

Abstract
Bacterial cell division is a highly complex process that requires tight coordination between septum formation and chromosome replication and segregation. In bacteria that divide by binary fission a single septum is formed at mid-cell, a process that is coordinated by the conserved cell division scaffold protein FtsZ. In contrast, during sporulation-specific cell division in streptomycetes, up to a hundred rings of FtsZ (Z rings) are produced almost simultaneously, dividing the multinucleoid aerial hyphae into long chains of unigenomic spores. This involves the active recruitment of FtsZ by the SsgB protein, and at the same time requires sophisticated systems to regulate chromosome dynamics. Here, we show that SepG is required for the onset of sporulation and acts by ensuring that SsgB is localized to future septum sites. Förster resonance energy transfer imaging suggests direct interaction between SepG and SsgB. The beta-lactamase reporter system showed that SepG is a transmembrane protein with its central domain oriented towards the cytoplasm. Without SepG, SsgB fails to localize properly, consistent with a crucial role for SepG in the membrane localization of the SsgB–FtsZ complex. While SsgB remains associated with FtsZ, SepG re-localizes to the (pre)spore periphery. Expanded doughnut-shaped nucleoids are formed in sepG null mutants, suggesting that SepG is required for nucleoid compaction. Taken together, our work shows that SepG, encoded by one of the last genes in the conserved dcw cluster of cell division and cell-wall-related genes in Gram-positive bacteria whose function was still largely unresolved, coordinates septum synthesis and chromosome organization in Streptomyces.
Keywords: cell division control, development, FtsZ, SsgA-like proteins
1. Introduction
Most unicellular bacteria grow and divide by binary fission, which involves an increase in cell length, chromosome replication and segregation, and septum formation, eventually resulting in two daughter cells that each inherits a single copy of the chromosome. Chromosome organization and segregation are tightly coordinated with the spatial and temporal initiation of cell division. The prokaryotic cell division scaffold is formed by the tubulin homologue FtsZ [1], which forms a contractile ring (or Z ring) that mediates recruitment of the cell division machinery to the mid-cell position (reviewed in [2,3]). In unicellular bacteria, septum-site localization and Z-ring stabilization is mediated by a number of proteins, including FtsA and ZipA [4–6], ZapA [7] and SepF [8,9]. The process of Z-ring (dis-)assembly during division is actively controlled (reviewed in [10]).
Streptomycetes are filamentous Gram-positive soil bacteria that have a complex multicellular life cycle [11,12], and produce over 60% of all known antibiotics and many other bioactive natural products [13,14]. Expression of these natural products is typically coordinated with the onset of spore development [15]. The vegetative mycelium consists of syncytial cells separated by widely spaced crosswalls [16]. During sporulation-specific cell division, FtsZ initially assembles in long filaments in the aerial hyphae, then as regular foci, to finally form a ladder of Z rings [17]. Eventually, cytokinesis results in long chains of spores, following a complex process of coordinated cell division and DNA segregation [18,19]. While ftsZ null mutants fail to produce septa and hence do not sporulate, cell division is not essential for growth of Streptomyces, which provides a unique system for the study of this process [19,20].
The mycelial lifestyle of streptomycetes imposes specific requirements for cell division control, in particular owing to the lack of a defined mid-cell position and the synchronous formation of multiple septa. The canonical model for the control of Z-ring formation involves the action of negative control systems such as Min, which prevents Z-ring assembly at the cell poles [21,22], and nucleoid occlusion, which prevents formation of the Z ring over non-segregated chromosomes [23–26]. However, in Streptomyces, septum-site localization is positively controlled, via the recruitment of FtsZ by SsgB, which in turn depends on the action of SsgA [27]. At the onset of sporulation-specific cell division, SsgA and SsgB briefly interact, but the precise role of SsgA in the recruitment of SsgB is still not well understood. The SsgA-like proteins are proteins that uniquely occur in actinomycetes [28,29]. This different mode of division control probably explains the absence of direct homologues of Min and Noc proteins and of the canonical Z-ring anchoring proteins. SepF and DivIVA are rare examples of cell division control proteins shared between Streptomyces and other bacteria, and of these, DivIVA is not involved in the control of division, but is instead required for driving apical (tip) extension and growth [30]. The concept of positive control of cell division is more widespread in bacteria, as it was also found in Myxococcus xanthus, where the ParA-like protein PomZ recruits FtsZ [31], whereas the recent discovery that Bacillus cells divide at mid-cell in the absence of Min and Noc may be explained by a positive control-like mechanism, although there the mechanism of septum positioning is unknown [32].
With the recruitment of FtsZ by SsgB, another important question is left unaddressed, namely how the highly symmetrical spacing between the sporulation septa is achieved; in other words, how is the localization of SsgB itself orchestrated? We previously suggested that the regularly spaced nucleoids might provide the boundaries for Z-ring assembly, in analogy with a nucleoid-occlusion mechanism [27]. One possible candidate is the conserved ylmG gene, which lies in the conserved dcw cluster for genes involved in division and cell-wall synthesis, and in-between sepF and divIVA. In firmicutes, SepF [8,9] and DivIVA [33] play an important role in the control of cell division in many Gram-positive bacteria, suggesting that SepG may also relate to the control of division. YlmG is especially found in actinobacteria, firmicutes and cyanobacteria, as well as in chloroplasts of photosynthetic eukaryotes. YlmG has been assigned to the YggT family of proteins (Pfam PF02325), which are named after Escherichia coli YggT and are a highly diverse family of membrane proteins found in bacteria and plastids [34]. Functional overlap between YlmG and the osmoregulation-related YggT is unlikely because of the lack of relevant sequence similarity and phylogenetic context. It was previously shown that the YlmG orthologue AtYLMG1-1 of the plant Arabidopsis thaliana is exclusively found in the membrane fraction [35]. AtYLMG1-1 also controls nucleoid morphology, whereby knockdown of the gene via expression of an antisense RNA impaired nucleoid partitioning and changed the morphology of nucleoids. Similar results were obtained in the cyanobacterium Synechococcus elongatus [35]. Additionally, deletion of ylmG in the cyanobacterium Synechocystis PCC6803 resulted in thinner cell-wall formation and slower growth rate, though no notable defects in nucleoid segregation was observed [36]. Similar minor effects on cell-wall thickness were observed during cell division in Streptococcus [37].
In this work, we show that after the initial localization of SsgB by SsgA at a central location in the hyphae, YlmG plays an important role in the stable localization of SsgB to future septum sites and consequently also in the recruitment of FtsZ. The protein was therefore renamed SepG. The SepG protein localizes to the periphery of the future spore compartments, where it contributes to maintenance of chromosome compaction. In the absence of SepG, the nucleoid expands to the outer limits of the spore compartments, compromising septum synthesis. Taken together, our data provide important new insights into the function of SepG that is well conserved in Gram-positive bacteria, and show that SepG is required for positive control of cell division and has a significant effect on nucleoid morphogenesis in Streptomyces.
2. Results
2.1. Ylmg (SepG) is a predicted membrane protein
To be able to recruit FtsZ, SsgB needs to dock to the membrane, and the lack of a membrane domain suggests that another protein ensures its membrane attachment. In our quest to identify possible partners for SsgB that may coordinate the membrane docking in streptomycetes, we considered the exceptional evolutionary conservation of the FtsZ-recruiting protein SsgB. SsgB is completely conserved in streptomycetes except for aa position 128, a variation which can be used as phylogenetic marker, dissecting the streptomycetes into two different subclades, namely those that sporulate in submerged cultures and those that fail to do so [38]. Conversely, conservation of the SsgB proteins between even related actinomycetes is surprisingly low. We therefore analysed the databases for membrane proteins with similar conservation. Using as criterion more than 90% aa identity between Streptomyces species and less than 70% aa identity between orthologues from other actinomycete genera, analysis of the around 1850 putative membrane proteins of S. coelicolor was narrowed down to 20 candidate proteins (table 1). Of these, the 94 aa protein YlmG (SCO2078) stood out, with its predicted non-transmembrane domain consisting of aa residues 26–72 showing a very similar conservation as SsgB, with near complete conservation between the Streptomyces orthologues (99–100%), but low conservation (50–60% aa identity) between Streptomyces and other actinomycetes. Interestingly, the two membrane-spanning domains are less well conserved; these data suggest possible covariation of the central (non-membrane) segment of YlmG and SsgB. Furthermore, ylmG lies in the dcw cluster, between sepF (SCO2079) and divIVA (SCO2077). SepF controls the polymerization of FtsZ and, whereas in Streptomyces DivIVA controls tip growth [29], it plays a major role in septum-site positioning in Bacillus subtilis [8,39]. Taken together, the phylogenetic data and genomic location suggest a role for YlmG in early cell division events and the possible linkage to SsgB was therefore investigated. Considering the genomic location and its potential role in cell division (see below), the protein was renamed SepG.
Table 1.
Membrane proteins with high conservation in streptomycetes but lower conservation between different actinomycete genera. The 20 best hits are listed.
| SCOa | protein | function | conservationb |
|---|---|---|---|
| SCO1541 | SsgBc | cell division protein | 99 |
| SCO4609 | HtpX | metallopeptidase | 97 |
| SCO2155 | Cox1 | cytochrome c oxidase subunit I | 96 |
| SCO2151 | Cox3 | cytochrome c oxidase subunit III | 96 |
| SCO2078 | YlmG | membrane protein | 96 |
| SCO2944 | sugar permease subunit | 95 | |
| SCO4602 | NuoH2 | NADH dehydrogenase subunit | 95 |
| SCO1389 | ClsA | cardiolipin synthase | 95 |
| SCO5118 | OppB | oligopeptide permease subunit | 95 |
| SCO3404 | FtsH2d | metalloprotease | 95 |
| SCO1527 | alcohol phosphatidyl transferase | 94 | |
| SCO2534 | hemolysin-like protein | 93 | |
| SCO1796 | stomatin-like protein | 93 | |
| SCO2150 | QcrC | cytochrome c heme-binding subunit | 93 |
| SCO5670 | putative polyamine permease subunit | 93 | |
| SCO2945 | sugar permease subunit | 93 | |
| SCO2148 | QcrB | cytochrome b subunit | 93 |
| SCO4722 | SecY | preprotein translocase subunit | 92 |
| SCO1215 | CtaG | cytochrome c oxidase assembly factor | 92 |
| SCO3945 | CydA | cytochrome ubiquinol oxidase subunit | 92 |
| SCO2087 | MurX | phospho-N-acetylmuramoyl-pentapeptide-transferase | 92 |
aAccording to the Streptomyces coelicolor database numbering.
bAverage aa identity (in %) between streptomycetes. For this comparison, the eight genomes of the streptomycetes in StrepDB were used. The value is on average 2–3% lower if S. lividans is not included.
cIncluded for reference purposes.
dDoes not occur in all streptomycetes.
