

Abstract
Protein synthesis by the ribosome can fail for numerous reasons including faulty mRNA, insufficient availability of charged tRNAs and genetic errors. All organisms have evolved mechanisms to recognize stalled ribosomes and initiate pathways for recycling, quality control and stress signaling. Here we review the discovery and molecular dissection of the eukaryotic ribosome-associated quality-control pathway for degradation of nascent polypeptides arising from interrupted translation.
Faithful interpretation of the genetic code is essential to make functional protein products that participate in all areas of cellular physiology. Hence, every step in the production of proteins not only is under tight regulatory control but also is monitored for errors. All the key ‘ingredients’ for translation are subject to quality control: cells have evolved pathways to degrade aberrant mRNAs1, to detect mutant or damaged rRNAs and ribosomes2, and to ensure appropriate tRNA aminoacylation3. Various steps during translation are also monitored, including kinetic proofreading during codon-anticodon recognition4,5, and several protein quality-control pathways check the folding of nascent polypeptides during and after synthesis6.
The importance of high-fidelity translation is evidenced by the numerous diseases associated with remarkably subtle deviations from normal. For example, defective editing activity of a single tRNA synthetase, whose intrinsic accuracy is already around 99%, leads to neurodegeneration in mice7. Similarly, mutation of only one isoacceptor tRNA out of six in mice can predispose cells toward ribosome stalling and neurodegeneration8. More broadly, mutations to myriad translation components are linked to disease9. Thus, cells devote considerable resources to defending their proteome from erroneous products whose accumulation induces stress responses10–12 and whose failed clearance causes an increasingly broad range of protein-misfolding diseases13.
In recent years, two seemingly different areas of quality control, mRNA surveillance and protein degradation, have intersected at the ribosome. The truncated protein products generated by ribosomes that stall on a defective mRNA have been found to be targeted for degradation by a specialized pathway that is initiated on the ribosome. In this Review, we discuss the discovery and mechanistic dissection of this ribosome-associated quality-control pathway in eukaryotes, highlight key areas for future investigation and speculate about its potentially broader roles in cellular physiology.
Why degrade nascent proteins?
At first glance, it is not intuitively obvious why a cell should target a polypeptide for degradation before it has an opportunity to fold. In hindsight, one answer is straightforward: the cell benefits from detecting and removing errors at the earliest opportunity. There are situations in which a nascent polypeptide on the ribosome can be deduced to have a low probability of acquiring a fully functional state. If a ribosome will never successfully reach the correct termination codon, the protein product is necessarily truncated and is very likely to be defective; even if this truncated polypeptide could fold into a stable protein, it might lack key downstream domains and hence be functionally impaired or have dominant-negative effects14,15. Thus, it would be advantageous for the cell to degrade these incomplete nascent chains by using the criteria of truncation rather than their capacity to fold.
Because the truncated polypeptide is essentially ‘captive’ on the ribosome, tagging it for degradation at this stage would ensure its rapid elimination and minimize inappropriate interactions in the bulk cytosol. Thus, ribosome-associated quality control eliminates the partially synthesized protein products from ribosomes that stall before reaching the stop codon. How nascent chains on actively elongating ribosomes are directly monitored for their folding status and are subjected to degradation is less well studied at this time (Box 1) and has been reviewed elsewhere16,17.
Box 1. Ribosome-associated versus cotranslational quality control.
Nascent chains can be tagged for destruction before they leave the ribosome. This can happen for two conceptually different reasons whose mechanistic underpinnings are probably distinct.
Cotranslational quality-control mechanisms
These mechanisms sense the folding or maturation state of nascent chains as they are translated and influence their fate16,17,77–81. For example, specialized ribosome-associated chaperones help nascent chains fold and insulate them from the rest of the cellular milieu79. General cytosolic protein quality-control mechanisms (for example, the N-end rule) can target nascent chains for destruction77,80,81. Cotranslational quality-control mechanisms are therefore used to handle and avoid intrinsic defects in protein quality and to cue the crucial decisions regarding the nascent chain itself.
Ribosome-associated quality-control mechanisms
These mechanisms sense the state of translation rather than the state of the nascent chain. When translation stalls, for example because of defective mRNA, are detected, the associated nascent chains26 and mRNA20 may be targeted for destruction. Like cotranslational quality control, ubiquitination of the nascent chain occurs at the ribosome. However, unlike cotranslational quality control, ribosome-associated quality control does not seem to depend on the state of the nascent chain; any nascent chain will be tagged for destruction if it is the product of defective translation. Thus, ribosome-associated quality control appears to make key fate decisions about the mRNA and nascent protein by monitoring the translation machinery, predominantly the ribosome31.
Translation can stall for several reasons, including truncated or damaged mRNA18,19, excessive mRNA secondary structure20, insufficient amounts of a particular amino acid or tRNA21,22 and translation of particular mRNA sequences including the poly(A) tail23–26 (Fig. 1). These unsuccessful translation cycles typically signify a problem and merit a response from the cell to resolve the issue and adapt accordingly. This response may include degrading the mRNA20,27,28, ribosome2 or nascent polypeptide chain26,29. Although most quality-control pathways recognize the target to be degraded30, the stalled nascent chain and corresponding mRNA do not have a direct role in recognition. Instead, the cell monitors the state of the ribosome31 and in cases of stalling ‘assumes’ that the protein, mRNA, and perhaps even the ribosome are likely to be corrupted and hence should be targeted for degradation.
Figure 1.

Causes of aberrant translation elongation. Top, normal translation involves initiation, elongation through the coding region, termination and recycling of ribosomal subunits. Bottom, four different situations that can cause ribosomal stalling before the stop codon is reached. In each case, stalling can initiate one or more downstream pathways that facilitate stall resolution and cellular adaptation. Figure adapted with permission from ref. 63, Elsevier. aa-tRNA, aminoacyl-tRNA; ORF, open reading frame.
mRNA surveillance necessitates nascent-chain degradation
The first link between failed translation and quality control in eukaryotes came from the study of defective mRNA. It was observed that mRNAs with premature stop codons are selectively degraded by nonsense-mediated decay (NMD)32–34. Using artificial model substrates (Box 2), it was later found that mRNAs that lack stop codons27,28, are truncated within the coding region19, contain strong secondary structure20, have rare codons20 or are damaged18 are all selectively degraded. Critically, these mRNA-surveillance mechanisms all require the mRNA to be translated. Thus, the idea emerged that mRNAs are ‘tested’ for their integrity by translation, and failure of this test initiates their degradation to prevent production of faulty proteins1,31.
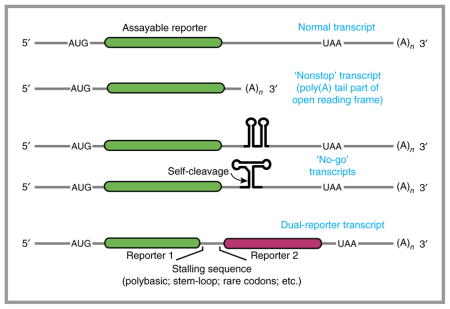
Box 2. Tools of the trade.
Model substrates have been instrumental in the investigation of ribosome-associated quality-control mechanisms. The original model substrate was a gene lacking a stop codon in any frame of the untranslated region (termed a ‘non-stop’ mRNA)27,28. This synthetic substrate mimics mRNAs subject to premature polyadenylation, a potentially common mRNA defect82. In this case, the ribosome reads into the poly(A) tail, which is interpreted as a tract of lysine residues. Later it was discovered that simply including a stretch of adenosine nucleotides anywhere in the coding sequence arrests translation and induces a ribosome-associated quality-control pathway that may be identical to the response induced by a native non-stop mRNA26. Stretches of basic residues (arginine or lysine), depending on the codon21, also trigger ribosome-associated quality control in yeast61, although this may not be mechanistically identical to the non-stop case.
