

Abstract
The MYC family of oncogenes encodes a set of three related transcription factors that are overexpressed in many human tumors and contribute to the cancer-related deaths of more than 70,000 Americans every year. MYC proteins drive tumorigenesis by interacting with co-factors that enable them to regulate the expression of thousands of genes linked to cell growth, proliferation, metabolism, and genome stability. One effective way to identify critical cofactors required for MYC function has been to focus on sequence motifs within MYC that are conserved throughout evolution, on the assumption that their conservation is driven by protein-protein interactions that are vital for MYC activity. In addition to their DNA-binding domains, MYC proteins carry five regions of high sequence conservation known as Myc boxes (Mb). To date, four of the Myc box motifs (MbI, MbII, MbIIIa, and MbIIIb) have had a molecular function assigned to them, but the precise role of the remaining Myc box, MbIV, and the reason for its preservation in vertebrate Myc proteins, is unknown. Here, we show that MbIV is required for the association of MYC with the abundant transcriptional coregulator host cell factor 1 (HCF-1). We show that the invariant core of MbIV resembles the tetrapeptide HCF-binding motif (HBM) found in many HCF-interaction partners, and demonstrate that MYC interacts with HCF in a manner indistinguishable from the prototypical HBM-containing protein VP16. Finally, we show that rationalized point mutations in MYC that disrupt interaction with HCF-1 attenuate the ability of MYC to drive tumorigenesis in mice. Together, these data expose a molecular function for MbIV and indicate that HCF-1 is an important co-factor for MYC.
Keywords: Cancer, chromatin, MYC, host cell factor
INTRODUCTION
The MYC family of oncogenes (c-, N-, and L-MYC) encodes a set of transcription factors that feature prominently in cancer1. Capable of acting as both transcriptional activators and repressors, MYC proteins drive tumorigenesis by controlling the expression of thousands of genes connected to cell growth, proliferation, metabolism, and genome stability. Recognition of target genes by MYC is dependent on interaction with its obligate partner, MAX2, which creates a stable DNA-binding domain (DBD) that recognizes E-box motifs (CACGTG) in promoters and enhancers of MYC-regulated genes. Once bound to its target sites, MYC interacts with sets of cofactors3 to modulate the gene expression patterns that ultimately lead to malignancy. Because of the pervasive involvement of MYC in human cancer, much interest is centered on identification of the partner proteins that MYC uses to control transcription, with the objective of identifying new interaction surfaces that can be pharmacologically exploited to kill cancer cells.
Across the evolutionary spectrum, MYC family members share a number of conserved sequence motifs that, where examined, play important roles in MYC function. Besides the basic- helix-loop-helix-leucine zipper domain that binds MAX, there are five short regions of homology among MYC proteins that have been termed “MYC boxes” (Mb)—MbI, MbII, MbIIIa, MbIIIb, and MbIV4 (Supplemental Figure 1a). Analysis of these MYC boxes has yielded insight into the actions and regulation of MYC. MbI is a major site of MYC regulation by phosphorylation and ubiquitin-mediated proteolysis5. MbII interacts with the histone acetyltransferase component TRRAP6 (and other co-factors) to stimulate MYC target gene induction. MbIIIa associates with the histone deactylase HDAC3 to repress transcription7. And MbIIIb binds directly to WDR5, which facilitates MYC recruitment to target genes in the context of chromatin8.
The only remaining MYC box for which a molecular function is undiscovered is MbIV, a ~20 residue motif found in all vertebrate MYC proteins. Deletion of MbIV reduces the ability of MYC to bind naked DNA, and is associated with decreased transforming potential in vitro9, but how MbIV functions in this capacity, and the important cofactors that interact with this element, are unknown. Here, we present evidence that MbIV mediates the interaction of MYC with host cell factor 1 (HCF-1), a conserved transcriptional cofactor that assembles into numerous transcriptional and chromatin-regulatory complexes10. We show that invariant residues in the MbIV core constitute a canonical HCF-binding motif (HBM) that is critical for the interaction of MYC with HCF-1. Alanine substitution mutations in MbIV that disrupt the MYC–HCF-1 interaction have no impact on the ability of MYC to bind naked DNA, but profoundly decrease its ability to drive tumorigenesis in mice. Our data establish HCF-1 as an important interaction partner for MYC and illuminate the molecular function of MbIV.