The phylogenetic distribution of sepG includes the phylum of the actinobacteria (high G+C Gram-positive bacteria), which encompasses both sporulating and non-sporulating genera, several Firmicutes (low G+C Gram-positives), including Bacillus and Streptococcus, as well as cyanobacteria and chloroplasts. A detailed phylogenetic tree was presented by Kabeya et al. [35]. The wide distribution of SepG suggests an ancient common ancestor. Its distant orthologue AtYLMG1-1 from A. thaliana was shown to be a membrane protein, and indeed, all prediction programmes strongly suggest that SepG has two membrane-spanning domains at its N- and C-termini, namely from aa 6–25 and from aa 73–90. The different prediction algorithms are uncertain as to the topology of the central domain, but our experiments show that it is most likely to reside inside the cytoplasm (see section Membrane topology of SepG at the end of the Results section).
2.2. sepG null mutants are compromised in sporulation
To study the function of sepG in Streptomyces, an in-frame deletion mutant was created in S. coelicolor M145. For this, gene replacement construct pGWS731 (see Material and methods section) was introduced into S. coelicolor M145 and colonies selected for apramycin resistance (marker for sepG disruption) and sensitivity to thiostrepton (marker for the vector). These colonies presumably had sepG replaced by the apramycin resistance cassette on the genome. Expression of the Cre recombinase in these cells resulted in a number of markerless deletion mutants, which all had very similar phenotypes with impaired sporulation as judged by the lack of grey pigmentation. After PCR verification of the deletion of sepG in these colonies, one representative colony was then selected and designated GAL1.
Growth on soya flour mannitol (SFM) agar plates showed that sepG mutants were impaired in development. While the parental strain S. coelicolor M145 developed normal grey-pigmented colonies indicative of sporulation after 3 days of incubation, the sepG null mutant GAL1 had a nearly white phenotype, eventually showing light grey pigmentation after elongated incubation (figure 1a). The non-sporulating ssgB null mutant was used as a reference (figure 1a). Sporulation of the strains was investigated by phase-contrast light microscopy of surface-grown colonies after 3 days of growth. The wild-type strain M145 produced abundant spore chains with evenly sized spores, whereas GAL1 produced far fewer spore chains with spores of variable sizes (figure 1b). Plasmid pGWS771, a low-copy-number vector expressing sepG behind the complete ftsZ promoter region (including three promoters and the ribosome binding site sequence), was introduced into strain GAL1 to generate strain GAL93. The restored sporulation of GAL93 indicates that the postponed sporulation was indeed owing to the deletion of sepG (figure 1b). Strain GAL7, which is GAL1 transformed with SepG localization construct pGWS755, also showed restored sporulation, suggesting that the chimeric SepG–eGFP is functional (figure 1b).
Figure 1.
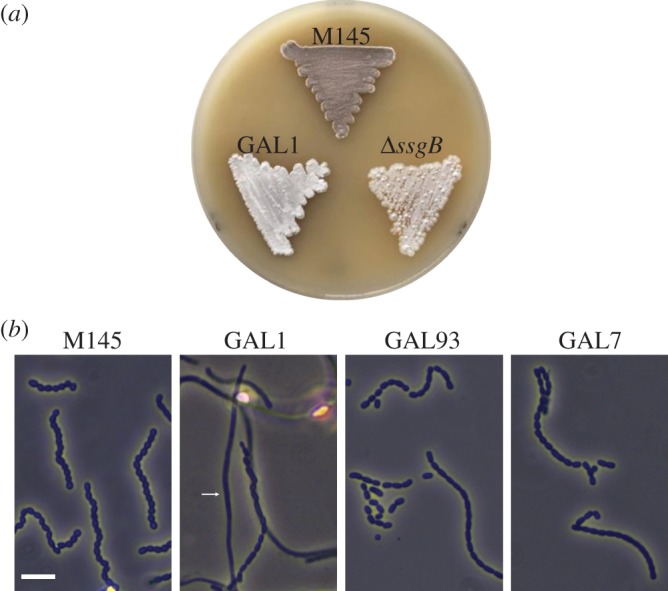
Compromised sporulation of the sepG null mutant of S. coelicolor. (a) Comparison of S. coelicolor M145 and its sepG null mutant GAL1. Note the lack of grey pigmentation of the sepG mutant owing to failure to produce the grey spore pigment. (b) Phase-contrast light micrographs of cultures grown on SFM agar against slides. Note that expression of wild-type SepG (GAL93) or SepG–eGFP (GAL7) restores sporulation to the ylmG mutant. The arrow indicates a straight aerial hypha in GAL1. All cultures were grown on SFM agar plates for 3 days at 30°C. Scale bar, 5 µm.
Closer inspection of the aerial hyphae and spores by cryoscanning electron microscopy (SEM) showed that where the parental strain S. coelicolor M145 produced abundant spiralling hyphae and well-developed chains of spores, the sepG null mutant predominantly produced straight aerial hyphae, whereby only very few chains of irregularly shaped spores could be identified (figure 2a). To quantify the size distribution, approximately 130 spores from the sepG mutant and the wild-type strain S. coelicolor M145 were measured on high-resolution TEM images (figure 2b). While wild-type spores showed the typical Gaussian distribution between 0.8 and 1.2 µm in length with an average size of 0.96 µm, sepG mutant spores showed a broader distribution, with an average size of 1.24 µm (t-test, p < 0.001); in other words, close to 0.3 µm longer on average than wild-type spores. Introduction of plasmid pGWS755 (SepG–eGFP) restored the spore size to 1.06 µm ± 0.05 (p > 0.95 compared with the parental strain, p < 0.001 compared with the sepG mutant). Taken together, these data identify sepG as a novel sporulation gene involved in sporulation-specific cell division.
Figure 2.
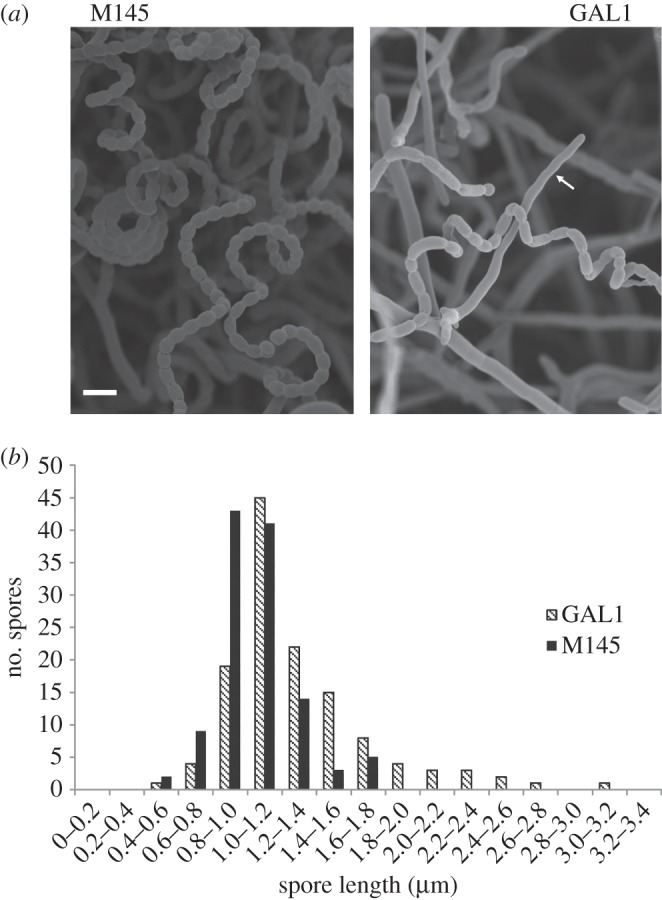
Comparison of spores produced by wild-type S. coelicolor M145 and its sepG null mutant. (a) Cryoscanning electron micrographs of wild-type and sepG mutant aerial hyphae. The parental strain sporulated abundantly, while occasional chains of irregularly sized spores were produced by the sepG mutant. The arrow indicates a straight aerial hypha in GAL1. Cultures were grown on SFM agar plates for 5 days at 30°C. Scale bar, 2 µm. (b) Size distribution of wild-type and sepG mutant spores. Spores were measured from a representative number of transmission electron micrographs, so that around 130 spores were measured for each strain. Note that a significant number of sepG mutant spores was unusually long.
2.3. SepG plays a role in spore-wall synthesis and nucleoid morphology
To obtain more detailed insights into spore morphology and spore-wall thickness, we performed high-resolution imaging using transmission electron microscopy (TEM) on thin sections. This again revealed the irregular sporulation of the sepG mutant. Furthermore, sepG mutant spores contained a considerably thinner, less well stained and therefore more electron-lucent cell wall (i.e. lighter in appearance), indicative of altered spore-wall synthesis (figure 3). Another major alteration observed in sepG mutant spores was the shape of the spore nucleoid. Rather than a well-condensed nucleoid in the centre of the spores as seen in wild-type spores, nucleoids of the sepG mutant spores were less well condensed and primarily localized close to the edge of the spores, with a pattern suggesting an unusual toroidal or doughnut shape (figure 3). Notably, such a toroidal shape is found routinely for nucleoids in developing or germinating spores of Bacillus [40]. The DNA was more centrally localized in complemented mutant GAL93, although it was less well condensed than in wild-type spores.
Figure 3.
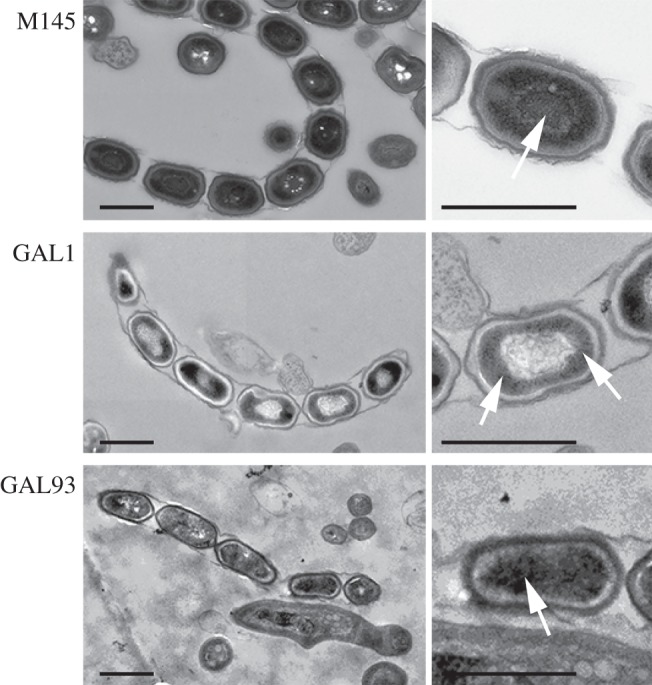
Transmission electron micrographs of wild-type, sepG mutant and complemented sepG mutant spores. Both a representative overview (left) and close-up (right) are presented. Arrows indicate nucleoids. Note that whereas the wild-type (M145) nucleoid is well condensed and located at the centre of the spores, that of the sepG mutant (GAL1) has an unusual distribution. DNA in the sepG mutant complemented with wild-type sepG (GAL93) was more centrally located than GAL1, although not as well condensed as in wild-type spores. Note the lighter appearance of the spore wall in the sepG mutant. Cultures were grown on SFM agar plates for 5 days at 30°C. Scale bar, 1 µm.
