

Abstract
The increasing incidence of diabetes mellitus (DM) and chronic periodontitis (CP) worldwide imposes a rethinking of individualized therapy for patients with both conditions. Central to bidirectional links between DM and CP is deregulated systemic inflammation and dysfunctional immune responses to altered-self and non-self. Control of blood glucose levels and metabolic imbalances associated with hyperglycemia in DM, and disruption of pathogenic subgingival biofilms in CP are currently the main therapeutic approaches for these conditions. Mounting evidence suggests the need to integrate immune modulatory therapeutics in treatment regimens that address the unresolved inflammation associated with DM and CP. The current review discusses the pathogenesis of DM and CP with emphasis on deregulated inflammation, current therapeutic approaches and the novel pro-resolution lipid mediators derived from Ω-3 polyunsaturated fatty acids.
Keywords: Diabetes mellitus, periodontitis, inflammation, lipid mediators, pro-resolution therapeutics
1. Introduction
Diabetes mellitus (DM) is a chronic condition characterized by chronic hyperglycemia that results from unregulated endocrine and metabolic pathways in glucose utilization. Diabetes-associated microvascular disease is a leading cause of blindness, renal failure and nerve damage, and significantly contributes to the increasing incidence of myocardial infarction and stroke in the developing world [1]. In 2010 more than 285 million people worldwide suffered from DM. The World Health Organization (WHO) predicted a 50% increase in DM incidence by 2030. Type I DM (T1DM), characterized by primary deficiency in insulin (INS) production due to pancreatic β-cell destruction leading to hypoinsulinemia, accounts for less than 10% of DM cases and has the highest incidence in northern European countries and US states with high Scandinavian descent. On the other hand, type II DM (T2DM) characterized by a peripheral INS resistance (IR) associated with hyperglycemia accounts for more than 90% of diabetes cases and its incidence around the world is increasing [2]. The differences in prevalence and distribution of T1DM and T2DM may be explained by the relative contribution of genetic and environmental factors in onset of disease.
About two-thirds of T1DM susceptibility is due to genetic factors primarily related to human leukocyte antigen (HLA) genes located on chromosome 6p21. More than 95% of people with T1DM possess HLA-DR3 or HLA-DR4 haplotype and individuals having any of the two concurrently with an DQ8 haplotype have the highest chance of inheriting this condition. The other third of susceptibility is related to environmental factors such as childhood infections that result in immune-mediated destruction of β-cells. Similarly, in T2DM both genetic and environmental factors contribute to IR and loss of β-cells. Although there is no HLA linkage in T2DM increasing evidence suggests that single nucleotide polymorphysims (SNPs) in several candidate diabetogenic and obesity-related genes play important roles in susceptibility to T2DM. Obesity is associated with T2DM in 60-70% of cases in North America, Europe and Africa, and in less than 30% of cases in Asia. Environmental factors contributing to onset of obesity and T2DM include decreased energy expenditure, increased caloric intake and
unbalanced nutrient composition. Diabetes-associated dyslipidemia characterized by high triglyceride levels, low high-density lipoprotein (HDL) and high low-density lipoprotein (LDL) levels are directly linked to IR. The altered glucose and lipid metabolism seen in T2DM leads to microvascular, neurologic and immunological changes that underlie diabetes-associated coronary artery disease, peripheral arterial disease, nephropathy, neuropathy, retinopathy, and periodontitis (Fig. 1A).
Periodontal diseases comprise of a group of conditions affecting the tooth supporting structures and ultimately leading to tooth loss when untreated. According to the WHO the prevalence of severe chronic periodontitis (CP) varies worldwide from 10 to 15% in adult populations whereas complete edentulism (no natural teeth) varies from 10 to 35% among countries depending on national income [3]. The most common forms of periodontal diseases are plaque-induced gingivitis and CP. Gingivitis is defined as an inflammation of the gingiva induced by bacteria located at the gingival margin. The host response to similar plaque levels varies significantly among patients with gingivitis and bacteria associated with disease progression are also present in health. Gingivitis is reversible upon removal of the etiologic biofilm but when untreated it progresses to CP in certain individuals. Several lines of evidence demonstrated that in some individuals gingivitis never progresses to CP, regardless of periodontal care [4,5]. CP commonly develops in the fourth decade of life, with highest prevalence in senior individuals. CP is characterized by extension of gingival inflammation to the alveolar bone, connective tissue degradation and net loss of tooth attachment to periodontium.
The main etiologic factor for these conditions is the bacterial biofilm. Although bacteria are required for disease to occur they are not sufficient. Several host and environmental factors such as genetic predisposition, lifestyle and oral hygiene, systemic conditions and stress contribute to onset and progression of CP [6] (Fig. 1B). Indeed certain pathogenic bacteria in subgingival biofilms produce specific virulence factors that facilitate tissue penetration, immune evasion and cause direct damage to periodontal tissues [7]. However, current evidence suggests that host factors constitute the major driver of periodontal tissue degradation in CP. These factors include over-expression of inflammatory cytokines, proteolytic enzymes, increased oxidative stress and failure to resolve inflammation [8]. The onset of CP in a systemic predisposing context has been extensively studied since immunity was found to be an essential contributor to both initiation and progression of periodontal destruction [9]. It became clear that both genetic and epigenetic factors contribute to the clinical phenotype seen in CP. Genetic traits such as specific polymorphisms in the interleukin (IL)-1 gene cluster and HLA-DR4 antigens impact the potency of immunity in an individual thus setting a threshold for severity of CP [10]. The relative contribution of common immune deregulation due to genetic factors to the links between CP and systemic conditions such as DM is incompletely understood. Therapeutic success depends on our understanding of spatial and temporal relationships between local and systemic events in onset of DM and related complications.
2. Pathogenic Mechanisms in DM and CP
2.1. Endocrine and Metabolic Deregulation in DM
Blood glucose levels are primarily maintained through a tightly regulated balance between glucose output by liver physiologically stimulated by glucagon and suppressed by INS, and glucose uptake by muscle physiologically stimulated by INS. The secretion of INS is increased when blood glucose levels exceeds 5.5 mmol/l and is reduced when they fall bellow 4.5 mmol/l. In adipose tissue INS stimulates the uptake and storage of fatty acids as triglycerides (TG) and inhibits the lipolysis of stored TG. Circulating INS has a half-live of 3-5 minutes. After being secreted into the portal circulation 60% of INS is cleared by the liver through its first-pass metabolism and 35-40% by the kidney. The brain has its own regulatory mechanisms of ensuring sufficient glucose levels at all times. The two main gluconeogenetic organs, kidney and liver maintain adequate blood glucose levels in prolonged fasting periods and during high-intensity exercise [11]. Furthermore, catecholamines, glucocorticoids, thyroid and growth hormones (GH) increase blood glucose levels mainly through stimulation of glycogenolysis and gluconeogenesis (catecholamines, glucocorticoids, GH), transcriptional regulation of genes involved in glucose homeostasis (glucocorticoids, thyroid hormones) and inhibition of INS secretion (GH) and action (glucocorticoids).
In 90% of T1DM cases reduction in islet mass is immune-mediated, patients having circulating antibodies to islet cells (ICA), INS (IAA), glutamic acid decarboxylase (GAD65) or tyrosine phosphatases (IA-2 and IA-2β) at the time of diagnosis and in 10% of the cases it is idiopathic (type 1B) with no evidence of autoimmunity leading to death of β-cells. The autoimmune response seen in T1DM results from a failure of self-tolerance in T-cells. Insulitis lesions contain a predominant CD8+ T cell population combined with macrophages (MΦ) (CD68+), CD4+ T cells, B lymphocytes (CD20+) and plasma cells (CD138+) [12]. The finding that IAA concentration correlates with the rate of progression to T1DM in children followed from birth and similar findings in studies on rodent models of T1DM suggest that proinsulin is a key autoantigen in the disease [13]. Further, the reduced pancreatic size preceding the onset of T1DM in the presence of autoantibodies and absence of insulitis suggests that multiple mechanisms lead to selective loss of β cells in the pathogenesis of T1DM [14].
T2DM is characterized by a peripheral IR, which leads to hyperinsulinemia and eventually to pancreatic β-cell exhaustion and apoptotic death. Impaired glucose tolerance (IGT) and impaired fasting plasma glucose (IFG) refer to intermediate metabolic stages between euglycemia and T2DM. Patients with IGT have normal blood glucose levels most of the time but become hyperglycemic after large glucose loads. On the other hand, patients with IFG have increased fasting blood glucose levels but are usually euglycemic after food intake. The pathophysiology of both IGT and IFG is related to some degree of primary IR. IGT and IFG are not clinical entities on their own but rather represent risk factors for development of T2DM. The exact mechanisms leading to development of IR are not fully understood. Activation of inflammatory pathways systemically or in adipose and other peripheral tissues has been correlated with IR [15]. There is increasing evidence that endoplasmic reticulum (ER) functions expand beyond post-translational protein processing into metabolic regulatory actions being considered a central nutrient sensor. Metabolic imbalances eventually induce ER stress that results in both inhibition of INS signaling and activation of pro-inflammatory pathways [16].
In both types of DM hyperglycemia is directly related to pancreatic dysfunction and ultimately leads to macro- and microvascular complications including atherosclerosis, ischemic heart disease, nephropathy, retinopathy, neuropathy and periodontal disease. Uncontrolled hyperglycemia is central to development of angiopathy through several biochemical and molecular mechanisms manifested in both intra- and extracellular compartments. The four major pathways through which hyperglycemia alters cellular physiology and extracellular matrix structure are the polyol pathway, the hexosamine pathway, activation of PKC, and advanced glycation end product (AGE) formation. The first three pathways generally alter cellular function by acting directly on intracellular pathways while AGE directly impact the extracellular matrix (ECM) quality and indirectly the normal cell function through specific receptors for AGE (RAGE). Increasing evidence suggests that excessive production of reactive oxygen species (ROS) by the mitochondrial electron transport chain in chronic hyperglycemia is the trigger point of glucose-induced metabolic alterations by inhibition of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) [17].
Overproduction of ROS by the mitochondrial electron transport chain was proposed as a unifying mechanism for diabetic microvascular complications as it relates to all metabolic changes that occur in DM [18]. The breakdown of glucose through glycolysis generates nicotinamide adenine dinucleotide (NADH) and flavin adenine dinucleotide (FADH2) to provide the energy necessary for producing adenosine triposphate (ATP) through oxidative phosphorylation by the mitochondrial electron transport chain. The latter uses the difference in electric potential at the inner mitochondrial membrane to generate high-energy bonds in ATP. High mitochondrial membrane potential due to intracellular excess of glucose leads to overproduction of ROS by increased half-life of the superoxide (O2−)-producing electron transport systems. High levels of O2− and consumption of NAD+ inhibit GAPDH resulting in accumulation of upstream intermediates in glycolysis that will be processed through pathways of glucose over-utilization such as polyol, hexozamine, de novo synthesis of diacylglycerol (DAG), a cofactor of PKC, and formation of AGEs from accumulating trioses (Fig. 2). Furthermore, ROS increase local production of TGFβ1, fibronectin and plasminogen activator inhibitor (PAI) 1 in renal cells playing a critical role in excessive ECM deposition [19].
Fig. (2).
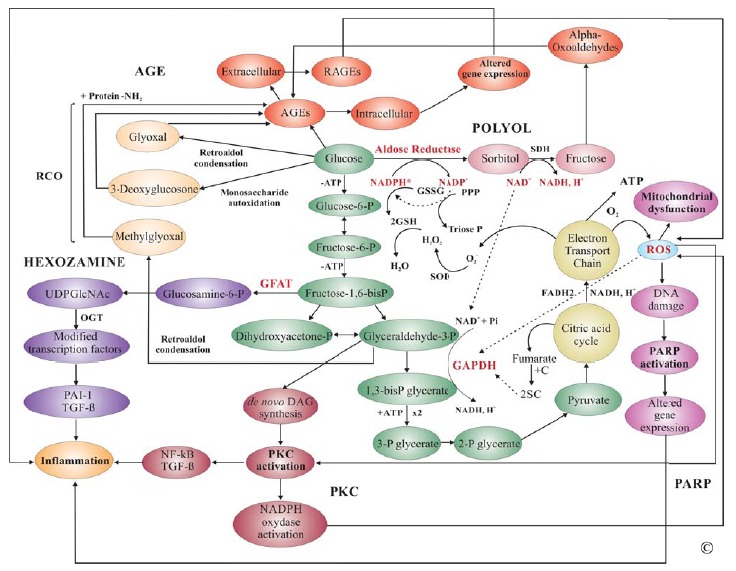
Biochemical Mechanisms Underlying Diabetic Complications. Degradation of glucose through glycolysis occurs in the cytoplasm and mitochondria of most eukaryotic cells. Pyruvate, the end product of glycolysis, enters the citric acid cycle (Krebs) that is coupled with the mitochondrial electron transport chain to produce high-energy bonds in ATP for use in cellular activities (green). Deregulation of glycolysis by excess intracellular glucose mainly resulting in overproduction of ROS leads to inhibition of GAPDH involved in a rate- limiting step of glycolysis. Subsequent accumulation of metabolites upstream of glyceraldehyde-3-phosphate leads to activation of polyol (pink), hexozamine (purple), AGE (orange) and PKC (red) pathways ultimately inducing a pro-inflammatory phenotype. Abbreviations: AGE, advanced glycation end product; SDH, Sorbitol dehydrogenase; RCO, reactive carbonyl compounds; GSSG, oxidized glutathione; GSH, reduced glutathione; P, phosphate; PPP, pyrophosphate; SOD, superoxide dismutase; GFAT, Glutamine fructose-6- phosphate amidotransferase; SOD, superoxide dismutase; O2-, superoxide anion; UDPGlcNAc, UDP-N-acetylglucosamine; OGT, O-linked GlcNAc transferase; PKC, protein kinase C; PARP, poly(ADP-ribose) polymerase; NF-kB, nuclear factor kappa B; TGFβ, transforming growth factor β; ROS, reactive oxygen species; C, cysteine; 2SC, S-(2succinyl)cysteine (2SC).
2.2. Diabetes-Associated Systemic Inflammation
A chronic low-grade inflammation is characteristic to the onset of T2DM in obese patients. Studies in specific MΦ knockout mice have shown that obesity does not correlate to IR in the absence of local inflammation triggered by resident MΦ [20,21]. The characterization of adipose tissue MΦ (ATM) and their relationship to adipocytes led to a better understanding of inflammatory mechanisms in T2DM. While bone marrow-derived MΦ make up ~10% of total adipose tissue in lean individuals they represent 40% of it in overweight individuals [22]. Recruitment of MΦ into obese adipose tissue seems to be mediated through secretion of MCP-1 by adipocytes in the context of high-fat diet [23,24]. It is now clear that in obesity ATM undergo a M1 type activation leading to cytokine (tumor necrosis factor alpha-TNFα, IL-6 and IL-1β)-mediated paracrine induction of inflammation and IR in INS-sensitive cells mainly through activation of JNK, inhibitor of κB kinase (IKK) β and interferon-regulatory factor (IRF) family members. Moreover, deregulated secretion of adipokines (PAI-1-increased) and adipose derived hormones (resistin-increased; adiponectin-decreased) in visceral fat adipocytes favors an inflammatory and pro-coagulant state [25,26].
Activation of ATM occurs mainly as a result of endoplasmic reticulum (ER) stress in response to fatty acids, excess of nutrients, improperly folded proteins, microhypoxia and lipopolysaccharide (LPS). ER stress in response to extracellular nutrients is central to inflammatory basis of IR and T2DM through three main ER membrane-bound proteins involved in the unfolded protein response (UPR): RNA-activated kinase (PKR)-like ER kinase (PERK), activating transcription factor-6 (ATF6) and inositol requiring enzyme 1 (IRE1). All three pathways enhance local inflammation and IR through activation of NF-κB and inhibition of INS receptor substrates (IRS1 and IRS2) by phosphorylation, respectively [27]. Moreover, several lymphocytes subtypes accumulate in obese adipose tissue and modify the MΦ behavior to promote IR. LPS, saturated and to a less extend polyunsaturated fatty acids activate ATM to a M1 phenotype via TLR4 while Ω-3 fatty acids are anti-inflammatory mediating activation of ATM to a M2 phenotype. Similarly, activated Kupffer cells induce IR in liver although hepatocytes are less dependent on INS for glucose uptake. MCP-1 knockout mice are protected from high-fat-induced hepatosteatosis indicating a role of monocytic cells in obesity-related liver dysfunction [24] (Fig. 3).
Fig. (3).
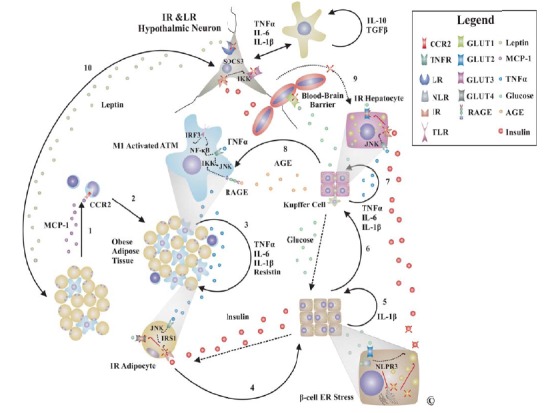
Inflammation in Pathogenesis of T2DM. Adipose tissue production of chemokines is considered one of the earliest events in pathogenesis of T2DM (1). Subsequent recruitment of mononuclear cells predominantly monocytes in adipose tissue (2) generates a local pro-inflammatory state (3) inducing IR in adipocytes that contributes to hyperglycemia and compensatory hyperinsulinemia ultimately favoring NLPR3 inflammasome activation and pancreatic stress (4,5). Pancreatic insufficiency generates chronic hypoinsulinemia that generally characterizes advanced stage T2DM (5, crossed INS). In response to hypoinsulinemia and activation of Kupffer cells with subsequent inflammatory-induced hepatocyte IR the liver releases massive amounts of glucose into the circulation (6,7). The resulting chronic hyperglycemia leads to AGE formation that further activates immune cells to a pro-inflammatory phenotype closing a negative self-amplifying vicious circle (8). IR and leptin resistance (LR) in hypothalamic neurons leads to loss of vagal inhibition of hepatic gluconeogenesis and increased appetite (9,10). Glial cells may contribute to LR during sustained low-grade inflammation through synthesis of cytokines for survival (IL-10) and inflammation (TNFα, IL-1β, IL-6) although it is still controversial. High-fat diet may also induce apoptosis of hypothalamic neurons through ER stress mediated mechanisms. Abbreviations: CCR2, chemokine C-C motif receptor; TNFR, TNF receptor; LR, leptin receptor; RAGE, receptor for AGE; TLR, Toll-like receptor; NLR, NOD-like receptor; IR, INS receptor; GLUT, glucose transporter; MCP-1, monocyte chemotactic protein-1 = CCL2; IRS1, INS receptor substrate 1; SOCS, Suppressor of cytokine signaling.
Several negative feedback loops to repress the M1 phenotype or mediate the switch to a M2 phenotype have recently been described including intracellular and intercellular mechanisms. The three main intracellular loops are IκBα-mediated κB pathway attenuation, activating transcription factor-3 (ATF3)-mediated pro-inflam-matory gene suppression and an orphan nuclear receptor Nurr1 (NR4A2)-mediated turnover of p65 component of NF-κB. Intercellular pathways are IL-10-mediated suppression of TNFα-induced IR, TAM group of receptor tyrosine kinases-mediated inhibition of TLR signaling and adiponectin-mediated suppression of MΦ LPS-induced responses and stimulation of IL-10 and IL-1 receptor antagonist production. On the other hand hyperglycemia as a result of either IR or β-cells destruction activates pro-inflammatory pathways through glycolysis-derived mediators and nutrient sensing mechanisms. ER stress-activated PERK and IRE1 pathways mediate apoptosis in several cell types including pancreatic β-cells through activation of pro-caspases [27]. TXNIP24- mediated NLRP3 inflammasome activation by excess glucose results in production of inflammatory mediators by pancreatic MΦ and apoptosis of β-cells leading to hypoinsulinemia. This closes a vicious cycle of inflammation-IR-hyperglycemia.