RESULTS AND DISCUSSION
MYC associates with Host Cell Factor 1
We recently identified WDR5 as a MYC-interaction partner8 by performing a proteomic screen for proteins that bind the “central portion” of c-MYC (residues 151–319), a region that includes MYC boxes IIIa, IIIb, and IV1 (Supplemental Figure 1a). We transiently-expressed FLAG-tagged Gal4-fusions carrying the MYC central portion, or the MYC activation domain, in HEK293 cells, recovered the fusions by immunoprecipitation, and analyzed immunoprecipitates using multidimensional protein identification technology (MudPIT). In addition to WDR5, this analysis identified HCF-1 as a protein that is enriched in the central portion pulldown, compared to the activation domain control (Supplemental Figure 1b). By co-immunoprecipitation coupled with immunoblotting (coIP), we confirmed that endogenous HCF-1 associates with the Gal4–MYC (151–319) fusion (Figure 1a). We also determined that endogenous HCF-1 associates with MYC stably expressed in HEK293 cells by retroviral transduction (Figure 1b), and that endogenous MYC associates with endogenous HCF-1 (Figure 1c) in this same cell type. These data are consistent with a recent proteomic survey of biotin-tagged MYC that identified HCF-1 as a MYC-associated protein in tumor xenografts11, and establish that HCF-1 is a bonafide MYC-interaction partner.
Figure 1.
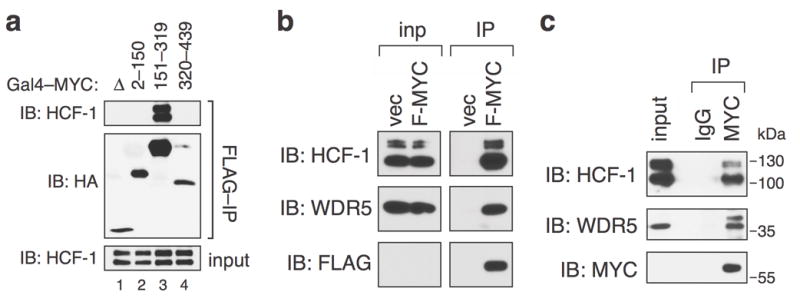
c-MYC associates with HCF-1. (a) The central portion of c-MYC interacts with HCF-1. HA- and FLAG-epitope-tagged Gal4 DNA-binding domain alone (Δ), or fused to c-MYC residues 2–150, 151–319, or 320–439, was transiently-expressed in human HEK293 cells from the vector pcDNA3.1 and recovered by immunoprecipitation (IP) on anti-FLAG resin (A2220; Sigma) as previously described8. Immunoprecipitates, and a sample of the input, were probed by immunoblotting (IB) with antibodies against the HA-epitope tag (α-HA-HRP; Roche) or endogenous HCF-1 (A301-399A; Bethyl). This experiment was replicated three times in the laboratory. HEK293 cells were obtained from the American Type Culture Collection (ATCC) and confirmed to be mycoplasma free. (b) Full-length c-MYC associates with HCF-1. HEK293 cells were engineered by retroviral transduction of pBabe-Hygro vectors8 to express FLAG-tagged full-length human c-MYC, or an empty vector (vec) control. Cells were lysed in buffer containing 50 mM Tris.Cl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, and protease inhibitor cocktail (Roche), briefly sonicated, and centrifuged at 12,000 g for 10 minutes at 4°C to precipitate debris. The supernatant was then subject to IP on anti-FLAG resin, and immunoprecipitates, or a sample of the input lysate (“inp”), probed by IB with antibodies against HCF-1, WDR5 (D9E1I; Cell Signaling), or FLAG (A8598; Sigma). Note that, due to low expression of FLAG-MYC in these cells, this protein is not visible in the input samples. This experiment was replicated five times in the laboratory.(c) Endogenous MYC binds endogenous HCF-1. Lysates were prepared from HEK293 cells treated for two hours with proteasome inhibitor MG132 (25 μM) to promote accumulation of MYC, subject to IP with the anti-MYC antibody N262 (Santa Cruz), or IgG control (Pierce) and immunoprecipitates (or a sample of the input) probed with antibodies against endogenous HCF-1, WDR5, and c-MYC (9E10; Roche). Location of molecular weight markers is presented in kilodaltons (kDa). This experiment was replicated three times in the laboratory.