Viability of the sepG mutant spores was tested by analysing heat resistance. For this, spores were incubated for 10 min at 60°C and plated onto SFM agar plates. While 45% of the wild-type spores survived the heat treatment, only 8% of the sepG mutant spores survived the heat treatment. The defect could be largely complemented by expressing wild-type SepG or SepG–eGFP: spores of GAL7 and GAL93, which are sepG mutants harbouring complementation construct pGWS771 and pGWS755, respectively, showed improved heat tolerance, with 32% and 36% of the spores surviving the heat treatment.
To better visualize the nucleoid distribution in wild-type and sepG mutant spores, the nucleoids were stained with DAPI and imaged with stimulated emission depletion (STED) microscopy (figure 4a). In wild-type spores, the nucleoids were seen as very bright dots that localized to the central part of the spores. Interestingly, in sepG null mutants, the nucleoids were less condensed and more dispersed, whereby the centres of the spores were largely devoid of DNA. Spores of strain GAL93, which is the sepG null mutant containing plasmid pGWS771 expressing wild-type sepG, no longer showed such doughnut-shaped DNA structures, although the nucleoids appeared less well condensed when compared with wild-type spores (figure 4a). To provide statistical insights into the frequency of occurrence of this altered nucleoid morphology in GAL1, the percentage of spores with rim-localized DNA in each strain was calculated (using more than 100 spores for each strain). Fourteen per cent of wild-type spores had dents in their intensity profile over a spore, which indicates that the DNA localizes to the spore periphery. For GAL1 that was 74%, while the complemented strain again showed the wild-type proportion of 1 out of 7 (13%) with rim-localized DNA. The average DNA distribution in 10 spores along a spore chain was plotted (figure 4b). The wild-type nucleoid profile showed one single peak for the DNA intensity with regular height and width, suggesting well-condensed DNA. Conversely, nucleoid profiles of GAL1 displayed a clear intensity drop in the middle of the spore, indicating that the DNA in each spore compartment localizes more towards the edge of the spores and away from the centre. DNA distribution in the complemented mutant GAL93 was largely restored, showing a single peak in the middle of the spores.
Figure 4.
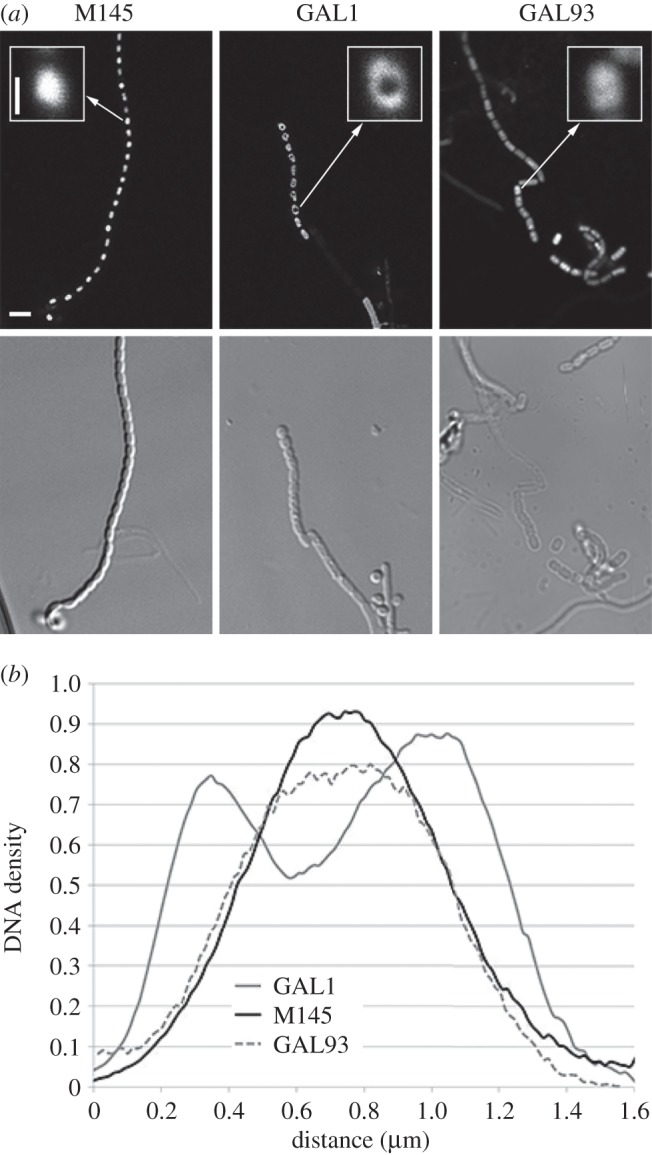
Aberrant nucleoid distribution in sepG mutant spores. (a) Fluorescence micrographs of nucleoid distribution in the spores; Top, STED images; bottom, light images. DNA staining of wild-type spores (M145, left) shows normal spore lengths and proper nucleoid condensation. In contrast, nucleoids of sepG null mutants (GAL1, middle) appear toroid and less well condensed. The aberrant nucleoid shape is complemented by a clone expressing sepG (strain GAL93; right), but DNA condensation is still affected. Inset: 7× magnification of representative nucleoids. Cultures were grown on SFM agar plates for 5 days at 30°C. Bar, 2 µm. Bar for inset, 500 nm. (b) DNA distribution along single spores in S. coelicolor M145, in GAL1 and in GAL93. The x-axis represents the distance between two poles of the spores, and the y-axis shows the DNA density. Note the double peak for GAL1, highlighting higher density of DNA to the spore periphery when sepG is absent, while in wild-type and GAL93 spores the nucleoid is localized to the centre.
Thus, TEM, STED and nucleoid distribution data strongly suggest that SepG is required for proper nucleoid compaction during sporulation. It is as yet unclear if this is a direct or indirect effect of the deletion of sepG. However, the defect can be complemented by introduction of wild-type copy of sepG, underlining that the absence of SepG is the primary cause of the defect.
2.4. Localization of SepG
To determine the localization of SepG in S. coelicolor, C-terminal fusions with either enhanced green fluorescent protein (eGFP) or (monomeric) mCherry were expressed from the ftsZ promoter region (from plasmids pGWS755 and pGWS791, respectively). Preliminary transcript analysis and DNA sequence analysis failed to identify promoter sequences immediately upstream of sepG, whereas promoter activity was identified inside of the upstream-located sepF (data not illustrated). To avoid including sepF sequences, we instead directed transcription of sepG from the promoter region of ftsZ, so as to coordinate transcription with cell division. The ftsZ promoter region consists of three promoters, one of which is expressed primarily during vegetative growth, one during sporulation and one is constitutive [41]. We will further refer to this as the ftsZ promoter region. Various independent DNA microarray analyses from our laboratory indicate that sepG and ftsZ have a similar expression profile throughout growth, whereby sepG is consistently expressed about 2–2.5 times higher than ftsZ in S. coelicolor M145 [42]; this implies that use of the ftsZ promoter region will not lead to overexpression of sepG, nor with low-copy-number vectors. Indeed, introduction of pGWS755 or pGWS791 restored sporulation to the sepG null mutant, showing that both SepG fusion proteins are functional and expressed properly in vivo. SepG–mCherry and SepG–eGFP had the same localization profile, providing further support for the functionality and reproducibility of the localization experiments (see also below).
Confocal fluorescence microscopy was applied to investigate SepG–eGFP localization in wild-type cells and the sepG mutant. The pattern of SepG–eGFP localization was dynamic and developmental stage-dependent. In young aerial hyphae, when the chromosomal DNA was still uncondensed and septal membrane synthesis had not yet initiated, SepG–eGFP formed distinct and widely spaced foci (figure 5a). Such a localization pattern is very similar to that of the initial stage of SsgB localization, prior to its septum-site localization [27] (figure 6). As development progressed, nucleoids segregated and septal ladders became apparent, whereas SepG–eGFP disappeared (figure 5b). During spore maturation the nucleoids condensed, and at this stage, SepG–eGFP relocalized to the sites of active cell-wall remodelling (figure 5c), with a ring of SepG surrounding the nucleoid (figure 5d). The fluorescence images strongly suggest that in sepG mutant spores, the nucleoid takes up the space normally occluded by a SepG-dependent mechanism (figure 5e). In mature spores, which are physically separated, no GFP signal could be observed, suggesting that SepG eventually disappears (not illustrated).
Figure 5.
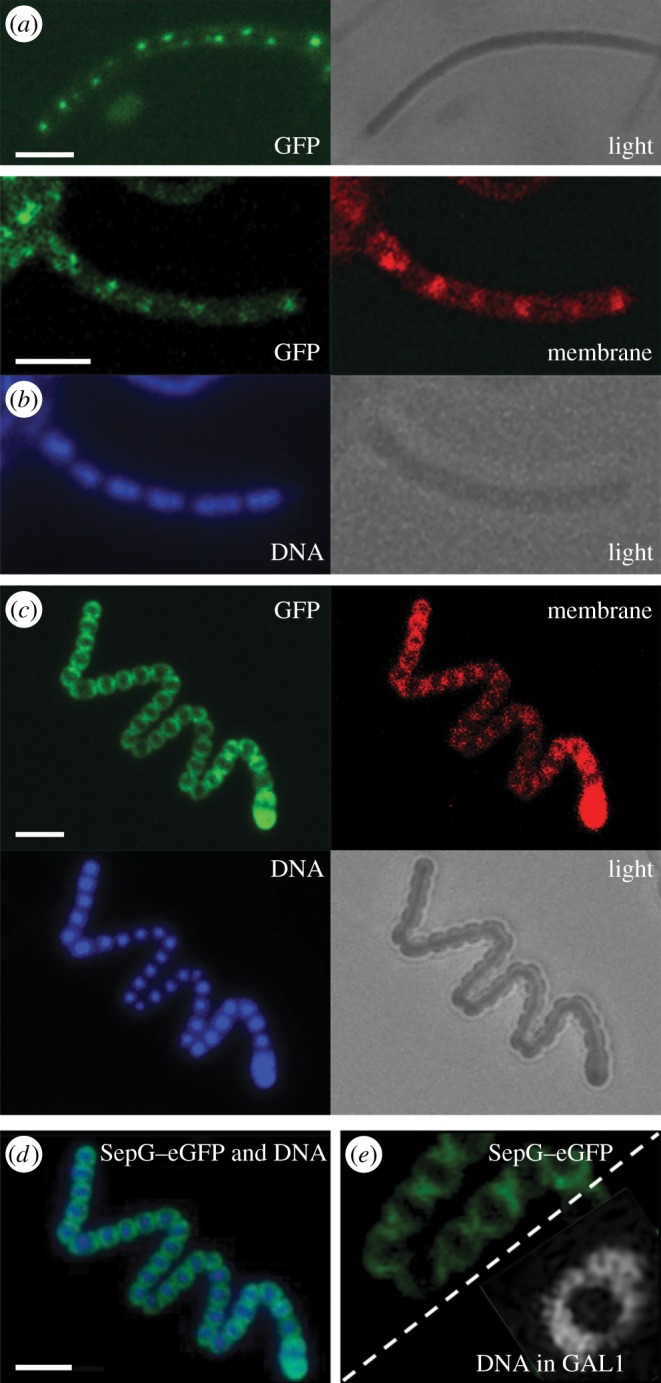
Localization of SepG in S. coelicolor. (a–c) Sporogenic aerial hyphae of S. coelicolor M145 were imaged by fluorescence microscopy visualizing SepG–eGFP, membrane (stained with FM5–95), DNA (stained with DAPI) and a light micrograph at the onset of sporulation in aerial hyphae (a), during sporulation (b) and spore maturation (c). (d) A merged image of SepG–eGFP and the DNA taken from (c). (e) SepG–eGFP in wild-type cells (strain GAL7; left) showing a similar localization in the spores as the DNA in the sepG null mutant (strain GAL93; right), suggesting that SepG aids in DNA condensation in the centre of the spores. Note that the images in panel (e) are only presented for comparison, as the sepG–GFP data were obtained from panel (d) (FM imaging), while the nucleoid was obtained from figure 4a (STED imaging). Scale bar, 3 µm.