2.3. Inflammatory Pathogenesis of CP
It is generally accepted that periodontal pathogens are required but not sufficient to induce loss of tooth attachment but rather that several host and environmental factors dramatically modify the expression of disease [28]. Progression of CP results from a failure of the immune system to clear infectious agents and to restore periodontal homeostasis [29]. Gingivitis, the initial reversible stage of CP is associated with inflammation of gingival tissues without detectable evidence of attachment loss. Histologically, gingivitis consists of a continuum of progressing inflammatory lesions associated with vascular changes and a cellular infiltrate of predominantly lymphocytes and polymorphonuclear neutrophils. The established and advanced lesions characterized by a local influx of plasma cells and connective tissue degradation represent chronic stages of gingivitis and the stage of transition to CP in susceptible individuals. Although in some individuals gingivitis never progresses to CP, existing evidence suggests that gingivitis always precedes CP, and more importantly it represents a risk factor for periodontal attachment loss [30]. CP lesions are characterized by complex myelo-lymphoid cell infiltrates associated with irreversible loss of alveolar bone. Host-derived factors including chemokines, cytokines and matrix-degrading enzymes (metalloproteases) contribute to tissue destruction and disease progression [31] (Fig. 4). Mounting evidence supports the major role of host responses conditioned by predisposing genetic factors as being the major determinants in pathogenesis of CP [32].
Fig. (4).
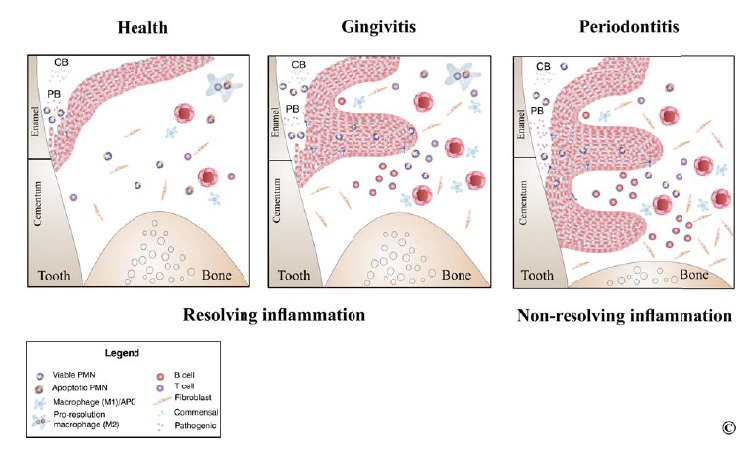
Inflammation in pathogenesis of periodontitis. In health, persistent low-abundance commensal bacteria in the subgingival biofilm trigger neutrophil recruitment to the periodontium and gingival crevicular fluid. In the gingival crevice, neutrophils form a “wall” between host tissues and bacteria colonizing subgingival areas contributing to control of biofilm pathogenicity. In gingivitis, neutrophils accumulate in higher numbers in the tissue and crevicular fluid due to increased biofilm mass, particularly in early stages of gingivitis. Epithelial and vascular proliferation characteristic to gingivitis are not associated with loss of tooth attachment to bony housing. In both health and gingivitis inflammation, resolution leads to restoration of homeostasis once neutrophils and tissue debris have been cleared by pro-resolution macrophages. In CP, non-resolved inflammation is characterized by a predominant lymphocyte infiltrate, pro-inflammatory cytokines and matrix degrading enzymes.
Although it was initially considered primarily an infectious disease CP results from a combination of specific bacterial colonization with subsequent formation of particular microniches as triggering factor, and local dysfunctional host immune responses to these pathogens. Several oral microorganisms including Candida albicans, Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans induce local synthesis of chemokines such as MCP-1 and IL-8 by endothelial cells and monocytes resulting in accumulation of mononuclear cells [33]. The inability of polymorphonuclear neutrophils and MΦ to phagocytose and neutralize pathogens results in the activation of an adaptive immune response accompanied by the production of interferon gamma (IFNγ), among other mediators. Secretion of CCL2 results in a significant influx of classical monocytes (pro-inflammatory), immature dendritic cells and T-helper (Th) cells to the site of inflammation. These newly recruited macrophages will polarize to an M1 phenotype (pro-inflammatory) in the context of increasing levels of pro-inflam-matory mediators. Dendritic cells and M1 MΦ produce IL-12, a cytokine critical for the initiation of adaptive immunity, through stimulation Th cell differentiation. In response to lipopolysaccharide, TNFα and IFNγ activated M1 macrophages produce IL-23, a cytokine that stimulates expansion of Th17 lymphocytes. The latter produce IL-17, a potent pro-inflammatory cytokine that stimulates polymorphonuclear neutrophil recruitment and activation, IL-1 β, IL-6, TNF, matrix metalloproteinases (MMPs) and RANKL, generating an inflammation amplification loop and resolution failure.
RANKL is an essential pro-osteclastic mediator, which, together with its decoy receptor osteoprotegerin, is critical for bone resorption-formation coupling. It is well established that pro-inflammatory cytokines are expressed at high levels in gingival tissues from patients with CP [34,35]. The significant impact these mediators have on tissue destruction was demonstrated in rodent and primate models of CP in which connective tissue attachment and alveolar bone loss was reduced through the use of inhibitors or knockout of genes encoding IL-1β, IL-6 and TNFα [36,37]. The major cellular source of gingival pro-inflammatory cytokines, particularly IL-1 β, IL-8 and TNFα, in severe to advanced CP, is the MΦ [38]. Several studies have found significantly increased levels of IL-12, IL-23, IL-23 receptor and IFNγ in periodontitis lesions [39]. A recent study has further suggested that a predominantly Th17-polarized cellular infiltrate in CP could be driven by IL-23-producing M1 macrophages [40]. However, it is widely accepted that inhibition of the host response may lead to a higher prevalence of more severe forms of CP. Polymorphonuclear neutrophil depletion, or functional impairments associated with leukocyte adhesion deficiency, Chediak-Higashi syndrome, Papillon-Lefèvre syndrome and AIDS result in enhanced alveolar bone loss, possibly through sustained M1 MΦ-governed inflammatory infiltrates and failure to resolve inflammation. These findings support the paradigm of an efficient innate immune response to subgingival biofilms being critical for limiting inflammation, allowing active resolution and coupled bone remodeling.
By contrast, the M2 (anti-inflammatory and pro-resolution) macrophage-derived anti-inflammatory cytokine IL-10, although widely expressed in inflamed periodontal tissues, is associated with decreased severity of periodontitis [41,42]. Its protective role was also observed in IL-10 knockout mice that are more susceptible to Porphyromonas gingivalis - induced alveolar bone loss [43]. Similarly, the concentration of IL-4 was found to decrease in the gingival crevicular fluid of patients with CP compared with controls [44]. Adoptive transfer of Th2 cells to nude rats attenuated the severity of periodontitis, and the predominance of Th2 cells in a mouse model of Porphyromonas gingivalis - induced periodontitis resulted in minimal lesions [45]. Transforming growth factor beta (TGFβ) may also play an important role in limiting periodontal tissue destruction through its regulatory actions on cell growth, differentiation, matrix production and immunosuppression, inhibiting pro-inflammatory factors. In active periodontal lesions, TGFβ is negatively correlated with RANKL levels, supporting its protective role against tissue breakdown [46,47]. It is still unclear whether a highly specific virulence of periodontal pathogens such as Porphyromonas gingivalis, Treponema denticola, Tannerella forsythia or Aggregatibacter actinomycetemcomitans, or an a-priori host-immune susceptibility to dysfunctional responses to these bacteria, is responsible for inefficient pathogen clearance and the failure to resolve periodontal inflammation. Although the initiation of local periodontal immune dysfunction that results in disease progression is unknown, mounting evidence suggests that periodontal pathogens have important immunomodulatory functions.
The potential of certain periodontal pathogens to stimulate M1 or M2 MΦ environments seems to depend on host susceptibility rather than their virulence. Existing data suggest that polymorphisms in genes transcribing IL-1β, IL-6, IL-10, Fc-gamma receptor, vitamin D receptor and CD14 may be associated with increased susceptibility to CP [48,49]. Regardless of the relative contribution of host predisposition and bacterial challenge, it appears that progression of tissue destruction is caused by a combination of several factors, including the presence of pathogenic bacteria, high levels of pro-inflammatory cytokines and prostaglandins, the production and activation of MMPs and RANKL, and the relatively low levels of IL-10, TGFβ, tissue inhibitors of MMPs and osteoprotegerin [37]. Disruption and reduction of subgingival biofilms and calculus may not be sufficient to arrest disease progression in the long term, although immediate maintainable results are obvious therapeutic. Modulation of host immune responses to pathogenic subgingival biofilms may prevent recurrence of disease or slow progression due to unresolved periodontal inflammation.
2.4. Bidirectional Links Between DM and CP
Multiple lines of evidence from in vitro, animal and human studies indicate that the links between DM and CP are bidirectional and likely centered on deregulated inflammation manifested systemically. Two major longitudinal studies with a median follow up of 6 and 11 years investigated the impact of CP on development cardiorenal complications in patients with T1DM and T2DM in Scandinavia and US (Pima Indians). The first study found that in patients with T1DM and severe CP compared with those with no or mild CP there was a higher incidence of proteinuria and cardiovascular complications, including angina, intermittent claudication, transient ischemic attack, MI, and stroke [50]. The second study found increased adjusted relative risk (3.2, 95% CI 1.1-9.3) of cardiorenal mortality in patients with severe CP compared with those who had no, mild, or moderate CP [51]. In line with these findings CP and edentulism predicted incident microalbuminuria and end stage renal disease in a sample of 529 patients with T2DM [52]. Observations from these studies support previous findings of increased risk of poor glycemic control at follow-up in patients with severe CP and DM. Similarly, two major studies on the risk of developing T2DM in patients with CP were carried during the first National Health and Nutrition Examination Survey (NHANES I) and its Epidemiological Follow-Up study (NHEFS), and in a Japanese population, which included 9296 and 5848 individuals respectively. The NHANES I/NHEFS found adjusted OR between 1.5-2.08 for incident DM in patients with high periodontal index scores or tooth loss at baseline. On the other hand, the Japanese study found no association between CP and incident DM despite statistically significant positive associations in the unadjusted analyses [53-55]. Results from these reports should be interpreted with caution because the measure of exposure (CP) has been imprecise and the lack of laboratory testing to exclude undiagnosed DM at baseline indicate a possible reverse causality.
In addition to a potential role of CP in onset of T2DM multiple studies found that treatment of CP particularly scaling and root planing (SRP), improves metabolic control by a significant reduction in HbA1c and inflammatory mediators (CRP, TNFα, IL-6, and fibrinogen), and increase in levels of adiponectin [56-58]. Data from 8 randomized controlled clinical trials and 2 controlled clinical trials on the effects of SRP on glycemic control, which included a total of 439 patients were assessed in two recent systematic reviews [59]. The sample sizes varied from 30 and 193 patients and the follow up range was 3-18 months. Both reviews included meta-analyses on 145 and 244 study participants and found a significant post-periodontal therapy HbA1c reduction of 0.40% (95% CI -0.77% to -0.04% and -0.78 to -0.01 respectively). Only 2 studies were included for both reviews but inclusion criteria were slightly different and more strict than a previous meta-analysis that found a 0.46% improvement in HbA1c levels following periodontal therapy [60,61]. This improvement in glycemic control may be significant considering that each percent reduction in HbA1c in patients with DM results in a 35% reduction in the risk for macrovascular complications and that HbA1c reduction by 0.2% in general population is associated with 10% reduction in mortality [62]. However, larger RCTs and a full appreciation of the role of antibiotics for CP treatment are needed to better understand the impact of periodontal therapy on glycemic control in patients with DM. Since inflammation can promote IR and favor poor metabolic control, the hypothesis that treatment of CP and the associated inflammation can improve glycemic control in DM seems biologically plausible.
Multiple investigations attempted to mechanistically link DM and CP focusing on potential explanations for the increased prevalence and severity of CP in patients with DM. Three major theories have emerged from this research: (1) the excessive inflammatory response to subgingival biofilms, (2) the uncoupling of bone resorption and apposition, and (3) the impact of AGEs on cellular and extracellular compartments in periodontal tissues. Further, CP may influence the course of DM likely through sustained systemic inflammation and bacteremia associated with systemic dissemination of periodontal pathogens that may contribute to insulin resistance, obesity and atheroma formation [63]. Central to relationships between DM and CP seems to be systemic inflammation measured as elevation in circulating pro-inflammatory markers. Several lines of evidence suggest that CP in otherwise healthy individuals is associated with increased levels of pro-inflammatory and pro-thrombotic mediators in serum and that periodontal therapy results in long-term reduction in such markers (CRP, TNFα, and PAI-1) and improvement of endothelial function (decreased the levels of soluble E-selectin) [64-66]. A dose-response relationship between CP severity and plasma levels of TNFα, a cytokine that promotes IR, was found in patients with T2DM [67]. Further, monocyte hyper-responsi-veness may be reversed in patients with DM by SRP resulting in reduced monocyte-derived TNFα, hs-CRP, and soluble E-selectin [64].
DM impacts periodontal tissues in multiple ways including immunological dysfunctions, microvascular alterations, and changes in extracellular matrix [68]. Impaired neutrophil margination along gingival endothelium, altered chemotaxis and phagocytosis, and monocyte/MΦ hyper-responsiveness in patients with DM may explain the higher prevalence and severity of CP in these individuals [69,70]. Since reduced neutrophil chemotaxis significantly contributes to pathogenesis of aggressive periodontitis and worsens phenotype in CP, and is also seen in patients with T1DM a plausible hypothesis that common HLA-DR antigens are involved in neutrophil dysfunction associated with periodontitis and hyperglycemia has been tested [71,72]. However, a study on 41 patients with T1DM has found reduced neutrophil chemotaxis with no correlation to HLA alleles suggesting a phenotypic modification of circulating neutrophils by metabolic imbalances characteristic to DM rather than HLA-mediated common predisposition altered neutrophil migration in DM and CP [73]. Although neutrophils from patients with DM appear to be primed for hyper-responsive superoxide production bacterial killing is often impaired. These alterations combined with increased expression of leukocyte adhesion molecules may lead to ectopic release of neutrophil superoxide, particularly at sites with high physiological neutrophil trafficking such as biofilm-bearing tissues. Increased leukocyte adhesion molecule expression and gingival microvascular permeability in DM in the absence of CP suggests an immune-vascular priming that predisposes to CP [74,75].
The imbalance between the receptor activator for nuclear factor κ B ligand (RANKL) and osteoprotegerin (OPG) is thought to be central to the inability of patients with DM to regenerate alveolar bone at sites with progressing CP. Inflammation-mediated uncoupling of bone formation and resorption is associated with hyperglycemia in the context of reduced osteoblast proliferation, differentiation, and collagen production that could be reversed with insulin treatment [76]. Uncontrolled DM may favor bone resorption by reducing periodontal OPG/RANKL ratio through down-regulation of OPG and through induction of apoptosis in bone lining cells resulting in reduction in bone repair following resorption [77,78]. Osteoblasts regulate osteoclast differentiation and activation through RANKL and its high-affinity decoy receptor OPG. It is widely accepted that the balance between RANKL and OPG determines the differentiation and the activity of osteoclasts [79]. Although osteoblasts are the major producers of RANKL, other cells can express this cytokine, including fibroblasts and activated T-cells. Further, pro-inflammatory cytokines such as IL-1β and TNFα also mediate bone uncoupling through osteoclast differentiation and activation at sites with CP. Alveolar bone resorption was reduced in experimental periodontitis by the use of IL-1β and TNFα antagonists, and in IL-1β or TNFα knockout mice [36,80]. DM seems to enhance TNFα-mediated apoptosis of matrix forming cells in CP contributing to impaired healing [81]. Therefore, diabetes-mediated altered immune responses to subgingival biofilms may be responsible for more severe and rapid bone loss in patients with DM through enhanced resorption and diminished formation.
The altered immune responses to subgingival biofilms are in part explained by AGE-mediated priming and extracellular matrix modification. The observation of high levels of albumin AGE in gingival tissues of diabetic mice support the hypothesis that AGE mediate activation of inflammatory pathways in periodontium through increasing susceptibility to CP. Immune cell priming for hyper-active responses likely occurs through activation of the receptor for AGE (RAGE) and subsequent pro-inflammatory cytokine production [82]. Nonetheless, AGE deposition in gingival tissues impairs fibroblast function by reducing their migratory and attaching properties and results in cross-linking matrix proteins such as collagen type I and fibronectin [83,84]. In support of these findings, a cross-sectional study including 69 patients with T2DM and CP AGE has found a significant association between serum AGE and severity of CP [85]. Zizzi et al. investigated the gingival expression of AGE in patients with T1DM or T2DM diagnosed with CP. A total of 64 subjects were included in this study: 16 systemically and periodontally healthy individuals, and 48 patients with generalized severe CP (16 with T1DM, 16 with T2DM, and 16 systemically healthy). It was found that patients with DM (both type I and II) have significantly higher levels of gingival AGE compared to healthy controls and that there is a positive statistically significant correlation between gingival AGE and length of time affected by DM [86]. No correlation was found between HbA1c, lipid profile, body mass index, or age and gingival AGE.
Transient bacteremia associated with CP, which could contribute to initiation and progression of atheroma plaques in arterial walls may also impact on the course of T2DM and metabolic syndrome. Subgingival persistence of Porphyromonas gingivalis more frequently in patients with T2DM having higher HbA1c levels after periodontal therapy indicates that CP may impact glycemic control in these patients [87]. This mechanism involves direct infectious pathogenicity of periodontal bacteria and indirect induction of systemic inflammation that correlates with atherosclerosis and cardiovascular diseases [88-90]. In support of this hypothesis periodontal pathogens Porphyromonas gingivalis, Prevotella intermedia, Bacteroides forsythus, and Actinobacillus aggregatibacter were found in significant quantities in atheromatous plaques suggesting colonization of disseminated oral bacteria in systemic vasculature [91,92]. One large study named the Oral Infections and Vascular Disease Epidemiology Study (INVEST) that investigated the relationship between oral microbiota and subclinical atherosclerosis, has analyzed 657 dentate subjects with a mean age of 69 years and no history of stroke of MI. Investigators found a positive correlation between the 4 bacterial species and carotid artery intima-media thickness (IMT) [93]. The same group has previously found a significant positive correlation between tooth loss, mostly due to periodontitis, and carotid artery atheroma prevalence in the same population [94]. More recently, the INVEST group reported a direct positive correlation between levels of subgingival bacteria and prevalence of hypertension [95].
3. Therapeutic Management
3.1. Therapeutic Management of DM
Control of blood glucose levels is the primary aim of DM management. The current therapeutic options for control of blood glucose are restricted diet, administration of INS, oral antidiabetic drugs grouped into INS secretagogues (sulfonylureas, meglitinides, d-phenylalanine derivatives and glucagon-like peptide-1 (GLP-1) receptor agonists), biguanidines, thiazolidinediones and α-glucosidase inhibitors [96]. Two newer classes of DM medications with differing actions on glycemia and weight gain include sodium-glucose cotransporter 2 (SGLT2) inhibitors that increase urinary glucose excretion and inhibitors of dipeptidyl peptidase 4 (DPP-4) that reduce both glucagon and glucose blood levels [97,98]. A diet with low glycemic index improves glycemic control and reduces the risk of hypoglycemic events [99]. Additionally, exercise combined with controlled diet reduces the incidence of DM in high risk individuals (IGT and IRS) and improves systolic and diastolic blood pressure (BP) levels [100]. Continuous subcutaneous INS infusion is better at controlling blood glucose levels and preventing severe hypoglycemia compared to multiple INS injections [101]. Long acting INS has similar benefits on overall glycemic control as intermediate acting INS but offers better control of nocturnal blood glucose levels. A presumed mitogenic action of long acting INS warrants caution in prescription until further evidence against this possibility is available [102].