Association of MYC with HCF-1 is mediated via Myc box IV
HCF-1 is an abundant nuclear protein12 that is an essential co-factor for transcription of herpes simplex virus immediate early genes. It is also a core component of multiple chromatin modulatory complexes13, including MLL/Set-type histone methyltransferases14 and MOF/NSL histone acetyltransferases15. Interestingly, both types of enzymes also contain WDR5, raising the possibility that the association we observe between MYC and HCF-1 reflects interaction of a WDR5/HCF-1-containing complex with MYC. To explore this possibility, we asked whether a triple point mutation in MbIIIb (“WBM”) that disrupts interaction of MYC with WDR58 has any effect on association of MYC with HCF-1. Remarkably, the WBM mutant interacts with HCF-1 at levels comparable to the WT MYC protein (Figure 2a), demonstrating that HCF-1 interacts with MYC independent of WDR5, and revealing that sequences outside of the conserved MbIIIb core mediate the MYC–HCF-1 association.
Figure 2.
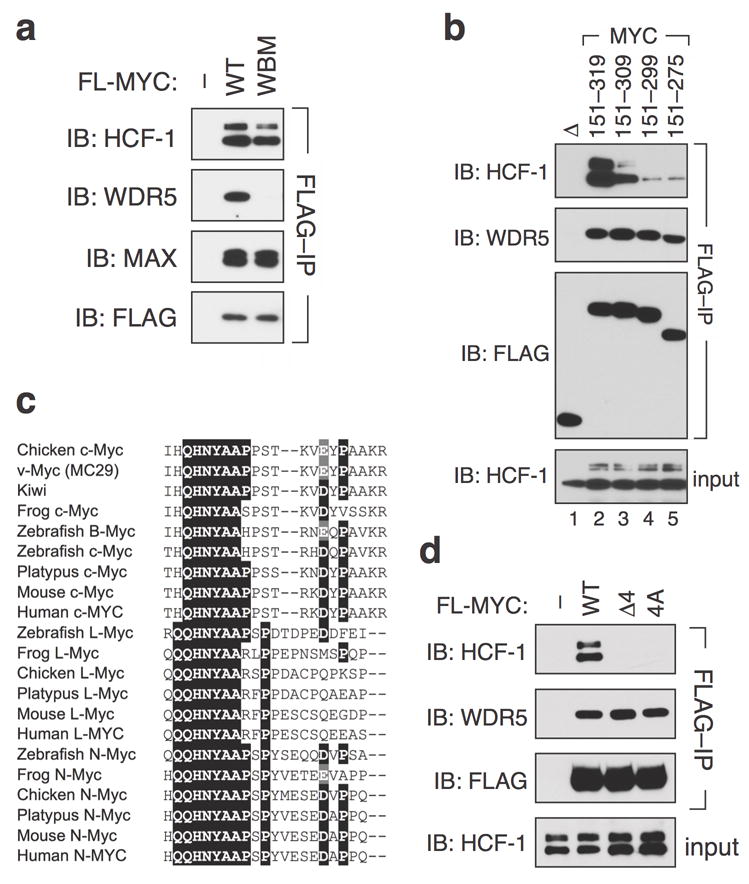
MYC box IV is required for the association of c-MYC with HCF-1. (a) HCF-1 and WDR5 interact independently with full-length MYC. HEK293 cells were engineered by retroviral transduction of pBabe-Hygro vectors8 to express FLAG-tagged full-length wild-type (WT) or WDR5-interaction-defective (WBM; I262E, V264E, V265E) c-MYC, or an empty vector control. Lysates were subject to IP with anti-FLAG resin, and immunoprecipitates probed with antibodies against FLAG or endogenous HCF-1, WDR5, or MAX (C-124; Santa Cruz). This experiment was replicated three times in the laboratory. (b) Residues 300–319 of MYC are required for robust association with HCF-1. FLAG-epitope-tagged Gal4 DNA-binding domain fusions carrying the indicated MYC residues, or the Gal4 DNA-binding domain alone (Δ), were transiently-expressed from the vector pcDNA3.18 in HEK293 cells and recovered by immunoprecipitation on anti-FLAG resin. Immunoprecipitates were probed with antibodies against HCF-1, WDR5, or FLAG. An IB of HCF-1 levels in the input of the IP reaction is also shown. This experiment was replicated four times in the laboratory.(c) QHNYAA is the invariant core of MbIV. Alignment of MYC box IV sequences (corresponding to residues 304–326 of human c-MYC) from the indicated family members and species. Residues conserved in more than 50% of the aligned sequences are boxed in black; conserved substitutions in these positions are boxed in gray. A more complete representation of the alignment, and relevant accession numbers is presented in Supplemental Figure S2. (d) The QHNY core of MbIV is required for association with HCF-1. Lysates from HEK293 cells transduced with pBabe-Hygro vectors8 to express FLAG-tagged full-length WT MYC, a MYC mutant in which these four residues have been deleted (Δ4), a MYC mutant in which these four residues are mutated to alanine (4A), or vector control (–), were subject to IP on anti-FLAG resin. Immunoprecipitates were probed with antibodies against FLAG, endogenous HCF-1, and WDR5. An IB of HCF-1 levels in the input to the IP reaction is also shown. This experiment was replicated four times in the laboratory.
