability of iTreg and Th17 cells. Our data demonstrate that adoptive transfer of M2a macrophages drives expansion of the iTreg-Th17 cell axis, which can contribute to reestablishing immune homeostasis in the gut.
MATERIALS AND METHODS
Mice
Rag1−/− C57BL/6, Rag1−/− BALB/c, GT(Rosa)26SorEYFP, and Lyz2Cre mice were purchased from the Jackson Laboratory. CD45.1 and CD45.2 Foxp3EGFP on the C57BL/6 background, and Foxp3EGFP on the BALB/c background were generated and screened as previously described (27). The Animal Resource Committee at the Medical College of Wisconsin approved all animal experiments, and all mice were maintained under SPF conditions. Both male and female mice were used in all experiments.
Antibodies
The following antibodies were used: anti-CD4 (clone RM4), anti- CD62L (clone MEL-14), anti-IFN-γ (clone XMG1.2), anti-I-Ab (clone AF6-120.1), anti-CD40 (clone 3/23), anti-Bcl-2 (clone 3F11) from BD Biosciences; anti-TCRβ (clone H57-597), anti-CD45RB (clone C363.16A), anti-KLRG1 (clone 2F1), anti-PDL2 (clone 122), anti-Ki67 (clone SolA15) from eBioscience; anti-CD45.1 (clone A20), anti-CD44 (clone IM7), anti-CD103 (clone 2E7), anti-IL-17a (clone TC11-18H10.1), anti-F4/80 (clone BM8), anti-CD11b (clone M1/70), anti-CD86 (clone GL-1), anti-CD80 (clone 16-10A1), anti-PDL1 (clone 10F.9G2) from BioLegend, and anti-CD25 (clone PC61 5.3) from Invitrogen.
Flow cytometry and cell sorting
Single cell suspensions were prepared from spleen (SP), mesenteric lymph nodes (MLN), small intestine (SI) and colon, and stained as indicated. Data was collected on a custom four-laser LSRII and analyzed using FlowJo (FlowJo, LLC) software. For sorting, pooled splenocytes and lymph node cells (axillary, brachial, inguinal and mesenteric) were stained with anti-CD4 and anti-CD45RB, and sorted on the basis of antibody and EGFP fluorescence. All sorting was done on a FACSAria (Becton-Dickenson). The average purity and viability of the sorted CD4+ populations was 99.7+/− 0.05 and 93.20+/− 0.32; n=68, respectively.
Intracellular cytokine staining and cell labeling
Intracellular cytokine staining was performed after 5-hours of re-stimulation with PMA (5ng/ml; Sigma-Aldrich) and ionomycin (0.5μM; Sigma-Aldrich) in the presence of Golgi Plug (1μl/ml; BD Biosciences). Surface staining of cells was performed using a modified FACS buffer containing 10μg/ml brefeldin A (Sigma-Aldrich). Cells were stained on ice for 20 minutes with the primary antibodies: anti-CD4 and anti-TCRβ, then washed with the modified FACS buffer and fixed with 1% paraformaldehyde overnight at 4 degrees. After this incubation, cells were washed with 1ml PBS and then permeabilized with 1ml 0.1% Triton-X buffer. Intracellular staining was performed for 30 minutes at room temperature with anti-IL-17A and anti-IFN-γ. CD4+EGFP− T cells were labeled with the CellTrace™ Violet Cell Proliferation kit as per manufacturer protocol. Cells were analyzed by flow cytometry.
Colitis induction
Colitis was induced in 7–8 week old Rag1−/− mice by intra-peritoneal injection of 4×105 CD4+EGFP−CD45RBhi cells from Foxp3EGFP mice. Following cell transfer, mice were weighed twice weekly.
Isolation of lamina propria lymphocytes
The entire colon and the distal 15 cm of the small intestine were used as a source of intra-epithelial lymphocytes (IEL) and lamina propria lymphocytes (LPL). IELs were removed by gentle shaking of 0.5 cm intestinal sections for 30 minutes in buffer containing 10% FCS, 1mM DTT (Sigma Aldrich) and 5mM EDTA. IELs were removed and the remaining intestinal sections were digested with Collagenase D (1mg/ml; Roche) in the presence of DNase I (50U/ml; Worthington Biochemical). A discontinuous Percoll gradient (67%, 44%; Sigma Aldrich) was used to isolate washed LPL.
Histology
Complete colons were fixed in formalin, processed, and stained with H&E using a histology core facility. Blinded sections from the entire colon were examined by a pathologist (N.H.S.) and large intestine colitis scores were determined for the following inflammatory changes on a 4-point semi-quantitative scale, with 0 representing no change (28). The following features were considered: severity, depth and chronic nature of the inflammatory infiltrate, crypt abscess formation, granulomatous inflammation, epithelial cell hyperplasia, mucin depletion, ulceration, and crypt loss.
Pre-treatment of mice with cytokines and macrophages
Mice were pre-treated with IL-4 (50μg/kg; Peprotech) and/or IL-13 (25μg/kg; Peprotech) at days -7, -5 and -3 by intra-peritoneal injection. On day 0, mice received naïve T cells from Foxp3EGFP mice. In other experiments, mice were pre-treated with 2×105 undifferentiated or differentiated bone marrow derived macrophages by intra-peritoneal injection on day -1 followed by adoptive transfer of naïve T cells on day 0.
Differentiation of bone marrow-derived macrophages
Bone marrow was isolated from femurs and tibias of 6–12 week old mice. Bone marrow cells were plated at 2×106 cells per ml in DMEM F12 media (supplemented with 10% FCS, 5% L- glutamine, 1% penicillin-streptomycin, 1% gentamicin) on 10-cm petri dishes. Cells were either left in media (M0) or differentiated into M1 macrophages with 100ng/ml recombinant IFN-γ (Peprotech) and 20ng/ml LPS (Sigma Aldrich) or M2 macrophages with 100ng/ml recombinant IL-4 (Peprotech) and 50ng/ml recombinant IL-13 (Peprotech) for 7 days. Macrophages were stained with anti-F4/80 and sorted on the basis of antibody expression and for some experiments EYFP expression. Sort purity and viability was 94.35+/− 0.58 and 77.85+/− 1.90; n=64, respectively.
In vitro iTreg cell conversion
Sorted CD4+EGFP− cells from Foxp3EGFP mice (1×106/ml) were cultured in anti-CD3 mAb (clone 14-2C11 at 2.5μg/ml) coated dishes in the presence of soluble anti-CD28 mAb (clone 37.51 at 1μg/ml), 100U/ml IL-2 and TGF-β1 (5ng/ml; R&D systems), or acidified M2a culture supernatant as indicated. At 72 hours, cells were analyzed by flow cytometry for CD4 and EGFP expression. In some experiments, a blocking antibody for TGF-β1 (1D11) was used.
Colitis treatment with Treg cells
In some experiments, mice with colitis were treated by IP injection with 1×106 CD4+EGFP+ nTreg cells either 10 days or 31 days after colitis initiation. In other experiments, mice with colitis were treated by IP injection with a mixture containing 5×105 CD4+EGFP+ nTreg cells and 5×105 CD4+EGFP+ iTreg cells derived in vitro 10 days after colitis induction. All nTreg cells were purified by cell sorting from the spleens of Foxp3EGFP mice. Cell sorting was also used to purify In vitro derived iTreg cells prior to adoptive transfer.
RNA and cDNA isolation
Macrophages were stained and sorted on the basis of F4/80 expression. Total RNA was extracted using the RNeasy Mini Kit (Qiagen) according to manufacturer’s protocol. cDNA was synthesized with the Superscript III First Strand Synthesis System and oligonucleotide (dT) primers (Invitrogen) according to manufacturer’s protocol. Isolated cDNA was used for quantitative PCR.
Gene expression analysis
TRIzol (Invitrogen) was used to isolate total RNA from M0, M1, and M2a bone marrow macrophages. Labeled target was prepared and hybridized to Affymetrix 430 2.0 GeneChips in accordance with the manufacturer’s protocol. A minimum of three biological replicates were performed and the results were averaged. The data was normalized with the Robust Mulit-array Analysis (RMA) algorithm derived by the Bioconductor group (http://www.bioconductor.org) (29). Probe sets that exhibited a twofold difference (|log2 ratio| > 1.0) relative to M0 cells were identified and those that possessed a false discovery rate (FDR) <10% by the non-parametric rank product test (30) were used in subsequent analyses. The microarray data are available in the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/gds) under the accession number GSE72518 (31).