Figure 6.

Effect of SepG on the localization of eGFP fusions of SsgA, SsgB and FtsZ. All proteins localize correctly in wild-type cells (M145, left). In contrast, SepG is required for the formation of ladders of FtsZ or for the localization of SsgB at division sites. SsgA has a normal focal pattern in the sepG mutant, whereas focal patterns for SsgB were seen in around 33% of the aerial hyphae (67% of the GAL1 aerial hyphae showed diffuse localization of SsgB); SsgB fails to localize to the future septum sites in sporogenic aerial hyphae of GAL1. The occasional Z rings formed in sepG null mutant GAL1 are also seen in ssgB mutants [27]. Cultures were grown on SFM agar plates for 5 days at 30°C. Scale bar, 1 µm (for all images).
2.5. Localization of SsgB to septum sites depends on SepG
We previously showed that FtsZ is directly recruited by SsgB in an SsgA-dependent manner, followed by FtsZ-dependent recruitment of the other divisome components [27]. The phenotype of the sepG null mutants prompted us to investigate whether the localization of one or more of these proteins was affected by the absence of SepG. To address this question, the integrative vectors pGWS116, pGWS526 or pKF41, expressing SsgA–eGFP, SsgB–eGFP or FtsZ–eGFP, respectively, were introduced into the sepG null mutant. While SsgA–eGFP localized normally in the mutant, SsgB–eGFP showed two patterns of localization, namely diffuse along the hyphal periphery or foci at what are most likely the future septum sites. The diffuse pattern was mainly observed in sepG null mutants, whereas foci were predominantly seen in the parental strain M145. In line with the delocalized pattern of the FtsZ-recruiting SsgB protein, the sepG mutant also failed to produce the typical ladder-like patterns of FtsZ–eGFP seen in wild-type hyphae (figure 6). Only occasional septa were formed, a phenotype that is also observed in ssgB null mutants and other sporulation (whi) mutants [27].
SsgB–eGFP still formed distinct foci, but mainly in the middle of young aerial hyphae (which can be distinguished from pre-sporulating aerial hyphae as they are narrower in width [18]), representing the first stage of SsgB localization (figure 6). However, in contrast to wild-type cells [27], in sepG null mutants the SsgB foci subsequently disappeared (figure 6), indicating that the stable localization of SsgB requires SepG. In wild-type cells, SsgB–eGFP foci appeared in 80% of the hyphae with around 1.25 ± 0.14 µm spacing between them. While in ssgA and sepG null mutants, SsgB–eGFP foci were only apparent in 30% and 33% of the hyphae, spaced by 0.53 ± 0.07 and 0.57 ± 0.04 µm, respectively. Thus, after the SsgA-mediated initial localization of SsgB, SepG directly or indirectly acts to ensure the localization of SsgB at future division sites at the membrane, which is a crucial step in initiating sporulation-specific cell division. This is consistent with SepG being required for the septum-site localization of SsgB after its initial localization by SsgA (figure 6).
This raised the question whether SepG directly interacts with SsgB. To investigate this in vivo, we applied Förster resonance energy transfer (FRET), which allows the identification of direct molecular interactions. As low-wavelength fluorophore, eGFP was used, whereas mCherry was used as longer-wavelength fluorophore. SepG–mCherry was introduced into S. coelicolor M145 expressing either SsgA–eGFP or SsgB–eGFP to generate strains GAL94 and GAL95, respectively; SepG–eGFP was introduced into S. coelicolor M145 expressing FtsZ–mCherry to generate strain GAL96. SepG–eGFP was also introduced in M145 expressing free mCherry under the control of the gap1 promoter (from SCO1947, the gene for glyceraldehyde-3-phosphate dehydrogenase) to generate strain GAL103. Strain GAL102, which is S. coelicolor M145 co-expressing SsgB–eGFP with FtsZ–mCherry, was used as positive control, considering the direct interaction between SsgB and FtsZ [27]. As negative control we used S. coelicolor K202, which expresses FtsZ–eGFP alone.
The data are summarized graphically in figure 7b, whereby the negative control was set to zero. GAL102 showed a FRET efficiency consistent with the short intermolecular distance between SsgB and FtsZ 1.08 ± 0.03 (table 2; p < 0.01 compared with SepG-free mCherry). The negative control FtsZ–eGFP showed a similar fluorescence intensity before and after acceptor photobleaching (intensity before bleach is set to 1) of 0.97 ± 0.02, consistent with a lack of interaction with an acceptor (table 2). Importantly, SepG and SsgB colocalized, as shown by the more than 90% overlap of their foci in approximately 100 hyphae (figure 7a), and the FRET data strongly suggest close interaction between them, with an increase in fluorescence intensity to 1.05 ± 0.02 (p < 0.01), an intermolecular distance close to that between the known partners SsgB and FtsZ. No interaction was seen between SepG and SsgA (no fluorescence intensity increase; 0.99 ± 0.03, p > 0.4), between SepG and FtsZ (0.90 ± 0.03, p > 0.1) or between SepG and free mCherry (0.97 ± 0.02).
Figure 7.
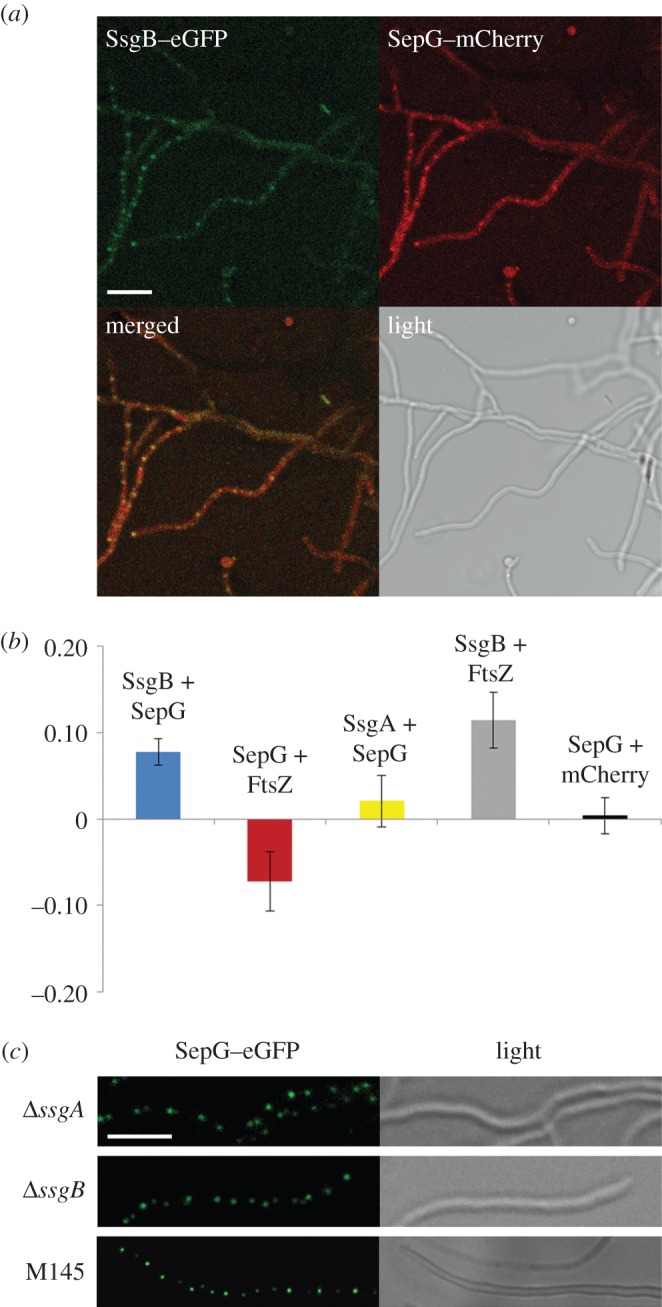
FRET imaging of the interaction between SepG and SsgB. (a) FM images showing the colocalization of SsgB and SepG. Scale bar, 5 µm. (b) Graphical representations of the FRET data. All efficiencies are represented relative to the control (which was set to zero). FRET data are summarized in table 2. (c) FM images showing localization of SepG–eGFP in ssgA- and ssgB-null mutants. Scale bar, 2 µm.
Table 2.
FRET efficiencies for different combinations of donor (eGFP) and acceptor (mCherry).
| GAL95 |
GAL96 |
GAL94 |
GAL102 |
GAL103 |
K202 |
|
|---|---|---|---|---|---|---|
| strain | SsgB + SepG | SepG + FtsZ | SsgA + SepG | SsgB + FtsZ | SepG + mcherry | FtsZ |
| average | 1.05 | 0.90 | 0.99 | 1.08 | 0.97 | 0.97 |
| standard deviation | 0.02 | 0.03 | 0.03 | 0.03 | 0.02 | 0.02 |
| n | 24 | 18 | 11 | 6 | 12 | 9 |
We then wondered if SepG could localize independent of SsgA or SsgB, which should be the case if SsgB localization depends on SepG but not vice versa. To test this, construct pGWS756, expressing SepG–eGFP, was introduced into the ssgA and ssgB null mutants. SepG formed distinct and widely spaced foci in the aerial hyphae in both ssgA and ssgB null mutants, with a pattern very similar to that in wild-type cells (figure 7c). SepG–eGFP also localized at the intersection of prespores in wild-type cells (figure 5); because ssgA and ssgB mutants do not produce spores, SepG–eGFP was only visualized as foci in these mutants.