To delineate which residues in the central portion of MYC are required for interaction with HCF- 1, we performed coIP experiments using a set of carboxy-terminal truncation mutants in the FLAG-Gal4–MYC (151–319) context, probing for their interaction with endogenous HCF-1. This analysis (Figure 2b) showed that deletion of MYC residues 310–319 reduces association with HCF-1 substantially, and that the interaction is further reduced by deletion of residues 300–319. Interestingly, residues 300–319 of c-MYC correspond closely to MbIV, a vertebrate-specific Myc box implicated in DNA-binding by MYC, as well as cellular transformation in vitro9. Alignment of Myc sequences from vertebrates spanning the evolutionary spectrum (Figure 2c and Supplemental Figure S2) reveals that conservation within MbIV centers on an invariant “QHNYAA” motif (residues 306–311), perfectly conserved in all Myc family members from all vertebrates. Consistent with the involvement of these residues in interaction with HCF-1, deletion of residues 306–309 (“Δ4”), or mutation of the same four residues to alanine (“4A”; QHNY to AAAA), perturbs association of full-length MYC with endogenous HCF-1, while leaving the MYC–WDR5 interaction unaffected (Figure 2d). Thus, the invariant QHNY motif within MbIV is necessary for interaction of MYC with HCF-1.
Myc box IV is an HCF-binding motif
HCF-1 is a collection of polypeptides derived by proteolytic cleavage of a 2035 residue precursor into amino- and carboxy-terminal fragments that remain non-covalently associated with one another12. Proteins that associate with HCF-1 often carry a tetrapeptide “HCF-binding motif” (HBM)16 that mediates interaction with an amino-terminal domain of HCF-1 called “HCFVIC”17. The consensus for the HBM is defined as D/E-H-X-Y18, but alignment of 14 validated HBM sequences (Figure 3a) indicates that only the histidine and tyrosine residues in the HBM are invariant, and that some flexibility is permitted in the first position of the HBM (D, E, G, or N). Notably, the deubiquitylating enzyme BAP119 carries the sequence “NHNY”, which is very similar in sequence and composition to the “QHNY” core of MbIV. In BAP1, mutation of these residues to alanine—analogous to our 4A mutation in MYC—disrupts interaction with HCF-119. The similar nature of the BAP1 HBM and MbIV sequences, and the similar effects of alanine- substitutions on HCF-1 interaction, raises the intriguing possibility that MbIV is an HBM.
Figure 3.
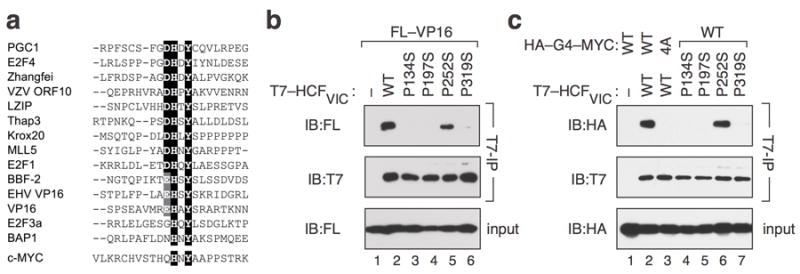
MbIV is an HBM. (a) Alignment of experimentally-validated HBM motifs from 14 known HCF-1-interaction partners. Residues conserved in more than 50% of the aligned sequences are boxed in black; conservative substitutions in these positions are boxed in gray. The MbIV sequence from human c-MYC is shown below. (b) Impact of mutations in HCF-1VIC on interaction with VP16. HEK293 cells were transiently transfected with plasmid vector control (–; pCGT12) or otherwise identical vectors expressing T7-tagged HCFVIC (2–380)—wild-type (WT) or the indicated mutants—together with a pcDNA3.18 vector expressing FLAG-tagged VP16 (FL–VP16). Lysates were prepared, subject to IP with an anti-T7 epitope antibody (70566; Novagen), and immunoprecipitates probed with anti-FLAG and anti-T7 antibodies. An IB of FL– VP16 levels in the input to the IP reaction is also shown. (c) MYC contains a canonical HBM. HEK293 cells were transiently transfected with plasmid vector control (–; pCGT12) or otherwise identical vectors expressing T7-tagged HCFVIC (2–380)—wild-type (WT) or the indicated mutants—together with a pcDNA3.18 vector expressing either the WT or 4A mutant version of FLAG-tagged Gal4–HA–MYC (151–319). Lysates were prepared, subject to IP with an anti-T7 epitope antibody, and immunoprecipitates probed with anti-HA and anti-T7 antibodies. An IB of FLAG-tagged Gal4–HA–MYC (151–319) levels in the input to the IP reaction is also shown. This experiment was replicated three times in the laboratory.