Of all oral antidiabetic drugs sulfonylureas, mainly second generation (glyburide, glipizide, glimepiride) and biguanidines including metformin are the treatment of choice for T2DM in addition to controlled diet. INS secretagogues act by binding to β-cells sulfonylurea receptors (SUR) associated with the inward rectifier ATP-sensitive K+ channel inhibiting the efflux of K+ ions. This results in membrane depolarization and opening of voltage-gated calcium (Ca2+) channels, influx of Ca2+ and subsequent release of preformed INS. Biguanidines act independent of β-cell function by reducing hepatic and renal gluconeogenesis, slowing glucose absorption from gastrointestinal tract, directly stimulating glycolysis in tissues and reducing the plasma glucagon levels. Thiazolidinediones decrease IR mainly by regulating genes involved in glucose and lipid metabolism, and adipocyte differentiation being ligands of peroxisome proliferator-activated receptor-gamma (PPAR-γ), part of the steroid and thyroid superfamily of nuclear receptors. Alpha-glucosidase inhibitors are competitive inhibitors of α-glucosidases in the small intestine reducing the postprandial digestion and absorption of starch and disaccharides.
Several clinical trials have been conducted with patients having either T1DM or T2DM to assess the benefits of glycemic control in DM. The Diabetes Control and Complications Trial (DCCT) was a randomized long-term prospective study involving 1441 patients with T1DM aged 13-39 years observed for 6.5 years between 1983 and 1989 [103]. Patients were divided into conventional therapy as defined by one or two daily INS injections with self-monitoring of urine and blood glucose without adjustment of INS dosage and intensive therapy as defined by three or more INS administrations daily with self-monitoring and adjustment of INS dosage accordingly. At the end of the study it was concluded that intensive INS therapy effectively controls BG, delays the onset and slows the progression of retinopathy, nephropathy and neuropathy in patients with T1DM. Based on these observations the DCCT research group introduced the concept of glycemic memory that includes the long-term benefits of significant periods of glycemic control.
Multiple clinical trials were similarly conducted for T2DM. The United Kingdom Prospective Diabetes Study (UKPDS) was a very large randomized prospective study including 3867 newly diagnosed T2DM patients aged 25-65 years observed over 10 years between 1977 and 1987 [104]. The average initial HbA1C levels
was 9.1%. A significant proportion of the patients were overweight and hypertensive. The aim of the study was to assess the impact of intensive glycemic control and of blood pressure (BP) control on risk of developing microvascular and macrovascular complications in T2DM. Treatment groups included control group on conventional therapy with controlled diet alone and separate groups on intensive therapy with either insulin or sulfonylureas (chlorpropamide, glyburide, or glipizide) alone. Metformin was added as a randomization option for patients responding unsatisfactorily to other therapies. A separate group included patients under tight control of BP with stepwise antihypertensive therapy including either an angiotensin-converting enzyme (ACE) inhibitor (captopril) or a β-blocker (atenolol). More drugs were added in the following stepwise sequence: a diuretic, slow-release nifedipine, methydopa, and prazosin to achieve tight BP control. At the end of the study the following were concluded: intensive therapy to reduce blood glucose reduces the risk of microvascular complications overall compared to conventional therapy; metformin alone significantly reduces the risk of macrovascular disease (myocardial infarction, stroke); tight BP control significantly reduced the risk of both microvascular and macrovascular complications with higher efficiency than reducing HbA1C levels to 7%.
A more recent double-blind randomized prospective clinical trial with a 2-by-2 factorial design called Nateglinide and Valsartan in IGT Outcomes Research (NAVIGATOR) included 9306 patients from 40 countries (average age 64 years) with IGT and established CVD or CV risk [105,106]. The study has assessed the impact of Nateglinide and Valsartan treatments in addition to lifestyle modification on DM incidence, DM related clinical parameters and CV events over 5 years. At the end of the study it was concluded that Nateglinide treatment three times daily with up to 60 mg/dose does not prevent CV events or DM onset while Valsartan treatment as single dose of up to 160 mg/day reduced the incidence of DM by 14% without significant impact on the rate of CV events. Other smaller studies including the Kumamoto study (110 patients, 6 years follow-up), the Steno-2 study (160 patients, 7.8 years follow-up) and the STOP-NIDDM (1429 patients, 3 years follow-up) have shown similar results to the DCCT and UKPDS studies indicating that control of blood glucose levels reduces the risk of developing and slows the progression of diabetic complications [107-109].
3.2. Therapeutic Management of CP
Conventional periodontal therapy for CP includes patient education on appropriate measures to reduce subgingival biofilms and to control modifiable risk factors for disease progression in addition to in-office SRP that focuses on removal of the etiologic biofilms and calculus. Further, therapy includes management of periodontal-systemic interrelationships by appropriate referral when indicated. In most cases this form of therapy allows for resolution of inflammation to occur and for periodontal tissues to heal [110]. However, disruption of subgingival biofilms is successful for only short periods of time because maturation of new pathogenic biofilms is favored by presence of periodontal pockets around teeth that cannot be adequately accessed through regular home care by patients [111]. Therefore maintenance therapy by SRP at regular 3-4 months intervals is indicated in cases of mild-to-moderate CP. The periodontal literature supports the concept of bacterial “critical mass” by assuming that the major goal of SRP is reduction in subgingival biofilm mass to a critical level that results in equilibrium between remaining bacteria and the host response [112]. At sites with severe loss of attachment that cannot predictably be managed by SRP due to poor access, surgical re-establishment of maintainable periodontal tissues generally follows SRP. If left untreated, CP results in pain, discomfort, tooth mobility and tooth loss, and may represent a risk factor for other chronic conditions such as DM and cardiovascular diseases by contributing to systemic inflammation and bacterial dissemination [66].
Since microbial irritants constitute a significant contributing factor to onset and progression of CP the use of antibiotics in conjunction with mechanical or surgical therapy is chosen for certain cases. This aims to reduce, eliminate or change the quality of microbial pathogens or to modify the host response to subgingival biofilms. Patients refractory to conventional therapy, suffering from aggressive forms of periodontitis or with medical conditions predisposing to periodontitis may benefit from systemic antibiotic therapy [113]. Since CP is associated with multiple potential periodontal pathogens i.e. Gram negative anaerobes, facultative anaerobes, Gram positive anaerobic cocci and rods antibiotics with broad spectrum of activity are usually chosen. In some cases, in order to broaden the spectrum of activity, combination drug therapies such as Amoxicillin and Metronidazole may be considered advantageous over single drug choices. Several studies demonstrated that in patients with advanced CP, systemic administration of Metronidazole plus Amoxicillin resulted in improvement in periodontal conditions, elimination/suppression of putative periodontal pathogens and reduction in the size of inflammatory lesions. The antibiotic regimen alone, however, was less effective than mechanical therapy with respect to clinical measures of inflammation and attachment levels [114,115]. Similar to systemic antibiotics, local delivery antimicrobial agents can be used as adjunct to SRP in isolated sites with CP. However, there is insufficient data to conclude that adjunctive use of such agents can either reduce the need for surgery or improve long-term tooth retention [116]. Therefore, clinical judgement is often used in selecting cases that may benefit from antimicrobials in addition to mechanical therapy.
In some instances patients with CP are refractory to conventional periodontal therapy and may be diagnosed with refractory CP [117]. Refractory CP patients have a recurring disease progression pattern and experience continuous loss of clinical attachment after treatment, which does not correlate with plaque levels, microbial burden, and treatment compliance [118]. Patients with refractory CP were demonstrated to lose a significantly higher number of teeth during periodontal maintenance when compared to non-refractory patients [119]. Management of disease in such patients is challenging and shows inconsistent results. Patients refractory to periodontal therapy or with recurrent disease seem to have distinct subgingival microbial profiles that may respond to additional antibiotic therapy [120]. However, in many cases subgingival bacteria in these patients develops antibiotic resistance particularly to β-lactam antibiotics [121]. Therefore, collection of subgingival microbial samples form sites with progressing disease, antibiotic-sensitivity testing and selection of an appropriate antibiotic regimen are indicated. In addition to atypical microbial profiles several lines of evidence suggest that patients with refractory periodontitis have a hyperactive neutrophil phenotype that may be contributing to increased oxidative stress in inflamed periodontal tissues, excess MMP release and failure to resolve inflammation [122]. Modulation of immune response to subgingival biofilms is desired to control pathogenicity, resolve inflammation and restore tissue homeostasis while limiting bacterial dissemination and maintaining low levels of inflammatory mediators that may impact systemic health.
While typical treatment protocols for CP aim at removing etiologic factors at regular intervals to prevent disease recurrence, immune modulation therapeutics show great promise in light of mounting evidence demonstrating altered inflammatory pathways in CP pathogenesis. As connective tissue degradation and irreversible bone resorption mark the onset of CP, regulating gingival enzymatic activity is one therapeutic target for immune modulation in long-term maintenance of tooth attachment levels. One class of such immune modulatory drugs targets tissue-degrading enzymes such as MMPs, which are abundant in periodontal tissues as a result of chronic un-resolved inflammation. Doxycycline, in addition to its antimicrobial ability, has demonstrated good anticollagenase activity even when sub-antimicrobial doses are used. In a randomized clinical trial on effects of sub-antimicrobial dose doxycycline (SDD) (20 mg x 2 times per day for 12 weeks) on gingival fluid collagenase and clinical attachment loss Golub et al found significant improvement in attachment levels associated with a decrease in collagenase activity [123]. Continuous drug therapy over the 12-week treatment period was needed to maintain and maximize improvement. In a large multi-centered, double-blind, randomized, placebo-controlled parallel group trial Canton et al assessed the efficacy and safety of SDD as an adjunct to SRP over a period of 9 months, in patients with CP. They found a modest but significant improvement in attachment levels compared to placebo without detrimental shifts in the normal periodontal flora or the acquisition of doxycycline or multiantibiotic resistance [124]. Similar findings were reported by Preshaw et al who conducted a multi-center doubleblind, randomized, placebo-controlled with parallel treatment groups that included 210 subjects [125]. To confirm the anti-MMP activity exerted by SDD, Emingil et al measured MMP-8 levels in gingival crevicular fluid along with clinical parameters for patients with CP under SDD therapy as adjunct to SRP for 3 months with a follow-up of 12 months [111]. Gingival MMP-8 levels were significantly reduced in the SDD group at 3 and 6 months compared to placebo but were insignificant compared to SRP alone at 9 and 12 months post-therapy. Altogether these findings support the use of SDD as adjunct to conventional periodontal therapy in select cases and over log periods of time to prevent relapse.
3.3. Management of Unresolved Inflammation in DM
Resolution of inflammation is a tightly regulated active process required for restoration of homeostasis after successful removal of altered-self or non-self. For resolution to occur pro-inflammatory pathways need to be switched off upon clearing local tissue debris by professional phagocytes. Neutrophils provide essential signals needed to initiate resolution processes. Following non-self recognition, uptake, neutralization and clearance, the neutrophil triggers self-destroying mechanisms in a non-flogistic manner by initiating apoptosis through sequential activation of caspases (3, 7, 8, 9), activation of calpains and ubiquitin- proteasome complexes [112-114]. This results in degradation of critical cytoplasmic proteins such as the ubiquitously present actin, and nuclear inter-nucleo-somal chromatin. Aged or dying neutrophils present on their surface phospholipids phosphatidycholine (PC), phosphatidylethanolamine (PE) and phosphatidylserine (PS), oxidized phospholipids and carbohydrates including fucose and N-acetyl-glucosamine [115]. Several pattern recognition molecules including thrombospondin 1, complement component C1q, mannose-binding lectin, surfactant proteins, pentraxins, and serum amyloid bind apoptotic cell surface markers and act as bridging molecules to enhance phagocytosis by macrophages [116,117]. Specific receptors on MΦ and dendritic cells including scavenger receptors A, B, CD36 and CD68, αvβ3 integrin, PS receptor and complement receptors (CR1, CR3, CR4) bind to bridging molecules resulting in activation for phagocytosis and clearance of dying cells from the tissue [118,119]. This physiological “start of the end” allows neutrophils to die silently and concomitantly send “find me” and “eat me” signals without releasing their toxic intracellular milieu into host tissues. In some instances a secondary apoptotic phenotype of neutrophils can be induced by persistence and increase in pro-inflammatory lipid mediators such as leukotriene B4. This late apoptotic phenotype characterized by selective leakage of intracellular content particularly DNA fragments is distinct from primary necrosis but it may lead to secondary necrosis in the absence of efficient resolving mechanisms [120].
Macrophages polarized to an anti-inflammatory and pro-resolution phenotype play critical roles in post-inflammatory healing and regeneration of damaged tissues. Compelling evidence showed that MΦ possess unique phenotypic plasticity and differentiation dynamics that change based on local microenvironmental signals. Pro-resolution macrophages particularly those stimulated by IL-4, IL-10, IL-13 and transforming growth factor beta (TGFβ) phagocytoses apoptotic cells and promote tissue remodeling and regeneration [123]. These MΦ produce low levels of pro-inflammatory cytokines (IL-1, IL-6, TNFα, IL-12, IL-23), high levels of anti-inflammatory cytokines (IL-10, IL-1 receptor antagonist), growth factors (TGFβ, VEGF, EGF) and chemokines CCL17, CCL22, CCL24 that bind to CCR3 and CCR4 promoting T-helper 2 cell responses and tissue repair/regeneration. In addition they produce arginase to metabolize arginine to ornithine and polyamines needed in the healing processes and up-regulate ferroportin while down-regulating H ferritin and hemeoxygenase, which favors enhanced release of iron, thus supporting cell proliferation. Nonetheless, they express high levels of scavenger, mannose and galactose receptors and of CD163 (the receptor for the hemoglobin-haptoglobin complexes) [124,125]. Upon clearing interstitial environment of redundant and extraneous cellular material, and stimulating healing of damaged tissues resolution macrophages drain via lymphatic vessels into the circulation to be cleared in the spleen [126,127]. Un-regulated MΦ polarization to a pro-resolution phenotype and induction of pro-inflammatory phenotypes are thought to be in part responsible for persistent inflammation associated with chronic conditions such as obesity, atherosclerosis and DM [128].
Obesity is associated with increased fat-tissue turnover and extensive adipocyte necrosis, which acts as a pro-inflammatory stimulus for ATM resulting in a progressive switch from pro-resolution to pro-inflammatory phenotype in these cells. Ultimately the obese adipose tissues throughout the body will sustain local and systemic inflammation, and IR [26,129]. The increased production of pro-thrombotic and pro-atherogenic factors by these MΦ will contribute to the cardiovascular risks identified in obesity-associated metabolic syndrome. Therefore, in addition to blood glucose normalizing agents therapy of DM and associated co-morbidities should address the un-regulated inflammatory responses to altered-self. Since recruitment of pro-inflammatory MΦ into adipose tissue and pancreatic islets contributes to IR and impaired INS secretion, chemokines and receptors involved in monocyte migration are potential therapeutic target for DM. Monocyte chemotactic protein 1 (MCP1, CCL2) is a chemokine involved in recruitment and tissue infiltration of MΦ in patients with T2DM favoring onset of IR [130-132]. CCX140-B, an orally available antagonist of MCP-1 receptor CCR2 was able to reduce HbA1C levels by 0.14% in patients with T2DM during a 4 week clinical trial [133]. The same antagonist reduced the number of pro-inflammatory adipose tissue macrophages, improved glycemic control and insulin sensitivity in diabetic mice [134]. These actions support a significant role of CCR2 in sustained adipose tissue inflammation mediated by MΦ as previously observed in animal studies [135].
NF-κB treatment strategy for DM emerged from compelling evidence that this pro-inflammatory pathway is central to pro-inflammatory M1 polarization of adipose tissues macrophages. TNFα and IL-1β are major cytokines involved in activation of NF-κB. Additionally, TNFα produced by pro-inflammatory adipose tissue MΦ demonstrated a significant role in induction of IR in rodents, which led to attempts to translate these findings in humans. Despite original conclusions from a limited number of underpowered clinical studies that TNFα does not have an important role in T2DM in humans, recent reports are in line with findings from animal studies [136]. One such study found that 6-month TNFα inhibition via entanercept in obese individuals without DM resulted in significant decrease in fasting plasma glucose and increase in adiponectin, probably reflecting improved INS sensitivity [137]. Additionally, TNFα inhibition in patients with rheumatoid arthritis results in improved INS sensitivity and blood glucose levels suggesting that TNFα may play an important role in pathogenesis of T2DM [138]. Similar results were found by antagonizing IL-1β with specific antibodies (gevokizumab or canakinumab) or anakinra, a recombinant human IL-1 receptor antagonist in pre-diabetic individuals and those with impaired glucose tolerance [139,140]. Authors found improved β-cell secretory function that may prevent onset of DM, improved INS sensitivity and glycemic control, and reduced systemic inflammatory markers. In support of a role of both IL-1 β and TNFα in pathogenesis of T2DM and glycemic control it was found that diacerein, which reduces the levels of both cytokines via unknown mechanisms, improved INS secretion and reduced HbA1C levels in drug-naive patients with T2DM [141]. Altogether, these findings support the two major pro-inflammatory cytokines TNFα and IL-1β as therapeutic targets for management of DM-associated inflammation in humans.
3.4. Management of Unresolved Inflammation in CP
Anti-cytokine therapy for management of inflammation in CP was studied in patients with rheumatoid arthritis and CP. To date, therapeutic blockade of TNFα was tested in three randomized controlled clinical trials that included patients with both rheumatoid arthritis patients and CP [142-144]. In the first study, infliximab prevented attachment loss and progressive bone loss although it tended to aggravate gingival inflammation [144]. In the second study, infliximab infusions reduced gingival crevicular fluid levels of TNFα, gingival inflammation and attachment loss in patients with rheumatoid arthritis patients compared with those untreated and with healthy controls [142]. The third study randomized 40 patients with moderately severe rheumatoid arthritis and severe CP to receive mechanical periodontal therapy or no therapy. All patients received disease-modifying anti-rheumatic drugs but half received anti-TNFα medication before randomization [143]. SRP alone or in conjunction with anti-TNFα therapy were associated with significant improvements in rheumatoid arthritis disease activity scores, serum TNFα and periodontal parameters although the post-therapy follow up 6 weeks was short. Altogether, these data indicate a potential improvement in periodontal condition when using anti-TNFα therapy in conjunction with mechanical debridement for CP. Therapeutic blockade of IL-1 receptor has only been investigated in animal models of periodontitis and showed significant reduction in local infiltration of inflammatory cells, reduced osteoclast activation and bone resorption [36,145]. Anakinra was proven to be only weakly effective in rheumatoid arthritis, despite experimental models suggesting that its anti-inflammatory and osteoclast inhibiting effects may improve systemic bone loss [146].