Quantitative PCR
Quantitative PCR used to measure macrophage gene products was performed in a StepOnePlus PCR System (Applied Biosystems) using the TaqMan Fast Universal PCR Master Mix (Life Technologies). Gapdh (Mm99999915–g1; Life Technologies) was used as a housekeeping gene. Pre-developed specific primers used were: Tgfb1 (Mm00441724-m1; Life Technologies); Il10 (Mm00439616-m1; Life Technologies); Arginase 1 (Mm00475988-m1; Life Technologies); Fizz1 (Mm00445109-m1; Life Technologies); Nos2 (Mm00440485-m1; Life Technologies); Ym1 (Mm00657889-m1; Life Technologies); Pdl1 (Mm00452054-m1; Life Technologies); and Pdl2 (Mm00451734-m1; Life Technologies).
The abundance of segmented filamentous bacteria (SFB) was estimated by real time qPCR as previously described (32) using the Applied Biosystems Step One Plus Real Time PCR System. Briefly, a short segment of the 16S rRNA gene (174 bp) was specifically amplified by real time PCR using a conserved 16S rRNA specific primer pair (Operon Technologies) to determine the total amount of bacteria in each sample. SFB abundance was determined using group specific primers (Operon Technologies, (32)). Bacterial numbers were estimated using standard curves constructed with reference bacterial DNA specific for SFB. Note that qPCR measures 16S gene copies per sample, not actual bacterial numbers or colony forming units.
Statistical analysis
Random coefficient models with unstructured variance covariance structure were used to compare the weight changes over time for different groups. Cubic curves were fitted to the data, and when the cubic term was non-significant, quadratic curves were used. Kaplan-Meier curves with Wilcoxon log-rank tests were used to compare the survival times. A nonparametric Kruskal-Wallis test was used for comparison of the other continuous variables. If the groups were significantly different, a Mann-Whitney test was used for pairwise comparisons. For variables with sample size n<5, ANOVA and t-test were used instead. Log transformations were applied to variables that were skewed before doing ANOVA or t-test. A p-value < 0.05 was considered significant. The SAS 9.4 software package was used for all analyses.
RESULTS
Strain-Dependent development of the iTreg-Th17 cell axis
The development of iTreg and Th17 cells is linked by a shared requirement for TGF-β1-mediated signals. At mucosal surfaces, iTreg cells play a specialized role in controlling Th2 responses (2), while Th17 cells commonly arise in associated with Th1-mediated inflammation. This suggests that a Th2 environment preferentially promotes iTreg cell development while a Th1 milieu skews toward Th17 cell development. To examine this hypothesis, we utilized a T cell transfer model of colitis to compare the development of iTreg and Th17 cells in BALB/c and C57BL/6 mice, the prototypical Th2/M2 and Th1/M1-prone strains.
Naïve CD45RBhi EGFP− CD4+ T cells were isolated by cell sorting from Foxp3EGFP mice and transferred into congenic Rag1−/− hosts, and the recipient mice were examined at regular intervals out to 40 days. The kinetics of weight loss, colitis development, and frequency of pro-inflammatory IFN-γ+ and IFN-γ+ IL-17A+ T cells were similar between the two strains (Figure S1A–L). However, the number of CD4+ T cells recovered from spleen and mesenteric lymph nodes was comparatively decreased by 4-fold in mice from the C57BL/6 background 40 days after the T cell transfer (Figure 1A). Consistent with this decrease, the frequency and number of Foxp3+ iTreg cells in the spleen, mesenteric lymph nodes, small intestine and colon of C57BL/6 mice were also significantly decreased, culminating in an overall 35-fold reduction in iTreg cells by 40 days (Figures 1B–F). Similarly, the frequency and number of CD4+IL-17A+ T cells that developed in the spleen, mesenteric lymph nodes, small intestine and colon lamina propria were decreased 8-fold in C57BL/6 mice when compared to BALB/c mice at the same time point (Figures 1G–J). These reductions were not likely the result of different rates of cell proliferation or apoptosis, as the frequency of Ki67+ and Bcl2+ iTreg cells was similar between the strains (Figure S1M). Together these data demonstrate that the C57BL/6 strain has a reduced capacity to induce the iTreg-Th17 cell axis following the transfer of naïve CD4+ T cells into Rag1−/− mice. The data raise the possibility that a Th2 milieu can support the development of CD4+ iTreg and IL-17A+ T cells, although in the absence of nTreg cells, the greater number of iTreg cells in BALB/c mice was not sufficient to slow the early course of disease progression.
Figure 1. Development of iTreg and Th17 cells during experimental colitis in Rag1−/− C57BL/6 and Rag1−/−BALB/c mice.
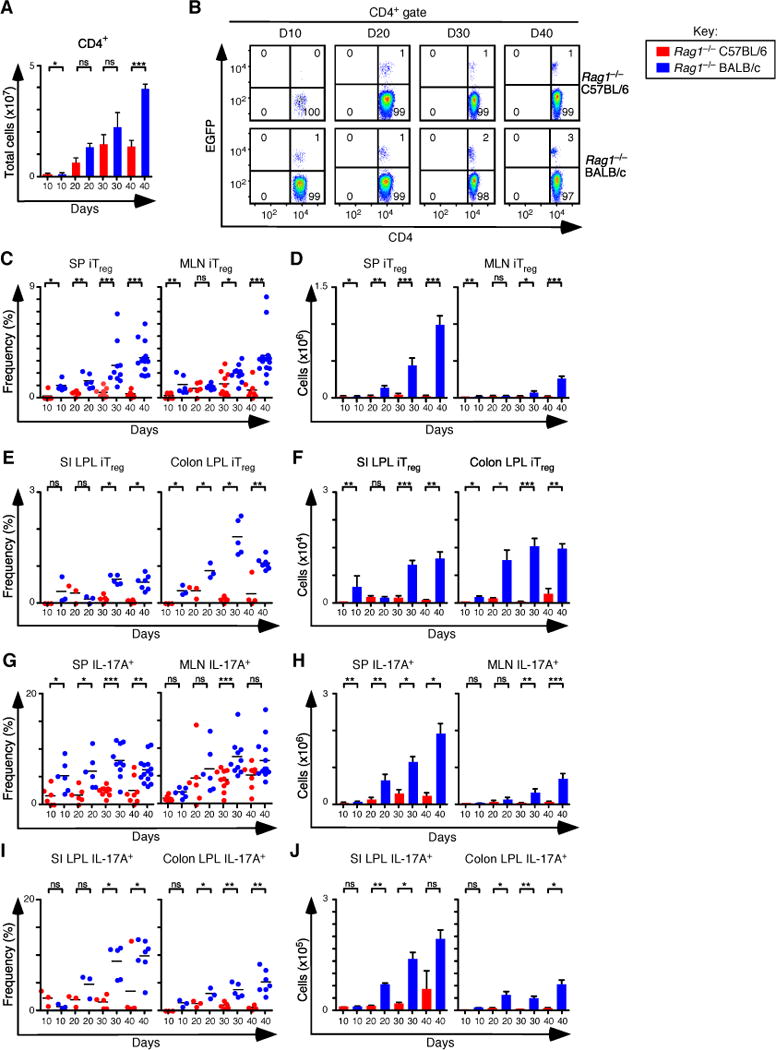
(A) Total number of CD4+ T cells recovered from spleen (SP) and mesenteric lymph nodes (MLN) of Rag1−/− C57BL/6 and Rag1−/− BALB/c at days 10 (n=6 for both), 20 (n=6 for both), 30 (n=10 for both), and 40 (n=8 Rag1−/− C57BL/6, and n=14 Rag1−/− BALB/c) after adoptive transfer of naïve T cells from Foxp3EGFP mice. (B) Representative flow cytometry analysis of CD4 and EGFP (Foxp3) expression to assess the frequency of iTreg cells in MLN for each group. Numbers in quadrants are averages. (C–D) Frequency (C) and number (D) of iTreg cells that develop in SP and MLN. (E–F) Frequency (E) and number (F) of iTreg cells that develop in small intestine (SI) and colon lamina propria at days 10 (n=3 for both), 20 (n=3 for both), 30 (n=5 for both), and 40 (n=4 for Rag1−/− C57BL/6 and n=7 for Rag1−/− BALB/c. (G–H) Frequency (G) and number (H) of Th17 cells that develop in SP and MLN. (I–J) Frequency (I) and number (J) of Th17 cells that develop in SI and colon lamina propria. Each symbol represents an individual mouse, and small horizontal lines represent the mean. Data are representative of 2–3 independent experiments, 2–7 mice per experiment. *p< 0.05, **p< 0.005, ***p<0.0005 and ns, not statistically different; Mann- Whitney test or t-test.