If MbIV is an HBM, we would expect that interaction of MYC with HCF-1 depends on residues in HCF-1 important for interaction with validated HBM-containing proteins. Specifically, we expect that the HCFVIC domain would suffice for interaction with MbIV-containing sequences, and that the interaction would be sensitive to mutations in HCFVIC that disrupt interaction with known HBM-containing proteins, such as the prototypical HBM-containing protein VP1616 (Figure 3b). Indeed, we observed that Gal4–MYC (151–319) interacts robustly with HCFVIC and in a manner that is disrupted by the 4A mutation in the Gal4–MYC fusion (Figure 3c, compare lanes 2 and 3). Moreover, three point mutations in HCFVIC that disrupt interaction with VP16 (Figure 3b; P134S, P197S, and P319S)20 also disrupt the interaction between HCFVIC and Gal4–MYC (Figure 3c, compare lanes 2 with 4, 5, and 7). And finally, a control mutation in HCFVIC that is permissive for interaction with VP16 (Figure 3b; P252S)20 is also permissive for interaction with Gal4–MYC (Figure 3c, compare lanes 2 and 6). Thus MYC interacts with HCF-1 in a manner that is indistinguishable from VP16. Based on these data, we conclude that MbIV is an HCF- binding motif. We also predict, based on the conservation of the HBM within MbIV, that all vertebrate MYC proteins can associate with HCF-1.
The HCF-binding motif in MYC contributes to its tumorigenic potential
What role does the HBM have in MYC biology? Complete deletion of MbIV within the context of N-MYC reduces its ability to bind naked DNA, and is associated with a reduction in the ability of N-MYC to promote anchorage-independent growth of fibroblasts in culture9. Because MbIV is in relatively close proximity to the DNA-binding domain of MYC1, however, it is possible that deletion of this region introduces structural change to the protein that perturb DNA recognition. To more precisely probe the role of the HBM in DNA-binding by MYC, we assayed the ability of the 4A MYC mutant to bind naked E-box containing DNA via electrophoretic mobility shift assay. This analysis (Figure 4a) showed that MYC/MAX complexes bearing the 4A mutation bind DNA in a manner indistinguishable from WT MYC (compare lanes 3 and 4). Thus, the HBM within MYC—and therefore invariant residues in MbIV—are not required for DNA binding by MYC.
Figure 4.