Several clinical trials have assessed the effects of adjunctive non-steroidal anti-inflammatory drugs (NSAIDs) delivered systemically or locally in addition to mechanical therapy [147-149]. Despite heterogeneity of data that emerged from these studies limited quantitative analyses tended to indicate a significant benefit for alveolar bone preservation and disease progression but not for clinical attachment levels when NSAIDs were associated with conventional periodontal therapy. This may be in part explained by inhibition of prostaglandin E2 (PGE2) inhibition, a lipid mediator that correlated with periodontal inflammation and bone resorption [150]. The effect of PGE2 in pathogenesis of CP is highly dependent on expression of PG receptors EP2 and EP4, which activate adenylate cyclase and protein kinase A signaling mediating bone resorption [151]. Unfortunately, non-selective NSAIDs seem to damage the mucosa of the gastrointestinal tract by interfering with the physiologic synthesis of PG (PGE2 and PGI2) that results in increased gastric acid secretion, reduction in blood flow through the gastric mucosa and inhibition of mucus production [152]. Similarly to non-selective NSAIDs, selective cycloxygenase 2 (COX-2) inhibitors showed promising results for the stabilization of periodontal conditions by reducing the rate of alveolar bone resorption. They also offer the advantage of reducing the above mentioned adverse systemic effects compared to non-selective NSAIDs but were shown to pose significant cardiovascular risks [153]. Further, because existing evidence shows that the effects of NSAIDs drop off rapidly after drug withdrawal and there aren’t yet any data from long-term, multi-center prospective clinical trials available we cannot determine whether these therapeutic effects can be retained on a long-term basis [152].
3.5. Lipid Mediators of Inflammation Resolution in DM and CP
3.5.1. Lipid Mediators of Inflammation Resolution
Lipid molecules play several roles in living organisms mainly through their amphipathic nature. They are major constituents of biological lipid bilayer membranes, serve as the most efficient energy source, are covalently or non-covalently linked to proteins by post-translational modifications to facilitate protein-protein and protein-lipid interactions and some lipid molecules serve as intercellular mediators in immune functions and maintenance of homeostasis. The latter category includes lipid mediators that are divided into several subclasses base on their structure: fatty acids (e.g. eicosanoids), phospholipids (e.g. platelet activating factor - PAF), lysophospholipids (e.g. lysophosphatidic acid), and others (e.g. ceramide) [154]. Lipid mediators are synthesized from cell membrane phospholipids with contribution of several key enzymes including 1) phospholipase A2 (PLA2) that releases arachidonic acid (AA) or another polyunsaturated fatty acid (PUFA) from position sn-2 of phospholipids and the remaining lysophospholipid, 2) acyl-CoA lysophospholipid acyltransferase (acyltransferase, LPLAT) that incorporates a PUFA in position sn-2 of glycerophospholipids, 3) acetyl-CoA lyso-PAFacetyltrasnferase (lysoPAFAT) that converts lysophospholipids with alkyl-ether bond at sn-1 to PAF and incorporates arachidonyl-CoA into lysophosphatidyilcholine (LPC) (LPCAT2), 4) cyclooxygenases (COX) that incorporates two oxygen molecules into AA to generate thromboxanes (TXs) and prostaglandin (PG) endoperoxydes subsequently converted to PGs by cell-specific PG synthases, and 5) lipoxygenases (LOX) that incorporates a single oxygen molecule into PUFA to generate hydroperoxyeicosatetraenoic (HpETEs) and hydroeicosatetraenoic acids (HETEs) precursors for leukotrienes (LT) and lipoxins (LX) respectively when PUFA is AA, and resolvins (Rv) of E and D series, protectins (PT) and maresins when PUFA is eiscosapentaenoic acid (EPA) or docosahexaenoic acid (DHA) (Fig. 5). Other enzymes also contribute to generation of lipid mediators: cytochrome P450 yielding epoxyeicosatrienoic acids (EETs), 19-HETE, 20-HETE, dihydroperoxytetraenoic acids (diHETEs), phospholipase C (PLC) yielding DAG, IP3 and D (PLD) yielding phosphatidic acid, and sphingomyelinase yielding ceramide. Phospholipases and lysophos-pholipid acyltrasferases facilitate membrane phospholipid recycling during generation of lipid mediators in Lands’ cycle [154,155] (Fig. 6).
Fig. (5).
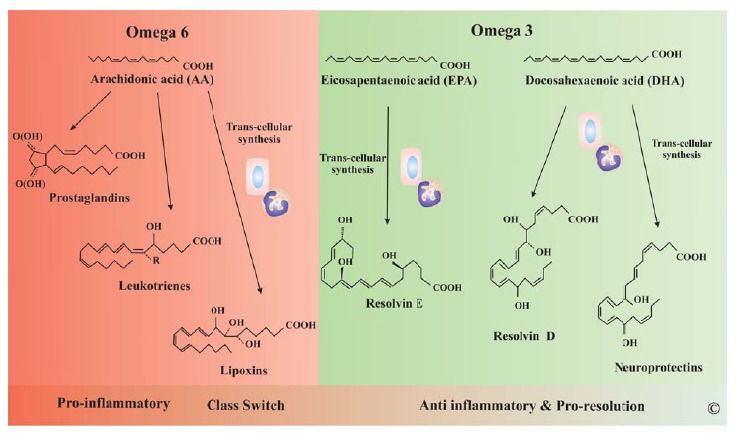
Lipid Mediators of Inflammation. During inflammation several lipid mediators are produced to enhance and then to reverse inflammation. Most mediators derived from AA promote inflammation (PG and LT) but some newly characterized molecules including lipoxins (LX) are anti-inflammatory. LX are considered the class switch molecules from pro to anti-inflammatory lipid mediators. The Ω-3 fatty acids EPA and DHA are processed to resolvins, protectins. Together with LX these lipid mediators are synthesized trough a trans-cellular process in which intermediates are synthesized by a cell type and taken by other cell types to produce the final active mediator.
Fig. (6).
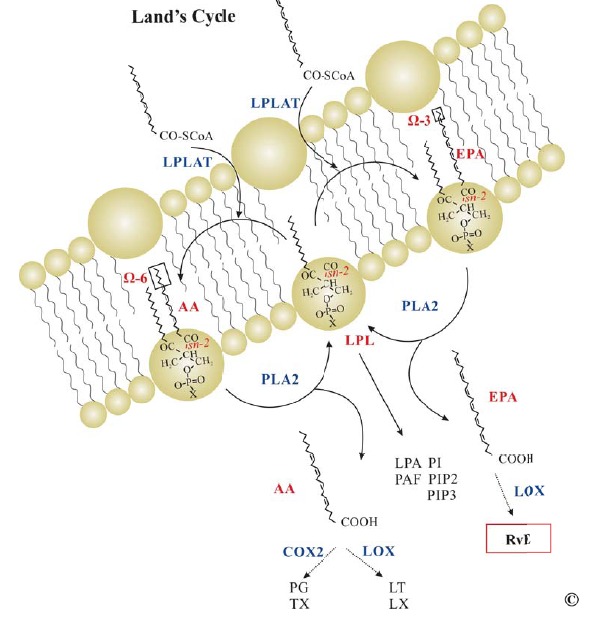
Lands’ Cycle. PUFAs in position sn-2 of membrane PL are released by PLA2 and further processed to lipid mediators. The remaining LPL depending on X in PO3X may generate LPA (X=O2-), PC or PAF32 (X=choline), PS (X=serine), PE (X=ethanolamine) or PI, PIP2, PIP3 (X=inositol). Alternatively LPLs may receive a new PUFA through LPLAT closing the Lands’ cycle. In general PC, PE and PS are not released from the membrane but may be exposed on the outer leaflet during apoptosis by and enzyme called flippase. Abbreviations: AA, arachidonic acid; EPA, eicosapentaenoic acid; PLA2, phospholipase A2; LPLAT, lysophospholipid (LPL) acyltransferase; LPA, lysophosphatidic acid; PAF, platelet activating factor; PI, phosphatidyl inositol; PIP, PI phosphate; PIP2; PI diphosphate; PIP3, PI triphosphate; PG, prostaglandin; TX, thomboxane; LT, leukotriene; LX, lipoxin; RvE, resolvin E series; CoA, coenzyme A.
There are two main COX isoforms in mammals; a constitutive form COX-1 and an inducible form COX-2 with their encoding genes located on chromosomes 9 and 1 respectively [156,157]. COX-1 is ubiquitously expressed in cells and helps maintain tissue homeostasis and COX-2 expression is induced during injury by LPS, IL-1β, TNFα, IFNγ to generate pro-inflammatory eicosanoids. Three main mechanisms contribute to induction of COX-2 expression: transcriptional regulation through NF-κB and CREB, mRNA stabilization through the proximal AT-rich sequence of the 3’-untranslated region, and expression of microRNAs. In recent years a third isoform acetaminophen-sensitive COX-3, which apparently is a splice variant of COX-2 has been reported [158]. This isoform has low sensitivity to non-steroidal anti-inflammatory (NSAIDs) drugs. Aspirin modifies both COX enzymes through acetylation of serine residues near the catalytic pocket. Although aspirin and other NSAIDs were previously thought to abolish the enzymatic activity of COX it became clear that they rather change COXs to a LOX-like acting enzyme generating precursors for different classes of lipid mediators with particular activities during the resolution phase of inflammation [159,160].
There are six main LOX enzymes coded by separate genes and named after the position of the carbon where the oxygen is inserted: 5-LOX (5S-LOX) mainly expressed in leukocytes, 8/12R-LOX mainly expressed in epidermis, two types of 12-LOX (12S-LOX and 12R-LOX) mainly expressed in megakariocytes/platelets (12S-LOX), and two types of 15-LOX (15-LOX-1 and 15-LOX-2) mainly expressed in erhythroid and epithelial cells respectively [161]. 12-LOX is up-regulated in islets of both T1DM and T2DM patients decreasing INS secretion and sensitivity, and increasing production of inflammatory mediators [162]. Further, hyperglycemia and inflammatory cytokines increase the expression of 12-LOX and pharmacological inhibition of this enzyme prevented development of DM in animal models [162-165]. Long-chain Ω-3 PUFA replace arachidonic acid as eicosanoid substrate for LOX thereby indirectly altering the expression of pro-inflammatory genes. In addition, they provide substrates for biosynthesis of resolvins (Rvs), protectins (PDs) and maresins (MaR). Long-chain Ω-3 PUFAs inhibit responses to LPS, TNFα and saturated fatty acids possibly through endogenously derived lipid mediators Rvs and PDs.
The eicosanoids derived from AA can be divided into two main classes based on their action: pro-inflammatory and anti-inflam-matory. The first category includes PGs of E series (PGE2) synthesized through the COX pathway and LTs (LTA4, LTB4, LTC4, LTD4 and LTE4) synthesized through LOX pathway. TXA2 and TXB2 result from COX processing and are potent vasoconstrictor and stimulators of platelet aggregation. PAF belongs to a third class of AA- derived mediators resulting from PLA2 processing of membrane phospholipids and mediates platelet aggregation, bronchocontriction and vasodilation. The second category includes PGs of G, F and I series synthesized through COX pathway, and lipoxins (LXs) and aspirin triggered 15-epi-LXs (ATLs) synthesized through both COX and LOX pathways during trans-cellular biosynthesis. AA-derived lipid mediators act through specific G-protein coupled receptors (GPCRs) that were phylogenetically grouped into five cluster groups: CL-1, CL-2, CL-3 lipid receptors, and CL-4, CL-5 peptide receptors [166]. These receptors are part of a larger phylogenetic tree that includes proton sensors, chemoattractant receptors, LPA/S1P receptors, prostanoid receptors and lipid/nucleotide receptors [154] (Table 1).
Table 1.
Eicosanoids and Their Actions.
| Pathway | Mediator | Receptor [cluster] | Actions |
|---|---|---|---|
| COX | PGD2 PGE2 PGF2α PGI2 PGJ2 15d-PGJ2 TXA2 TXB2 |
DP1
[CL-1], DP2
[CL-1] EP1 [CL-2]; EP2 [CL-1] EP3 [CL-3]; EP4 [CL-1] FP [CL-2] IP [CL-1] DP1 [CL-1] PPARγ, DP2 [CL-1] TP- (TXA2) [CL-2] TP- (TXB2) [CL-2] |
Bronchoconstriction, vasodilation ↑cell survival, ↓DC chemotaxis/IL-12 synthesis, ↓EO recruitment Vasodilation, fever, GIT-SM contraction/relaxation, ↓gastric acid secretion, ↑mucus secretion, ↑T-cell chemotaxis, maintains PDA Bronchoconstriction, uterus contraction Bronchodilation, vasodilation, ↓platelet aggregation Vasodilation ↓iNOS, ↓MMP, ↑PMN apoptosis (PPARγ) ↑chemotaxis, ↑CAM expression (DP2) Vasoconstriction, ↑platelet aggregation Vasoconstriction |
| LOX | 5(S)-HETE 12(S)-HETE 15(S)-HETE LTA4 LTB4 LTC4, D4, E4 LXA2 LXB2 RvE1 RvE2 |
BLT2 [CL-5] BLT2 [CL-5] BLT2 [CL-5] Cys-LT1, Cys-LT2 BLT1, BLT2 Cys-LT1, Cys-LT2 ALX [CL-5] ALX [CL-5] ChemR23 [CL-5] BLT1 [CL-5] ChemR23, BLT1 |
↑PMN chemotaxis, constriction, ↑mucus (L) ↑PMN chemotaxis ↑PMN chemotaxis Intermediate compound ↑PMN chemotaxis, degranulation, O2− SRS-A Vasodilation, ↓PMN chemotaxis, ↓neovascularization ↑iNOS, ↑PGI2, ↑MΦ→PMN clearance ↓PMN, EO chemotaxis, ↑ CCR5 on PMN, ↓DC chemotaxis/IL-12 Equivalent to RvE1 in low doses |
| CYT P450 | EETs 11,12 diHETE 19-HETE 20-HETE |
TP- (TXA2)[CL-2] | Vasodilation Vasodilation? Vasodilation by ↓20-HEPE Vasoconstriction |
LXs are the first class of anti-inflammatory and pro-resolving lipid mediators identified in vivo. LXA4 and LXB4 result from platelet 12-LOX processing of LTA4 synthesized in leukocytes via 5-LOX. Alternatively, both LX can result from leukocyte 5-LOX processing of 15(S)-HpETE synthesized in epithelial cells via 15-LOX pathway [167]. Similarly, aspirin- modified COX in EC acting as 15-LOX generates 15-R-HETE that is further processed by leukocyte 5-LOX to yield 15-epi-LX [168]. LXs have potent anti-inflammatory actions by down-regulating neutrophil transmigration [169, 170], reducing vascular permeability [171], cytokine release and function [172], pain signals [173]. LXs also have pro-resolving actions in vivo by promoting phagocytosis and removal of apoptotic neutrophils by macrophages and inhibiting fibrosis [174, 175]. Nonetheless, LXs induces prostacyclin (PGI2) and nitric oxide synthesis by endothelial cells suggesting a direct impact on maintaining vascular homeostasis [176,177]. In recent years a new class of lipid mediators with potent anti-inflammatory and pro-resolution properties have been isolated and characterized by Serhan et al. These mediators are endogenously synthesized from Ω-3 fatty acids including DHA and EPA through trans-cellular biosynthesis involving aspirin acetylated-COX-2 and several LOX. RvE series are derived from EPA through epithelial or EC (acetylated-COX-2 or 15-LOX) crosstalk with leukocytes (5-LOX). Similarly, PGE3 and LTB5 can be synthesized from EPA. RvD series (RvD1, RvD2, RvD3, RvD4), PDs or neuroprotectins (NPDs) when produced by neural tissues and MaR are derived from DHA through several LOX processing steps in leukocytes and other cells [182-185].
RvE1 was first identified in vivo in dorsal air pouch exudates of mice treated with aspirin and EPA. The endothelial cell-neutrophil trans-cellular biosynthesis was confirmed by treating human microvascular endothelial cells with aspirin and EPA, and subsequent incubation of resulting products with human neutrophils [159]. RvE1 results from EPA modification by aspirin acetylated COX-2 or CYT P450 or 15-LOX-1 in endothelial cells and uptake of 18R-HEPE and further processing in leukocytes through 5-LOX pathway (Fig. 7). RvEs were characterized and tested in vivo and in vitro demonstrating potent anti-inflammatory and pro-resolution properties [184]. RvE1 (5S, 12R, 18R-trihydroxy-eicosapentaenoic acid) is structurally defined as a Ω-3 eicosanoid with 5 double bonds grouped as one conjugated diene and one conjugated triene that confer molecular stability. Three hydroxyl groups are sequentially added to EPA after removal from the membrane PL by PLA2 in a trans-cellular biosynthetic process [183]. Enzymatic conversion of RvE1 generates inactive or less potent compounds with one exception, 20-hydroxy-RvE1, in blood and other tissues: 20-carboxy-RvE1, 20-hydroxy-RvE1 (major metabolite in neutrophils, as potent as RvE1), 19-hydroxy-RvE1, 18-oxo-RvE1, 12-oxo-RvE1, 11,12-dihydro-RvE1 and 11,12-dihydro-12-oxo-RvE1. Three compounds namely 20-hydroxy- RvE1, 18-oxo-RvE1 and 11,12-dihydro-RvE1 were the major metabolites in human blood [186]. Two GPCR receptors ChemR23 (now named ERV1) and BLT1 have been shown to signal in response to high affinity binding by RvE1. ChemR23 binding reduced IL-12 production by dendritic cells while BLT1 binding competitively blocked LTB4 suggesting a potential mechanism for its anti-inflammatory action [187,188]. RvE1 significantly reduced neutrophil infiltration during acute peritonitis and up-regulated CCR5 expression on late apoptotic neutrophils [189,190].
Fig. (7).
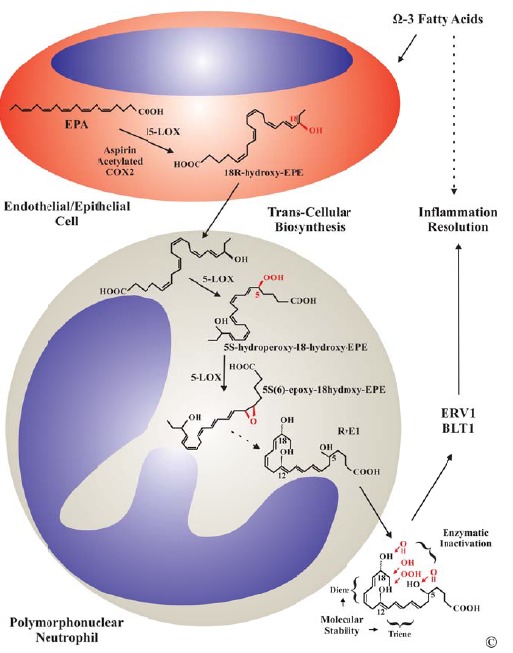
RvE1 Biosynthesis. RvE1 is synthesized through neutrophil-endothelial cell crosstalk from EPA. Endothelial cells produces an RvE1 intermediate, 18R-hydroxy-EPA, through the 15-LOX pathway (either a 15-LOX or an acetylated COX-2 by aspirin). The leukocyte processes 18R-hydroxy-EPA through the 5-LOX pathway to produce RvE1, the final active mediator that is released to act locally having a short half-life. Synthetic RvE1, which has a longer half-life, inhibited neutrophil infiltration in different disease models including peritonitis, colitis, acute lung injury, asthma and periodontal disease. It also promoted molecular pathways characteristic to the resolution phase of inflammation.
3.5.2. Low-Dose Aspirin in DM
The prevention of primary MI in patients with DM with daily aspirin was first addressed in a randomized, double blind, placebo-controlled trial by the Physicians’ Health Study Research Group including 22,071 participants with an average of 5 years follow-up completed in 1989. The study had found a 44% reduction in MI risk but a slightly increased risk of hemorrhagic stroke. Reduction in MI risk was apparent only in patients over 50 years of age [191]. Based on these observations and a few supporting studies that followed the American Diabetes Association strongly recommended long term daily aspirin for primary prevention of MI in DM. The efficiency of daily aspirin (75-325 mg/dose/day) and other anticoagulants alone or in combination with statins (HMG-CoA10 reductase inhibitors) on prevention of CV events and stroke in patients with DM has since been tested in large trials [192].