Pre-conditioning with IL-4 and IL-13 promotes iTreg and Th17 cell development
As BALB/c mice are Th2/M2-prone, we speculated that the canonical Th2 cytokines IL-4 and IL-13 might impact iTreg and Th17 cell differentiation in C57BL/6 mice. To examine this hypothesis, we pre-treated Rag1−/− C57BL/6 mice with IL-4 and IL-13 on days -7, -5, and-3, followed by adoptive transfer of naïve T cells on day 0. Control groups were not pre-treated or pre-treated with either IL-4 or IL-13 alone. In these pretreatment experiments, only the innate immune cells in Rag1−/− mice are influenced by the IL-4 and IL-13 cytokines. In the absence of nTreg cells, all mice lose weight and develop colitis as expected. However, pre-treatment of mice with the combination of IL-4 and IL-13 resulted in delayed weight loss and increased survival when compared to all the other groups (Figures 2A, B). The overall number of CD4+ T cells was also increased (Figure 2C). The frequency and the total number of iTreg cells increased up to 19 fold when compared to no pre-treatment controls (Figures 2D–F). Independent of pre-treatment regimens, all mice developed colitis as shown by the colitis scores of H&E-stained colon sections (Figures 2G, H). In parallel to iTreg cell increase, the frequency and total number of CD4+IL-17A+ T cells increased up to 5 fold when compared to the no pre-conditioning control group (Figures 2I–K). As noted, in these experiments nTreg cells were not added, so all mice eventually developed colitis and about half of the T cells in the spleen and about 1/3 of the T cells in the MLN expressed IFN-γ or both IFN-γ and IL-17A (Figures 2L–O). These data are consistent with the hypothesis that innate immune cells stimulated by IL-4 in combination with IL-13 develop an enhanced capacity to support iTreg and Th17 cell development.
Figure 2. Pre-treatment of Rag1−/− C57BL/6 mice with IL-4 and IL-13 enhances the development of the iTreg-Th17 cell axis.

(A) Weight change analysis of mice of Rag1−/− C57BL/6 mice not pre-treated or pre-treated with IL-4, IL-13, IL-4 and IL-13 in combination. Each line in the left and center panels represents an individual recipient mouse (None n=73, IL-4 n=15, IL-13 n=10 and IL-4+IL13 n=15). (B) Kaplan-Meier survival curves of mice in (A). (C) Quantification of donor CD4+ T cells from SP and MLN for each condition (None n=47, IL-4 n=11, IL-13 n=7, IL-4+IL-13 n=13). (D) Representative flow cytometry analysis of CD4 and EGFP (Foxp3) expression to assess the frequency of iTreg cells in MLN for each group. Numbers in quadrants are averages. (E–F) Frequency (E) and number (F) of iTreg cells in the SP and MLN for each pre-conditioning regimen. (G–H) Colitis scores (G) and representative H&E stained sections (H) from mice where tissues were taken for histology (None n=15, IL-4 n=11, IL-13 n=10 and IL-4+IL13 n=10). (I) Representative flow cytometry analysis of IL-17A and IFN-γ expression to assess the frequency of IFN-γ+, IL-17A+ and IFN-γ+IL-17A+ cells in MLN for each group. Numbers in quadrants are averages. (J–K) Frequency (J) and number (K) of CD4+ IL-17A+ T cells in the SP and MLN for each group. (L, M) Frequency (L) and number (M) of CD4+ IFN-γ+ T cells in the SP and MLN for each group. (N–O) Frequency (N) and number (O) of CD4+ IFN-γ+ IL-17A+ T cells in the SP and MLN for each group. Each symbol represents a mouse, and small horizontal bars represent the mean. Data are from 3–17 independent experiments, 1–5 mice per experiment. *p< 0.05, **p<0.005, ***p<0.0005; Mann- Whitney test.
Pre-treatment with M2a macrophages expands the iTreg-Th17 cell axis
Like the cytokines IL-4 and IL-13, alternatively activated M2a macrophages are also associated with Th2 immune responses and secrete several anti-inflammatory cytokines including TGF-β1 and IL-10. Both TGF-β1 and IL-10 cytokines stabilize Treg cells and help maintain their function. To test the role of different macrophage polarization states on the development of the iTreg-Th17 cell axis, we cultured bone marrow macrophages in the presence of IFN-γ and LPS (M1), IL-4 and IL-13 (M2a), or none of these cytokines (M0). We then confirmed macrophage polarization by transcriptional profiling, rt-PCR of characteristic transcripts, and by FACS (Figure 3 and Figure S2). From the gene expression profiles of M1 and M2a macrophages, we identified 1891 probe sets that were differentially regulated relative to M0 macrophages. Of these differentially regulated probe sets, 497 were unique to M2a cells, 1018 were unique to M1 cells and 376 probe sets were shared (Figures 3A, B). Data files are available through the National Center for Biotechnology Information Gene Expression Omnibus (accession number GSE72518, http://www.ncbi.nlm.nih.gov/geo/). By gene array, expression of Arg1 was increased more than 200 fold in M2a cells relative to M0 cells. Similarly, expression of Mgl2, Mmp12, Mmp13, Itgax, Egr2, Cdh1, Retnla, Chi3l3, and Klf9 was upregulated in M2a cells but not M1 cells (Figure 3C). Differential expression of several canonical M2a or M1 genes, including Arg1, Retnla (Fizz), Chi3l3 (YM1), Nos2 and the cytokines genes Tgfb1 and Il10 was confirmed by qPCR (Figure 3D). FACS analysis revealed that both M1 and M2a macrophages expressed F4/80 and CD11b, confirming their identity as macrophages. (Figure 3E). Finally, intraperitoneal injection of IL-4+IL-13 following the protocol used in Figure 2 resulted in an increase in Arg1 expression and a decrease in Nos2 expression by PECs (Figure 3F). This suggests pretreatment with these cytokines has similar effects on macrophage polarization in vitro and in vivo.
Figure 3. Transcriptional and phenotypic profile of M1 and M2a polarized macrophages.

(A) Venn diagram showing commonly and uniquely regulated probe sets found in M1 and M2a polarized macrophages. Probe sets that revealed a twofold or greater difference (|log2 ratio| > 1.0) and rank product FDR <10% relative to M0 cells are shown. Data is averaged from 3–5 arrays for each subset. (B) Heatmap showing the fold change in expression of the 1891 differentially regulated probe sets identified in A. Genes are organized into unique and co-regulated gene clusters as indicated. (C) Annotated heat map showing the expression levels of select differentially regulated probe sets. For B and C the scale (–64.0 fold to +64 fold) represents the fold change relative to the mean normalized intensity value (log2 ratio) in M0 cells (n=5). (D) q-PCR analysis of select gene expression in M0, M1, and M2 cells (n=3 for each gene). (E) Representative FACS analysis of M0, M1, and M2 macrophages (n=3) derived from bone marrow cells compared to PECs. (F) q-PCR analysis of Arginase 1 and NOS2 expression in PECs from mice pre-treated with IL-4 and IL-13 or PBS controls (n=4 for each group). (G) Representative flow cytometry analysis of F4/80 and YFP expression of recovered PECs, 48 hours after pre-treatment of Rag1−/− C57BL/6 with YFP+ M2a macrophages and frequency of F4/80+YFP+ PECs (n=3). (H) q-PCR analysis of Arginase 1 and NOS2 expression in PECs from mice in (G). F4/80+ YFP+ cells are donor M2a macrophages while F4/80+ YFP− cells are recipient mouse PECs.