Other mRNA sequences that induce translational stalling also induce ribosome-associated quality-control pathways. These include stem-oops20, rare codon tracts62 and mRNAs truncated within the coding region19. An advantage of encoding an arrest-inducing sequence in the middle of a gene is that an additional readout can be used past the arrest point. For example, encoding fluorescent proteins before and after a stall site allows a quantitative, ratiometric readout of translation arrest and nascent-chain degradation43. Other assays used to measure processing of ribosome-associated quality-control substrates include measuring mRNA levels (for example, by northern blotting20,26,29,61) and protein fragments (for example, by western blotting26,61), and ribosome footprinting59.
This model of mRNA surveillance has two key implications. First, ribosomes involved in detecting a defective mRNA would never reach a stop codon, the signal that normally terminates translation and initiates recycling of the ribosome. Therefore, these ribosomes need to be recycled by a noncanonical pathway or degraded to avoid accumulation of nonfunctional complexes. Second, although mRNA degradation limits subsequent rounds of defective protein translation, detection of a defective mRNA necessarily produces a partially synthesized and potentially defective protein. Prescient studies by Inada and colleagues26,29 have used cleverly designed assays (Box 2) to rigorously demonstrate that such polypeptides are rapidly degraded by the proteasome. Thus, ribosome rescue and polypeptide degradation are now understood to necessarily accompany most or all mRNA decay pathways, but the molecular mechanisms have only recently come into focus.
The initial insight into eukaryotic ribosome rescue came from genetic studies in yeast implicating Dom34 (Pelota in mammals) and Hbs1 in no-go decay (NGD)20, a situation in which ribosomes are translationally stalled. The homology of Dom34 and Hbs1 to release factors eRF1 and eRF3 indicates that they function at the ribosome. Reconstitution studies in a purified system have shown that Dom34 and Hbs1 are able to split the subunits of stalled ribosomes35, a reaction later found to also use the ATPase Rli1 (ABCE1 in mammals)36,37. Splitting would permit the 60S and 40S subunits to be reused for translation, similarly to the analogous recycling reaction that follows normal translation termination (mediated by eRF1, eRF3 and Rli1 (refs. 37,38)).
The experiments reconstituting the recycling of stalled ribosomes contained a nascent peptidyl-tRNA that was only a few amino acids long and thus could ‘drop off’ upon ribosome splitting35,37. Stalled ribosomes in a physiologic context would typically contain much longer nascent chains, and drop-off would be unlikely to occur. How such polypeptides are resolved was unclear until the crucial discovery by Bengtson and Joazeiro of a ubiquitin ligase that polyubiquitinates stalled truncated polypeptides and facilitates their rapid degradation39. The ligase, termed Ltn1 (originally Rkr1), is homologous to mammalian Listerin, which is encoded by a gene identified in a forward genetic screen for neurodegenerative disease40. The role of Ltn1 will be discussed in detail below.
Thus, the study of eukaryotic mRNA surveillance pathways led to an appreciation of a concurrent need for ribosome recycling and nascent polypeptide degradation, which are also presumably linked to mRNA degradation (Fig. 1). This is perhaps not surprising in hindsight, given how bacteria deal with the problem: they use an entirely different system to simultaneously target defective mRNA for degradation, terminate translation, recycle ribosomes and tag the truncated nascent chain for destruction41,42 (Box 3).
Box 3. The prokaryotic solution: tmRNA.
Bacteria have evolved a distinct mechanism to resolve translational stalling at the end of a truncated mRNA lacking a stop codon. This situation results in a trapped mRNA, partially synthesized nascent chain and ribosome. All these three issues are resolved by the molecule tmRNA83–86, an RNA with both tRNA- and mRNA-like properties. The tRNA-like domain of tmRNA encodes an alanine codon that can bind at the empty A site of the stalled ribosome and restart translation; the adjacent mRNA-like region is used to translate a short degradation tag before a stop codon is reached. Thus, the nascent chain terminates and is released, but it is short lived, owing to the C-terminal degradation tag. The ribosome is recycled by the usual termination and recycling pathway, and the defective mRNA is released so that it can be degraded by its exposed 3′ end.
As elegant as the tmRNA system is, it is limited to truncated mRNAs that leave an empty A site in the mRNA channel. Thus, other types of stalling, e.g., those resulting from mRNA secondary structure or amino acid insufficiency, would need a nuclease to remove mRNA from the A site87. The greater diversity of potential clients in eukaryotes might have driven the evolution of more elaborate surveillance and degradation systems. Nevertheless, eukaryotic and prokaryotic ribosome-associated quality control share some key features. Both systems ensure efficient nascent-chain and mRNA degradation, and both permit the addition of an artificial tag to the C terminus. However, the eukaryotic mechanism performs ribosome splitting, nascent-chain tagging and nascent-chain extraction in separate steps—a modular design that may allow regulation and accommodate a more diverse array of substrates.
The factors involved in ribosome-associated quality control
The discovery of Ltn1’s function in degradation of stalled translation products facilitated the placement of other genes found by subsequent work into a single pathway. Two parallel studies in yeast, investigating different problems, converged on the set of factors that currently define ribosome-associated quality control. The first study used genetic interaction maps in yeast to find factors that modulate the cytosolic heat-shock response43. Among this data set were two factors with very similar profiles of genetic interactions with translation-related genes: one was Ltn1, and the other was named Rqc1. Affinity purification of Rqc1 copurified not only Ltn1 but also Rqc2 (termed Tae2 at the time), the entire 60S ribosomal subunit, and the AAA+ ATPase Cdc48 with its cofactors Npl4 and Ufd1. In parallel, a search for genetic interactions with Ltn1 and mRNA decay pathways uncovered Rqc1, Rqc2 and the Cdc48 complex, all of which again were seen physically associated with 60S subunits44. Thus, these factors together compose the 60S-associated ribosome quality-control complex (RQC).
Using reporters for defective mRNAs and translational stalling, these and subsequent studies have shown that each RQC component is required for efficient degradation of stalled protein products43–45. The presence of polyubiquitin in purified RQC is completely dependent on Ltn1 and, to a lesser degree, Rqc2; in contrast, Rqc1 and the Cdc48 complex are dispensable for this phenotype43. Recruitment of the Cdc48 complex to the ribosome requires both Rqc1 and nascent-chain ubiquitination43,44. These observations, together with nascent-chain interaction analysis39,43, have suggested a model wherein Ltn1 and Rqc2 facilitate nascent-chain ubiquitination. Subsequently, the Cdc48 complex, recruited in part via Rqc1, mediates extraction of ubiquitinated nascent chains from the ribosome for degradation (Fig. 2). In agreement with this model, in the absence of functional Cdc48, stalling reporters have been observed in ribosomal fractions with a peptidyl-tRNA still attached45.
Figure 2.

Primary steps and factors of ribosome-associated quality control. A stalled ribosome is recognized and acted upon by ribosome-recycling factors that split the ribosomal subunits. Removal of the 40S subunit exposes the peptidyl-tRNA and intersubunit interface on the 60S subunit. These cues are recognized by RQC components, whose assembly on the 60S subunit permits ubiquitination of nascent polypeptides. The polyubiquitinated complex is then disassembled, thus allowing recycling of the factors and degradation of the nascent chain. Shown below are the homologous yeast and mammalian factors implicated at each step. Question marks indicate factors implicated by homology whose direct role remains to be examined experimentally. Figure adapted with permission from ref. 57, Elsevier.