Despite the initial results supporting the CV preventive action of aspirin in DM a meta-analysis of randomized trials of antiplatelet therapy for prevention of life-threatening events in high-risk patients has found a modest overall 7% reduction in CV events in the subpopulation of patients with DM. On the other hand it was concluded that daily low doses of aspirin (LDA) (75-150 mg/day) are as efficient as higher doses in preventing stable angina, intermittent claudication and atrial fibrillation in patients with high risk [193]. A couple important negative trials that followed, namely the POPADAD (Prevention of Progression Of Arterial Disease and Diabetes) and the JPAD (Japanese Primary Prevention of Atherosclerosis With Aspirin for Diabetes) have questioned the highly efficient preventive action of aspirin in DM although these latter trials were criticized to be underpowered. Indeed both trials have used soft end points including the development of peripheral arterial disease and stable angina as primary events rather than harder end points like MI and CV death [194-196].
Altogether, to date, there is insufficient evidence to support aspirin’s action to prevent CV events in patients with DM more efficiently than in individuals without. In line with these observations the 2010 position statement of the American Diabetes Association recommends daily LDA (75-162 mg/day) for prevention of primary CV events in patients with DM and no previous history of vascular disease who are at increased risk (10 year risk of CVD events over 10%) and who are not at increased risk for bleeding. Adults with increased CVD risk are most men over age 50 years and women over age 60 years diagnosed with DM who have one or more additional major risk factors including smoking, hypertension, dyslipidemia, family history of premature CVD, and albuminuria. Patients with intermediate risk are younger patients with one or more risk factors and older patients with no risk factors. They may be prescribed LDA for prevention of primary CV events until further evidence is available. Aspirin should not be recommended for CV prevention for patients with DM at low risk including those who do not fall into high or intermediate risk categories [197].
3.5.3. Ω-3 PUFAs and Derived Mediators for Resolution of Inflammation in DM
The use of Ω-3 PUFAs in patients with vascular disease gained increasing interest since it was found that Greenland Inuits had a lower prevalence of coronary heart disease compared to Scandinavian control subjects [198,199]. It was later shown that Inuits also had reduced platelet reactivity and prolonged bleeding time compared to Scandinavian controls [200]. This was attributed to the Eskimo diet, which has an extremely high content of fish abundant in Ω-3 PUFAs, mainly EPA and DHA [201]. Subsequent observations have indicated a significant role of dietary Ω-6-to-Ω-3 PUFAs ratio in the incidence of DM in different populations [202]. Numerous studies have shown significant beneficial effects of Ω-3 PUFAs in patients with DM regarding leukocyte function, glucose metabolism, IR and diabetic complications [203]. It is believed that Ω-3 PUFAs modulate inflammation by partially replacing cell membrane-bound Ω-6 PUFAs when the former are in high enough concentration in diet, subsequently generating anti-inflammatory and pro-resolution lipid mediators. Although, the exact mechanisms of this pro-resolving loop in DM are unclear, Ω-3 PUFAs seem to prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome in MΦ [204].
Since mammals cannot naturally produce Ω-3 PUFAs they must rely on dietary supply to maintain a balanced Ω-6-to-Ω-3 ratio [205,206]. Further, the average low purity of Ω-3 PUFAs in commercial preparations and the limited rate of gut absorption in the context of relatively high doses necessary to achieve some of the biological effects observed make the administration pharmacological doses difficult [207]. Kang et al. have generated a mouse that carries fat-1, an enzyme expressed by Caenorhabditis elegans, which adds a double bond into an unsaturated fatty-acid hydrocarbon chain converting Ω-6 to Ω-3 PUFAs [208]. This allowed further investigations on Ω-3 PUFAs roles in development of DM. Fat-1 transgenic mice were protected from becoming diabetic in response to streptozocin through regulation of inflammation by reducing levels of IL-1β, TNFα, NF-κB and 12-HETE [209]. In humans, genetic variance in the fatty acid desaturase (FADS) gene cluster (FADS1 and FADS2) seems to regulate the fatty acid composition in phospholipids [210,211]. This suggests potential genetic basis for differential processing of PUFAs to pro- and anti-inflammatory lipid mediators. Therefore current evidence points to two major rate limiting steps in metabolism of Ω-3 PUFAs in humans: absorption in the intestine and enzymatic processing to active metabolites.
As terminal active metabolites of Ω-3 PUFAs the pro-resolution lipid mediators LXs, Rvs PDs and MaRs show great promise as novel therapeutics for modulating unregulated inflammation associated with DM. As opposed to their precursors DHA and EPA, these mediators exert their biological actions at nanomolar range. Interestingly, obesity significantly decreased DHA-derived 17-hydroxyDHA (RvD1 precursor) and PD1 levels in murine adipose tissue while dietary EPA/DHA treatment restored endogenously synthesized Ω-3-derived mediators, reduced adipose tissue inflammation and improved INS sensitivity in obese mice [212]. In diabetic fat-1 mice PGE2 and 12-HETE were decreased while LXA4 and 18-HETE (precursor for RvE1) were highly increased [209]. Interestingly, pancreatic tissues of fat-1 mice exposed to streptozocin produced high amounts of LXA4 and did not develop DM. Additionally, in vitro and in vivo animal studies on LXA4 have demonstrated potent actions to reduce inflammation, promote resolution and increase INS sensitivity in DM [213,214]. These actions were associated with decrease in pro-inflammatory (IL-6, TNFα, IFNγ) and increase in pro-resolution cytokines (IL-10), and increased GLUT-4 and INS receptor substrate 1 (IRS-1) [215]. Similarly, Rvs, including RvE1, RvD1 and RvD2 counteracted IR adipokine production and monocyte accumulation in adipose tissue, promoted M2 polarization of MΦ, improved INS sensitivity, decreased hepatic steatosis and suppressed pro-inflammatory mediators in pancreatic β-cells [216-221]. One of these studies found increased INS sensitivity through up regulation of GLUT-4, IRS-1 and PPARγ after treatment of ob/ob mice with 1.2 ng/g body weight RvE1 every 24 h for 4 days [220]. In addition EPA, the ω-3 fatty acid precursor for RvE1, was found to up regulate PPARγ in spleen CD4 T cells [222]. Altogether, these findings suggest that DHA/EPA-derived lipid mediators can promote DM-associated inflammation and improve INS sensitivity.
3.5.4. Ω-3 PUFAs and Derived Mediators for Resolution of Inflammation in CP
Several reports from animal studies investigating the immune modulatory actions of Ω-3 PUFAs in CP suggested a significant role in reduction of inflammatory mediators, oxidative stress and alveolar bone loss when DHA and/or EPA were added to the diet [223-225]. In line with these findings a recent human RCT on the effects of fish oils (30% EPA/DHA) combined with low dose aspirin (81 mg) and SRP compared to SRP and placebo on periodontal parameters in CP found a significant reduction in probing depths, inflammatory mediators and a gain in clinical attachment in the treatment group [226]. Similar findings were reported in a Japanese RCT that investigated the effects of dietary Ω-3 PUFAs on progression of CP over 5 years in senior individuals [227]. High dietary intake of DHA and to a lesser degree EPA were associated with lower prevalence of CP in a representative US population sample from the 1999-2004 National Health and Nutrition Examination Survey (NHANES) [228]. These studies indicated that Ω-3 PUFAs and derived pro-resolution mediators can modulate periodontal inflammation and promote healing and regeneration.
Adapted from various sources: Review PGD2 and PGJ2 [178]; lipid mediator receptors [154,179]; CYT P450 metabolites [180]; RvE review [181]. Abbreviations: CAM, cell adhesion molecule; DC, dendritic cell; EO, eosinophil; GIT-SM, gastrointestinal tract smooth muscle; L, lung; MΦ, macrophage; PDA, patent ductus arteriosus; PMN, polymorphonuclear neutrophil; SRS-A, slow-reacting substance of anaphylaxis; Note: CYT P450 enzymes may also generate precursors for synthesis of LX, Rv and PD.
In animal models of periodontitis RvE1 was shown to protect from inflammation-mediated alveolar bone loss and promote bone regeneration when used as topical single therapy [229-231]. Using a critical size defect craniotomy mouse model of bone healing and regeneration Gao et al have shown RvE1 enhances new bone formation through its receptor ChemR23 (ERV1) [230]. Similarly, in a rabbit model of Porphyromonas gingivalis-induced CP Hasturk et al found that topical RvE1 can reduce alveolar bone loss and enhance bone regeneration [229]. Interestingly, local RvE1 treatment attenuated atherogenesis induced by periodontitis in the same animal model and prevented vascular inflammation and atherogenesis in healthy rabbits [232]. RvE1 seems to protect periodontal tissues from inflammation-mediated destruction mainly by modulating neutrophil infiltration and function locally [233]. RvE1 enhanced phagocytosis of Porphyromonas gingivalis by neutrophils in part through modulating the phosphorylation of Akt, the downstream ribosomal protein S6 and mitogen-activated protein kinases (MAPK) [234]. Similarly, LXA4 topical in micromolar concentration promoted alveolar bone regeneration in an minipig model of periodontitis by activating osteogenic pathways and promoting endogenous synthesis pro-resolution mediators [235]. RvD1 enhanced wound closure by promoting proliferation and migration of periodontal ligament fibroblasts and induced a significant decrease in PGE2 levels in these cells [236]. The different actions of pro-resolution mediators derived from Ω-3 PUFAs is explained in part by the specific receptors used by individual LXs and Rvs, and receptor distribution in tissues [237]. There are currently clinical trials in progress evaluating the benefits of resolution molecules (e.g. lipoxin analogue BLXA4-ME) in the oral cavity, but no data is available to date [238].
4. Future Perspectives
In light of compelling evidence that inflammation plays crucial roles in onset and progression of DM and CP resolution therapeutics needs to be incorporated in individualized treatment regimens for patients suffering from these conditions. While management of hyperglycemia in DM and pathogenic sugbgingival biofilms in CP are crucial for preventing disease progression and onset of complications, resolving local and systemic inflammation is key to maintaining a healthy immune response to non-self and restoring body homeostasis. Since MΦ are central players in homeostasis and host defense possessing remarkable plasticity in different tissue environments, modulation of MΦ polarization to a pro-resolution M2 phenotype seems to address the un-regulated inflammation associated with DM and CP [239]. One approach to regulate MΦ polarization is systemic delivery of immune modulatory drugs such as thiazolidinediones (PPARγ agonists). Interestingly, PPARγ-mediated M2 polarization seems to occur systemically as thiazolidinediones up-regulate M2 markers in monocytes but not resident MΦ [240]. The other approach is to polarize M2 MΦ ex vivo for therapeutic systemic delivery. M2 macrophages that maintain their phenotype for up to 4 weeks were safely delivered and improved murine colitis and pancreatic and renal injury in DM [241,242]. Importantly, the Ω-3-derived pro-resolution mediators have been shown to skew adipose tissue MΦ toward an M2 phenotype [243]. Therefore, a third approach to modulation of MΦ polarization is the use of nano-proresolving lipid therapeutics in combination with blood glucose lowering medicines. This approach seems feasible in light of emerging evidence that individualized combined therapies in the form of polypills can be successful in managing complex conditions such as cardiovascular diseases and DM [244-246].
Conclusion
Management of de-regulated inflammation associated with DM and CP can be addressed through an integrative approach that addresses systemic immune responses to altered self and non-self. As central regulators of innate and adaptive immunity MΦ are an important therapeutic target for immune modulation. Increasing evidence form in vitro and in vivo animal and human studies suggests that pro-resolution therapeutics derived from Ω-3 PUFAs have the potential to resolve inflammation and restore homeostasis in DM and CP in part through regulation neutrophil recruitment and MΦ polarization.
Fig. (1).
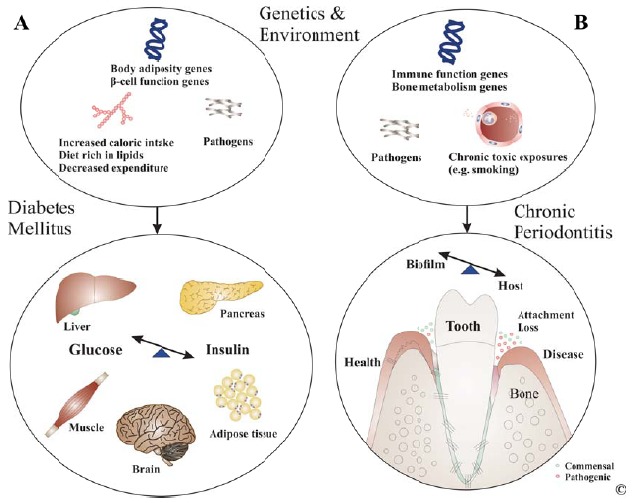
Diabetes Mellitus and Chronic Periodontitis. The interplay between genetics and the environment in pathogenesis of both DM and CP is subject to intense investigation. A: Regardless of mechanisms of onset elevated blood glucose levels characterize both types of DM. Immune- or pathogen-mediated destruction of -cells and the associated hypoinsulinemia is central to T1DM. A combination between predisposition to deregulated body adiposity or -cell function and excessive caloric intake and/or decreased caloric expenditure are central to T2DM. The liver physiologically maintains blood glucose levels through glycogenolysis (main) and gluconeogenesis (secondary) in the first phase of fasting (4-16 h postprandial). When exogenous carbohydrates become available the liver reduces release of stored and de novo synthesized glucose and delivers absorbed glucose from intestine resulting in increased glycemia that stimulates -cells to release INS while glucagon secretion is inhibited to ensure uptake and use of glucose in peripheral tissues, particularly muscle and brain. Excess carbohydrates lead to sustained hyperglycemia resulting in increased INS release into the circulation. Development of IR in peripheral tissues prevents adequate uptake and use/storage of circulating glucose resulting in hyperglycemia and compensatory hyperinsulinemia. Continuous supra-solicitation of - cells is accompanied by their apoptotic death mostly due to endoplasmic reticulum stress leading to hypoinsulinemia. Deregulation of lipid, carbohydrate and protein metabolism and IR in peripheral organs, liver and the hypothalamus perturb hepatic glucose regulation resulting in massive glucose release into the circulation. B: Host-biofilm disequilibria are central to pathogenesis of CP. While putative periodontal pathogens such as Porphyromonas gingivalis, Treponema denticola, Tannerela forsythia and Aggregatibacter actinomycetemcomitans are associated with periodontitis, they are not sufficient for disease onset. Multiple host and environmental factors contribute to host-biofilm imbalance that results in inflammation-mediated loss of tooth attachment to periodontium. Immune function genes such as IL-1, IL-6, IL-10, CD14 and bone metabolism genes such as vitamin D receptor were linked to CP. Environmental factors such as smoking and chronic conditions such as DM significantly increase the risk for CP and affect its course and response to therapy..
ACKNOWLEDGEMENTS
Supported in part by the National Institute of Dental and Craniofacial Research through USPHS grants K99DE024575 (CS) and DE25030 (TVD).