In order to identify transferred macrophages, we generated bone marrow derived M2a macrophages from ROSA26EYFP × Lyz2Cre mice in vitro. Using this congenically marked system, we transferred 2×105 YFP+ M2a cells into C57BL6 Rag1−/− mice by i.p. injection. We looked for the M2a cells in the peritoneal cavity 2 days after transfer and found that about 1/3 of the PECs were YFP+, indicating they were transferred macrophages (Figure 3G). This population was largely gone by day 7 (data not shown). We also found that following M2a transfers, host YFP− macrophages have increased expression of Arg1 but not Nos2 (Figure 3H). These later data suggest that the transferred M2a macrophages may also act by polarizing the host macrophage compartment.
Next, Rag1−/− C57BL/6 mice were pre-treated with the different types of macrophages, followed by colitis induction with naïve T cells isolated from Foxp3EGFP mice. Control mice were not pre-treated with macrophages and received naïve T cells. Weight change over time and survival data were consistent with the delayed onset of colitis in mice pre-treated with M2a macrophages when compared to M1 pre-treated mice, or to mice with no macrophage pretreatment (Figures 4A, B). Up to a 5 fold expansion of CD4+ T cells was observed in mice pre-treated with M2a macrophages compared to controls (Figure 4C), and the frequency and number of iTreg cells in the spleen and mesenteric lymph nodes was increased up to 16-fold (Figures 4D–F) and reached a maximum value by about 40 days after T cell transfer (Figure 4G). Histological sections of the colons from affected mice showed severe colitis (Figures 4H, I). In parallel to iTreg cells, IL-17A producing CD4+ T cells showed a similar increase in the M2a pre-conditioned mice when compared to the other groups (Figures 4J–L). These pre-conditioning experiments were similar to those in Figure 2 in that nTreg cells were not added, so all mice eventually developed colitis, despite the expansion of the iTreg-Th17 cell axis, and the majority of T cells in the spleen and MLN expressed IFN-γ and/or IL-17A (Figures 4M–P). In individual M2a pre-treated mice, there was a direct correlation between the frequency of iTreg cells and the frequency of Th17 cells (Figure 4Q).
Figure 4. Pre-Treatment of Rag1−/− C57BL/6 mice with M2a macrophages boosts iTreg-Th17 cell axis development in the spleen and MLN.
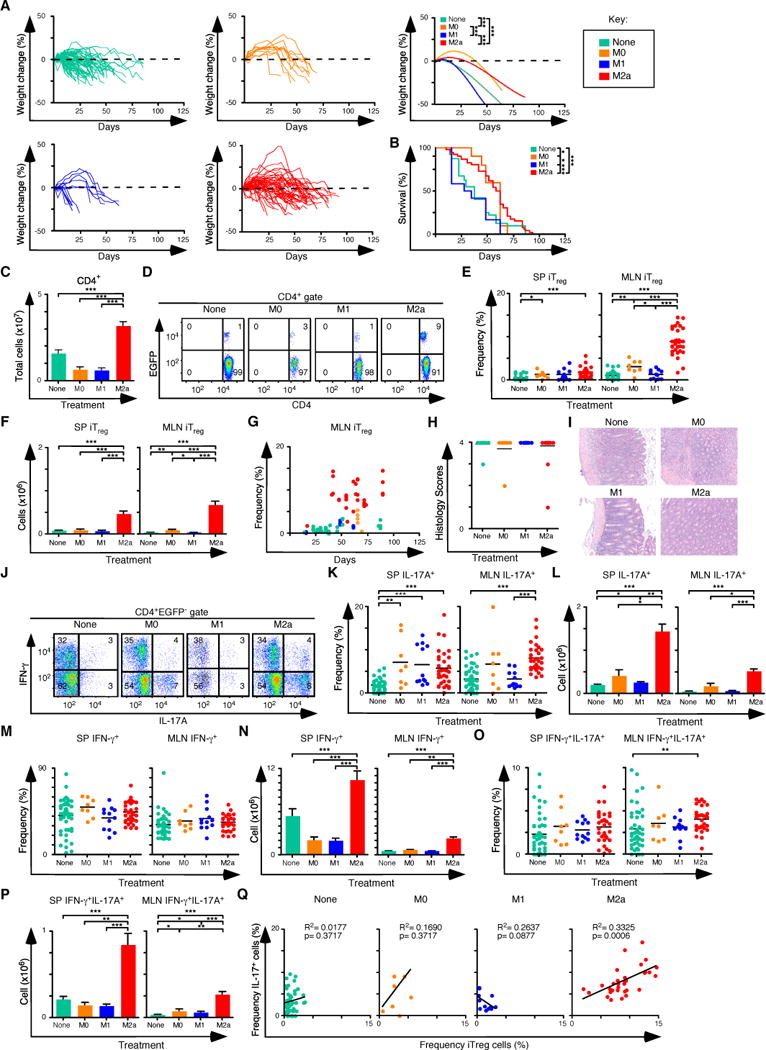
(A) Weight change analysis of Rag1−/− C57BL/6 mice pre-treated with M0, M1, M2a macrophages or not pre-treated. Each line in the left and center panels represents an individual recipient mouse (none n=73, M0 n=11, M1 n=13, M2 n=52). (B) Kaplan-Meier survival curves of mice in (A). (C) Quantification of donor CD4+ T cells from SP and MLN for each pre-condition treatment (None n=47, M0 n=8, M1 n=12, M2 n=32). (D) Representative flow cytometry analysis of CD4 and EGFP (Foxp3) expression to assess the frequency of iTreg cells in MLN for each group. Numbers in quadrants are averages. (E–F) Frequency (E) and number (F) of iTreg cells in the SP and MLN. (G) Comparison of the frequency of iTreg cells in MLN and days where mice were taken for all groups. (H–I) Colitis scores (H) and representative H&E stained sections (I) from mice where tissues were taken for histology (none n=15, M0 n=7, M1 n=12, M2 n=26). (J) Representative flow cytometry analysis of IL-17A and IFN-γ expression to assess the frequency of IFN-γ+, IL-17A+ and IFN-γ+ IL-17A+ cells in MLN for each group. Numbers in quadrants are averages. (K–L) Frequency (K) and number (L) of CD4+ IL-17A+ T cells in the SP and MLN for each group. (M–N) Frequency (M) and number (N) of CD4+ IFN-γ+ T cells in the SP and MLN for each group. (O–P) Frequency (O) and number (P) of CD4+ IFN-γ+ IL-17A+ T cells in the SP and MLN for each group. (Q) Linear regression analysis comparing the frequency of MLN iTreg and CD4+IL-17A+ T cells from each group. (E, G, K, O, Q) Each symbol represents a mouse, and small horizontal bars represent the mean. Data are from 3–17 independent experiments, 1–5 mice per experiment. *p< 0.05, **p<0.005, ***p<0.0005; Mann- Whitney test.
Consistent with data from the spleen and MLN, increases in the frequency and number of iTreg and Th17 cells in the SI and colons of M2a pretreated mice were also observed (Figures 5A–F). In the SI, the frequency of both IFN-γ+ cells and IL-17A+ cells following M2a transfer was similar to controls. In contrast, the frequency of both IFN-γ+ cells and IL-17A+ cells in colons was increased following M2a pretreatment (Figures 5G–J). There are two caveats to this comparison. First M2a treatments delay weight loss by about 3 weeks (Figure 4A), so the time at which mice are analyzed is different between the two groups. Second, no nTreg therapy is provided in these experiments, so all mice develop strong Th1 responses leading to progressive colitis. Thus in this system, changes in CD4+ T cell populations observed in the colon are reflected in the MLN. Together, our results demonstrate that M2a macrophages support CD4+ T cell homeostatic expansion, characterized by the development of the iTreg-Th17 cell axis, even during a dominant Th1 inflammatory response.
Figure 5. Pre-Treatment of Rag1−/−C57BL/6 mice with M2a macrophages boosts iTreg-Th17 cell axis development in the small intestine and colon.