These functional assignments matched with Ltn1 being a predicted ubiquitin ligase and the capacity of the Cdc48 complex to impart force on polyubiquitinated clients in other systems46. However, the functional roles of Rqc1 and Rqc2 were unclear. Another important observation was the association of all of these factors primarily with the 60S subunit39,43,44 but not with 80S ribosomes or polysomes. This finding linked the action of these factors with splitting of the ribosome, although the order of events and their role in splitting, if any, remained unclear. Thus, with a solid parts list and several basic features of this pathway in hand (Fig. 2), the next challenge was to understand how these components work to mediate quality control.
The order of events in the RQC pathway
An important advance toward dissection of the RQC pathway was the serendipitous observation of polyubiquitinated nascent chains on stalled ribosomes produced by in vitro translation in reticulocyte lysates47. The protein translocation field had long used the trick of translating a truncated mRNA to produce stalled ribosome–nascent chain complexes48. This permits the generation of defined-length translation complexes, depending on the point of truncation, to trap putative intermediates in a cotranslational process such as protein translocation into an organelle. The realization that ubiquitination of a small proportion of these nascent chains might represent a physiologically relevant degradation pathway suggested an experimentally tractable route to its mechanistic study.
Characterization of the reticulocyte lysate in vitro system provided several pieces of evidence suggesting that this system reflected the mammalian correlate of the yeast Ltn1 pathway47. First, ubiquitination occurred on nascent chains that remained ribosome associated and covalently attached to a peptidyl-tRNA. Second, multiple types of ribosome stalling led to ribosome-associated nascent-chain ubiquitination. Third, ubiquitination was dependent on the mammalian Ltn1 homolog, Listerin, which associated with 60S subunits as seen in yeast. Hence, the polyubiquitinated nascent chain cofractionated with 60S subunits. What remained uncertain was whether ribosome splitting preceded or rapidly followed ubiquitination: both possibilities would result in the same endpoint of ubiquitinated nascent chain–60S complexes.
This question was resolved by the demonstration that inhibition of splitting (by depleting Hbs1 or using a dominant-negative mutant) precluded Listerin recruitment and nascent-chain ubiquitination47. Conversely, artificially splitting the ribosomal subunits permitted Listerin to mediate ubiquitination of the nascent chain. Furthermore, an unstructured nascent chain was shown to drop off with its pepti-dyl-tRNA, in a process that was dependent on subunit splitting and could not have occurred if the polypeptide were first ubiquitinated. Thus, ribosome splitting not only precedes but also is required for nascent-chain ubiquitination. Subsequent reconstitution of nascent-chain ubiquitination with recombinant splitting factors and Listerin confirmed this conclusion and showed that Listerin alone can discriminate between stalled nascent chains on 80S versus 60S ribosomes and preferentially ubiquitinate the latter49. Because discrimination occurred regardless of how 80S ribosomes were split, Listerin appeared to identify intrinsic features specific to 60S–nascent chain complexes, a conclusion later supported by the structural studies described below.
The reconstitution experiments47,49, together with the earlier yeast studies39,43,44, segregated the pathway into three discrete and successive phases (Fig. 2): (i) splitting of a stalled ribosome into subunits; (ii) RQC assembly and nascent-chain ubiquitination; and (iii) nascent-chain extraction and degradation. This framework provisionally assigns splitting factors the task of recognizing a stalled ribosome; Ltn1 and Rqc2 the task of nascent-chain ubiquitination; and Rqc1 the task of Cdc48 recruitment for nascent-chain extraction.
Recognition of a stalled ribosome
Because stalling can occur for many reasons (Fig. 1), the configuration of the nascent chain–ribosome–mRNA complex can differ in key ways. This includes the presence or absence of mRNA in the ribosomal aminoacyl (A) site, the codon identity of the A site and the conformational state of the ribosome. It is unclear whether each of these states is recognized and split by a unified mechanism or whether different states require specific factors50,51. We discuss below how different types of stalling might be handled.
At present, the best-studied system that mediates preferential splitting of stalled ribosomes is the Hbs1–Dom34–Rli1 pathway, and its best-characterized target is a ribosome stalled at the 3′ end of a truncated mRNA. Hbs1 is a member of the translational GTPase family, whose members bind at the GTPase center near the A site of the ribosome52. The family includes eukaryotic elongation factor 1 (eEF1A), which delivers tRNAs to the ribosome, and eRF3, which delivers eRF1 to the ribosome for termination53,54; another member, eEF2, mediates ribosome translocation. These proteins, either alone (eEF2) or in complex with a partner (eEF1A with tRNA; eRF3 with eRF1; and Hbs1 with Dom34) bind in a GTP-dependent manner to distinct states of the ribosome: eEF2 probably prefers the hybrid state of the ribosome, whereas the other complexes favor the nonrotated (or canonical) state54. In addition, the binding partners (tRNA and eRF1) impart further specificity on the basis of the mRNA codon in the A site55,56. Thus, the translational GTPases can be conceptualized as monitoring the state of the ribosome and initiating the appropriate downstream reaction54.
From the available information, the simplest model to explain Dom34–Hbs1 specificity for stalled ribosome complexes is one in which the crucial cue is a failure to be promptly engaged by either aminoacyl-tRNA–eEF1A or eRF1–eRF3 complexes (Fig. 3). This is easiest to understand for the situation in which a ribosome translates to the end of a truncated mRNA: the A site is unoccupied by mRNA, thus precluding recognition by either the tRNA–eEF1A or eRF1–eRF3 complex, both of which rely on A-site codon interactions. In mammalian in vitro systems, both purified or in lysate, a stalled truncated complex is efficiently split by Pelota–Hbs1–ABCE1 (refs. 35,37 49, 5, 7). In yeast, resolution of this situation is dependent on Dom34–Hbs1–Rli1 (refs. 35,36). In the absence of Dom34, the mRNA is not degraded efficiently because the 3′ end is protected by a stalled ribosome from the RNA-degradation machinery20, and the protein is not produced efficiently because the mRNA cannot be translated repeatedly58. Ribosome profiling in the presence and absence of Dom34 has shown that an endogenous substrate for this pathway is Hac1, a cytosolically spliced mRNA whose incorrect ligation results in an mRNA truncated in the coding region59. Thus, Dom34–Hbs1–Rli1 is both necessary and sufficient for resolving stalling on truncated mRNAs in vitro and in vivo.
Figure 3.

Working model for recognition of a stalled ribosome by recycling factors. Top left (green background), a simplified translation elongation cycle is shown. A translating ribosome in the nonrotated state (center) engages the tRNA–eEF1A–GTP ternary complex in response to a sense codon in the A site. Codon recognition by the tRNA triggers GTP hydrolysis by eEF1A, release of the latter from the ribosome and accommodation of the tRNA to catalyze peptide-bond formation. The ribosome is then translocated by one codon via the action of eEF2 to complete the cycle. Top right (white background), when a stop codon enters the A site, it is recognized by an eRF1–eRF3–GTP complex that functions analogously to the elongation complex. Upon accommodation of eRF1, the ATPase Rli1 (ABCE1 in mammals) is recruited, and peptidyl-tRNA is hydrolyzed, thus releasing the nascent protein. The ribosomal subunits are separated by the action of the eRF1–Rli1 complex. Bottom (pink background), failure to be engaged by either the eEF1 or eRF1 complex permits ‘default’ engagement by the Dom34–Hbs1–GTP complex, which does not exhibit codon specificity. These factors act similarly to the homologous eRF1–eRF3 complex, with the exception that Dom34 (Pelota in mammals) does not catalyze peptidyl-tRNA hydrolysis. Thus, subunit separation results in a 60S–peptidyl-tRNA complex that is targeted by the RQC. T, GTP; D, GDP; E, exit tunnel.