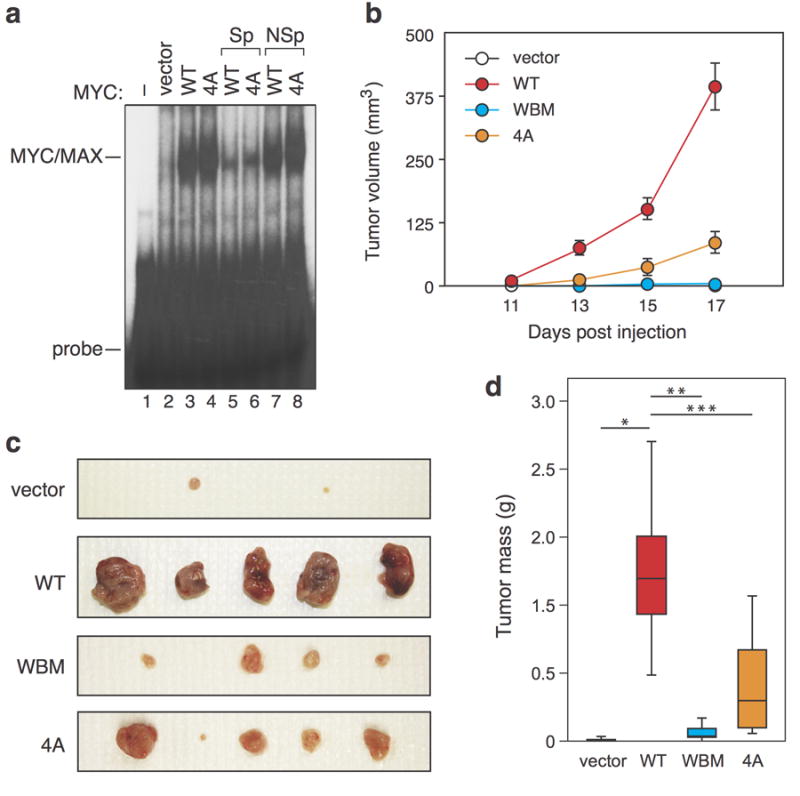
The c-MYC HBM is dispensable for binding to naked DNA but required for the full tumorigenic potential of MYC in mice. (a) The HBM is not required for MYC/MAX dimers to bind DNA in vitro. Electrophoretic mobility shift assays were performed as described8. In brief, HEK293 cells were transiently transfected with the indicated Flag-tagged full length MYC proteins in pcDNA3.1-Puro8. Nuclear extracts were prepared by cytoplasmic lysis in buffer containing 10 mM HEPES pH 7.4, 10 mM KCl, 0.1 mM EDTA, 0.1 MM EGTA, 0.4% NP-40. 15 μg of lysate was incubated for 30 min at room temperature with 21 fmol double-stranded, 32P end labelled DNA probe carrying an E-box (5’-gatccgtaagacCACGTGgtcgtcaggat-3’), followed by separation on a 5% acrylamide 0.5 x Tris-borate-EDTA–polyacrylamide gel. To confirm the specificity of the interaction, indicated reactions also contained a 10-fold molar excess of unlabeled specific competitor DNA (“Sp”, lanes 5–6) or a non-specific competitor where the E- box had been mutated to ACATGT (“NSp”, lanes 7–8). This experiment was replicated four times in the laboratory. (b–d) The HBM contributes to the oncogenic activity of MYC in vivo. Murine NIH3T3 fibroblasts were stably transduced with pBabe-Hyrgo vectors8 to express the indicated FLAG-tagged MYC proteins, or empty vector control. These fibroblasts were assayed for MYC expression and the ability to drive anchorage-independent growth in soft-agar (see Supplemental Figure S3). The same cells (7.5 × 106 of each) were then injected into the flanks of 5–6 week old female athymic nude mice (Foxn1nu; Harlan Sprague Dawley) for this analysis. No randomization was used. (b) Ellipsoid tumor volume was monitored (measured with calipers; investigator blinded to sample identity) at the indicated times post-injection (n=10 mice for each experimental arm). Data are presented as mean +/- SEM. Sample size was chosen based on sample size needed in previous comparable experiments, and by performing a power analysis for a p-value of 0.05, power of 0.8, 25% difference, 25% sigma (std dev), which showed the minimum number of mice needed to be eight. (c) Tumors were excised and photographed 17 days after injection. Representative tumors from five injection sites from vector control, WT MYC, and each of the MYC mutants are shown. (d) Tumors were weighed after excision and masses presented as a box and whisker plot. For Student’s t-test (two-tailed): vector versus WT, p=0.000025 (*); WT versus WBM, p=0.000029 (**); WT versus 4A, p=0.00041 (***). All mouse experiments were performed in compliance with US Federal laws and with approval of the Vanderbilt University School of Medicine Animal Care and Use Committee.
Next, we asked whether the 4A mutation alters the ability of MYC to promote anchorage- independent growth of NIH3T3 fibroblasts in soft agar (Supplemental Figure S3). In this assay, the 4A mutation had no detrimental effect on MYC expression or activity; indeed the 4A MYC mutant had increased colony forming potential, relative to the wild-type protein. We have previously found that a WDR5-interaction-defective MYC mutant (WBM) retains activity in soft- agar assays, but is defective for tumorigenesis in mice8. To probe the effects of the 4A mutation in a more tumor-relevant context, therefore, we next injected the same engineered fibroblasts used in the soft agar assay into the flanks of athymic nude mice and followed tumor formation over time. We also compared the 4A mutant with the previously characterized WBM mutant. Under these conditions, the 4A MYC mutant was significantly less active than the WT MYC protein, producing slower-growing tumors (Figure 4b) that were, on average, 68% smaller by mass after resection (Figure 4c–d). As expected8, the WBM mutant was also severely impaired in this assay (Figure 4b–d). The reduced tumorigenic activity of the 4A MYC protein in this assay reveals that the HCF-binding motif in MYC has an important role in MYC-driven tumorigenesis.