Taken together, our data show that the stable localization of the FtsZ-recruiting SsgB requires SepG, and not vice versa. FRET experiments suggest that SepG and SsgB directly interact, whereas the lack of interaction of SepG with SsgA or FtsZ is consistent with the relocation of SepG prior to assembly of the other divisome members. We also sought to confirm this by two-hybrid experiments using the central (cytoplasmic) domain of SepG as the bait, but—like for many other cell division proteins—this failed to provide conclusive data, both in direct studies with SepG and SsgB, or in a library screen with (partial) SepG as the bait (now shown). The precise way that SsgB and SepG interact is under investigation.
2.6. Membrane topology of SepG
All membrane prediction programs (e.g. DAS, TMPred) predict with high probability scores that SepG is a membrane protein with two TM domains. To establish if indeed SepG is integrated in the membrane, and the SepG–eGFP fusion protein is stable, we first estimated the mobility of SepG–eGFP using fluorescence recovery after photobleaching (FRAP). Freely mobile fusion proteins would have a half time of recovery of around 0.03 s in the cytoplasm, whereas for membrane-associated fusions, the recovery should be much slower (in the order of seconds) [43]. In Streptomyces, the half time of recovery for freely mobile eGFP with egfp transcribed from gap promoter proteins was around 0.8 s, whereas for SepG–eGFP it was 42.3 ± 3.3 s. TatA–eGFP was included as a positive control for a membrane protein, and showed a half time of recovery of 82.3 ± 11.5 s. These data clearly show that SepG–eGFP recovers much slower than free eGFP proteins, consistent with SepG–eGFP localized inside the membrane (and no evidence of free eGFP such as after inadvertent cleavage of the SepG–eGFP fusion protein in vivo).
To obtain further experimental evidence that SepG is a membrane protein and to establish whether the central domain is localized inside the cytoplasm or outside the cell, we used the β-lactamase reporter system for transmembrane proteins, an elegant system developed originally by Broome-Smith et al. [44] that allows establishing the orientation of transmembrane (TM) domains. BlaM is a β-lactamase that is very efficiently translocated over the membrane, and if it is fused behind a TM domain with an inside–outside orientation, it will result in BlaM translocation over the membrane and thus convey ampicillin resistance to the cells [44–46]. Construct pGWS755, which expresses SepG–eGFP, was used as the basis for further derivatives. While the construct is made for expression in Streptomyces, introduction of pGWS755 in E. coli JM109 also results in the production of SepG–eGFP, as shown by fluorescence imaging. This indicates that at least one of the S. coelicolor ftsZ promoters is actively transcribed in E. coli. Interestingly, the fluorescent fusion protein localized specifically to the poles of E. coli cells (data not illustrated). Derivatives with various parts of sepG fused in front of blaM were made as described in the Material and methods section. These allow the expression in E. coli of the following fusion products: SepG-BlaM (to probe full-length SepG; pGWS793), SepG1–40-BlaM (TM domain 1; pGWS794) and SepG41–94-BlaM (TM domain 2; pGWS795). In constructs pGWS793, pGWS794 and pGWS795, only the part of blaM encoding the mature protein (starting with HPETLVK) was used. As a positive control BlaM including its signal peptide was expressed from construct pGWS796 under the control of the ftsZ promoter region. Low-copy-number plasmid pHJL401 (which carries blaM as resistance marker) was also used as a positive control.
Constructs were introduced into E. coli JM109 and transformants tested for their ability to grow on ampicillin (see Material and methods). All transformants grew equally well on LB agar plates without ampicillin (figure 8a). The negative control (JM109 harbouring pGWS755 expressing SepG–eGFP) failed to grow on plates containing ampicillin, whereas the positive controls, JM109 with either plasmid pHJL401 or with a construct expressing BlaM with signal sequence from the S. coelicolor ftsZ promoter region (pGWS796), grew well under all conditions (figure 8a). This again shows that at least one of the S. coelicolor ftsZ promoters results in active transcription in E. coli. E. coli cells expressing BlaM fused behind the N-terminal part of SepG (i.e. the first TM domain, pGWS794) failed to grow on ampicillin, whereas constructs where BlaM was fused behind full-length SepG (pGWS793) or the C-terminal part of SepG (containing only the second TM domain; pGWS795) conveyed ampicillin resistance, albeit with smaller colonies in the case of pGWS795 (figure 8a). These results strongly suggest that the N-terminal domain of SepG is oriented from outside to inside the cell, whereas the C-terminal domain is oriented towards the outside of the cell. At higher concentrations of ampicillin (20 µg ml−1), E. coli cells expressing SepG–BlaM did grow, but those containing only the C-terminal TM domain in front of SepG–BlaM failed to grow, perhaps because only a smaller fraction of the proteins is correctly oriented in the membrane when a single TM domain is present.
Figure 8.

Membrane topology of SepG. (a) Growth of E. coli JM109 carrying plasmids expressing different BlaM fusions, namely full-length SepG–BlaM (pGWS793); N-terminal SepG1-40-BlaM (pGWS794; first TM domain of SepG); C-terminal SepG41–94-BlaM (pGWS795; second TM domain of SepG); BlaM with signal sequence expressed from the ftsZ promoter(s) (pGWS796; positive control); BlaM with signal sequence expressed from its native promoter (pHJL401; positive control); and pGWS755 expressing SepG–eGFP (pGWS755; negative control). Transformants were grown overnight at 37°C on LB agar plates with 0, 10 or 20 µg ml−1 ampicillin. (b) Topological model of SepG. Left, wild-type SepG, with the topology model generated using TMRPRES2D [47]. Right, model of SepG fused to eGFP, drawn based on the wild-type SepG model.
Growth of all transformants was then analysed in liquid LB with or without ampicillin, using a Bioscreen C automated microwell plate reader with continuous shaking and temperature control (three independent colonies and three replicates per colony). These data were fully consistent with the observations made when the transformants were grown on LB agar plates. E. coli expressing BlaM fused behind full-length SepG grew very well at all ampicillin concentrations, as did transformants harbouring the positive control plasmids pGWS796 and pHJL401, which express blaM from the S. coelicolor ftsZp and from the native blaMp, respectively (electronic supplementary material, figure S1). Conversely, E. coli cells expressing N-terminal SepG1–40-BlaM or SepG–eGFP failed to grow in the presence of ampicillin. Transformants expressing BlaM fused behind the C-terminal part SepG41–94 did grow at 10 µg ml−1 ampicillin, but only after prolonged incubation, demonstrating that a single TM domain is not sufficient to provide stable integration of proteins in the membrane (electronic supplementary material, figure S1).
3. Discussion
The control of cell division in the multicellular streptomycetes is very different from that in for example E. coli and B. subtilis. During sporulation, the aerial hyphae differentiate into long chains of spores, and the more or less simultaneous formation of many septa results in spectacular ladders of Z rings [17]. The canonical control systems and septum-localizing proteins (Min, Noc, FtsA, ZipA, ZapA, EzrA, etc.) found in bacteria that divide by binary fission are all absent in streptomycetes, and instead actinomycete-specific proteins control the localization of the septa. In streptomycetes, cell division is positively controlled, whereby FtsZ is actively recruited to septum sites by the action of the actinomycete-specific SsgB [27].
In this work, we show that ylmG (sepG), which lies between sepF and divIVA, and is one of the last genes in the dcw cluster whose function had not yet been established, plays a major role in the control of sporulation-specific cell division in S. coelicolor. SepG is a predicted membrane protein, and the distant SepG orthologue AtYLMG1-1 of A. thaliana is exclusively found in membrane fractions [35]. Our data show that this is also the case in S. coelicolor (see below). In Streptomyces, SepG helps to orchestrate the earliest stage of sporulation-specific cell division, as it is required for the correct localization of SsgB to future septum sites. After the initial localization of SsgB by SsgA to a central position in the aerial hyphae, this is the next crucial step in the positive control of cell division in Streptomyces, followed by the recruitment of FtsZ by SsgB. In addition, at a later stage of spore development, SepG helps maintaining nucleoid shape, either directly or indirectly. In wild-type cells, SepG localizes in a ring at the periphery of the spores and surrounding the nucleoid, whereas in sepG null mutants, large doughnut-shaped (toroid) nucleoids are formed accompanied by thinner spore walls, suggesting imperfect cell-wall synthesis, which is consistent with the observed reduced heat resistance of the spores. One explanation is that the expanded nucleoids in spores of sepG null mutants enforce the formation of larger spores, and hence with thinner cell walls assuming that the same amount of cell-wall material is available per spore. In A. thaliana, the SepG orthologue AtYLMG1-1 also plays a role in controlling nucleoid morphology, whereby knockdown of the gene impaired nucleoid partitioning and changed the shape of the nucleoid. Similar results were obtained in the cyanobacterium S. elongatus [35]. This therefore is most likely to be an inherent feature of SepG that remained conserved through evolution. After the localization of SsgB to the future sites of division, SepG continues to follow the spore-wall synthetic machinery, and ensures the coordination of spore-wall synthesis with nucleoid compaction by a mechanism that as yet remains unresolved. In line with its more prominent role during sporulation-specific cell division, previous microarray data showed that, as for ssgB, the transcription of sepG is enhanced at the onset of sporulation [42,48].
The fact that SepG localizes normally in ssgA and ssgB mutants, whereas conversely, SsgB fails to localize properly in sepG mutants, supports a model wherein SepG ensures the correct localization of the FtsZ-recruiting SsgB protein. The failure of SsgB to find the septum sites thereby explains the mislocalization of FtsZ in sepG null mutants. As an alternative to recruitment, SepG may also be involved in stabilizing SsgB complexes in the sporogenic hyphae. A question to answer is whether SepG directly interacts with SsgB. FRET data provide in vivo evidence that supports a direct interaction between SepG and SsgB during the onset of cell division. In further support of this notion, specific FRET signals were only obtained when SepG and SsgB were analysed, but not for SepG with SsgA, FtsZ or free mCherry. Interestingly, the central domain of SepG shows a similar conservation as SsgB, with extremely high conservation in streptomycetes, but low conservation between different actinomycete genera. Such a conservation pattern is not evident in the predicted TM domains, and the suggestive covariation in terms of aa conservation in different bacteria between the SepG cytoplasmic domain and SsgB provides supporting phylogenetic evidence consistent with possible interaction between SsgB and SepG. No other interaction partners were identified in a library screen with SepG as the bait. Furthermore, no interaction was observed between SepG and SsgA. The way in which SepG and SsgB interact is currently under investigation in our laboratory.