Dom34–Hbs1–Rli1 can also resolve stalling in which mRNA is present in the A site. In vitro in both mammalian and yeast systems, stalling due to aminoacyl-tRNA unavailability, poly(A) translation or a stem-loop can all be engaged by Dom34–Hbs1 (refs. 35,37,49,52). Furthermore, Hbs1 depletion or excess GTPase-deficient Hbs1 partially inhibits nascent-chain ubiquitination of stalls in a poly(A) tail, similarly to the results seen with a truncated mRNA47. Thus, it can be inferred that, at least in vitro, the Dom34–Hbs1 complex can bind to a stalled ribosome independently of codon identity in the A site. This notion is consistent with a moderate-resolution cryo-EM structure showing Dom34–Hbs1 bound to a nonrotated ribosome stalled by a stem-loop52. In vivo evidence for a Dom34–Hbs1 requirement in splitting internally stalled ribosomes is less complete but is supported by the finding of blocked translocons in yeast strains that lack Dom34 or Hbs1 and express a stalled protein targeted to the endoplasmic reticulum or mitochondria. Furthermore, the tRNA-linked product of a reporter protein stalled in a poly(A) tail has been shown to be stabilized comparably in Dom34- or Ltn1-deleted yeast45. Thus, it can provisionally be assumed that the Dom34–Hbs1–Rli1 system is at least one pathway for resolving stalling in which mRNA is in the A site. How might specificity for stalls be determined in this case?.
After initial binding of a GTP–Hbs1–Dom34 complex to a stalled ribosome, GTP hydrolysis by Hbs1 is crucial for Hbs1 dissociation, which allows accommodation of Dom34 into the A site of the ribosome35 (Fig. 3). The accommodated Dom34 recruits Rli1, which catalyzes subunit separation through a poorly understood mechanism36,37. Until accommodation occurs, the binding is presumably labile, thereby permitting kinetic proofreading. It is therefore attractive to think that the time needed for GTP hydrolysis by Hbs1 represents a window for competition by tRNA–eEF1A or eRF1–eRF3 complexes. Competition is feasible only if the appropriate codon is in the A site, and the suitable complex is available. Otherwise, Hbs1 hydrolyzes its GTP, thereby initiating downstream steps in splitting. Thus, if a suitable aminoacylated tRNA is unavailable (for example, in the case of rare codons or amino acid deficiency) or the ribosome cannot elongate, owing to a physical block, Dom34–Hbs1 would eventually access the ribosome and initiate splitting.
Less clear is the situation in which the A-site codon has a suitable aminoacyl-tRNA. This might occur with stalling at a polybasic coding sequence such as the poly(A) tail. Perhaps competition with aminoacyl-tRNA–eEF1A does occur in these situations, but elongation is not possible, owing to unfavorable architecture around the peptidyl transferase center, as has been observed in other cases of peptide-induced stalling23,60. In this case, Dom34–Hbs1 would eventually act, albeit more slowly.
Alternatively, other factors may participate in this circumstance. This possibility has been suggested by the observation that deleting the 40S ribosomal protein Asc1 or the ubiquitin ligase Hel2 leads to increased protein synthesis downstream of polybasic stretches43,61,62. How polybasic regions selectively recruit Hel2 or why its presumptive ubiquitination activity toward the ribosome causes translation to be aborted remain to be examined. It is important to understand this step in molecular detail because the outcomes of both mRNA degradation and protein degradation are ultimately decided by the irreversible decision to split or not to split a translation complex.
Assembly and structure of the ubiquitination complex
The immediate consequence of removing the small subunit from a nascent chain–80S complex is the exposure of the intersubunit interface of the nascent chain–60S complex (Fig. 2). If the nascent chain is relatively short, it can drop off and leave behind an empty 60S that presumably can reenter the translation cycle35,37,47. However, longer nascent chains would be trapped, thereby exposing the attached peptidyl (P)-site tRNA at the interface side of the 60S. It has therefore been speculated that the interface and/or tRNA might provide the cue for Ltn1 recruitment47,63, thereby explaining why Ltn1 is seen only on 60S complexes and does not promiscuously target translating ribosomes39,43,47. Indeed, the initial low-resolution structure of a reconstituted nascent chain–60S–Listerin complex has shown that Listerin’s position substantially clashes with the 40S49.
However, such reconstituted complexes rapidly reassociate with free 40S ribosomal subunits, and the basis for Listerin specificity toward nascent chain–containing 60S over free 60S is unclear57. By contrast, 60S–peptidyl-tRNA particles produced in a cytosolic translation extract contain stably bound Listerin and do not reassociate with free 40S subunits47. This suggests that Listerin acts with cofactors that stabilize its 60S association and prevent 40S binding. One such cofactor has been found to be NEMF, the mammalian homolog of Rqc2. Inclusion of NEMF in the reconstituted in vitro reaction has been shown to inhibit 40S rebinding to 60S–nascent chain complexes and stabilize the Listerin-60S interaction, thereby improving ubiquitination efficiency57. Order of addition experiments have indicated that NEMF can be recruited to nascent chain–60S complexes first, and Listerin recruitment follows (Fig. 4a).
Figure 4.

Steps of RQC assembly on 60S–peptidyl-tRNA complexes. (a) Upon subunit separation, the exposed interface of the 60S subunit has high affinity for both the 40S subunit and the factor NEMF (Rqc2 in yeast). NEMF binding via both the tRNA and 60S interface effectively precludes 40S reassociation and facilitates binding of Listerin (Ltn1 in yeast). Listerin’s RING domain is positioned near the polypeptide exit tunnel, thus facilitating nascent-chain ubiquitination. Figure adapted with permission from ref. 57, Elsevier. (b) Intersubunit view of the assembled mammalian 60S–peptidyl-tRNA–RQC complex. Teal, NEMF; purple, P-site tRNA; orange, Listerin; gray, ribosome. The direct interaction between NEMF and Listerin is shown. (c) Cutaway view illustrating the direct recognition of P-site tRNA by NEMF and the proximity of Listerin to the polypeptide exit tunnel over 100 Å away.
Structures of the yeast and mammalian 60S–RQC by cryo-EM have not only corroborated these biochemical conclusions but also defined the binding sites and locations of these factors57,64,65 (Fig. 4b,c). To isolate the yeast 60S–RQC, the complex was assembled in vivo and stabilized by use of a strain in which the RING ligase domain of Ltn1 is deleted (thereby precluding progression beyond the step of RQC assembly64,65); the complex was then affinity isolated via an epitope-tagged Rqc1 or Ltn1. The mammalian complex was instead assembled in vitro by incubation of a purified stalled 80S ribosome–nascent chain complex with recombinant splitting factors (Pelota, Hbs1 and ABCE1), NEMF and Listerin49,57. Thus, both the yeast and mammalian complexes represent the step immediately preceding nascent-chain ubiquitination.