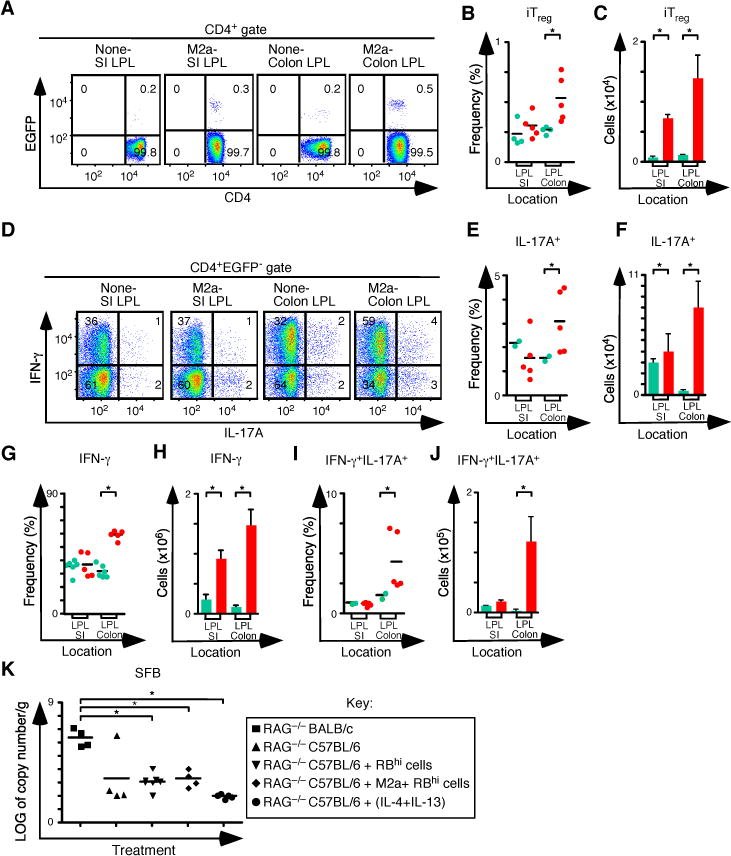
(A) Representative flow cytometry analysis of CD4 and EGFP (Foxp3) expression to assess the frequency of iTreg cells in SI and colon lamina propria for each group. Numbers in quadrants represent the average. (B–C) Frequency (B) and number (C) of iTreg cells in the SI and colon (None n=4 and M2a n=5). (D) Representative flow cytometry analysis of IL-17A and IFN-γ expression to assess the frequency of IFN-γ+, IL-17A+ and IFN-γ+IL-17A+ T cells in MLN for each group. Numbers in quadrants are averages. (E–F) Frequency (E) and number (F) of CD4+ IL-17A+ T cells in the SP and MLN for each group. (G–H) Frequency (G) and number (H) of CD4+ IFN-γ+ T cells in the SP and MLN for each group. (I–J) Frequency (I) and number (J) of CD4+ IFN-γ+ IL-17A+ T cells in the SP and MLN for each group. (K) Log of copy number/g of SFB for each group. Each symbol represents a mouse, and small horizontal bars represent the mean. Data are from 2–3 independent experiments, 1–2 mice per experiment. *p< 0.05; Mann- Whitney test.
Development of iTreg and Th17 responses can be influenced by the composition of the microbiome. For example polysaccharide A produced by B. fragilis can increase the production of iTreg cells, and segmented filamentous bacteria (SFB) are associated with Th17 cell development. We used qPCR to analyze SFB in the distal small intestines, and we compared pretreated C57BL6 Rag1−/− mice with controls. Although BALB/c Rag1−/− mice had higher SFB levels than C57BL/6 Rag1−/− mice, SFB levels in C57BL/6 Rag1−/− mice did not change following colitis induction or as a result of either pretreatment regimen (Figure 5K). These data indicate that the observed increase in Th17 cell development following either cytokine or M2a pretreatment did not depend on an increase in SFB content of the distal SI.
M2a macrophages enhance nTreg cell immunotherapy
The capacity of M2a macrophages to expand the iTreg-Th17 cell axis when nTreg cells were absent (Figure 4) suggested their potent therapeutic potential when combined with nTreg cell immunotherapy. We evaluated this possibility by using the M2a macrophages in two settings. First, we pre-treated Rag1−/− mice with M2a macrophages, then induced colitis by transferring CD45.2+ naïve T cells from Foxp3EGFP mice. Mice were then treated with 1×106 sorted CD45.1+ nTreg cells on day 10, the time point when the mice with the most aggressive disease have lost 2–5% of their initial body weight. In the second experimental condition, we induced colitis then treated symptomatic mice that survived to 30 days with M2a macrophages, followed by CD45.1+ nTreg cells on day 31. Control groups did not receive any macrophage transfers but were treated with 1×106 sorted CD45.1+ nTreg cells. Strikingly, mice that received M2a macrophages either as pre-treatment or as treatment for established disease gained weight and survived to the completion of the experiment at 125 days (Figures 6A–C). Although the number of CD4+ T cells recovered was similar or reduced between treated mice and controls (Figure 6D), the frequency and number of iTreg and nTreg cells recovered was increased 2–3 fold in mice that received M2a macrophages, and there was preferential accumulation of both iTreg and nTreg subsets in the MLN (Figures 6E–I). In addition to weight gain, other signs of colitis improved in mice that received both M2a macrophages and nTreg cells. The inflammatory infiltrates and colitis scores were decreased when compared to control mice (Figures 6J, K). The frequency and number of CD4+IL-17A+ T cells was also increased 2–4 fold, indicating expansion of the iTreg-Th17 axis, even when nTreg cells were also present (Figures 6L–N). The number of cells producing IFN-γ was decreased when compared to control mice (Figures 6O,P), although there was a modest increase in the number of cells producing IFN-γ and IL-17A in the MLN (Figures 6Q, R). The cell surface phenotypes of the iTreg and nTreg populations in macrophage-treated mice were similar (Figure 6S). These data demonstrate that in a model of inflammatory colitis dependent upon IFN-γ, transfer of M2a macrophages can be used as a cellular therapeutic adjuvant to treat established disease. The macrophages increased the number of Treg and Th17 cells recovered from treated mice.
Figure 6. M2a macrophages improve the treatment of colitis with nTreg cells.