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
REFERENCES
- 1.Fowler M.J. Microvascular and macrovascular complications of diabetes. Clin. Diabetes. 2008;26(2):77–82. [Google Scholar]
- 2.Wild S., Roglic G., Green A., Sicree R., King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27(5):1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 3.Petersen P.E., Ogawa H. The global burden of periodontal disease: towards integration with chronic disease prevention and control. Periodontol. 2000. 2012;60(1):15–39. doi: 10.1111/j.1600-0757.2011.00425.x. [DOI] [PubMed] [Google Scholar]
- 4.Schätzle M. LOE H, Lang NP, Bürgin W, Anerud A, Boysen H. The clinical course of chronic periodontitis. J Clin Periodontol. Wiley Online Library. 2004;31(12):1122–1127. doi: 10.1111/j.1600-051X.2004.00634.x. [DOI] [PubMed] [Google Scholar]
- 5.Löe H., Anerud A., Boysen H., Morrison E. Natural history of periodontal disease in man. Rapid, moderate and no loss of attachment in Sri Lankan laborers 14 to 46 years of age. J. Clin. Periodontol. 1986;13(5):431–445. doi: 10.1111/j.1600-051x.1986.tb01487.x. [DOI] [PubMed] [Google Scholar]
- 6.Genco RJ, Borgnakke WS. Risk factors for periodontal disease. 2000 doi: 10.1111/j.1600-0757.2012.00457.x. [DOI] [PubMed] [Google Scholar]
- 7.Darveau R.P. Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 2010;8(7):481–490. doi: 10.1038/nrmicro2337. [DOI] [PubMed] [Google Scholar]
- 8.Bartold P.M., Cantley M.D., Haynes D.R. Mechanisms and control of pathologic bone loss in periodontitis. Periodontol. 2010;53(1):55–69. doi: 10.1111/j.1600-0757.2010.00347.x. [DOI] [PubMed] [Google Scholar]
- 9.Van Dyke TE, Lester MA, Shapira L. The role of the host response in periodontal disease progression: implications for future treatment strategies. 1993 doi: 10.1902/jop.1993.64.8s.792. [DOI] [PubMed] [Google Scholar]
- 10.Nares S. The genetic relationship to periodontal disease. Periodontol. 2000. 2002;32:36–49. doi: 10.1046/j.0906-6713.2002.03204.x. [DOI] [PubMed] [Google Scholar]
- 11.Wasserman D.H., Wasserman D.H. Regulation of glucose fluxes during exercise in the postabsorptive state. Annu. Rev. Physiol. 1995;57(1):191–218. doi: 10.1146/annurev.ph.57.030195.001203. [DOI] [PubMed] [Google Scholar]
- 12.Atkinson M.A., Eisenbarth G.S., Michels A.W. Type 1 diabetes. Lancet. 2014;383(9911):69–82. doi: 10.1016/S0140-6736(13)60591-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kent S.C., Chen Y., Bregoli L., et al. Expanded T cells from pancreatic lymph nodes of type 1 diabetic subjects recognize an insulin epitope. Nature. 2005;435(7039):224–228. doi: 10.1038/nature03625. [DOI] [PubMed] [Google Scholar]
- 14.Gaglia J.L., Guimaraes A.R., Harisinghani M., et al. Noninvasive imaging of pancreatic islet inflammation in type 1A diabetes patients. J. Clin. Invest. 2011;121(1):442–445. doi: 10.1172/JCI44339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shoelson S.E. Inflammation and insulin resistance. J. Clin. Invest. 2006;116(7):1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hotamisligil G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140(6):900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 18.Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;•••:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- 19.Lee H.B., Yu M-R., Yang Y., Jiang Z., Ha H. Reactive oxygen species-regulated signaling pathways in diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14(8) Suppl. 3:S241–S245. doi: 10.1097/01.asn.0000077410.66390.0f. [DOI] [PubMed] [Google Scholar]
- 20.Hotamisligil G.S., Shargill N.S., Spiegelman B.M. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1992;259(5091):87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 21.Solinas G., Karin M. JNK1 and IKK: molecular links between obesity and metabolic dysfunction. FASEB J. 2010;24(8):2596–2611. doi: 10.1096/fj.09-151340. [DOI] [PubMed] [Google Scholar]
- 22.Weisberg S.P., Hunter D., Huber R., et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen A., Mumick S., Zhang C., et al. Diet induction of monocyte chemoattractant protein-1 and its impact on obesity. Obesity (Silver Spring) 2005;13(8):1311–1320. doi: 10.1038/oby.2005.159. [DOI] [PubMed] [Google Scholar]
- 24.Kanda H. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 2006;116(6):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hirosumi J., Tuncman G., Chang L., et al. A central role for JNK in obesity and insulin resistance. Nature. 2002;420(6913):333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 26.Olefsky J.M., Glass C.K. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol. 2010;72(1):219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 27.Eizirik D.L., Cardozo A.K., Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr. Rev. 2007;29(1):42–61. doi: 10.1210/er.2007-0015. [DOI] [PubMed] [Google Scholar]
- 28.Offenbacher S. Periodontal diseases: pathogenesis. Ann. Periodontol. 1996;1(1):821–878. doi: 10.1902/annals.1996.1.1.821. [DOI] [PubMed] [Google Scholar]
- 29.Van Dyke T.E. Control of inflammation and periodontitis. Periodontol. 2007;45(1):158–166. doi: 10.1111/j.1600-0757.2007.00229.x. [DOI] [PubMed] [Google Scholar]
- 30.Lang N.P., Schätzle M.A., Löe H. Gingivitis as a risk factor in periodontal disease. J. Clin. Periodontol. 2009;36:3–8. doi: 10.1111/j.1600-051X.2009.01415.x. [DOI] [PubMed] [Google Scholar]
- 31.Page R.C., Offenbacher S., Schroeder H.E., Seymour G.J., Kornman K.S. Advances in the pathogenesis of periodontitis: summary of developments, clinical implications and future directions. Periodontol. 1997;14(1):216–248. doi: 10.1111/j.1600-0757.1997.tb00199.x. [DOI] [PubMed] [Google Scholar]
- 32.Bartold P.M., Van Dyke T.E. Periodontitis: a host-mediated disruption of microbial homeostasis. Unlearning learned concepts. Periodontol. 2000. 2013;62(1):203–217. doi: 10.1111/j.1600-0757.2012.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang Y., Russell T.R., Graves D.T., Cheng H., Nong S.H., Levitz S.M. Monocyte chemoattractant protein 1 and interleukin-8 production in mononuclear cells stimulated by oral microorganisms. Infect. Immun. 1996;64(11):4450–4455. doi: 10.1128/iai.64.11.4450-4455.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gorska R., Gregorek H., Kowalski J., Laskus-Perendyk A., Syczewska M., Madalinski K. Relationship between clinical parameters and cytokine profiles in inflamed gingival tissue and serum samples from patients with chronic periodontitis. J. Clin. Periodontol. 2003;30(12):1046–1052. doi: 10.1046/j.0303-6979.2003.00425.x. [DOI] [PubMed] [Google Scholar]
- 35.Stashenko P., Van Dyke T., Tully P., Kent R., Jr, Sonis S., Tanner A.C. Inflammation and genetic risk indicators for early periodontitis in adults. J. Periodontol. 2011;82(4):588–596. doi: 10.1902/jop.2010.100443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Assuma R., Oates T., Cochran D., Amar S., Graves D.T. IL-1 and TNF antagonists inhibit the inflammatory response and bone loss in experimental periodontitis. J. Immunol. 1998;160(1):403–409. [PubMed] [Google Scholar]
- 37.Graves D. Cytokines that promote periodontal tissue destruction. J. Periodontol. 2008;79(8) Suppl.:1585–1591. doi: 10.1902/jop.2008.080183. [DOI] [PubMed] [Google Scholar]
- 38.Matsuki Y., Yamamoto T., Hara K. Interleukin-1 mRNA-expressing macrophages in human chronically inflamed gingival tissues. Am. J. Pathol. 1991;138(6):1299. [PMC free article] [PubMed] [Google Scholar]
- 39.Ohyama H., Kato-Kogoe N., Kuhara A., et al. The involvement of IL-23 and the Th17 pathway in periodontitis. J. Dent. Res. 2009;88(7):633–638. doi: 10.1177/0022034509339889. [DOI] [PubMed] [Google Scholar]
- 40.Allam J-P., Duan Y., Heinemann F., et al. IL-23-producing CD68(+) macrophage-like cells predominate within an IL-17-polarized infiltrate in chronic periodontitis lesions. J. Clin. Periodontol. 2011;38(10):879–886. doi: 10.1111/j.1600-051X.2011.01752.x. [DOI] [PubMed] [Google Scholar]
- 41.Garlet G.P. Destructive and protective roles of cytokines in periodontitis: a re-appraisal from host defense and tissue destruction viewpoints. J. Dent. Res. 2010;89(12):1349–1363. doi: 10.1177/0022034510376402. [DOI] [PubMed] [Google Scholar]
- 42.Lappin D.F., MacLeod C.P., Kerr A., Mitchell T., Kinane D.F. Anti-inflammatory cytokine IL-10 and T cell cytokine profile in periodontitis granulation tissue. Clin. Exp. Immunol. 2001;123(2):294–300. doi: 10.1046/j.1365-2249.2001.01448.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sasaki H., Okamatsu Y., Kawai T., Kent R., Taubman M., Stashenko P. The interleukin-10 knockout mouse is highly susceptible to Porphyromonas gingivalis-induced alveolar bone loss. J. Periodontal Res. 2004;39(6):432–441. doi: 10.1111/j.1600-0765.2004.00760.x. [DOI] [PubMed] [Google Scholar]
- 44.Pradeep A.R., Roopa Y., Swati P.P. Interleukin-4, a T-helper 2 cell cytokine, is associated with the remission of periodontal disease. J. Periodontal Res. 2008;43(6):712–716. doi: 10.1111/j.1600-0765.2007.01079.x. [DOI] [PubMed] [Google Scholar]
- 45.Stashenko P., Goncalves R.B., Lipkin B., Ficarelli A., Sasaki H., Campos-Neto A. Th1 immune response promotes severe bone resorption caused by Porphyromonas gingivalis. Am. J. Pathol. 2007;170(1):203–213. doi: 10.2353/ajpath.2007.060597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dutzan N., Vernal R., Hernandez M., et al. Levels of interferon-gamma and transcription factor T-bet in progressive periodontal lesions in patients with chronic periodontitis. J. Periodontol. 2009;80(2):290–296. doi: 10.1902/jop.2009.080287. [DOI] [PubMed] [Google Scholar]
- 47.Steinsvoll S., Halstensen T.S., Schenck K. Extensive expression of TGF-beta1 in chronically-inflamed periodontal tissue. J. Clin. Periodontol. 1999;26(6):366–373. doi: 10.1034/j.1600-051x.1999.260606.x. [DOI] [PubMed] [Google Scholar]
- 48.Yoshie H., Kobayashi T., Tai H., Galicia J.C. The role of genetic polymorphisms in periodontitis. Periodontol. 2000. 2007;43:102–132. doi: 10.1111/j.1600-0757.2006.00164.x. [DOI] [PubMed] [Google Scholar]
- 49.Kinane D.F., Hart T.C. Genes and gene polymorphisms associated with periodontal disease. Crit. Rev. Oral Biol. Med. 2003;14(6):430–449. doi: 10.1177/154411130301400605. [DOI] [PubMed] [Google Scholar]
- 50.Thorstensson H., Kuylenstierna J., Hugoson A. Medical status and complications in relation to periodontal disease experience in insulin-dependent diabetics. J. Clin. Periodontol. 1996;23(3 Pt 1):194–202. doi: 10.1111/j.1600-051x.1996.tb02076.x. [DOI] [PubMed] [Google Scholar]
- 51.Saremi A., Nelson R.G., Tulloch-Reid M., et al. Periodontal disease and mortality in type 2 diabetes. Diabetes Care. 2005;28(1):27–32. doi: 10.2337/diacare.28.1.27. [DOI] [PubMed] [Google Scholar]
- 52.Shultis W.A., Weil E.J., Looker H.C., et al. Effect of periodontitis on overt nephropathy and end-stage renal disease in type 2 diabetes. Diabetes Care. 2007;30(2):306–311. doi: 10.2337/dc06-1184. [DOI] [PubMed] [Google Scholar]
- 53.Taylor GW, Burt BA, Becker MP, et al. Severe periodontitis and risk for poor glycemic control in patients with non-insulin-dependent diabetes mellitus. 1996. [DOI] [PubMed]
- 54.Ide R., Hoshuyama T., Wilson D., Takahashi K., Higashi T. Periodontal disease and incident diabetes: a seven-year study. J. Dent. Res. 2011;90(1):41–46. doi: 10.1177/0022034510381902. [DOI] [PubMed] [Google Scholar]
- 55.Demmer R.T., Jacobs D.R., Desvarieux M. Periodontal disease and incident type 2 diabetes: results from the first national health and nutrition examination survey and its epidemiologic follow-up study. Diabetes Care. 2008;31(7):1373–1379. doi: 10.2337/dc08-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson T.C., Needleman I., Wild S.H., Moles D.R., Mills E.J. Treatment of periodontal disease for glycaemic control in people with diabetes. Cochrane Database Syst. Rev. 2010;(5):CD004714. doi: 10.1002/14651858.CD004714.pub2. [DOI] [PubMed] [Google Scholar]
- 57.Katagiri S., Nitta H., Nagasawa T., et al. Multi-center intervention study on glycohemoglobin (HbA1c) and serum, high-sensitivity CRP (hs-CRP) after local anti-infectious periodontal treatment in type 2 diabetic patients with periodontal disease. Diabetes Res. Clin. Pract. 2009;83(3):308–315. doi: 10.1016/j.diabres.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 58.O'Connell P.A., Taba M., Nomizo A., et al. Effects of periodontal therapy on glycemic control and inflammatory markers. J. Periodontol. 2008;79(5):774–783. doi: 10.1902/jop.2008.070250. [DOI] [PubMed] [Google Scholar]
- 59.Teeuw W.J., Gerdes V.E., Loos B.G. Effect of Periodontal Treatment on Glycemic Control of Diabetic Patients: A systematic review and meta-analysis. Diabetes Care. 2010;33(2):421–427. doi: 10.2337/dc09-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kiran M., Arpak N., Unsal E., Erdoğan M.F. The effect of improved periodontal health on metabolic control in type 2 diabetes mellitus. J. Clin. Periodontol. 2005;32(3):266–272. doi: 10.1111/j.1600-051X.2005.00658.x. [DOI] [PubMed] [Google Scholar]
- 61.Jones J.A., Miller D.R., Wehler C.J., et al. Does periodontal care improve glycemic control? The Department of Veterans Affairs Dental Diabetes Study. J. Clin. Periodontol. 2007;34(1):46–52. doi: 10.1111/j.1600-051X.2006.01002.x. [DOI] [PubMed] [Google Scholar]
- 62.Khaw K-T., Wareham N., Bingham S., Luben R., Welch A., Day N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann. Intern. Med. 2004;141(6):413–420. doi: 10.7326/0003-4819-141-6-200409210-00006. [DOI] [PubMed] [Google Scholar]
- 63.Hayashi C., Gudino C.V., Gibson F.C., III, Genco C.A. Review: Pathogen-induced inflammation at sites distant from oral infection: bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol. Oral Microbiol. 2010;25(5):305–316. doi: 10.1111/j.2041-1014.2010.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lalla E., Kaplan S., Yang J., Roth G.A., Papapanou P.N., Greenberg S. Effects of periodontal therapy on serum C-reactive protein, sE-selectin, and tumor necrosis factor-? secretion by peripheral blood-derived macrophages in diabetes. A pilot study. J. Periodontal Res. 2007;42(3):274–282. doi: 10.1111/j.1600-0765.2006.00945.x. [DOI] [PubMed] [Google Scholar]
- 65.Tonetti M.S., D'Aiuto F., Nibali L., et al. Treatment of periodontitis and endothelial function. N. Engl. J. Med. 2007;356(9):911–920. doi: 10.1056/NEJMoa063186. [DOI] [PubMed] [Google Scholar]
- 66.Genco R.J., Van Dyke T.E. Prevention: Reducing the risk of CVD in patients with periodontitis. Nat. Rev. Cardiol. 2010;7(9):479–480. doi: 10.1038/nrcardio.2010.120. [DOI] [PubMed] [Google Scholar]
- 67.Engebretson S., Chertog R., Nichols A., Hey-Hadavi J., Celenti R., Grbic J. Plasma levels of tumour necrosis factor-alpha in patients with chronic periodontitis and type 2 diabetes. J. Clin. Periodontol. 2007;34(1):18–24. doi: 10.1111/j.1600-051X.2006.01017.x. [DOI] [PubMed] [Google Scholar]
- 68.Mealey B.L., Rose L.F. Diabetes mellitus and inflammatory periodontal diseases. Curr. Opin. Endocrinol. Diabetes Obes. 2008;15(2):135–141. doi: 10.1097/MED.0b013e3282f824b7. [DOI] [PubMed] [Google Scholar]
- 69.Manouchehr-Pour M., Spagnuolo P.J., Rodman H.M., Bissada N.F. Impaired neutrophil chemotaxis in diabetic patients with severe periodontitis. J. Dent. Res. 1981;60(3):729–730. doi: 10.1177/00220345810600031101. [DOI] [PubMed] [Google Scholar]
- 70.Graves D.T., Liu R., Alikhani M., Al-Mashat H., Trackman P.C. Diabetes-enhanced inflammation and apoptosis--impact on periodontal pathology. J. Dent. Res. 2005;85(1):15–21. doi: 10.1177/154405910608500103. [DOI] [PubMed] [Google Scholar]
- 71.Kjersem H., Hilsted J., Madsbad S., Wandall J.H., Johansen K.S., Borregaard N. Polymorphonuclear leucocyte dysfunction during short term metabolic changes from normo- to hyperglycemia in type 1 (insulin dependent) diabetic patients. Infection. 1988;16(4):215–221. doi: 10.1007/BF01650754. [DOI] [PubMed] [Google Scholar]
- 72.Takashiba S., Ohyama H., Oyaizu K., Kogoe Kato N., Murayama Y. HLA genetics for diagnosis of susceptibility to early‐onset periodontitis. J. Periodontal Res. 1999;34(7):374–378. doi: 10.1111/j.1600-0765.1999.tb02269.x. [DOI] [PubMed] [Google Scholar]
- 73.Gustke C.J., Stein S.H., Hart T.C., et al. HLA-DR alleles are associated with IDDM, but not with impaired neutrophil chemotaxis in IDDM. J. Dent. Res. 1998;77(7):1497–1503. doi: 10.1177/00220345980770070401. [DOI] [PubMed] [Google Scholar]
- 74.Gyurko R., Siqueira C.C., Caldon N., Gao L., Kantarci A., Van Dyke T.E. Chronic hyperglycemia predisposes to exaggerated inflammatory response and leukocyte dysfunction in Akita mice. J. Immunol. 2006;177(10):7250–7256. doi: 10.4049/jimmunol.177.10.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sima C., Rhourida K., Van Dyke T.E., Gyurko R. Type 1 diabetes predisposes to enhanced gingival leukocyte margination and macromolecule extravasation in vivo. J. Periodontal Res. 2010;45(6):748–756. doi: 10.1111/j.1600-0765.2010.01295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Beam H.A., Parsons J.R., Lin S.S. The effects of blood glucose control upon fracture healing in the BB Wistar rat with diabetes mellitus. J. Orthop. Res. 2002;20(6):1210–1216. doi: 10.1016/S0736-0266(02)00066-9. [DOI] [PubMed] [Google Scholar]
- 77.Graves D.T., Li J., Cochran D.L. Inflammation and uncoupling as mechanisms of periodontal bone loss. J. Dent. Res. 2011;90(2):143–153. doi: 10.1177/0022034510385236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He H., Liu R., Desta T., Leone C., Gerstenfeld L.C., Graves D.T. Diabetes causes decreased osteoclastogenesis, reduced bone formation, and enhanced apoptosis of osteoblastic cells in bacteria stimulated bone loss. Endocrinology. 2004;145(1):447–452. doi: 10.1210/en.2003-1239. [DOI] [PubMed] [Google Scholar]
- 79.Javed A., Chen H., Ghori F.Y. Genetic and Transcriptional Control of Bone Formation. Oral Maxillofac. Surg. Clin. North Am. 2010;22(3):283–293. doi: 10.1016/j.coms.2010.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Graves D.T., Cochran D. The contribution of interleukin-1 and tumor necrosis factor to periodontal tissue destruction. J. Periodontol. 2003;74(3):391–401. doi: 10.1902/jop.2003.74.3.391. [DOI] [PubMed] [Google Scholar]
- 81.Liu R., Bal H.S., Desta T., et al. Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. J. Dent. Res. 2006;85(6):510–514. doi: 10.1177/154405910608500606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lalla E., Lamster I.B., Feit M., et al. Blockade of RAGE suppresses periodontitis-associated bone loss in diabetic mice. J. Clin. Invest. 2000;105(8):1117–1124. doi: 10.1172/JCI8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Murillo J., Wang Y., Xu X., et al. Advanced glycation of type I collagen and fibronectin modifies periodontal cell behavior. J. Periodontol. 2008;79(11):2190–2199. doi: 10.1902/jop.2008.080210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ren L., Fu Y., Deng Y., Qi L., Jin L. Advanced glycation end products inhibit the expression of collagens type I and III by human gingival fibroblasts. J. Periodontol. 2009;80(7):1166–1173. doi: 10.1902/jop.2009.080669. [DOI] [PubMed] [Google Scholar]
- 85.Takeda M., Ojima M., Yoshioka H., et al. Relationship of serum advanced glycation end products with deterioration of periodontitis in type 2 diabetes patients. J. Periodontol. 2006;77(1):15–20. doi: 10.1902/jop.2006.77.1.15. [DOI] [PubMed] [Google Scholar]