Assignment of density to Ltn1 versus Rqc2 was based on difference maps from structures obtained from individual deletion strains, the expected shape for Ltn1 according to its negative-stain structure in isolation65 and an earlier low-resolution Listerin structure bound to the 60S49. Similarly, assignments in the mammalian structure were based on density relative to a structure lacking NEMF and, in areas with sufficient resolution, by direct building of atomic models. These analyses collectively showed that Rqc2 and NEMF are in approximately the same position, occupying a large proportion of the 60S surface that would ordinarily bind the 40S subunit. This binding position has provided a mechanism for Rqc2’s and NEMF’s specificity for 60S and for how they prevent 40S-subunit reassociation after 80S splitting.
The architecture of NEMF consists of N- and C-terminal globular lobes that are positioned near each other at the ribosomal P site and connected by a long outstretched middle region extending to the sarcin-ricin loop (SRL). The N- and C-terminal globular domains cradle the P-site tRNA57,64,65 (Fig. 4c), thus explaining the factor’s specificity for nascent chain–containing 60S complexes over empty 60S57. Of note, free tRNA can compete for NEMF binding to its target, but only at concentrations exceeding that of cytosolic tRNA57. Thus, NEMF uses coincidence detection of both tRNA and 60S to identify their targets, and the multitude of interactions provide a specific high-avidity interaction.
The middle domain of NEMF that interacts with the SRL and ribosomal P stalk is precisely where the N terminus of Listerin interacts with both the ribosome and NEMF (Fig. 4b). The ensuing middle part of Listerin contains HEAT repeats that adopt a superhelical structure extending over 100 Å toward the exit tunnel at the other side of the 60S. This is followed by an RWD domain whose interaction with the ribosome57 positions the C-terminal RING domain very close to the exit tunnel. Thus, the specificity of Listerin for stalled nascent chains is imparted at two levels. First, the N terminus of Ltn1 and Listerin clashes with the region where the 40S subunit would be, thereby excluding promiscuous ubiquitination of translating polypeptides. Second, the reliance of Listerin on NEMF for part of the 60S interaction probably helps Listerin find 60S subunits whose nascent-chain occupancy has already been vetted via the P-site tRNA. The importance of these key interactions for ubiquitination efficiency have been validated by structure-guided point mutations in the mammalian system57.
An additional tRNA was unexpectedly observed in the yeast, but not mammalian, 60S–RQC structure64 (Fig. 5a). This difference is probably because the mammalian complex came from a purified in vitro reaction lacking free tRNA, whereas the yeast complex was assembled in vivo. The extra tRNA is in nearly the same position as tRNAs bound to the A site of translating 80S ribosomes. In the absence of 40S or mRNA, this A-site tRNA in the RQC structure is held in place by interactions with Rqc2 (Fig. 5a,b). We will discuss the implications and repercussions of this finding in the next section.
Figure 5.

CAT-tail formation by Rqc2p. (a) Architecture of the Rqc2–60S complex bound to both A- and P-site tRNAs, illustrating that the two amino acid–attachment sites are juxtaposed at the peptidyl transferase center (PTC). (b) Recognition of the A-site tRNA via the anticodon loop and D loop is thought to provide specificity for alanine and threonine tRNAs. (c) A speculative elongation cycle in which a 60S–RQC complex with P-site tRNA interacts with a threonine- or alanine-charged tRNA in the A site. This brings the charged amino acid into the peptidyl transferase center, thereby facilitating attack of the ester bond (arrow) on the peptidyl-tRNA. Transfer of the nascent chain to the A-site tRNA frees the P-site tRNA, which over time dissociates. The A-site peptidyl-tRNA can then engage the P site, which might be a higher-affinity site, to complete the cycle. Figure adapted with permission from ref. 64, AAAS.
CAT tails and induction of a stress response
The saturation of most biosynthetic or quality-control processes triggers stress responses that facilitate restoration of homeostasis. Prominent examples include the unfolded-protein responses of the endoplasmic reticulum11 and mitochondria12, and the heat-shock response in the cytosol10. Perhaps similarly, disruption of Ltn1 or Rqc1 induces activation of heat-shock factor 1 (Hsf1) in yeast43; however, disruption of Rqc2 not only fails to induce Hsf1 but also abrogates the response induced by deleting Ltn1 or Rqc1. Although the RQC-dependent mechanism of induction of Hsf1 remains unknown, the observation of an A-site tRNA bound to Rqc2 in the cryo-EM structure (Fig. 5a,b) has led to new insights that might hold the key to understanding this stress response.
Sequencing of tRNAs from the purified 60S–RQC complexes has revealed that they are markedly overrepresented by alanyl- and threonyl-tRNAs64. Alanine and threonine are also enriched in total amino acid analysis of the stalled nascent polypeptide. Furthermore, the molecular weight of stalled model polypeptides has been found to be slightly larger than predicted in earlier studies43, a discrepancy that has proven to be a C-terminal extension64. This extra polypeptide is dependent on Rqc2 and is particularly prominent when resolution of the stalled nascent chain is inhibited by preventing its ubiquitination or extraction. These observations have led to a model wherein charged A-site tRNA in the 60S–RQC structure represents a snapshot of polypeptide elongation preferentially with alanine and threonine (Fig. 5c), in an unusual reaction that is not dependent on either the 40S subunit or mRNA. Such extensions have been dubbed C-terminal alanine and threonine (CAT) tails64.
Direct sequencing of CAT tails from a stalled polypeptide has shown roughly equal amounts of alanine and threonine yet without a specific sequence. The preference for these residues is apparently imparted by the specificity of Rqc2 for alanyl- and threonyl-tRNA (Fig. 5b), both of which feature the modified base inosine in the wobble position of the anticodon loop66. The 60S–RQC structure suggests that Rqc2 facilitates positioning of the charged A-site tRNA sufficiently close to the peptidyl transferase center to permit attack of the peptidyl-tRNA ester bond64 (Fig. 5a). Once this bond is formed, the untethered P-site tRNA can dissociate, thereby permitting the adjacent A-site tRNA to occupy the P site (which may be the higher-affinity site). This would make the A site available to bind the next aminoacyl-tRNA (Fig. 5c). Reconstitution of CAT-tail elongation in vitro will enable this and other mechanistic models to be tested.
At least one purpose of CAT tails may be to extend or reposition the nascent polypeptide relative to the exit tunnel when a suitable ubiquitination site is not readily available. Given that lysines are relatively common, and stalling in a poly(A) tail presumably incorporates at least one lysine, most stalled complexes are likely to have a lysine inside the 35-residue-long ribosomal tunnel. Thus, CAT-tail elongation (which can proceed for at least this length64) may facilitate exposure of this lysine for ubiquitination by Ltn1. Such a function might be especially important when the stalled complex is being translocated across a membrane, and most of the nascent polypeptide is not available to Ltn1 (ref. 67). In cases in which the nascent chain does not include lysine, the threonines in CAT tails might provide alternative residues for ubiquitination.
CAT tails appear to be required for activating Hsf1 when the RQC is compromised. CAT tails and Hsf1 induction are both dependent on Rqc2 (refs. 43,64). Furthermore, point mutations in conserved Rqc2 residues that abolish CAT-tail formation while preserving degradation of model stalled polypeptides abolish RQC-induced Hsf1 signaling64. CAT tails, which are likely to contain homopolymers that are aggregation prone68, may disrupt protein homeostasis69 and therefore indirectly activate Hsf1. Alternatively, CAT tails may signal more directly to Hsf1. An important immediate goal is to rigorously establish the causal link between CAT tails and Hsf1 activation, after which the mechanism of activation can be investigated.