MYC and HCF-1
Based on our observations, we conclude that: (i) HCF-1 is a MYC-interaction partner, (ii) HCF-1 associates with MYC via conserved residues in the MbIV core, (iii) MbIV functions as a classical HBM, and (iv) the HBM within MbIV—and thus interaction of MYC with HCF-1—is required for the full tumorigenic potential of MYC in vivo. Our data reveal that HCF-1 is an important co- factor for MYC, and imply that the frank evolutionary conservation of MbIV in vertebrate MYC proteins is driven by the importance of the MYC–HCF-1 interaction.
Understanding the role of MYC boxes has played a major part in illuminating the functions of MYC, as well as its key interaction partners1. Our demonstration that MbIV is an HBM, and that this motif is required for the full-tumorigenic potential of MYC, reveals that HCF-1 is an important and conserved MYC-interaction partner. Supporting this notion, Dingar et al.,11, who identified HCF-1 as a MYC-associated protein in their proteomic survey, analyzed chromatin immunoprecipitation-sequencing (ChIP-seq) data generated by the ENCODE consortium21 and discovered that ~80% of MYC binding sites in human K562 cells coincide with a binding site for HCF-1. This level of coincident binding is similar to what we have observed with MYC and WDR58, and suggests that MYC interacts extensively with HCF-1 on chromatin.
Although MbIV is vertebrate-specific, it is interesting that Drosophila MYC (dMyc) associates with Drosophila HCF, and that this association contributes to Myc-dependent gene regulation and to the ability of dMyc to promote cell growth in flies22. Drosophila HCF also binds with dMyc on target gene chromatin. Curiously, although dMyc carries a perfect HBM consensus, this region is dispensable for association with HCF22, revealing that Drosophila HCF has a different (or expanded) modality for associating with partner proteins, and one that does not rely solely on HBM recognition. Evolutionary preservation of the MYC–HCF interaction in light of this HBM- independent binding mechanism provides further support for the importance of HCF to MYC function, and raises the intriguing possibility that association of MYC with HCF across the evolutionary spectrum is more widespread than predicted by the presence of MbIV.
Given the known functions of HCF-110, it is most likely that it interacts with MYC as part of a multisubunit chromatin regulatory complex. In this regard, it is intriguing that many complexes containing HCF-1 also include WDR5, and yet we are able to show that HCF-1 and WDR5 can interact independently and with different regions of the MYC protein (MbIV versus MbIIIb). This finding has implications for understanding the context of the HCF-1 and WDR5 complexes at work, as it excludes both proteins from associating with MYC as part of an MLL/SET or MOF/NSL complex. By this criterion, the ATAC histone acetyltransferase23 is also excluded. A number of HCF-1 complexes bereft of WDR5 have been described, but further work will be required to determine the precise biochemical context in which HCF-1 recognizes MYC. Our demonstration that MbIV functions as a canonical HBM provides a tractable entry point for further biochemical and genetic dissection of the role of HCF-1 in MYC biology.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank S. Dey, S. Hiebert, H. McDonald, L. Neitzel, and A. Wilson for reagents, experimental assistance, and advice. This work was supported by the Edward P. Evans Foundation (to W.P.T.), the Melanoma Research Foundation (to W.P.T.), the Vanderbilt Ingram Cancer Center Support grant (NIH: CA68485), the NCI SPORE in Breast Cancer (P50 CA098131), the Cellular, Biochemical, and Molecular Sciences Training Program (T32 GM008554), the Integrated Biological Systems Training in Oncology Program (T32 CA119925), the National Center for Advancing Translational Sciences (UL1 TR000445), the Vanderbilt International Scholar Program, and grants R01CA148950 (to C.M.E.) and F30AG039164 (to. B.C.G.) from the National Institutes of Health.
Footnotes
CONFLICT OF INTEREST
The authors declare no conflict of interest.