Thus, in terms of the control of cell division, our data are consistent with a model wherein SepG recruits SsgB to the future septum sites, and SsgB subsequently recruits FtsZ. While SsgB and FtsZ remain together a divisome protein, SepG does not become part of the divisome, but instead moves away from SsgB, whereby our data suggest that it may subsequently relocate to the cell-wall synthetic machinery. While relatively little is known of nucleoid dynamics in Streptomyces, we can try to glean information from the well-studied B. subtilis, although Bacillus sepG null mutants have no apparent phenotype [8]. During vegetative growth the nucleoid localizes close to mid-cell in B. subtilis, but its shape changes during the onset of sporulation, and eventually a doughnut-shaped ring is formed similar to that found in S. coelicolor sepG mutants, with a diameter of around 1 µm [40]. During germination, these rings disappear and the nucleoids take up the diffuse lobular shape typical of vegetative cells [49]. The expansion of the nucleoid towards the spore periphery probably disturbs the cell cycle, which may trigger a nucleoid-occlusion-like mechanism to prevent septum synthesis close to the nucleoid; however, such a mechanism has not yet been identified in streptomycetes, neither in wild-type cells nor in mutants disturbed in DNA partitioning, such as parA and parB mutants of S. coelicolor.
An important question to resolve was what the membrane topology is of SepG, and in particular whether the small central (soluble) region is oriented towards the inside or the outside of the cell. The slow FRAP recovery, with a half time of recovery in the order of a minute, is consistent with SepG being a membrane protein, as freely diffusible GFP/mCherry would have a half time in the millisecond range. For the localization, studies both SepG–GFP and SepG–mCherry fusion proteins were used, and either protein is functional, because they could restore sporulation to the sepG null mutant. Furthermore, our FRET data on the C-terminal fusion proteins (either mCherry or GFP derivatives) show that the fluorophore is located inside the cytoplasm, and that most if not all of the fusion proteins in the cell are intact. Membrane topology analysis based on the BlaM reporter system [44] showed that the central part of SepG is located inside the cell. The first TM domain was able to direct BlaM to the cytoplasm, whereas fusion behind full-length SepG or behind the second TM domain directed BlaM to the periplasm, strongly suggesting that the first TM domain of SepG is oriented from outside to inside, whereas the second TM domain is oriented from inside to outside. A topological model for SepG is presented in figure 8b, including a suggested topology for the SepG–eGFP fusion protein. The orientation of the central domain towards the inside of the cell is consistent with a model wherein SepG interacts with SsgB and then recruits SsgB to the membrane. In the chimeric SepG–eGFP protein, eGFP will probably prevent full membrane spanning of the C-terminal TM domain of SepG, which may also explain why the fusion protein can only partially restore sporulation to sepG mutants.
Finally, the principles discovered in this work for Streptomyces may well translate to lower Gram-positives such as Bacillus and Streptococcus. This is among others based on strong phylogenetic evidence, because the location of sepG between divIVA and sepF was maintained during several hundred millions years of evolution, whereby ylmH was lost in actinomycetes. This suggests functional overlap between the Bacillus and Streptomyces orthologues, despite the apparent absence of SsgB in firmicutes. A key observation may well be that, as in Streptomyces, a cell-division control mechanism that is independent of the negative control systems Min and Noc has recently been identified in B. subtilis. It was shown that the Z ring can be positioned at division sites in the absence of Min and Noc, and that in Noc− cells the Z rings have a preference for the mid-cell position between the two nucleoids [32]. Conceivably, therefore, SepG may have a similar role in division-site selection in B. subtilis as in Streptomyces, but such a role would only be obvious if sepG were to be studied—and, if at all possible, mutated—in the absence of Noc. Further molecular insights into the function of SepG in streptomycetes will shed more light on the way the positive control of cell division is governed in these multicellular bacteria, as well as on the yet poorly understood mechanisms that prevent nucleoid damage during septum synthesis and cell-wall remodelling.
4. Material and methods
4.1. Bacterial strains and media
The bacterial strains used in this work are listed in electronic supplementary material, table S1. E. coli strains JM109 [50] and ET12567 [51] were used for routine cloning and for isolation of non-methylated DNA, respectively. E. coli transformants were selected on LB agar media containing the relevant antibiotics and grown O/N at 37°C. S. coelicolor A3(2) M145 [52] was used as parental strain to construct mutants. The ssgA null mutant GSA3 [53] and ssgB null mutant GSB1 [54] of S. coelicolor M145 were obtained from the Leiden University strain collection. All media and routine Streptomyces techniques are described in the Streptomyces manual [52]. Yeast extract-malt extract and tryptone soy broth with 10% sucrose were the liquid media for standard cultivation. Regeneration agar with yeast extract was used for regeneration of protoplasts and with appropriate antibiotics for selection of recombinants [52]. SFM agar plates were used to grow Streptomyces strains for preparing spore suspensions and for morphological characterization and microscopy.
4.2. Plasmids and constructs
All plasmids and constructs described in this work are listed in electronic supplementary material, table S2 and oligonucleotides in electronic supplementary material, table S3. PCRs were carried out with Phusion enzyme (Finnzymes, Bioké, Leiden, The Netherlands) as previously described [55].
4.2.1. Constructs for gene disruption
The strategy for creating knock-out mutants is based on the unstable multi-copy vector pWHM3 [56], essentially as described previously [48]. The −1442/+6 and +277/+1521 regions relative to the translational start of ylmG (sepG; SCO2078; 285 nt total) were amplified by PCR from the S. coelicolor M145 genome using primer pairs ylmG_LF_1442 and ylmG_LR + 6, and ylmG_RF + 277 and ylmG_RR + 1521, respectively (electronic supplementary material, table S3). Fragments were then cloned into EcoRI/BamHI-digested pGWS725, with the oligonucleotides designed such as to create a unique XbaI site in-between the flanking regions. The apramycin resistance cassette aac(3)IV flanked by loxP sites was then cloned via the engineered XbaI site to generate knock-out construct pGWS731. The presence of loxP sites allows the efficient removal of the apramycin resistance cassette from the chromosome following the introduction of plasmid pUWLCRE that expresses the Cre recombinase [57]. For complementation of the sepG null mutant the coding sequence (+1/+294 region) of sepG under control of the ftsZ promoters (pGWS771) was inserted into the integrative vector pSET152 [58], which integrates at the attachment site for bacteriophage ΦC31 on the S. coelicolor genome.
4.2.2. Constructs for enhanced green fluorescent protein and mCherry fusion proteins
To obtain a construct expressing SepG–eGFP, we used plasmid pKF41, which expresses FtsZ–eGFP from the native ftsZ promoter region [59]. The insert was excised using restriction enzymes BglII and NotI, cloned into the integrative vector pSET152. To replace the ftsZ coding region by that of sepG, the construct was digested with StuI and BamHI, and the sepG gene was PCR-amplified from the genome of S. coelicolor using primer pair ylmG_F + 1 and ylmG_R + 282. The PCR product was subsequently cloned as a StuI–BamHI fragment in between ftsZp and egfp to generate pGWS755. The insert of pGWS755 was then moved as an EcoRI–XbaI fragment into the low-copy-number vector pHJL401 [60] to generate pGWS756. Vector pHJL401 is a stable vector with (very) low-copy-number (1–5 copies per chromosome), which is very well suited for complementation studies [61]. To generate similar constructs expressing SepG–mCherry, the egfp gene in pGWS755 was removed by digestion with BamHI and NotI and replaced by the gene for mCherry to generate pGWS791.
4.2.3. Constructs for β-lactamase experiments
Various constructs were created to study the transmembrane topology of SepG in E. coli using BlaM (β-lactamase) as a reporter, according to a system that was described previously [44]. The gene for eGFP in pGWS755 was replaced by the blaM gene without the sequence encoding the signal peptide (i.e. from HPETLVK to the end) to generate construct pWGS793. The blaM gene was amplified by PCR from pHJL401 using primers BlaM_F_EB and BlaMR_XH, adding 36 nt encoding a 12 amino acid linker in primer BlaM_EB as described [44]. The resulting construct pGWS793 expresses SepG-BlaM hybrid protein from the ftsZ promoter region. The part of sepG encoding the N-terminal 40 aa including the first TM domain was amplified from S. coelicolor M145 genomic DNA using primers YlmG_F + 1 and YlmG_R + 120. The fragment was cloned into pGWS793 as a StuI–BamHI fragment to replace full-length sepG, resulting in pGWS794. Similarly, the part of sepG encoding aa 41–94 of SepG including the second TM domain was amplified from S. coelicolor M145 genomic DNA using primers YlmG_F + 121 and YlmG_R + 282 and cloned into pGWS793, to replace full-length sepG, resulting in pGWS795. Full-length blaM (including the sequence encoding the signal sequence for secretion) was amplified by PCR from pHJL401 using primers BlaM_F + 1_ES and BlaM_R_XH. This DNA fragment was cloned into pGWS793 as a StuI–XbaI fragment so as to replace sepG-blaM, resulting in construct pGWS796, which expresses full-length BlaM.
4.3. Microscopy
Sterile coverslips were inserted at an angle of 45° into SFM agar plates, and spores of S. coelicolor and derivatives were carefully inoculated at the intersection angle. After incubation at 30°C for 3–5 days, coverslips were positioned on a microscope slide pre-wetted with 5 µl of 1× PBS. Fluorescence and corresponding light micrographs were obtained with a Zeiss Axioscope A1 upright fluorescence microscope (with an Axiocam Mrc5 camera at a resolution of 37.5 nm pixel−1). The green fluorescent images were created using 470/40 nm band pass excitation and 525/50 band pass detection, whereas for the red channel 550/25 nm band pass excitation and 625/70 nm band pass detection was used [62]. DAPI was detected using 370/40 nm excitation with 445/50 nm emission band filter. For membrane staining, FM5–95 was used and for DNA staining DAPI (all obtained from Molecular Probes). To obtain a sufficiently dark background, all images were corrected by setting the signal outside the hyphae to zero. These corrections were made using Adobe Photoshop CS4.
Chromosome distribution in spores of S. coelicolor strains was studied using stimulated emission depletion (STED) microscopy. For this, patches of the different Streptomyces strains were grown on SFM agar plates and incubated at 30°C for 5 days. Live cells were stained with Syto 9 (0.5 mM) for 5 min, after which STED was performed with a Leica TCS STED CW system with 488 nm excitation (5% laser power) and depletion at 592 nm (30% laser power).
Morphological studies on surface-grown aerial hyphae and/or spores by SEM were performed using a JEOL JSM6700F scanning electron microscope as described previously [63]. For thin sections, square 2 mm blocks were cut from 6-day-old cultures and fixed with 1.5% glutaraldehyde in 0.1 M cacodylate buffer for 1 h, rinsed with 0.1 M cacodylate buffer, and post-fixed with osmium tetroxide for 1 h. Subsequently, samples were dehydrated (70% ethanol 15 min, 80% ethanol 15 min, 90% ethanol 15 min, 100% ethanol 15 min, 100% propylene oxide 1 h). Subsequently, samples were embedded in Epon resin by infiltrating for 1 h in a 1 : 1 Epon : propylene oxide mixture followed by 2 days incubation at 60°C for Epon polymerization. Sections (70 nm) were prepared and stained with uranyl acetate (10 min) and lead citrate (10 min). TEM for the analysis of cross sections of hyphae and spores was performed with a FEI Tecnai 12 BioTwin transmission electron microscope [64].