Stress and translational stalling may be related in the reverse direction as well. Ribosome profiling experiments have indicated that heat shock can lead to pervasive translation stalling ~60 codons from the initiation site70,71. This position is noteworthy because it is the point at which chaperones are likely to first engage the nascent polypeptide. Thus, there may be mechanisms by which chaperone availability is communicated to the translation apparatus to reduce synthesis. Similarly, stress in the endoplasmic reticulum has recently been observed to trigger regulatory ubiquitination on the ribosome, perhaps to modulate translation72. Whether or how these stress-triggered effects on translation initiate mRNA73,74 or polypeptide degradation remains to be studied in depth, but the observations highlight the intimate and emerging relationships between translation elongation and protein homeostasis pathways.
Nascent-chain extraction and degradation
Although a number of mechanistic issues remain to be resolved, the steps leading to a polyubiquitinated nascent chain–60S complex are reasonably well defined and have now been reconstituted with purified factors49,57. In contrast, the subsequent steps culminating in nascent-chain degradation and recycling of the 60S–RQC remain obscure. The central challenge is probably to free the nascent chain from the 60S–RQC within which it is embedded. The nascent chain is threaded through the very narrow 60S exit tunnel with bulky elements on either side: a polyubiquitinated N-terminal domain with potential folded regions and a covalently attached C-terminal tRNA.
Genetic studies have suggested that the Cdc48 complex is required for nascent-chain extraction43–45,75. Because neither Rqc1 nor Cdc48 is needed for nascent-chain ubiquitination in vitro57, both appear to act after Ltn1. This scenario is supported by the dependence of Cdc48 recruitment on both Ltn-mediated polyubiquitination and Rqc1 (refs. 43,44). Because Rqc1 is not needed for Ltn1-mediated ubiquitination43, the current model posits that the Cdc48 complex is recruited by a bivalent interaction with Rqc1 and polyubiquitin, after which it uses its ATP-powered activity to drive downstream steps. The established ‘separase’ activity of Cdc48 (ref. 46) may apply force to the nascent chain and/or RQC and hence facilitate extraction of the ubiquitinated substrate from the 60S ribosome.
Notably, once the ester bond between the tRNA and nascent chain is broken, there may be relatively little impedance to polypeptide release. Thus, hydrolysis of this bond is likely to be the key step in freeing both the nascent chain and the tRNA. One model suggests that Cdc48 pulls the nascent chain, via its polyubiquitin, from the mouth of the exit tunnel. This might reposition the ester bond slightly to a location that is more favorable for its hydrolysis. Perhaps the ability of Rqc2 to bind tRNA in the A site64 allows uncharged tRNA or eRF1 to somehow act in this hydrolysis reaction. It is noteworthy that eRF1 has the approximate shape of a tRNA76 and could conceivably interact with Rqc2 or NEMF in the same way as alanyl- or threonyl-tRNA. The main advantage of coupling tRNA hydrolysis with Cdc48 activity is that the released nascent chain can be delivered promptly to the proteasome by Cdc48.
Open questions and future challenges
Although knowledge of ribosome-associated protein quality control has been improving rapidly, many questions remain. The key open questions include the following: (i) What are the endogenous substrates of RQC? It is not clear what fraction of translation is flagged as defective, what the identities of these endogenous substrates are and whether they change under different conditions or between different cell types. (ii) Why does translation of poly(A) lead to stalling? The notion of ‘clogging’ the exit tunnel with basic residues does not explain why deleting the small-subunit protein Asc1 or the ubiquitin ligase Hel2 leads to increased protein synthesis past the site of stalling. (iii) How exactly do the ribosome-splitting factors Dom34 and Hbs1 recognize a stalled ribosome, and how do they outcompete translation factors? A key issue is whether the mRNA needs to be endonucleolytically processed for this recognition and if so, how this processing occurs. (iv) What is the molecular mechanism of CAT-tail elongation? At present, the only known components are Rqc2 and alanyl- and threonyl-tRNAs. Future studies will be required to determine whether other factors are required, where the energy for elongation comes from and how tRNA binding, peptide-bond formation and translocation are performed without the canonical translation factors. (v) What is the mechanism of nascent-chain extraction from the 60S subunit? Nothing is known about Rqc1, how it might help to recruit Cdc48, what part of the nascent chain or ribosome Cdc48 acts upon and what the energy from its ATP hydrolysis is needed for. (vi) How do defects in ribosome-associated quality-control pathways cause disease? Mutations that lead to neurodegeneration could act by a number of mechanisms, including stabilization of defective protein products, accumulation of CAT-tailed proteins, aberrant stress signaling or disruptions to ribosome homeostasis.
In the future, diverse approaches, from genomics to in vitro reconstitution to clinical studies, will be necessary to answer these questions, thus providing a better understanding of the mechanism, scope and consequences of ribosome-associated quality control.
Acknowledgments
We thank S. Shao for comments on this manuscript and A. Frost and P. Shen for help with figure preparation. This work was supported by the UK Medical Research Council (MC_UP_A022_1007 to R.S.H.), Stanford University (O.B.) and the US National Institutes of Health (1R01GM115968-01 to O.B.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.van Hoof A, Wagner EJ. A brief survey of mRNA surveillance. Trends Biochem Sci. 2011;36:585–592. doi: 10.1016/j.tibs.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. A late-acting quality control process for mature eukaryotic rRNAs. Mol Cell. 2006;24:619–626. doi: 10.1016/j.molcel.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 3.Yadavalli SS, Ibba M. Quality control in aminoacyl-tRNA synthesis its role in translational fidelity. Adv Protein Chem Struct Biol. 2012;86:1–43. doi: 10.1016/B978-0-12-386497-0.00001-3. [DOI] [PubMed] [Google Scholar]
- 4.Hopfield JJ. Kinetic proofreading: a new mechanism for reducing errors in biosynthetic processes requiring high specificity. Proc Natl Acad Sci USA. 1974;71:4135–4139. doi: 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaher HS, Green R. Fidelity at the molecular level: lessons from protein synthesis. Cell. 2009;136:746–762. doi: 10.1016/j.cell.2009.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wolff S, Weissman JS, Dillin A. Differential scales of protein quality control. Cell. 2014;157:52–64. doi: 10.1016/j.cell.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 7.Lee JW, et al. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature. 2006;443:50–55. doi: 10.1038/nature05096. [DOI] [PubMed] [Google Scholar]
- 8.Ishimura R, et al. RNA function: ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science. 2014;345:455–459. doi: 10.1126/science.1249749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scheper GC, van der Knaap MS, Proud CG. Translation matters: protein synthesis defects in inherited disease. Nat Rev Genet. 2007;8:711–723. doi: 10.1038/nrg2142. [DOI] [PubMed] [Google Scholar]
- 10.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 11.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 12.Pellegrino MW, Nargund AM, Haynes CM. Signaling the mitochondrial unfolded protein response. Biochim Biophys Acta. 2013;1833:410–416. doi: 10.1016/j.bbamcr.2012.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gregersen N, Bross P, Vang S, Christensen JH. Protein misfolding and human disease. Annu Rev Genomics Hum Genet. 2006;7:103–124. doi: 10.1146/annurev.genom.7.080505.115737. [DOI] [PubMed] [Google Scholar]