References
- 1.Tansey WP. Mammalian MYC proteins and cancer. New Journal of Science. 20142013:1–27. [Google Scholar]
- 2.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 3.Thomas LR, Tansey WP. MYC and Chromatin. The Open Access Journal of Science and Technology. 2015;3:101124. [Google Scholar]
- 4.Meyer N, Penn LZ. Reflecting on 25 years with MYC. Nature reviews Cancer. 2008;8:976–990. doi: 10.1038/nrc2231. [DOI] [PubMed] [Google Scholar]
- 5.Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, et al. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation- dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004;101:9085–9090. doi: 10.1073/pnas.0402770101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c- Myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 7.Kurland JF, Tansey WP. Myc-mediated transcriptional repression by recruitment of histone deacetylase. Cancer Res. 2008;68:3624–3629. doi: 10.1158/0008-5472.CAN-07-6552. [DOI] [PubMed] [Google Scholar]
- 8.Thomas LR, Wang Q, Grieb BC, Phan J, Foshage AM, Sun Q, et al. Interaction with WDR5 Promotes Target Gene Recognition and Tumorigenesis by MYC. Mol Cell. 2015;58:440–452. doi: 10.1016/j.molcel.2015.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cowling VH, Chandriani S, Whitfield ML, Cole MD. A conserved Myc protein domain, MBIV, regulates DNA binding, apoptosis, transformation, and G2 arrest. Mol Cell Biol. 2006;26:4226–4239. doi: 10.1128/MCB.01959-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zargar Z, Tyagi S. Role of host cell factor-1 in cell cycle regulation. Transcription. 2012;3:187–192. doi: 10.4161/trns.20711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dingar D, Kalkat M, Chan PK, Srikumar T, Bailey SD, Tu WB, et al. BioID identifies novel c-MYC interacting partners in cultured cells and xenograft tumors. Journal of proteomics. 2014 doi: 10.1016/j.jprot.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 12.Wilson AC, LaMarco K, Peterson MG, Herr W. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell. 1993;74:115–125. doi: 10.1016/0092-8674(93)90299-6. [DOI] [PubMed] [Google Scholar]
- 13.Vogel JL, Kristie TM. The dynamics of HCF-1 modulation of herpes simplex virus chromatin during initiation of infection. Viruses. 2013;5:1272–1291. doi: 10.3390/v5051272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, et al. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai Y, Jin J, Swanson SK, Cole MD, Choi SH, Florens L, et al. Subunit composition and substrate specificity of a MOF-containing histone acetyltransferase distinct from the male-specific lethal (MSL) complex. J Biol Chem. 2010;285:4268–4272. doi: 10.1074/jbc.C109.087981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freiman RN, Herr W. Viral mimicry: common mode of association with HCF by VP16 and the cellular protein LZIP. Genes Dev. 1997;11:3122–3127. doi: 10.1101/gad.11.23.3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Goto H, Motomura S, Wilson AC, Freiman RN, Nakabeppu Y, Fukushima K, et al. A single-point mutation in HCF causes temperature-sensitive cell-cycle arrest and disrupts VP16 function. Genes Dev. 1997;11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- 18.Luciano RL, Wilson AC. HCF-1 functions as a coactivator for the zinc finger protein Krox20. J Biol Chem. 2003;278:51116–51124. doi: 10.1074/jbc.M303470200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Misaghi S, Ottosen S, Izrael-Tomasevic A, Arnott D, Lamkanfi M, Lee J, et al. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol Cell Biol. 2009;29:2181–2192. doi: 10.1128/MCB.01517-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mahajan SS, Wilson AC. Mutations in host cell factor 1 separate its role in cell proliferation from recruitment of VP16 and LZIP. Mol Cell Biol. 2000;20:919–928. doi: 10.1128/mcb.20.3.919-928.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Furrer M, Balbi M, Albarca-Aguilera M, Gallant M, Herr W, Gallant P. Drosophila Myc interacts with host cell factor (dHCF) to activate transcription and control growth. J Biol Chem. 2010;285(3):9623–39636. doi: 10.1074/jbc.M110.140467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang YL, Faiola F, Xu M, Pan S, Martinez E. Human ATAC Is a GCN5/PCAF- containing acetylase complex with a novel NC2-like histone fold module that interacts with the TATA-binding protein. J Biol Chem. 2008;283(3):3808–33815. doi: 10.1074/jbc.M806936200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.