- 86.Zizzi A, Tirabassi G, Aspriello SD, Piemontese M, Rubini C, Lucarini G. Gingival advanced glycation end-products in diabetes mellitus-associated chronic periodontitis: an immunohistochemical. 2012 doi: 10.1111/jre.12007. [DOI] [PubMed] [Google Scholar]
- 87.Makiura N., Ojima M., Kou Y., et al. Relationship of Porphyromonas gingivalis with glycemic level in patients with type 2 diabetes following periodontal treatment. Oral Microbiol. Immunol. 2008;23(4):348–351. doi: 10.1111/j.1399-302X.2007.00426.x. [DOI] [PubMed] [Google Scholar]
- 88.Davé S., Van Dyke T. The link between periodontal disease and cardiovascular disease is probably inflammation. Oral Dis. 2008;14(2):95–101. doi: 10.1111/j.1601-0825.2007.01438.x. [DOI] [PubMed] [Google Scholar]
- 89.Beck J., Garcia R., Heiss G., Vokonas P.S., Offenbacher S. Periodontal disease and cardiovascular disease. J. Periodontol. 1996;67(10) Suppl.:1123–1137. doi: 10.1902/jop.1996.67.10s.1123. [DOI] [PubMed] [Google Scholar]
- 90.Friedewald V.E., Kornman K.S., Beck J.D., et al. The American J Cardiology and J Periodontology Editors' Consensus: periodontitis and atherosclerotic cardiovascular disease. Am. J. Cardiol. 2009:59–68. doi: 10.1016/j.amjcard.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 91.Nakano K., Nemoto H., Nomura R., et al. Detection of oral bacteria in cardiovascular specimens. Oral Microbiol. Immunol. 2009;24(1):64–68. doi: 10.1111/j.1399-302X.2008.00479.x. [DOI] [PubMed] [Google Scholar]
- 92.Koren O., Spor A., Felin J., et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA. 2011;108(Suppl. 1):4592–4598. doi: 10.1073/pnas.1011383107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Desvarieux M. Periodontal microbiota and carotid intima-media thickness: The oral infections and vascular disease epidemiology study (INVEST). Circulation. 2005;111(5):576–582. doi: 10.1161/01.CIR.0000154582.37101.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Desvarieux M. Relationship between periodontal disease, tooth loss, and carotid artery plaque: the oral infections and vascular disease epidemiology study (INVEST). Stroke. 2003;34(9):2120–2125. doi: 10.1161/01.STR.0000085086.50957.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Desvarieux M., Demmer R.T., Jacobs D.R., Jr, et al. Periodontal bacteria and hypertension: the oral infections and vascular disease epidemiology study (INVEST). J. Hypertens. 2010;28(7):1413–1421. doi: 10.1097/HJH.0b013e328338cd36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Katzung B., Trevor A. USA: McGraw Hill Professional; 2014. Basic and Clinical Pharmacology. [Google Scholar]
- 97.Drucker D.J. Dipeptidyl peptidase-4 inhibition and the treatment of type 2 diabetes: preclinical biology and mechanisms of action. Diabetes Care. Am Diabetes Assoc. 2007;30(6):1335–1343. doi: 10.2337/dc07-0228. [DOI] [PubMed] [Google Scholar]
- 98.Ghosh R.K., Ghosh S.M., Chawla S., Jasdanwala S.A. SGLT2 inhibitors: a new emerging therapeutic class in the treatment of type 2 diabetes mellitus. J. Clin. Pharmacol. 2012;52(4):457–463. doi: 10.1177/0091270011400604. [DOI] [PubMed] [Google Scholar]
- 99.Thomas D., Elliott E.J. Low glycaemic index, or low glycaemic load, diets for diabetes mellitus. Cochrane Database Syst. Rev. 2009;(1):CD006296. doi: 10.1002/14651858.CD006296.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Orozco L.J., Buchleitner A.M., Gimenez-Perez G., et al. Exercise or exercise and diet for preventing type 2 diabetes mellitus. Cochrane Database Syst. Rev. 2008;(3):CD003054. doi: 10.1002/14651858.CD003054.pub3. [DOI] [PubMed] [Google Scholar]
- 101.Misso M.L., Egberts K.J., Page M., O'Connor D., Shaw J. Continuous subcutaneous insulin infusion (CSII) versus multiple insulin injections for type 1 diabetes mellitus. Cochrane Database Syst. Rev. 2010;(1):CD005103. doi: 10.1002/14651858.CD005103.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vardi M., Vardi M., Jacobson E., et al. 2008. Intermediate acting versus long acting insulin for type 1 diabetes mellitus. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.The Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N. Engl. J. Med. 1993;329(14):977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 104.UKDPS RG. UK Prospective Diabetes Study Group Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. UK Prospective Diabetes Study Group. BMJ. 1998;317(7160):703–713. [PMC free article] [PubMed] [Google Scholar]
- 105.NAVIGATOR Study Group Effect of nateglinide on the incidence of diabetes and cardiovascular events. N. Engl. J. Med. 2010;362(16):1463–1476. doi: 10.1056/NEJMoa1001122. [DOI] [PubMed] [Google Scholar]
- 106.NAVIGATOR Study Group Effect of valsartan on the incidence of diabetes and cardiovascular events. N. Engl. J. Med. 2010;362(16):1477–1490. doi: 10.1056/NEJMoa1001121. [DOI] [PubMed] [Google Scholar]
- 107.Shichiri M., Kishikawa H., Ohkubo Y., Wake N. Long-term results of the Kumamoto Study on optimal diabetes control in type 2 diabetic patients. Diabetes Care. 2000;23(Suppl. 2):B21–B29. [PubMed] [Google Scholar]
- 108.Gæde P., Lund-Andersen H., Parving H-H., Pedersen O. Effect of a multifactorial intervention on mortality in type 2 diabetes. N. Engl. J. Med. 2008;358(6):580–591. doi: 10.1056/NEJMoa0706245. [DOI] [PubMed] [Google Scholar]
- 109.Chiasson J.L., Gomis R., Hanefeld M., Josse R.G., Karasik A., Laakso M. The STOP-NIDDM Trial: an international study on the efficacy of an alpha-glucosidase inhibitor to prevent type 2 diabetes in a population with impaired glucose tolerance: rationale, design, and preliminary screening data. Study to Prevent Non-Insulin-Dependent Diabetes Mellitus. Diabetes Care. 1998;21(10):1720–1725. doi: 10.2337/diacare.21.10.1720. [DOI] [PubMed] [Google Scholar]
- 110.Pihlstrom B.L., Michalowicz B.S., Johnson N.W. Periodontal diseases. Lancet. 2005;366(9499):1809–1820. doi: 10.1016/S0140-6736(05)67728-8. [DOI] [PubMed] [Google Scholar]
- 111.Emingil G., Atilla G., Sorsa T., Luoto H., Kirilmaz L., Baylas H. The effect of adjunctive low-dose doxycycline therapy on clinical parameters and gingival crevicular fluid matrix metalloproteinase-8 levels in chronic periodontitis. J. Periodontol. 2004;75(1):106–115. doi: 10.1902/jop.2004.75.1.106. [DOI] [PubMed] [Google Scholar]
- 112.Jesenberger V., Jentsch S. Deadly Encounter: Ubiquitin Meets Apoptosis. Nat. Rev. Mol. Cell Biol. 2002;3(2):112–121. doi: 10.1038/nrm731. [DOI] [PubMed] [Google Scholar]
- 113.Tait S.W., Green D.R. Caspase-independent cell death: leaving the set without the final cut. Oncogene. 2008;27(50):6452–6461. doi: 10.1038/onc.2008.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Villa P.G., Henzel W.J., Sensenbrenner M., Henderson C.E., Pettmann B. Calpain inhibitors, but not caspase inhibitors, prevent actin proteolysis and DNA fragmentation during apoptosis. J. Cell Sci. 1998;111(Pt 6):713–722. doi: 10.1242/jcs.111.6.713. [DOI] [PubMed] [Google Scholar]
- 115.Roos A., Xu W., Castellano G., et al. Mini-review: A pivotal role for innate immunity in the clearance of apoptotic cells. Eur. J. Immunol. 2004;34(4):921–929. doi: 10.1002/eji.200424904. [DOI] [PubMed] [Google Scholar]
- 116.Savill J., Dransfield I., Gregory C., Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2002;2(12):965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 117.Benoit M.E., Clarke E.V., Morgado P., Fraser D.A., Tenner A.J. Complement protein C1q directs macrophage polarization and limits inflammasome activity during the uptake of apoptotic cells. J. Immunol. 2012;188(11):5682–5693. doi: 10.4049/jimmunol.1103760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Savill J., Hogg N., Ren Y., Haslett C. Thrombospondin cooperates with CD36 and the vitronectin receptor in macrophage recognition of neutrophils undergoing apoptosis. J. Clin. Invest. 1992;90(4):1513–1522. doi: 10.1172/JCI116019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Canton J., Neculai D., Grinstein S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013;13(9):621–634. doi: 10.1038/nri3515. [DOI] [PubMed] [Google Scholar]
- 120.Hébert M.J., Takano T., Holthöfer H., Brady H.R. Sequential morphologic events during apoptosis of human neutrophils. Modulation by lipoxygenase-derived eicosanoids. J. Immunol. 1996;157(7):3105–3115. [PubMed] [Google Scholar]
- 121.Sica A., Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 2012;122(3):787–795. doi: 10.1172/JCI59643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ivashkiv L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013;34(5):216–223. doi: 10.1016/j.it.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Martinez F.O., Helming L., Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 2009;27(1):451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 124.Biswas S.K., Chittezhath M., Shalova I.N., Lim J-Y. Macrophage polarization and plasticity in health and disease. Immunol. Res. 2012;53(1-3):11–24. doi: 10.1007/s12026-012-8291-9. [DOI] [PubMed] [Google Scholar]
- 125.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8(12):958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Savill J.S., Wyllie A.H., Henson J.E., Walport M.J., Henson P.M., Haslett C. Macrophage phagocytosis of aging neutrophils in inflammation. Programmed cell death in the neutrophil leads to its recognition by macrophages. J. Clin. Invest. 1989;83(3):865–875. doi: 10.1172/JCI113970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Serhan C.N., Savill J. Resolution of inflammation: the beginning programs the end. Nat. Immunol. 2005;6(12):1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 128.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat. Rev. Immunol. 2010;10(2):117. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Arkan M.C., Hevener A.L., Greten F.R., et al. IKK-β links inflammation to obesity-induced insulin resistance. Nat. Med. 2005;11(2):191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 130.Sartipy P., Loskutoff D.J. Monocyte chemoattractant protein 1 in obesity and insulin resistance. Proc. Natl. Acad. Sci. USA. 2003;100(12):7265–7270. doi: 10.1073/pnas.1133870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kanda H., Tateya S., Tamori Y., et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Invest. 2006;116(116(6)):1494–1505. doi: 10.1172/JCI26498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Weisberg S.P., Hunter D., Huber R., et al. CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J. Clin. Invest. 2006;116(1):115–124. doi: 10.1172/JCI24335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hanefeld M., Schell E., Gouni-Berthold I., Melichar M., Vesela I., et al. Orally-Administered Chemokine Receptor CCR2 Antagonist CCX140-B in type 2 diabetes: a pilot double-blind, randomized clinical trial. J. Diabetes Metab. 2013;3:225. [Google Scholar]
- 134.Sullivan T., Miao Z., Dairaghi D.J., et al. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. Am. J. Physiol. Renal Physiol. 2013;305(9):F1288–F1297. doi: 10.1152/ajprenal.00316.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kurihara T., Warr G., Loy J., Bravo R. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 1997;186(10):1757–1762. doi: 10.1084/jem.186.10.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Donath M.Y. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat. Rev. Drug Discov. 2014;13(6):465–476. doi: 10.1038/nrd4275. [DOI] [PubMed] [Google Scholar]
- 137.Stanley T.L., Zanni M.V., Johnsen S., et al. TNF-α Antagonism with Etanercept Decreases Glucose and Increases the Proportion of High Molecular Weight Adiponectin in Obese Subjects with Features of the Metabolic Syndrome. J clinical endocrinology and metabolism. Endocrine Soc. 2013;96(1):E146–E150. doi: 10.1210/jc.2010-1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gonzalez-Gay M.A., De Matias J.M., Gonzalez-Juanatey C., et al. Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2006;24(1):83–86. [PubMed] [Google Scholar]
- 139.Larsen C.M., Faulenbach M., Vaag A., et al. Interleukin-1-receptor antagonist in type 2 diabetes mellitus. N. Engl. J. Med. 2007;356(15):1517–1526. doi: 10.1056/NEJMoa065213. [DOI] [PubMed] [Google Scholar]
- 140.Yazdani Biuki B., Stelzl H., Brezinschek H.P., et al. Improvement of insulin sensitivity in insulin resistant subjects during prolonged treatment with the anti‐TNF‐α antibody infliximab. Eur. J. Clin. Invest. 2004;34(9):641–642. doi: 10.1111/j.1365-2362.2004.01390.x. [DOI] [PubMed] [Google Scholar]
- 141.Hensen J., Howard C.P., Walter V., Thuren T. Impact of interleukin-1β antibody (canakinumab) on glycaemic indicators in patients with type 2 diabetes mellitus: Results of secondary endpoints from a randomized, placebo-controlled trial. Diabetes Metab. 2013;39(6):524–531. doi: 10.1016/j.diabet.2013.07.003. [DOI] [PubMed] [Google Scholar]
- 142.Mayer Y., Balbir-Gurman A., Machtei E.E. Anti-tumor necrosis factor-alpha therapy and periodontal parameters in patients with rheumatoid arthritis. J. Periodontol. 2009;80(9):1414–1420. doi: 10.1902/jop.2009.090015. [DOI] [PubMed] [Google Scholar]
- 143.Ortiz P., Bissada N.F., Palomo L., et al. Periodontal therapy reduces the severity of active rheumatoid arthritis in patients treated with or without tumor necrosis factor inhibitors. J. Periodontol. 2009;80(4):535–540. doi: 10.1902/jop.2009.080447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Pers J-O., Saraux A., Pierre R., Youinou P. Anti-TNF-alpha immunotherapy is associated with increased gingival inflammation without clinical attachment loss in subjects with rheumatoid arthritis. J. Periodontol. 2008;79(9):1645–1651. doi: 10.1902/jop.2008.070616. [DOI] [PubMed] [Google Scholar]
- 145.Delima A.J., Oates T., Assuma R., et al. Soluble antagonists to interleukin-1 (IL-1) and tumor necrosis factor (TNF) inhibits loss of tissue attachment in experimental periodontitis. J. Clin. Periodontol. 2001;28(3):233–240. doi: 10.1034/j.1600-051x.2001.028003233.x. [DOI] [PubMed] [Google Scholar]
- 146.Nam J.L., Winthrop K.L., van Vollenhoven R.F., et al. Current evidence for the management of rheumatoid arthritis with biological disease-modifying antirheumatic drugs: a systematic literature review informing the EULAR recommendations for the management of RA. Ann. Rheum. Dis. 2010;69(6):976–986. doi: 10.1136/ard.2009.126573. [DOI] [PubMed] [Google Scholar]
- 147.Heasman P.A., Benn D.K., Kelly P.J., Seymour R.A., Aitken D. The use of topical flurbiprofen as an adjunct to non-surgical management of periodontal disease. J. Clin. Periodontol. 1993;20(6):457–464. doi: 10.1111/j.1600-051x.1993.tb00389.x. [DOI] [PubMed] [Google Scholar]
- 148.Reddy M.S., Palcanis K.G., Barnett M.L., Haigh S., Charles C.H., Jeffcoat M.K. Efficacy of meclofenamate sodium (Meclomen) in the treatment of rapidly progressive periodontitis. J. Clin. Periodontol. 1993;20(9):635–640. doi: 10.1111/j.1600-051x.1993.tb00708.x. [DOI] [PubMed] [Google Scholar]
- 149.Howell T.H., Williams R.C. Nonsteroidal antiinflammatory drugs as inhibitors of periodontal disease progression. Crit. Rev. Oral Biol. Med. 1993;4(2):177–196. doi: 10.1177/10454411930040020301. [DOI] [PubMed] [Google Scholar]
- 150.Offenbacher S., Heasman P.A., Collins J.G. Modulation of host PGE2 secretion as a determinant of periodontal disease expression. J. Periodontol. 1993;64(5) Suppl.:432–444. doi: 10.1902/jop.1993.64.5s.432. [DOI] [PubMed] [Google Scholar]
- 151.Suzawa T., Miyaura C., Inada M., et al. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the respective EPs. Endocrinology. 2000;141(4):1554–1559. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- 152.Salvi G., Lang N. The effects of non-steroidal anti-inflammatory drugs (selective and non-selective) on the treatment of periodontal diseases. Curr. Pharm. Des. 2005;11(14):1757–1769. doi: 10.2174/1381612053764878. [DOI] [PubMed] [Google Scholar]
- 153.Funk C.D., FitzGerald G.A. COX-2 inhibitors and cardiovascular risk. J. Cardiovasc. Pharmacol. 2007;50(5):470–479. doi: 10.1097/FJC.0b013e318157f72d. [DOI] [PubMed] [Google Scholar]
- 154.Shimizu T. Lipid mediators in health and disease: enzymes and receptors as therapeutic targets for the regulation of immunity and inflammation. Annu. Rev. Pharmacol. Toxicol. 2009;49(1):123–150. doi: 10.1146/annurev.pharmtox.011008.145616. [DOI] [PubMed] [Google Scholar]
- 155.O'Donnell V.B. Ben Maskrey, Taylor GW. Eicosanoids: generation and detection in mammalian cells. Methods Mol. Biol. 2008;462:5–23. [PubMed] [Google Scholar]
- 156.Robertson R.P. Dominance of cyclooxygenase-2 in the regulation of pancreatic islet prostaglandin synthesis. Diabetes. 1998;47(9):1379–1383. doi: 10.2337/diabetes.47.9.1379. [DOI] [PubMed] [Google Scholar]
- 157.Rosen G.D., Birkenmeier T.M., Raz A., Holtzman M.J. Identification of a cyclooxygenase-related gene and its potential role in prostaglandin formation. Biochem. Biophys. Res. Commun. 1989;164(3):1358–1365. doi: 10.1016/0006-291x(89)91819-6. [DOI] [PubMed] [Google Scholar]
- 158.Simmons D.L., Botting R.M., Robertson P.M., Madsen M.L., Vane J.R. Induction of an acetaminophen-sensitive cyclooxygenase with reduced sensitivity to nonsteroid antiinflammatory drugs. Proc. Natl. Acad. Sci. USA. 1999;96(6):3275–3280. doi: 10.1073/pnas.96.6.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Serhan C.N., Clish C.B., Brannon J., Colgan S.P., Chiang N., Gronert K. Novel functional sets of lipid-derived mediators with antiinflammatory actions generated from omega-3 fatty acids via cyclooxygenase 2-nonsteroidal antiinflammatory drugs and transcellular processing. J. Exp. Med. 2000;192(8):1197–1204. doi: 10.1084/jem.192.8.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Serhan C.N., Haeggström J.Z., Leslie C.C. Lipid mediator networks in cell signaling: update and impact of cytokines. FASEB J. 1996;10(10):1147–1158. doi: 10.1096/fasebj.10.10.8751717. [DOI] [PubMed] [Google Scholar]
- 161.Kühn H., Thiele B.J. The diversity of the lipoxygenase family. Many sequence data but little information on biological significance. FEBS Lett. 1999;449(1):7–11. doi: 10.1016/s0014-5793(99)00396-8. [DOI] [PubMed] [Google Scholar]
- 162.Dobrian A.D., Lieb D.C., Cole B.K., Taylor-Fishwick D.A., Chakrabarti S.K., Nadler J.L. Functional and pathological roles of the 12- and 15-lipoxygenases. Prog. Lipid Res. 2011;50(1):115–131. doi: 10.1016/j.plipres.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Ma K., Nunemaker C.S., Wu R., Chakrabarti S.K., Taylor-Fishwick D.A., Nadler J.L. 12-Lipoxygenase Products Reduce Insulin Secretion and β-Cell Viability in Human Islets. J. Clin. Endocrinol. Metab. 2010;95(2):887–893. doi: 10.1210/jc.2009-1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sears D.D., Miles P.D., Chapman J., et al. 2009. [Google Scholar]
- 165.Tersey S.A., Carter J.D., Rosenberg L., Taylor-Fishwick D.A., Mirmira R.G., Nadler J.L. Amelioration of type 1 diabetes following treatment of non-obese diabetic mice with INGAP and lisofylline. J. Diabetes Mellitus. 2012;02(02):251–257. doi: 10.4236/jdm.2012.22040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Toh H., Ichikawa A., Narumiya S. Molecular evolution of receptors for eicosanoids. FEBS Lett. 1995;361(1):17–21. doi: 10.1016/0014-5793(95)00129-w. [DOI] [PubMed] [Google Scholar]