- 14.Roman C, Cohn L, Calame K. A dominant negative form of transcription activator mTFE3 created by differential splicing. Science. 1991;254:94–97. doi: 10.1126/science.1840705. [DOI] [PubMed] [Google Scholar]
- 15.Ishigame H, Mosaheb MM, Sanjabi S, Flavell RA. Truncated form of TGF-βRII, but not its absence, induces memory CD8+ T cell expansion and lymphoproliferative disorder in mice. J Immunol. 2013;190:6340–6350. doi: 10.4049/jimmunol.1300397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pechmann S, Willmund F, Frydman J. The ribosome as a hub for protein quality control. Mol Cell. 2013;49:411–421. doi: 10.1016/j.molcel.2013.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang F, Canadeo LA, Huibregtse JM. Ubiquitination of newly synthesized proteins at the ribosome. Biochimie. 2015;114:127–133. doi: 10.1016/j.biochi.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simms CL, Hudson BH, Mosior JW, Rangwala AS, Zaher HS. An active role for the ribosome in determining the fate of oxidized mRNA. Cell Reports. 2014;9:1256–1264. doi: 10.1016/j.celrep.2014.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meaux S, Van Hoof A. Yeast transcripts cleaved by an internal ribozyme provide new insight into the role of the cap and poly(A) tail in translation and mRNA decay. RNA. 2006;12:1323–1337. doi: 10.1261/rna.46306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440:561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Letzring DP, Dean KM, Grayhack EJ. Control of translation efficiency in yeast by codon-anticodon interactions. RNA. 2010;16:2516–2528. doi: 10.1261/rna.2411710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lareau LF, Hite DH, Hogan GJ, Brown PO. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 2014;3:e01257. doi: 10.7554/eLife.01257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson DN, Beckmann R. The ribosomal tunnel as a functional environment for nascent polypeptide folding and translational stalling. Curr Opin Struct Biol. 2011;21:274–282. doi: 10.1016/j.sbi.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Cao J, Geballe AP. Inhibition of nascent-peptide release at translation termination. Mol Cell Biol. 1996;16:7109–7114. doi: 10.1128/mcb.16.12.7109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu J, Deutsch C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J Mol Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ito-Harashima S, Kuroha K, Tatematsu T, Inada T. Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 2007;21:519–524. doi: 10.1101/gad.1490207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Frischmeyer PA, et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science. 2002;295:2258–2261. doi: 10.1126/science.1067338. [DOI] [PubMed] [Google Scholar]
- 28.van Hoof A, Frischmeyer PA, Dietz HC, Parker R. Exosome-mediated recognition and degradation of mRNAs lacking a termination codon. Science. 2002;295:2262–2264. doi: 10.1126/science.1067272. [DOI] [PubMed] [Google Scholar]
- 29.Dimitrova LN, Kuroha K, Tatematsu T, Inada T. Nascent peptide-dependent translation arrest leads to Not4p-mediated protein degradation by the proteasome. J Biol Chem. 2009;284:10343–10352. doi: 10.1074/jbc.M808840200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 31.Shoemaker CJ, Green R. Translation drives mRNA quality control. Nat Struct Mol Biol. 2012;19:594–601. doi: 10.1038/nsmb.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Losson R, Lacroute F. Interference of nonsense mutations with eukaryotic messenger RNA stability. Proc Natl Acad Sci USA. 1979;76:5134–5137. doi: 10.1073/pnas.76.10.5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Popp MWL, Maquat LE. Organizing principles of mammalian nonsense-mediated mRNA decay. Annu Rev Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maquat LE, Kinniburgh AJ, Rachmilewitz EA, Ross J. Unstable beta-globin mRNA in mRNA-deficient beta o thalassemia. Cell. 1981;27:543–553. doi: 10.1016/0092-8674(81)90396-2. [DOI] [PubMed] [Google Scholar]
- 35.Shoemaker CJ, Eyler DE, Green R. Dom34:Hbs1 promotes subunit dissociation and peptidyl-tRNA drop-off to initiate no-go decay. Science. 2010;330:369–372. doi: 10.1126/science.1192430. This study together with ref. 37 provides rigorous biochemical evidence that the Dom34–Hbs1 complex mediates the splitting of translationally stalled ribosomes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shoemaker CJ, Green R. Kinetic analysis reveals the ordered coupling of translation termination and ribosome recycling in yeast. Proc Natl Acad Sci USA. 2011;108:E1392–E1398. doi: 10.1073/pnas.1113956108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pisareva VP, Skabkin MA, Hellen CUT, Pestova TV, Pisarev AV. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 2011;30:1804–1817. doi: 10.1038/emboj.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pisarev AV, et al. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol Cell. 2010;37:196–210. doi: 10.1016/j.molcel.2009.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bengtson MH, Joazeiro CAP. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature. 2010;467:470–473. doi: 10.1038/nature09371. This paper reports the identification of Ltn1 as a ubiquitin ligase that may target nascent proteins on stalled ribosomes for proteasomal degradation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu J, et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc Natl Acad Sci USA. 2009;106:2097–2103. doi: 10.1073/pnas.0812819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Himeno H, Nameki N, Kurita D, Muto A, Abo T. Ribosome rescue systems in bacteria. Biochimie. 2015;114:102–112. doi: 10.1016/j.biochi.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 42.Karzai AW, Roche ED, Sauer RT. The SsrA–SmpB system for protein tagging, directed degradation and ribosome rescue. Nat Struct Biol. 2000;7:449–455. doi: 10.1038/75843. [DOI] [PubMed] [Google Scholar]
- 43.Brandman O, et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell. 2012;151:1042–1054. doi: 10.1016/j.cell.2012.10.044. This paper, together with ref. 44, used different genetic strategies to identify many of the key factors involved in the RQC pathway and link them to the 60S ribosomal subunit. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Defenouillère Q, et al. Cdc48-associated complex bound to 60S particles is required for the clearance of aberrant translation products. Proc Natl Acad Sci USA. 2013;110:5046–5051. doi: 10.1073/pnas.1221724110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verma R, Oania RS, Kolawa NJ, Deshaies RJ. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife. 2013;2:e00308. doi: 10.7554/eLife.00308. This study provides evidence that the AAA+ ATPase Cdc48 facilitates extraction of stalled protein products from the ribosome for subsequent proteasomal degradation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stolz A, Hilt W, Buchberger A, Wolf DH. Cdc48: a power machine in protein degradation. Trends Biochem Sci. 2011;36:515–523. doi: 10.1016/j.tibs.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 47.Shao S, von der Malsburg K, Hegde RS. Listerin-dependent nascent protein ubiquitination relies on ribosome subunit dissociation. Mol Cell. 2013;50:637–648. doi: 10.1016/j.molcel.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perara E, Rothman RE, Lingappa VR. Uncoupling translocation from translation: implications for transport of proteins across membranes. Science. 1986;232:348–352. doi: 10.1126/science.3961485. [DOI] [PubMed] [Google Scholar]
- 49.Shao S, Hegde RS. Reconstitution of a minimal ribosome-associated ubiquitination pathway with purified factors. Mol Cell. 2014;55:880–890. doi: 10.1016/j.molcel.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chiabudini M, et al. Release factor eRF3 mediates premature translation termination on polylysine-stalled ribosomes in Saccharomyces cerevisiae. Mol Cell Biol. 2014;34:4062–4076. doi: 10.1128/MCB.00799-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiabudini M, Conz C, Reckmann F, Rospert S. Ribosome-associated complex and Ssb are required for translational repression induced by polylysine segments within nascent chains. Mol Cell Biol. 2012;32:4769–4779. doi: 10.1128/MCB.00809-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Becker T, et al. Structure of the no-go mRNA decay complex Dom34–Hbs1 bound to a stalled 80S ribosome. Nat Struct Mol Biol. 2011;18:715–720. doi: 10.1038/nsmb.2057. This study reports the cryo-EM structure of a stalled ribosome bound to the Dom34–Hbs1 complex involved in its recognition and splitting. [DOI] [PubMed] [Google Scholar]