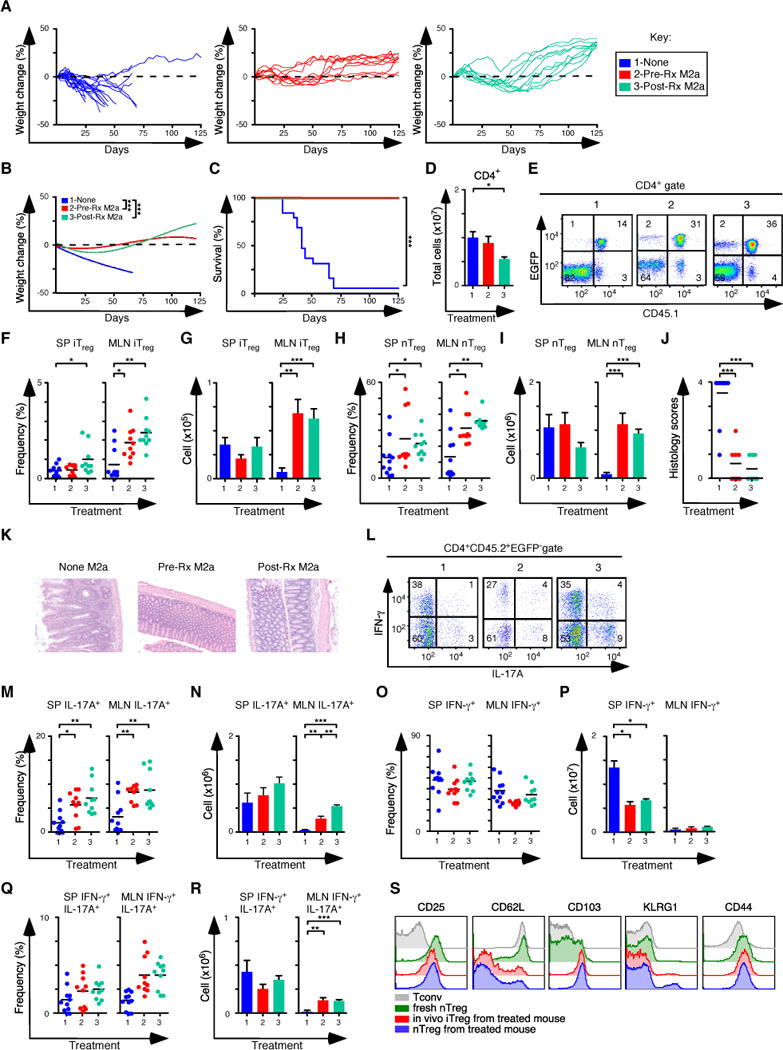
(A, B) Weight change analysis of mice not pre-treated (None), pre-treated (Pre-Rx M2a) or treated (Post-Rx M2a) with M2a macrophages at day 30 after establishment of colitis (None n=12; Pre-Rx n=10; Post-Rx M2a n=10). Note that all groups also received nTreg cells transfers on d31. (C) Kaplan-Meier survival curves of the mice in A. (D) Quantification of donor CD4+ T cells from SP and MLN for each experimental condition in A. (E) Representative flow cytometry analysis of CD45.1 and EGFP (Foxp3) expression to assess the frequency of in situ iTreg (CD45.1− EGFP+) and nTreg (CD45.1+ EGFP+) cells in MLN for each group. Numbers in quadrants are averages. (f–G) Frequency (F) and number (G) of iTreg cells in the SP and MLN. (H–I) Frequency (H) and number (I) of nTreg cells in the SP and MLN. (J–K) Colitis scores (J) and representative H&E stained sections (K) from mice where tissues were taken for histology (None M2a n=11; Pre-Rx M2a n=10; Post-Rx M2a n=10). (l) Representative flow cytometry analysis of IL-17A and IFN-γ expression to assess the frequency of IFN-γ+, IL-17A+ and IFN-γ+IL-17A+ cells in MLN for each group. Numbers in quadrants are averages. (M–N) Frequency (M) and number (N) of CD4+ IL-17A+ T cells in the SP and MLN for each group. (O–P) Frequency (O) and number (P) of CD4+ IFN-γ+ T cells in the SP and MLN for each group. (Q–R) Frequency (Q) and number (R) of CD4+ IFN-γ+ IL-17A+ T cells in the SP and MLN for each group. (S) Representative histograms comparing the expression levels of several proteins associated with Treg function. Tconv cells and nTreg cells were isolated from naïve mice, while in vivo derived iTreg cells and nTreg cells were isolated from mice treated with M2a + nTreg cells. Each symbol represents a mouse and small horizontal bars represent means. Data are from 2–6 independent experiments, 1–5 mice per experiment. *p< 0.05, **p<0.005, ***p<0.0005; Mann- Whitney test.
M2a macrophages stabilize in vitro-derived iTreg cells
Previous work has shown that in vitro-derived iTreg cells readily lose Foxp3 expression in adoptive transfer immunotherapy experiments (4). We examined the capacity of M2a macrophages to promote iTreg stability by pre-treating Rag1−/− mice with M2a macrophages, followed by the induction of colitis with naïve CD4+ T cells from rescued CD45.2+ Foxp3ΔEGFP mice, which carry a non-functional Foxp3 allele and cannot become iTreg cells after transfer. On day 10, mice were treated with 5×105 CD45.2 nTreg cells and 5×105 CD45.1 iTreg cells generated in vitro. Mice that received M2a macrophages gained weight and survived to completion of the experiment, while control mice that were not pre-treated failed to thrive (Figures 7A, B). In M2a pre-treated mice the number of CD4+ T cells recovered was decreased 3-fold, consistent with their clinical recovery (Figure 7C). Importantly the frequency of both Treg subsets was higher, and the number of iTreg and nTreg cells in the MLN was increased 2- to 3-fold following M2a pre-treatment (Figures 7D–H). The number of CD45.1+ Foxp3− (ex-iTreg) cells in the SP and MLN was also decreased 7-fold and colitis scores were lower in M2a pretreated mice (Figures 7I–L). Consistent the lower colitis scores, the frequency and number of IL-17A+, IFN-γ+, and IL-17A+IFN-γ+ ex-iTreg cells generally either trended downward or was reduced in the spleens and MLN of M2a pretreated mice (Figures 7M–S). These data indicate that transferred M2a macrophages promote the preferential accumulation of both Treg subsets in the MLN and influence the stability of Foxp3 expression in the iTreg subset.
Figure 7. M2a macrophages enhance the stability of iTreg cells.
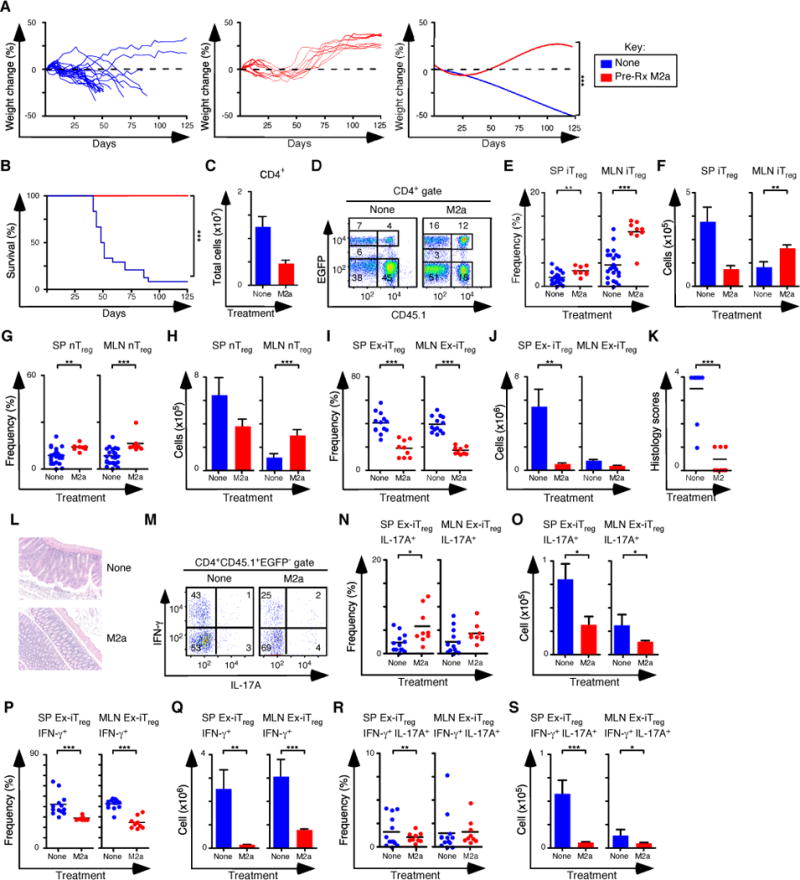
(A) Weight change analysis of mice not pre-treated or pre-treated M2a macrophages followed by colitis induction with naïve CD4+ T cells from rescued Foxp3ΔEGFP mice. All mice were treated with nTreg cells and in vitro derived iTreg cells (None n=24, M2a n=9). (B) Kaplan-Meier survival curves of mice not pre-treated or pre-treated with M2a macrophages (None n=24, M2a n=9). (C) Quantification of donor CD4+ T cells from SP and MLN for each pre-condition treatment. (D) Flow cytometry analysis of CD45.1 and EGFP (Foxp3) expression to assess the frequency of nTreg (CD45.1−EGFP+) and in vitro iTreg (CD45.1+EGFP+) cells in MLN for each group. Numbers in quadrants are averages. (E–F) Frequency (E) and number (F) of iTreg cells in the SP and MLN. (G–H) Frequency (G) and number (H) of nTreg cells in the SP and MLN. (I–J) Frequency (I) and number (J) of ex-iTreg cells in the SP and MLN. (K–L) Colitis scores (K) and representative H&E stained sections (L) from mice where tissue was taken for histology (None n=10, M2a n=9). (M) Representative flow cytometry analysis of IL-17A and IFN-γ expression to assess the frequency of IFN-γ+, IL-17A+ and IFN-γ+ IL-17A+ ex-iTreg cells from the MLN for each group. Numbers in quadrants are averages. (N–O) Frequency (N) and number (O) of IL-17A+ ex-iTreg cells in the SP and MLN for each group. (P–Q) Frequency (P) and number (Q) of IFN-γ+ ex-iTreg cells in the SP and MLN for each group. (R–S) Frequency (R) and number (S) of IFN-γ+ IL-17A+ ex-iTreg cells in the SP and MLN for each group. Each symbol represents a mouse, and small horizontal bars represent the mean. Data are from 2–12 independent experiments, 1–5 mice per experiment. *p< 0.05, **p<0.005, ***p<0.0005; Mann- Whitney test.