Acceptor photobleaching was performed with a Zeiss Imager. eGFP was excited with a 488 nm laser (5% intensity), and emission was detected from 505 to 530 nm. mCherry was excited with 543 nm laser (5% intensity) and detected with a 560 long-pass. Both channels were recorded sequentially to prevent signal cross-bleeding. Bleaching was performed with the 543 nm laser at maximum intensity for 25 iterations. For all experiments, the original intensity was determined as the average of three pre-bleach frames, and the post-bleaching intensity was set as the average of the first three post-bleach frames. For SsgA–eGFP with SepG–mCherry (no interaction), 11 independent experiments were carried out, for FtsZ–mCherry and SepG–eGFP 18, and for SsgB–eGFP and SepG–mCherry 24 (table 2). The average post-bleach intensity was calculated relative to the pre-bleach intensity, including standard error. All images were collected in a 512 × 512 pixel format with a 63 × 1.4 NA oil objective.
For FRAP experiments, post-bleach intensity was followed for 15 s. Intensity measurements were corrected for image bleaching by examining the loss of fluorescence intensity in non-bleached regions. Images and bleaches were performed with similar settings as for the FRET experiments.
4.4. Heat resistance test
Fresh spores were obtained from SFM agar plates after 5 days of incubation at 30°C. Spore suspensions were diluted in water and heat treated at 60°C for 10 min. Dilutions were plated on SFM agar plates, incubated at 30°C and survival rates determined. Experiments were carried out in triplicate.
4.5. Membrane topology analysis of SepG
Escherichia coli JM109 cells were transformed with constructs expressing different BlaM-fusion proteins. Transformants were grown in 1 ml LB media containing apramycin 50 µg ml−1 for 4 h to reach OD600 0.4–0.6. Cells were washed twice with LB to remove accumulated β-lactamases and resuspended in 1 ml LB + 10% glycerol. 0.5 µl cell suspension was spotted onto LB agar plates with different concentrations of ampicillin, and growth was observed after overnight incubation at 37°C. To investigate the growth rate of JM109 carrying different constructs, 3 µl of prewashed cell suspension was inoculated into 200 µl LB with or without ampicillin. The optical density was measured every 30 min for 12 h using an automated Bioscreen optical plate reader (Oy Growth Curves Ab Ltd, Finland), with settings of 37°C, continuous shaking, OD wideband filter and a start OD programmatically set to 0.100 for easier comparison. All samples were carried out using three independent transformants, and each of these was analysed in triplicate.
4.6. Computer analysis
Analysis of Streptomyces genes and proteins was done at the StrepDB database (http://strepdb.streptomyces.org.uk/). For phylogenetic analysis and correlations, the String engine (http://string.embl.de) was used, whereas putative transmembrane domains were identified using transmembrane prediction server DAS [65]. The topological model was generated using program TMRPRES2D [47]. Amino acid sequence alignment was done using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/clustalo).
Supplementary Material
Acknowledgements
We are very grateful to Leendert Hamoen for discussions and for comments on the manuscript.
Competing interests
We declare we have no competing interests.
Funding
The work was supported by the Netherlands Organization for Scientific Research (NWO) via ECHO grant 711.011.008 and VICI grant no. 10379 to G.P.v.W. and VIDI grant no. 12957 to D.C.
References
- 1.Bi EF, Lutkenhaus J. 1991. FtsZ ring structure associated with division in Escherichia coli. Nature 354, 161–164. (doi:10.1038/354161a0) [DOI] [PubMed] [Google Scholar]
- 2.Goehring NW, Beckwith J. 2005. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr. Biol. 15, R514–R526. (doi:10.1016/j.cub.2005.06.038) [DOI] [PubMed] [Google Scholar]
- 3.Adams DW, Errington J. 2009. Bacterial cell division: assembly, maintenance and disassembly of the Z ring. Nat. Rev. Microbiol. 7, 642–653. (doi:10.1038/nrmicro2198) [DOI] [PubMed] [Google Scholar]
- 4.Hale CA, de Boer PAJ. 1997. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell 88, 175–185. (doi:10.1016/S0092-8674(00)81838-3) [DOI] [PubMed] [Google Scholar]
- 5.RayChaudhuri D. 1999. ZipA is a MAP-Tau homolog and is essential for structural integrity of the cytokinetic FtsZ ring during bacterial cell division. EMBO J. 18, 2372–2383. (doi:10.1093/emboj/18.9.2372) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pichoff S, Lutkenhaus J. 2002. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J. 21, 685–693. (doi:10.1093/emboj/21.4.685) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gueiros-Filho FJ, Losick R. 2002. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 16, 2544–2556. (doi:10.1101/gad.1014102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hamoen LW, Meile JC, de Jong W, Noirot P, Errington J. 2006. SepF, a novel FtsZ-interacting protein required for a late step in cell division. Mol. Microbiol. 59, 989–999. (doi:10.1111/j.1365-2958.2005.04987.x) [DOI] [PubMed] [Google Scholar]
- 9.Ishikawa S, Kawai Y, Hiramatsu K, Kuwano M, Ogasawara N. 2006. A new FtsZ-interacting protein, YlmF, complements the activity of FtsA during progression of cell division in Bacillus subtilis. Mol. Microbiol. 60, 1364–1380. (doi:10.1111/j.1365-2958.2006.05184.x) [DOI] [PubMed] [Google Scholar]
- 10.Romberg L, Levin PA. 2003. Assembly dynamics of the bacterial cell division protein FtsZ: poised at the edge of stability. Annu. Rev. Microbiol. 57, 125–154. (doi:10.1146/annurev.micro.57.012903.074300) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Claessen D, Rozen DE, Kuipers OP, Sogaard-Andersen L, van Wezel GP. 2014. Bacterial solutions to multicellularity: a tale of biofilms, filaments and fruiting bodies. Nat. Rev. Microbiol. 12, 115–124. (doi:10.1038/nrmicro3178) [DOI] [PubMed] [Google Scholar]
- 12.Flärdh K, Buttner MJ. 2009. Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat. Rev. Microbiol. 7, 36–49. (doi:10.1038/nrmicro1968) [DOI] [PubMed] [Google Scholar]
- 13.Barka EA, Vatsa P, Sanchez L, Gavaut-Vaillant N, Jacquard C, Klenk HP, Clément C, Oudouch Y, van Wezel GP. 2016. Taxonomy, physiology, and natural products of the Actinobacteria. Microbiol. Mol. Biol. Rev. 80, 1–43. (doi:10.1128/MMBR.masthead.80-1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hopwood DA. 2007. Streptomyces in nature and medicine: the antibiotic makers. New York, NY: Oxford University Press. [Google Scholar]
- 15.van Wezel GP, McDowall KJ. 2011. The regulation of the secondary metabolism of Streptomyces: new links and experimental advances. Nat. Prod. Rep. 28, 1311–1333. (doi:10.1039/c1np00003a) [DOI] [PubMed] [Google Scholar]
- 16.Wildermuth H, Hopwood D. 1970. Septation during sporulation in Streptomyces coelicolor. J. Gen. Microbiol. 60, 51–59. (doi:10.1099/00221287-60-1-51) [DOI] [PubMed] [Google Scholar]
- 17.Schwedock J, McCormick JR, Angert ER, Nodwell JR, Losick R. 1997. Assembly of the cell division protein FtsZ into ladder like structures in the aerial hyphae of Streptomyces coelicolor. Mol. Microbiol. 25, 847–858. (doi:10.1111/j.1365-2958.1997.mmi507.x) [DOI] [PubMed] [Google Scholar]
- 18.Jakimowicz D, van Wezel GP. 2012. Cell division and DNA segregation in Streptomyces: how to build a septum in the middle of nowhere? Mol. Microbiol 85, 393–404. [DOI] [PubMed] [Google Scholar]
- 19.McCormick JR, Losick R. 1996. Cell division gene ftsQ is required for efficient sporulation but not growth and viability in Streptomyces coelicolor A3(2). J. Bacteriol. 178, 5295–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCormick JR, Su EP, Driks A, Losick R. 1994. Growth and viability of Streptomyces coelicolor mutant for the cell division gene ftsZ. Mol. Microbiol. 14, 243–254. (doi:10.1111/j.1365-2958.1994.tb01285.x) [DOI] [PubMed] [Google Scholar]
- 21.Marston AL, Thomaides HB, Edwards DH, Sharpe ME, Errington J. 1998. Polar localization of the MinD protein of Bacillus subtilis and its role in selection of the mid-cell division site. Genes Dev. 12, 3419–3430. (doi:10.1101/gad.12.21.3419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raskin DM, de Boer PAJ. 1997. The MinE ring: an FtsZ-independent cell structure required for selection of the correct division site in E. coli. Cell 91, 685–694. (doi:10.1016/S0092-8674(00)80455-9) [DOI] [PubMed] [Google Scholar]
- 23.Bernhardt TG, de Boer PAJ. 2005. SlmA, a nucleoid-associated, FtsZ binding protein required for blocking septal ring assembly over chromosomes in E. coli. Mol. Cell 18, 555–564. (doi:10.1016/j.molcel.2005.04.012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Woldringh CL, Mulder E, Huls PG, Vischer N. 1991. Toporegulation of bacterial division according to the nucleoid occlusion model. Res. Microbiol. 142, 309–320. (doi:10.1016/0923-2508(91)90046-D) [DOI] [PubMed] [Google Scholar]
- 25.Wu LJ, Errington J. 2004. Coordination of cell division and chromosome segregation by a nucleoid occlusion protein in Bacillus subtilis. Cell 117, 915–925. (doi:10.1016/j.cell.2004.06.002) [DOI] [PubMed] [Google Scholar]
- 26.Wu LJ, Errington J. 2012. Nucleoid occlusion and bacterial cell division. Nat. Rev. Microbiol. 10, 8–12. [DOI] [PubMed] [Google Scholar]
- 27.Willemse J, Borst JW, de Waal E, Bisseling T, van Wezel GP. 2011. Positive control of cell division: FtsZ is recruited by SsgB during sporulation of Streptomyces. Genes Dev. 25, 89–99. (doi:10.1101/gad.600211) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Traag BA, van Wezel GP. 2008. The SsgA-like proteins in actinomycetes: small proteins up to a big task. Antonie Van Leeuwenhoek 94, 85–97. (doi:10.1007/s10482-008-9225-3) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Dissel D, Claessen D, Van Wezel GP. 2014. Morphogenesis of Streptomyces in submerged cultures. Adv. Appl. Microbiol. 89, 1–45. (doi:10.1016/B978-0-12-800259-9.00001-9) [DOI] [PubMed] [Google Scholar]