- 167.Serhan C.N., Hamberg M., Samuelsson B. Lipoxins: novel series of biologically active compounds formed from arachidonic acid in human leukocytes. Proc. Natl. Acad. Sci. USA. 1984;81(17):5335–5339. doi: 10.1073/pnas.81.17.5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Serhan C.N. Lipoxins and novel aspirin-triggered 15-epi-lipoxins (ATL): a jungle of cell-cell interactions or a therapeutic opportunity? Prostaglandins. 1997;53(2):107–137. doi: 10.1016/s0090-6980(97)00001-4. [DOI] [PubMed] [Google Scholar]
- 169.Pouliot M., Clish C.B., Petasis N.A., Van Dyke T.E., Serhan C.N. Lipoxin A(4) analogues inhibit leukocyte recruitment to Porphyromonas gingivalis: a role for cyclooxygenase-2 and lipoxins in periodontal disease. Biochemistry. 2000;39(16):4761–4768. doi: 10.1021/bi992551b. [DOI] [PubMed] [Google Scholar]
- 170.Devchand P.R., Arita M., Hong S., et al. Human ALX receptor regulates neutrophil recruitment in transgenic mice: roles in inflammation and host defense. FASEB J. 2003;17(6):652–659. doi: 10.1096/fj.02-0770com. [DOI] [PubMed] [Google Scholar]
- 171.Takano T., Clish C.B., Gronert K., Petasis N., Serhan C.N. Neutrophil-mediated changes in vascular permeability are inhibited by topical application of aspirin-triggered 15-epi-lipoxin A4 and novel lipoxin B4 stable analogues. J. Clin. Invest. 1998;101(4):819–826. doi: 10.1172/JCI1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Hachicha M., Pouliot M., Petasis N.A., Serhan C.N. Lipoxin (LX)A4 and aspirin-triggered 15-epi-LXA4 inhibit tumor necrosis factor 1alpha-initiated neutrophil responses and trafficking: regulators of a cytokine-chemokine axis. J. Exp. Med. 1999;189(12):1923–1930. doi: 10.1084/jem.189.12.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Svensson C.I., Zattoni M., Serhan C.N. Lipoxins and aspirin-triggered lipoxin inhibit inflammatory pain processing. J. Exp. Med. 2007;204(2):245–252. doi: 10.1084/jem.20061826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Mitchell S., Thomas G., Harvey K., et al. Lipoxins, aspirin-triggered epi-lipoxins, lipoxin stable analogues, and the resolution of inflammation: stimulation of macrophage phagocytosis of apoptotic neutrophils in vivo. J. Am. Soc. Nephrol. 2002;13(10):2497–2507. doi: 10.1097/01.asn.0000032417.73640.72. [DOI] [PubMed] [Google Scholar]
- 175.Godson C., Mitchell S., Harvey K., Petasis N.A., Hogg N., Brady H.R. Cutting edge: lipoxins rapidly stimulate nonphlogistic phagocytosis of apoptotic neutrophils by monocyte-derived macrophages. J. Immunol. 2000;164(4):1663–1667. doi: 10.4049/jimmunol.164.4.1663. [DOI] [PubMed] [Google Scholar]
- 176.Paul-Clark M.J. 15-epi-lipoxin A4-mediated induction of nitric oxide explains how aspirin inhibits acute inflammation. J. Exp. Med. 2004;200(1):69–78. doi: 10.1084/jem.20040566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Brezinski M.E., Gimbrone M.A., Nicolaou K.C., Serhan C.N. Lipoxins stimulate prostacyclin generation by human endothelial cells. FEBS Lett. 1989;245(1-2):167–172. doi: 10.1016/0014-5793(89)80214-5. [DOI] [PubMed] [Google Scholar]
- 178.Herlong J.L., Scott T.R. Positioning prostanoids of the D and J series in the immunopathogenic scheme. Immunol. Lett. 2006;102(2):121–131. doi: 10.1016/j.imlet.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 179.Chiang N. The lipoxin receptor ALX: potent ligand-specific and stereoselective actions in vivo. Pharmacol. Rev. 2006;58(3):463–487. doi: 10.1124/pr.58.3.4. [DOI] [PubMed] [Google Scholar]
- 180.Roman R.J. P-450 metabolites of arachidonic acid in the control of cardiovascular function. Physiol. Rev. 2002;82(1):131–185. doi: 10.1152/physrev.00021.2001. [DOI] [PubMed] [Google Scholar]
- 181.Kohli P., Levy B.D. Resolvins and protectins: mediating solutions to inflammation. Br. J. Pharmacol. 2009;158(4):960–971. doi: 10.1111/j.1476-5381.2009.00290.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Hong S. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. autacoids in anti-inflammation. J. Biol. Chem. 2003;278(17):14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 183.Serhan C.N. Resolution phase of inflammation: novel endogenous anti-inflammatory and proresolving lipid mediators and pathways. Annu. Rev. Immunol. 2007;25(1):101–137. doi: 10.1146/annurev.immunol.25.022106.141647. [DOI] [PubMed] [Google Scholar]
- 184.Serhan C.N., Hong S., Gronert K., et al. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter proinflammation signals. J. Exp. Med. 2002;196(8):1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Serhan C.N., Yang R., Martinod K., et al. Maresins: novel macrophage mediators with potent antiinflammatory and proresolving actions. J. Exp. Med. 2009;206(1):15–23. doi: 10.1084/jem.20081880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Hong S., Porter T.F., Lu Y., Oh S.F., Pillai P.S., Serhan C.N. Resolvin E1 metabolome in local inactivation during inflammation-resolution. J. Immunol. 2008;180(5):3512–3519. doi: 10.4049/jimmunol.180.5.3512. [DOI] [PubMed] [Google Scholar]
- 187.Arita M., Ohira T., Sun Y-P., Elangovan S., Chiang N., Serhan C.N. Resolvin E1 selectively interacts with leukotriene B4 receptor BLT1 and ChemR23 to regulate inflammation. J. Immunol. 2007;178(6):3912–3917. doi: 10.4049/jimmunol.178.6.3912. [DOI] [PubMed] [Google Scholar]
- 188.Arita M. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005;201(5):713–722. doi: 10.1084/jem.20042031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Bannenberg G.L., Chiang N., Ariel A., et al. Molecular circuits of resolution: formation and actions of resolvins and protectins. J. Immunol. 2005;174(7):4345–4355. doi: 10.4049/jimmunol.174.7.4345. [DOI] [PubMed] [Google Scholar]
- 190.Ariel A., Fredman G., Sun Y-P., et al. Apoptotic neutrophils and T cells sequester chemokines during immune response resolution through modulation of CCR5 expression. Nat. Immunol. 2006;7(11):1209–1216. doi: 10.1038/ni1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Final report on the aspirin component of the ongoing Physicians“ Health Study. Steering Committee of the Physicians” Health Study Research Group. Mass Med Soc. 1989;321(3):129–135. doi: 10.1056/NEJM198907203210301. [DOI] [PubMed] [Google Scholar]
- 192.Calvin A.D., Aggarwal N.R., Murad M.H., et al. Aspirin for the Primary Prevention of Cardiovascular Events: A systematic review and meta-analysis comparing patients with and without diabetes. Diabetes Care. 2009;32(12):2300–2306. doi: 10.2337/dc09-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Collaboration A.T. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324(7329):71–86. doi: 10.1136/bmj.324.7329.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Belch J, MacCuish A, Campbell I, et al. The prevention of progression of arterial disease and diabetes (POPADAD) trial: factorial randomised placebo controlled trial of aspirin and antioxidants in patients with diabetes and asymptomatic peripheral arterial disease. 2007:337–a1840. doi: 10.1136/bmj.a1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Farkouh M.E., Fuster V. Diabetes and aspirin: beware of underpowered negative trials. Nat. Clin. Pract. Cardiovasc. Med. 2009;6(1):1–1. doi: 10.1038/ncpcardio1433. [DOI] [PubMed] [Google Scholar]
- 196.Ogawa H., Nakayama M., Morimoto T., et al. Low-dose aspirin for primary prevention of atherosclerotic events in patients with type 2 diabetes: a randomized controlled trial. JAMA. 2008;300(18):2134–2141. doi: 10.1001/jama.2008.623. [DOI] [PubMed] [Google Scholar]
- 197.Pignone M., Alberts M.J., Colwell J.A., et al. Aspirin for primary prevention of cardiovascular events in people with diabetes: a position statement of the american diabetes association, a scientific statement of the american heart association, and an expert consensus document of the american college of cardiology foundation. Diabetes Care. 2010;33(6):1395–1402. doi: 10.2337/dc10-0555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Schmidt E.B. n-3 fatty acids and the risk of coronary heart disease. Dan. Med. Bull. 1997;44(1):1–22. [PubMed] [Google Scholar]
- 199.Bjerregaard P., Mulvad G., Pedersen H.S. Cardiovascular risk factors in Inuit of Greenland. Int. J. Epidemiol. 1997;26(6):1182–1190. doi: 10.1093/ije/26.6.1182. [DOI] [PubMed] [Google Scholar]
- 200.Dyerberg J., Bang H.O. Haemostatic function and platelet polyunsaturated fatty acids in Eskimos. Lancet. 1979;2(8140):433–435. doi: 10.1016/s0140-6736(79)91490-9. [DOI] [PubMed] [Google Scholar]
- 201.Bang H.O., Dyerberg J., Hjøorne N. The composition of food consumed by Greenland Eskimos. Acta Med. Scand. 1976;200(1-2):69–73. doi: 10.1111/j.0954-6820.1976.tb08198.x. [DOI] [PubMed] [Google Scholar]
- 202.Simopoulos A.P. The importance of the ratio of omega-6/omega-3 essential fatty acids. Biomed. Pharmacother. 2002;56(8):365–379. doi: 10.1016/s0753-3322(02)00253-6. [DOI] [PubMed] [Google Scholar]
- 203.De Caterina R., Madonna R., Bertolotto A., Schmidt E.B. n-3 Fatty acids in the treatment of diabetic patients: Biological rationale and clinical data. Diabetes Care. 2007;30(4):1012–1026. doi: 10.2337/dc06-1332. [DOI] [PubMed] [Google Scholar]
- 204.Yan Y., Jiang W., Spinetti T., et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity. 2013;38(6):1154–1163. doi: 10.1016/j.immuni.2013.05.015. [DOI] [PubMed] [Google Scholar]
- 205.Simopoulos AP. Basel:: Karger Publisher; 1998. Overview of Evolutionary Aspects of 3 Fatty Acids in the Diet. The Return of w3 Fatty Acids into the Food Supply. [Google Scholar]
- 206.Simopoulos A.P. Evolutionary aspects of diet, the omega-6/omega-3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed. Pharmacother. 2006;60(9):502–507. doi: 10.1016/j.biopha.2006.07.080. [DOI] [PubMed] [Google Scholar]
- 207.Barber M.D., Fearon K.C. Tolerance and incorporation of a high-dose eicosapentaenoic acid diester emulsion by patients with pancreatic cancer cachexia. Lipids. 2001;36(4):347–351. doi: 10.1007/s11745-001-0726-4. [DOI] [PubMed] [Google Scholar]
- 208.Kang J.X., Wang J., Wu L., Kang Z.B. Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature. 2004;427(6974):504–4. doi: 10.1038/427504a. [DOI] [PubMed] [Google Scholar]
- 209.Bellenger J., Bellenger S., Bataille A., et al. High pancreatic n-3 fatty acids prevent STZ-induced diabetes in fat-1 mice: inflammatory pathway inhibition. Diabetes. Am Diabetes Assoc. 2011;60(4):1090–1099. doi: 10.2337/db10-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Schaeffer L. Common genetic variants of the FADS1 FADS2 gene cluster and their reconstructed haplotypes are associated with the fatty acid composition in phospholipids. Hum. Mol. Genet. 2006;15(11):1745–1756. doi: 10.1093/hmg/ddl117. [DOI] [PubMed] [Google Scholar]
- 211.Rzehak P., Rzehak P., Heinrich J., et al. Evidence for an association between genetic variants of the fatty acid desaturase 1 fatty acid desaturase 2 (FADS1 FADS2) gene cluster and the fatty acid composition of erythrocyte membranes. Br. J. Nutr. 2009;101(1):20–26. doi: 10.1017/S0007114508992564. [DOI] [PubMed] [Google Scholar]
- 212.Neuhofer A., Zeyda M., Mascher D., et al. Impaired local production of proresolving lipid mediators in obesity and 17-HDHA as a potential treatment for obesity-associated inflammation. Diabetes. 2013;62(6):1945–1956. doi: 10.2337/db12-0828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Börgeson E., Godson C. Resolution of inflammation: therapeutic potential of pro-resolving lipids in type 2 diabetes mellitus and associated renal complications. Front. Immunol. 2012;3:318. doi: 10.3389/fimmu.2012.00318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Das U.N. Arachidonic acid and lipoxin A4 as possible endogenous anti-diabetic molecules. Prostaglandins Leukot. Essent. Fatty Acids. 2013;88(3):201–210. doi: 10.1016/j.plefa.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 215.Börgeson E., McGillicuddy F.C., Harford K.A., et al. Lipoxin A4 attenuates adipose inflammation. FASEB J. 2012;26(10):4287–4294. doi: 10.1096/fj.12-208249. [DOI] [PubMed] [Google Scholar]
- 216.Lund T., Mangsbo S.M., Scholz H., et al. Resolvin E1 reduces proinflammatory markers in human pancreatic islets in vitro. Exp. Clin. Endocrinol. Diabetes. 2010;118(04):237–244. doi: 10.1055/s-0029-1241825. [DOI] [PubMed] [Google Scholar]
- 217.Claria J., Dalli J., Yacoubian S., Gao F., Serhan C.N. Resolvin D1 and resolvin D2 govern local inflammatory tone in obese fat. J. Immunol. 2012;189(5):2597–2605. doi: 10.4049/jimmunol.1201272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Hellmann J., Tang Y., Kosuri M., Bhatnagar A., Spite M. Resolvin D1 decreases adipose tissue macrophage accumulation and improves insulin sensitivity in obese-diabetic mice. FASEB J. 2011;25(7):2399–2407. doi: 10.1096/fj.10-178657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Clària J., González-Périz A., López-Vicario C., Rius B., Titos E. New insights into the role of macrophages in adipose tissue inflammation and Fatty liver disease: modulation by endogenous omega-3 Fatty Acid-derived lipid mediators. Front. Immunol. 2011;2:49. doi: 10.3389/fimmu.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.González-Périz A., Horrillo R., Ferré N., et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: a role for resolvins and protectins. FASEB J. 2009;23(6):1946–1957. doi: 10.1096/fj.08-125674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Titos E., Rius B., González-Périz A., et al. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J. Immunol. 2011;187(10):5408–5418. doi: 10.4049/jimmunol.1100225. [DOI] [PubMed] [Google Scholar]
- 222.Unoda K., Doi Y., Nakajima H., et al. Eicosapentaenoic acid (EPA) induces peroxisome proliferator-activated receptors and ameliorates experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2013;256(1-2):7–12. doi: 10.1016/j.jneuroim.2012.12.003. [DOI] [PubMed] [Google Scholar]
- 223.Kesavalu L., Vasudevan B., Raghu B., et al. Omega-3 fatty acid effect on alveolar bone loss in rats. J. Dent. Res. 2006;85(7):648–652. doi: 10.1177/154405910608500713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kesavalu L., Bakthavatchalu V., Rahman M.M., et al. Omega‐3 fatty acid regulates inflammatory cytokine/mediator messenger RNA expression in Porphyromonas gingivalis‐induced experimental periodontal disease. Oral Microbiol. Immunol. 2007;22(4):232–239. doi: 10.1111/j.1399-302X.2007.00346.x. [DOI] [PubMed] [Google Scholar]
- 225.Bendyk A., Marino V., Zilm P.S., Howe P., Bartold P.M. Effect of dietary omega‐3 polyunsaturated fatty acids on experimental periodontitis in the mouse. J. Periodontal Res. 2009;44(2):211–216. doi: 10.1111/j.1600-0765.2008.01108.x. [DOI] [PubMed] [Google Scholar]
- 226.El-Sharkawy H., Aboelsaad N., Eliwa M., et al. Adjunctive treatment of chronic periodontitis with daily dietary supplementation with omega-3 Fatty acids and low-dose aspirin. J. Periodontol. 2010;81(11):1635–1643. doi: 10.1902/jop.2010.090628. [DOI] [PubMed] [Google Scholar]
- 227.Iwasaki M., Yoshihara A., Moynihan P., Watanabe R., Taylor G.W., Miyazaki H. Longitudinal relationship between dietary ω-3 fatty acids and periodontal disease. Nutrition. 2010;26(11-12):1105–1109. doi: 10.1016/j.nut.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 228.Naqvi A.Z., Buettner C., Phillips R.S., Davis R.B., Mukamal K.J. n-3 fatty acids and periodontitis in US adults. J. Am. Diet. Assoc. 2010;110(11):1669–1675. doi: 10.1016/j.jada.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hasturk H., Kantarci A., Goguet-Surmenian E., et al. Resolvin E1 regulates inflammation at the cellular and tissue level and restores tissue homeostasis in vivo. J. Immunol. 2007;179(10):7021–7029. doi: 10.4049/jimmunol.179.10.7021. [DOI] [PubMed] [Google Scholar]
- 230.Gao L., Faibish D., Fredman G., et al. Resolvin E1 and chemokine-like receptor 1 mediate bone preservation. J. Immunol. 2013;190(2):689–694. doi: 10.4049/jimmunol.1103688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Van Dyke T.E. Proresolving lipid mediators: potential for prevention and treatment of periodontitis. J. Clin. Periodontol. 2011;38:119–125. doi: 10.1111/j.1600-051X.2010.01662.x. [DOI] [PubMed] [Google Scholar]
- 232.Hasturk H., Abdallah R., Kantarci A., et al. Resolvin E1 (RvE1) Attenuates Atherosclerotic Plaque Formation in Diet and Inflammation-Induced Atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2015;35(5):1123–1133. doi: 10.1161/ATVBAHA.115.305324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Fredman G., Oh S.F., Ayilavarapu S., Hasturk H., Serhan C.N., Van Dyke T.E. Impaired phagocytosis in localized aggressive periodontitis: rescue by Resolvin E1. PLoS One. 2011;6(9):e24422. doi: 10.1371/journal.pone.0024422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Herrera B.S., Hasturk H., Kantarci A., et al. The impact of resolvin E1 on murine neutrophil phagocytosis in type 2 diabetes. Infect Immun. Am Soc Microbiol. IAI; 2014. pp. 02444–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Van Dyke T.E., Hasturk H., Kantarci A., et al. Proresolving nanomedicines activate bone regeneration in periodontitis. J. Dent. Res. 2015;94(1):148–156. doi: 10.1177/0022034514557331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Mustafa M., Zarrough A., Bolstad A.I., et al. Resolvin D1 protects periodontal ligament. Am J Physiol. Cell Physiol Am Physiol Soc. 2013;305(6):C673–C679. doi: 10.1152/ajpcell.00242.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Freire MO, Van Dyke TE. Natural resolution of inflammation. 2000 doi: 10.1111/prd.12034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. https://clinicaltrials.gov/ct2/show/NCT02342691.
- 239.Sima C, Glogauer M. Macrophage subsets and osteoimmunology: tuning of the immunological recognition and effector systems that maintain alveolar bone. 2000 doi: 10.1111/prd.12032. [DOI] [PubMed] [Google Scholar]
- 240.Bouhlel M.A., Derudas B., Rigamonti E., et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137–143. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 241.Hutchinson J.A., Riquelme P., Sawitzki B., et al. Cutting Edge: Immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J. Immunol. 2011;187(5):2072–2078. doi: 10.4049/jimmunol.1100762. [DOI] [PubMed] [Google Scholar]
- 242.Wang Y., Wang Y.P., Zheng G., et al. Ex vivo programmed macrophages ameliorate experimental chronic inflammatory renal disease. Kidney Int. 2007;72(3):290–299. doi: 10.1038/sj.ki.5002275. [DOI] [PubMed] [Google Scholar]
- 243.Spite M., Clària J., Serhan C.N. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab. 2014;19(1):21–36. doi: 10.1016/j.cmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bell D.S., Dharmalingam M., Kumar S., Sawakhande R.B. Triple oral fixed‐dose diabetes polypill versus insulin plus metformin efficacy demonstration study in the treatment of advanced type 2 diabetes (TrIED study‐II). Diabetes Obes. Metab. 2011;13(9):800–805. doi: 10.1111/j.1463-1326.2011.01408.x. [DOI] [PubMed] [Google Scholar]
- 245.Gale E. The Polypill and Type 2 diabetes. Diabet. Med. 2004;21(s1):8–10. doi: 10.1111/j.1466-5468.2004.1180d.x. [DOI] [PubMed] [Google Scholar]
- 246.Nguyen C., Cheng-Lai A. The Polypill: A Potential Global Solution to Cardiovascular Disease. Cardiol. Rev. 2013;21(1):49–54. doi: 10.1097/CRD.0b013e3182755429. [DOI] [PubMed] [Google Scholar]