DISCUSSION
The central finding of this report is that in a colitis model where iTreg cell production is deficient, adoptively transferred M2a macrophages support the directionally concordant expansion of iTreg -Th17 cell axis, which is associated with reestablishing gastrointestinal tolerance, repair of mucosal injury and recovery from disease. Together, these data suggest that macrophage polarization is a critical point of intersection between innate and adaptive immunity that controls the inducible component of the gastrointestinal regulatory environment.
M2a macrophage polarization requires the STAT6-dependent cytokines IL-4 and IL-13 (33). In contrast, studies have also shown that STAT6 inhibits the development of iTreg cells by preventing the expression of Foxp3, and that STAT6 suppresses the expression of IL-17 by Th17 cells (34–36). Thus the effects of STAT6 on macrophage polarization and on the development of the iTreg-Th17 cell axis are dichotomous and at some level mutually exclusive. In the colitis model used in our studies, we avoided these potential blocks by preconditioning host macrophages in Rag1−/− mice with IL-4 and IL-13 or by polarizing macrophages with these cytokines in vitro. This experimental design bypassed any potential developmental block due to STAT6 phosphorylation. However, we also found that contrary to our expectations, the Th2-prone BALB/c mice expand the iTreg/Th17 cell axis to a greater degree than the Th1-prone C57BL/6 strain after T cell transfer. Others have reported that GATA binding protein 3 (GATA3), the master regulator of Th2 differentiation, is important for both Foxp3 expression and the function of Treg cells (37–39). Indeed, iTreg cells that express GATA3 are potent suppressors of Th2 inflammatory responses (38). In models of food allergy, iTreg cells are readily produced in the gut, and in humans, certain therapeutic options under consideration for IBD involve engendering Th2 responses (15, 40). Thus Th2 responses and a Th2-prone environment need not abrogate development of the iTreg-Th17 cell axis per se, as integration of cytokine signals may occur by direct competition for STAT binding sites in the Foxp3 and RORγt promoters. In any event, the iTreg-Th17 cell axis in the Th1-dependent model of colitis used here is readily modified by the transferred M2a macrophages.
In the T cell transfer model of colitis, expansion of CD4+ T cells is driven by the lymphopenic environment of Rag1−/− recipient mice and by the gut microbiome. M2a cells had an impact on the process as they improved the overall recovery of CD4+ T cells and CD4+ Teff cells producing IFN-γ by ~2-fold. In contrast, the number of iTreg cells increased ~13-fold (39-fold in MLN), and IL-17A+ cells increased ~8-fold. These data indicate that M2a cells have a preferential effect on the development of the iTreg-Th17 cell axis, albeit one that occurs in the setting of a more global effect on CD4+ T cell homeostatic and spontaneous proliferation.
Directionally concordant expansion of the iTreg-Th17 cell axis was not predicted by some current models, as Th17 cells mediate inflammation while Treg cells act to restrain these same responses. While both share certain developmental cues such as the requirement for TGF-β, cell fate decisions ultimately rest on competing environmental and metabolic factors that act in opposition. For example IL-6, IL-21 and IL-23 activate STAT3, which up-regulates ROR-γt and is required for IL-17 transcription (41). In comparison, IL-2 activates STAT5, which promotes Foxp3 expression, Treg differentiation, and transcriptional repression at the IL-17 locus (42). STAT3 and STAT5 compete for overlapping binding sites in both the Foxp3 and Rorc promoters (42–44). In the current study, the unique capacity of M2a macrophages to coordinate simultaneous expansion of both cell lineages indicates that iTreg and Th17 developmental pathways need not be mutually exclusive. Similar areas of intersection include the beneficial effects of retinoic acid on both iTreg and Th17 cell development, as well as the role of mammalian target of rapamycin complex 1 activation in Th17 cell differentiation and in Treg cell suppressive function. The effect of M2a macrophages on pathways that regulate T cell nutrient status and metabolism remain to be explored.
Th17 cells can be broadly divided into pathogenic and non-pathogenic subsets (22). Different combinations of TGF-β1, TGF-β3, IL-1β, IL-6 and IL-23 induce pathogenic Th17 cells, and they participate in inflammation by producing IL-17 family cytokines, TGF-β3, IFN-γ and IL 22. In contrast, non-pathogenic Th17 cells are induced by TGF-β1 and IL-6, and they produce IL-17 family cytokines plus IL-10 (22). Although M2a transfers expanded the iTreg-Th17 cell axis, they also decreased the proportion of pathogenic IL-17A+IFN-γ+ cells in the Th17 cell compartment. There are several plausible explanations for proposing a positive impact of IL17A on mucosal tolerance. IL-17R signaling in epithelial cells activates host defense pathways including the expression of antimicrobial peptides and chemokines involved in neutrophil and lymphocyte recruitment, such as CCL20. Antimicrobial peptides clearly shape the microbiome, which impacts iTreg development and mucosal tolerance. In addition, both Th17 and iTreg cells express CCR6 (receptor for CCL20), which mediates their migration. Thus expression of IL-17A by Th17 cells might promote the expression CCL20 by intestinal epithelial cells, which in turn recruits iTreg cells and a population of Th17 cells enriched in the non-pathogenic IL-10-producing subset (45).
In the current study, the sites of macrophage action and the cytokines mediating their beneficial effects were not explored. In Rag1−/− mice, macrophages are abundant in the peritoneal cavity, the lamina propria of the colon, and in the spleen. The transferred M2a cells could influence T cell differentiation in all 3 sites, and our experiments do not distinguish between the possibilities. M2a cells are also reported to produce TGF-β1 and IL-10, and both cytokines could influence the host innate immune compartment as well as the differentiation of effector and regulatory T cells.
In summary, the adoptive transfer of polarized M2a macrophages promotes gastrointestinal tolerance through preferential expansion of the iTreg-Th17 cell axis. In the setting of a strong Th1 response, co-transfer strategies specifically designed to enhance and stabilize the regulatory environment may optimize Treg cell immunotherapy.
Supplementary Material
Acknowledgments
We thank Tamara Nelson for assistance with cell sorting and James Verbsky for critical reading of the manuscript.