- 53.Jackson RJ, Hellen CUT, Pestova TV. Termination and post-termination events in eukaryotic translation. Adv Protein Chem Struct Biol. 2012;86:45–93. doi: 10.1016/B978-0-12-386497-0.00002-5. [DOI] [PubMed] [Google Scholar]
- 54.Dever TE, Green R. The elongation, termination, and recycling phases of translation in eukaryotes. Cold Spring Harb Perspect Biol. 2012;4:a013706. doi: 10.1101/cshperspect.a013706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–732. doi: 10.1016/s0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- 56.Brown A, Shao S, Murray J, Hegde RS, Ramakrishnan V. Structural basis for stop codon recognition in eukaryotes. Nature. 2015;524:493–496. doi: 10.1038/nature14896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shao S, Brown A, Santhanam B, Hegde RS. Structure and assembly pathway of the ribosome quality control complex. Mol Cell. 2015;57:433–444. doi: 10.1016/j.molcel.2014.12.015. This study reconstituted RQC assembly and ubiquitination with purified factors, ordered the steps in the reaction and determined the cryo-EM structure of the 60S–NEMF–Listerin complex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsuboi T, et al. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′end of aberrant mRNA. Mol Cell. 2012;46:518–529. doi: 10.1016/j.molcel.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 59.Guydosh NR, Green R. Dom34 rescues ribosomes in 3′untranslated regions. Cell. 2014;156:950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bhushan S, et al. Structural basis for translational stalling by human cytomegalovirus and fungal arginine attenuator peptide. Mol Cell. 2010;40:138–146. doi: 10.1016/j.molcel.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 61.Kuroha K, et al. Receptor for activated C kinase 1 stimulates nascent polypeptide-dependent translation arrest. EMBO Rep. 2010;11:956–961. doi: 10.1038/embor.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Letzring DP, Wolf AS, Brule CE, Grayhack EJ. Translation of CGA codon repeats in yeast involves quality control components and ribosomal protein L1. RNA. 2013;19:1208–1217. doi: 10.1261/rna.039446.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodrigo-Brenni MC, Hegde RS. Design principles of protein biosynthesis-coupled quality control. Dev Cell. 2012;23:896–907. doi: 10.1016/j.devcel.2012.10.012. [DOI] [PubMed] [Google Scholar]
- 64.Shen PS, et al. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science. 2015;347:75–78. doi: 10.1126/science.1259724. This study determined the structure of the 60S–RQC by cryo-EM, discovered CAT tails, and demonstrated their synthesis by an mRNA- and 40S-independent Rqc2-driven process. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lyumkis D, et al. Structural basis for translational surveillance by the large ribosomal subunit-associated protein quality control complex. Proc Natl Acad Sci USA. 2014;111:15981–15986. doi: 10.1073/pnas.1413882111. This study reports the cryo-EM structure of the 60S–RQC, in which the position of Rqc2 was first identified at the 60S interface bound to a P-site tRNA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gerber AP, Keller W. An adenosine deaminase that generates inosine at the wobble position of tRNAs. Science. 1999;286:1146–1149. doi: 10.1126/science.286.5442.1146. [DOI] [PubMed] [Google Scholar]
- 67.von der Malsburg K, Shao S, Hegde RS. The ribosome quality control pathway can access nascent polypeptides stalled at the Sec61 translocon. Mol Biol Cell. 2015;26:2168–2180. doi: 10.1091/mbc.E15-01-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Oma Y, Kino Y, Toriumi K, Sasagawa N, Ishiura S. Interactions between homopolymeric amino acids (HPAAs) Protein Sci. 2007;16:2195–2204. doi: 10.1110/ps.072955307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Satyal SH, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2000;97:5750–5755. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Shalgi R, et al. Widespread regulation of translation by elongation pausing in heat shock. Mol Cell. 2013;49:439–452. doi: 10.1016/j.molcel.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Liu B, Han Y, Qian SB. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol Cell. 2013;49:453–463. doi: 10.1016/j.molcel.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Higgins R, et al. The Unfolded protein response triggers site-specific regulatory ubiquitylation of 40S ribosomal proteins. Mol Cell. 2015;59:35–49. doi: 10.1016/j.molcel.2015.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Merret R, et al. Heat-induced ribosome pausing triggers mRNA co-translational decay in Arabidopsis thaliana. Nucleic Acids Res. 2015;43:4121–4132. doi: 10.1093/nar/gkv234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Merret R, et al. XRN4 and LARP1 are required for a heat-triggered mRNA decay pathway involved in plant acclimation and survival during thermal stress. Cell Reports. 2013;5:1279–1293. doi: 10.1016/j.celrep.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 75.Matsuda R, Ikeuchi K, Nomura S, Inada T. Protein quality control systems associated with no-go and nonstop mRNA surveillance in yeast. Genes Cells. 2014;19:1–12. doi: 10.1111/gtc.12106. [DOI] [PubMed] [Google Scholar]
- 76.Song H, et al. The crystal structure of human eukaryotic release factor eRF1--mechanism of stop codon recognition and peptidyl-tRNA hydrolysis. Cell. 2000;100:311–321. doi: 10.1016/s0092-8674(00)80667-4. [DOI] [PubMed] [Google Scholar]
- 77.Wang F, Durfee LA, Huibregtse JM. A cotranslational ubiquitination pathway for quality control of misfolded proteins. Mol Cell. 2013;50:368–378. doi: 10.1016/j.molcel.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lykke-Andersen J, Bennett EJ. Protecting the proteome: eukaryotic cotranslational quality control pathways. J Cell Biol. 2014;204:467–476. doi: 10.1083/jcb.201311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Preissler S, Deuerling E. Ribosome-associated chaperones as key players in proteostasis. Trends Biochem Sci. 2012;37:274–283. doi: 10.1016/j.tibs.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 80.Turner GC, Varshavsky A. Detecting and measuring cotranslational protein degradation in vivo. Science. 2000;289:2117–2120. doi: 10.1126/science.289.5487.2117. [DOI] [PubMed] [Google Scholar]
- 81.Duttler S, Pechmann S, Frydman J. Principles of cotranslational ubiquitination and quality control at the ribosome. Mol Cell. 2013;50:379–393. doi: 10.1016/j.molcel.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kaida D, et al. U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 2010;468:664–668. doi: 10.1038/nature09479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Himeno H, Kurita D, Muto A. tmRNA-mediated trans-translation as the major ribosome rescue system in a bacterial cell. Front Genet. 2014;5:66. doi: 10.3389/fgene.2014.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Komine Y, Kitabatake M, Yokogawa T, Nishikawa K, Inokuchi H. A tRNA-like structure is present in 10Sa RNA, a small stable RNA from Escherichia coli. Proc Natl Acad Sci USA. 1994;91:9223–9227. doi: 10.1073/pnas.91.20.9223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ushida C, Himeno H, Watanabe T, Muto A. tRNA-like structures in 10Sa RNAs of Mycoplasma capricolum and Bacillus subtilis . Nucleic Acids Res. 1994;22:3392–3396. doi: 10.1093/nar/22.16.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moore SD, Sauer RT. The tmRNA system for translational surveillance and ribosome rescue. Annu Rev Biochem. 2007;76:101–124. doi: 10.1146/annurev.biochem.75.103004.142733. [DOI] [PubMed] [Google Scholar]
- 87.Ivanova N, Pavlov MY, Felden B, Ehrenberg M. Ribosome rescue by tmRNA requires truncated mRNAs. J Mol Biol. 2004;338:33–41. doi: 10.1016/j.jmb.2004.02.043. [DOI] [PubMed] [Google Scholar]