- 30.Flärdh K. 2003. Essential role of DivIVA in polar growth and morphogenesis in Streptomyces coelicolor A3(2). Mol. Microbiol. 49, 1523–1536. (doi:10.1046/j.1365-2958.2003.03660.x) [DOI] [PubMed] [Google Scholar]
- 31.Treuner-Lange A, et al. 2013. PomZ, a ParA-like protein, regulates Z-ring formation and cell division in Myxococcus xanthus. Mol. Microbiol. 87, 235–253. (doi:10.1111/mmi.12094) [DOI] [PubMed] [Google Scholar]
- 32.Rodrigues CD, Harry EJ. 2012. The Min system and nucleoid occlusion are not required for identifying the division site in Bacillus subtilis but ensure its efficient utilization. PLoS Genet. 8, e1002561 (doi:10.1371/journal.pgen.1002561) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards DH, Errington J. 1997. The Bacillus subtilis DivIVA protein targets to the division septum and controls the site specificity of cell division. Mol. Microbiol. 24, 905–915. (doi:10.1046/j.1365-2958.1997.3811764.x) [DOI] [PubMed] [Google Scholar]
- 34.Ito T, Uozumi N, Nakamura T, Takayama S, Matsuda N, Aiba H, Hemmi H, Yoshimura T. 2009. The implication of YggT of Escherichia coli in osmotic regulation. Biosci. Biotechnol. Biochem. 73, 2698–2704. (doi:10.1271/bbb.90558) [DOI] [PubMed] [Google Scholar]
- 35.Kabeya Y, Nakanishi H, Suzuki K, Ichikawa T, Kondou Y, Matsui M, Miyagishima S-y. 2010. The YlmG protein has a conserved function related to the distribution of nucleoids in chloroplasts and cyanobacteria. BMC Plant Biol. 10, 57 (doi:10.1186/1471-2229-10-57) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marbouty M, Saguez C, Cassier-Chauvat C, Chauvat F. 2009. ZipN, an FtsA-like orchestrator of divisome assembly in the model cyanobacterium Synechocystis PCC6803. Mol. Microbiol. 74, 409–420. (doi:10.1111/j.1365-2958.2009.06873.x) [DOI] [PubMed] [Google Scholar]
- 37.Fadda D, Pischedda C, Caldara F, Whalen MB, Anderluzzi D, Domenici E, Massidda O. 2003. Characterization of divIVA and other genes located in the chromosomal region downstream of the dcw cluster in Streptococcus pneumoniae. J. Bacteriol. 185, 6209–6214. (doi:10.1128/JB.185.20.6209-6214.2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Girard G, Traag BA, Sangal V, Mascini N, Hoskisson PA, Goodfellow M, van Wezel GP. 2013. A novel taxonomic marker that discriminates between morphologically complex actinomycetes. Open Biol. 3, 130073 (doi:10.1098/rsob.130073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duman R, Ishikawa S, Celik I, Strahl H, Ogasawara N, Troc P, Löwe J, Hamoen LW. 2013. Structural and genetic analyses reveal the protein SepF as a new membrane anchor for the Z ring. Proc. Natl Acad. Sci. USA 110, E4601–E4610. (doi:10.1073/pnas.1313978110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pogliano K, Harry E, Losick R. 1995. Visualization of the subcellular location of sporulation proteins in Bacillus subtilis using immunofluorescence microscopy. Mol. Microbiol. 18, 459–470. (doi:10.1111/j.1365-2958.1995.mmi_18030459.x) [DOI] [PubMed] [Google Scholar]
- 41.Flärdh K, Leibovitz E, Buttner MJ, Chater KF. 2000. Generation of a non-sporulating strain of Streptomyces coelicolor A3(2) by the manipulation of a developmentally controlled ftsZ promoter. Mol. Microbiol. 38, 737–749. (doi:10.1046/j.1365-2958.2000.02177.x) [DOI] [PubMed] [Google Scholar]
- 42.Swiatek-Polatynska MA, Bucca G, Laing E, Gubbens J, Titgemeyer F, Smith CP, Rigali S, van Wezel GP. 2015. Genome-wide analysis of in vivo binding of the master regulator DasR in Streptomyces coelicolor identifies novel non-anonical targets. PLoS ONE 10, e0122479 (doi:10.1371/journal.pone.0122479) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mullineaux CW, Nenninger A, Ray N, Robinson C. 2006. Diffusion of green fluorescent protein in three cell environments in Escherichia coli. J. Bacteriol. 188, 3442–3448. (doi:10.1128/JB.188.10.3442-3448.2006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Broome-Smith JK, Tadayyon M, Zhang Y. 1990. Beta-lactamase as a probe of membrane protein assembly and protein export. Mol. Microbiol. 4, 1637–1644. (doi:10.1111/j.1365-2958.1990.tb00540.x) [DOI] [PubMed] [Google Scholar]
- 45.Tadayyon M, Zhang Y, Gnaneshan S, Hunt L, Mehraein-Ghomi F, Broome-Smith JK. 1992. Beta-lactamase fusion analysis of membrane protein assembly. Biochem. Soc. Trans. 20, 598–601. (doi:10.1042/bst0200598) [DOI] [PubMed] [Google Scholar]
- 46.Mueller KE, Fields KA. 2015. Application of beta-lactamase reporter fusions as an indicator of effector protein secretion during infections with the obligate intracellular pathogen Chlamydia trachomatis. PLoS ONE 10, e0135295 (doi:10.1371/journal.pone.0135295) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spyropoulos IC, Liakopoulos TD, Bagos PG, Hamodrakas SJ. 2004. TMRPres2D: high quality visual representation of transmembrane protein models. Bioinformatics 20, 3258–3260. (doi:10.1093/bioinformatics/bth358) [DOI] [PubMed] [Google Scholar]
- 48.Swiatek MA, Tenconi E, Rigali S, van Wezel GP. 2012. Functional analysis of the N-acetylglucosamine metabolic genes of Streptomyces coelicolor and role in the control of development and antibiotic production. J. Bacteriol. 194, 1136–1144. (doi:10.1128/JB.06370-11) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ragkousi K, Cowan AE, Ross MA, Setlow P. 2000. Analysis of nucleoid morphology during germination and outgrowth of spores of Bacillus species. J. Bacteriol. 182, 5556–5562. (doi:10.1128/JB.182.19.5556-5562.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sambrook J, Fritsch E, Maniatis T. 1989. Molecular cloning: a laboratory manual. New York, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- 51.MacNeil DJ, Gewain KM, Ruby CL, Dezeny G, Gibbons PH, Maeneil T. 1992. Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68. (doi:10.1016/0378-1119(92)90603-M) [DOI] [PubMed] [Google Scholar]
- 52.Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. 2000. Practical Streptomyces genetics. Norwich, UK: John Innes Foundation. [Google Scholar]
- 53.van Wezel GP, van der Meulen J, Kawamoto S, Luiten RG, Koerten HK, Kraal B. 2000. ssgA is essential for sporulation of Streptomyces coelicolor A3(2) and affects hyphal development by stimulating septum formation. J. Bacteriol. 182, 5653–5662. (doi:10.1128/JB.182.20.5653-5662.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keijser BJ, Noens EE, Kraal B, Koerten HK, van Wezel GP. 2003. The Streptomyces coelicolor ssgB gene is required for early stages of sporulation. FEMS Microbiol. Lett. 225, 59–67. (doi:10.1016/S0378-1097(03)00481-6) [DOI] [PubMed] [Google Scholar]
- 55.Colson S, Stephan J, Hertrich T, Saito A, van Wezel GP, Titgemeyer F, Rigali S. 2007. Conserved cis-acting elements upstream of genes composing the chitinolytic system of streptomycetes are DasR-responsive elements. J. Mol. Microbiol. Biotechnol. 12, 60–66. (doi:10.1159/000096460) [DOI] [PubMed] [Google Scholar]
- 56.Vara J, Lewandowska-Skarbek M, Wang YG, Donadio S, Hutchinson CR. 1989. Cloning of genes governing the deoxysugar portion of the erythromycin biosynthesis pathway in Saccharopolyspora erythraea (Streptomyces erythreus). J. Bacteriol. 171, 5872–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fedoryshyn M, Welle E, Bechthold A, Luzhetskyy A. 2008. Functional expression of the Cre recombinase in actinomycetes. Appl. Microbiol. Biotechnol. 78, 1065–1070. (doi:10.1007/s00253-008-1382-9) [DOI] [PubMed] [Google Scholar]
- 58.Bierman M, Logan R, O'Brien K, Seno ET, Nagaraja Rao R, Schoner BE. 1992. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49. (doi:10.1016/0378-1119(92)90627-2) [DOI] [PubMed] [Google Scholar]
- 59.Grantcharova N, Lustig U, Flärdh K. 2005. Dynamics of FtsZ assembly during sporulation in Streptomyces coelicolor A3(2). J. Bacteriol. 187, 3227–3237. (doi:10.1128/JB.187.9.3227-3237.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Larson JL, Hershberger CL. 1986. The minimal replicon of a streptomycete plasmid produces an ultrahigh level of plasmid DNA. Plasmid 15, 199–209. (doi:10.1016/0147-619X(86)90038-7) [DOI] [PubMed] [Google Scholar]
- 61.van Wezel GP, White J, Hoogvliet G, Bibb MJ. 2000. Application of redD, the transcriptional activator gene of the undecylprodigiosin biosynthetic pathway, as a reporter for transcriptional activity in Streptomyces coelicolor A3(2) and Streptomyces lividans. J. Mol. Microbiol. Biotechnol. 2, 551–556. [PubMed] [Google Scholar]
- 62.Willemse J, van Wezel GP. 2009. Imaging of Streptomyces coelicolor A3(2) with reduced autofluorescence reveals a novel stage of FtsZ localization. PLoS ONE 4, e4242 (doi:10.1371/journal.pone.0004242) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colson S, van Wezel GP, Craig M, Noens EEE, Nothaft H, Mommaas AM, Titgemeyer F, Joris B, Rigali S. 2008. The chitobiose-binding protein, DasA, acts as a link between chitin utilization and morphogenesis in Streptomyces coelicolor. Microbiology 154, 373–382. (doi:10.1099/mic.0.2007/011940-0) [DOI] [PubMed] [Google Scholar]
- 64.Piette A, et al. 2005. From dormant to germinating spores of Streptomyces coelicolor A3(2): new perspectives from the crp null mutant. J. Proteome Res. 4, 1699–1708. (doi:10.1021/pr050155b) [DOI] [PubMed] [Google Scholar]
- 65.Cserzo M, Eisenhaber F, Eisenhaber B, Simon I. 2004. TM or not TM: transmembrane protein prediction with low false positive rate using DAS-TMfilter. Bioinformatics 20, 136–137. (doi:10.1093/bioinformatics/btg394) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.