ABBREVIATIONS
- Treg
regulatory T cell
- iTreg
induced regulatory T cell
- nTreg
natural regulatory T cell
- Th17
T helper 17
- Tconv
conventional T cell
- EGFP
enhanced green fluorescent protein
- Foxp3
forkhead box P3
References
- 1.Haribhai D, Williams JB, Jia S, Nickerson D, Schmitt EG, Edwards B, Ziegelbauer J, Yassai M, Li SH, Relland LM, Wise PM, Chen A, Zheng YQ, Simpson PM, Gorski J, Salzman NH, Hessner MJ, Chatila TA, Williams CB. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity. 2011;35:109–122. doi: 10.1016/j.immuni.2011.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Josefowicz SZ, Niec RE, Kim HY, Treuting P, Chinen T, Zheng Y, Umetsu DT, Rudensky AY. Extrathymically generated regulatory T cells control mucosal TH2 inflammation. Nature. 2012;482:395–399. doi: 10.1038/nature10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haribhai D, Lin W, Edwards B, Ziegelbauer J, Salzman NH, Carlson MR, Li SH, Simpson PM, Chatila TA, Williams CB. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J Immunol. 2009;182:3461–3468. doi: 10.4049/jimmunol.0802535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmitt EG, Haribhai D, Williams JB, Aggarwal P, Jia S, Charbonnier LM, Yan K, Lorier R, Turner A, Ziegelbauer J, Georgiev P, Simpson P, Salzman NH, Hessner MJ, Broeckel U, Chatila TA, Williams CB. IL-10 Produced by Induced Regulatory T Cells (iTregs) Controls Colitis and Pathogenic Ex-iTregs during Immunotherapy. Journal of immunology. 2012;189:5638–5648. doi: 10.4049/jimmunol.1200936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hsieh CS, Liang Y, Tyznik AJ, Self SG, Liggitt D, Rudensky AY. Recognition of the peripheral self by naturally arising CD25+ CD4+ T cell receptors. Immunity. 2004;21:267–277. doi: 10.1016/j.immuni.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. Peripheral education of the immune system by colonic commensal microbiota. Nature. 2011;478:250–254. doi: 10.1038/nature10434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Relland LM, Williams JB, Relland GN, Haribhai D, Ziegelbauer J, Yassai M, Gorski J, Williams CB. The TCR repertoires of regulatory and conventional T cells specific for the same foreign antigen are distinct. J Immunol. 2012;189:3566–3574. doi: 10.4049/jimmunol.1102646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shih FF, Mandik-Nayak L, Wipke BT, Allen PM. Massive thymic deletion results in systemic autoimmunity through elimination of CD4+ CD25+ T regulatory cells. J Exp Med. 2004;199:323–335. doi: 10.1084/jem.20031137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annu Rev Immunol. 2012;30:733–758. doi: 10.1146/annurev-immunol-020711-075043. [DOI] [PubMed] [Google Scholar]
- 10.Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, Caccamo M, Oukka M, Weiner HL. Control of T(reg) and T(H)17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 11.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 12.Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F. A functionally specialized population of mucosal CD103+ DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med. 2007;204:1757–1764. doi: 10.1084/jem.20070590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Apostolou I, von Boehmer H. In vivo instruction of suppressor commitment in naive T cells. J Exp Med. 2004;199:1401–1408. doi: 10.1084/jem.20040249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Curotto de Lafaille MA, Lino AC, Kutchukhidze N, Lafaille JJ. CD25- T cells generate CD25+Foxp3+ regulatory T cells by peripheral expansion. J Immunol. 2004;173:7259–7268. doi: 10.4049/jimmunol.173.12.7259. [DOI] [PubMed] [Google Scholar]
- 15.Mucida D, Kutchukhidze N, Erazo A, Russo M, Lafaille JJ, Curotto de Lafaille MA. Oral tolerance in the absence of naturally occurring Tregs. J Clin Invest. 2005;115:1923–1933. doi: 10.1172/JCI24487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beres A, Komorowski R, Mihara M, Drobyski WR. Instability of Foxp3 expression limits the ability of induced regulatory T cells to mitigate graft versus host disease. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:3969–3983. doi: 10.1158/1078-0432.CCR-10-3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Floess S, Freyer J, Siewert C, Baron U, Olek S, Polansky J, Schlawe K, Chang HD, Bopp T, Schmitt E, Klein-Hessling S, Serfling E, Hamann A, Huehn J. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci U S A. 1993;90:770–774. doi: 10.1073/pnas.90.2.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramalingam R, Larmonier CB, Thurston RD, Midura-Kiela MT, Zheng SG, Ghishan FK, Kiela PR. Dendritic cell-specific disruption of TGF-beta receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol. 2012;189:3878–3893. doi: 10.4049/jimmunol.1201029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, Awasthi A, Yosef N, Quintana FJ, Xiao S, Peters A, Wu C, Kleinewietfeld M, Kunder S, Hafler DA, Sobel RA, Regev A, Kuchroo VK. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 2012;13:991–999. doi: 10.1038/ni.2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Denning TL, Wang YC, Patel SR, Williams IR, Pulendran B. Lamina propria macrophages and dendritic cells differentially induce regulatory and interleukin 17-producing T cell responses. Nature immunology. 2007;8:1086–1094. doi: 10.1038/ni1511. [DOI] [PubMed] [Google Scholar]
- 24.Murai M, Turovskaya O, Kim G, Madan R, Karp CL, Cheroutre H, Kronenberg M. Interleukin 10 acts on regulatory T cells to maintain expression of the transcription factor Foxp3 and suppressive function in mice with colitis. Nat Immunol. 2009;10:1178–1184. doi: 10.1038/ni.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 26.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. Journal of immunology. 2000;164:6166–6173. doi: 10.4049/jimmunol.164.12.6166. [DOI] [PubMed] [Google Scholar]
- 27.Lin W, Haribhai D, Relland LM, Truong N, Carlson MR, Williams CB, Chatila TA. Regulatory T cell development in the absence of functional Foxp3. Nat Immunol. 2007;8:359–368. doi: 10.1038/ni1445. [DOI] [PubMed] [Google Scholar]
- 28.Leach MW, Bean AG, Mauze S, Coffman RL, Powrie F. Inflammatory bowel disease in C.B-17 scid mice reconstituted with the CD45RBhigh subset of CD4+ T cells. Am J Pathol. 1996;148:1503–1515. [PMC free article] [PubMed] [Google Scholar]
- 29.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J. RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioinformatics. 2006;22:2825–2827. doi: 10.1093/bioinformatics/btl476. [DOI] [PubMed] [Google Scholar]
- 31.Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salzman NH, Hung K, Haribhai D, Chu H, Karlsson-Sjoberg J, Amir E, Teggatz P, Barman M, Hayward M, Eastwood D, Stoel M, Zhou Y, Sodergren E, Weinstock GM, Bevins CL, Williams CB, Bos NA. Enteric defensins are essential regulators of intestinal microbial ecology. Nature immunology. 2010;11:76–83. doi: 10.1038/ni.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012;122:787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takaki H, Ichiyama K, Koga K, Chinen T, Takaesu G, Sugiyama Y, Kato S, Yoshimura A, Kobayashi T. STAT6 Inhibits TGF-beta1-mediated Foxp3 induction through direct binding to the Foxp3 promoter, which is reverted by retinoic acid receptor. J Biol Chem. 2008;283:14955–14962. doi: 10.1074/jbc.M801123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, Mitsdoerffer M, Strom TB, Elyaman W, Ho IC, Khoury S, Oukka M, Kuchroo VK. IL-4 inhibits TGF-beta-induced Foxp3+ T cells and, together with TGF-beta, generates IL-9+ IL-10+ Foxp3(−) effector T cells. Nat Immunol. 2008;9:1347–1355. doi: 10.1038/ni.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cooney LA, Towery K, Endres J, Fox DA. Sensitivity and resistance to regulation by IL-4 during Th17 maturation. J Immunol. 2011;187:4440–4450. doi: 10.4049/jimmunol.1002860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu F, Sharma S, Edwards J, Feigenbaum L, Zhu J. Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat Immunol. 2015;16:197–206. doi: 10.1038/ni.3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Su MA, Wan YY. An essential role of the transcription factor GATA-3 for the function of regulatory T cells. Immunity. 2011;35:337–348. doi: 10.1016/j.immuni.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, Hall JA, Yagi R, Naik S, Bhairavabhotla R, Paul WE, Bosselut R, Wei G, Zhao K, Oukka M, Zhu J, Belkaid Y. GATA3 controls Foxp3(+) regulatory T cell fate during inflammation in mice. J Clin Invest. 2011;121:4503–4515. doi: 10.1172/JCI57456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heylen M, Ruyssers NE, Gielis EM, Vanhomwegen E, Pelckmans PA, Moreels TG, De Man JG, De Winter BY. Of worms, mice and man: an overview of experimental and clinical helminth-based therapy for inflammatory bowel disease. Pharmacology & therapeutics. 2014;143:153–167. doi: 10.1016/j.pharmthera.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 41.Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, O’Shea JJ. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity. 2010;32:605–615. doi: 10.1016/j.immuni.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O’Shea JJ, Laurence A. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol. 2011;12:247–254. doi: 10.1038/ni.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burchill MA, Yang J, Vogtenhuber C, Blazar BR, Farrar MA. IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells. Journal of immunology. 2007;178:280–290. doi: 10.4049/jimmunol.178.1.280. [DOI] [PubMed] [Google Scholar]
- 44.Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Positive and negative transcriptional regulation of the Foxp3 gene is mediated by access and binding of the Smad3 protein to enhancer I. Immunity. 2010;33:313–325. doi: 10.1016/j.immuni.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O’Connor W, Jr, Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA. Control of TH17 cells occurs in the small intestine. Nature. 2011;475:514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.