

Abstract
Building on the pioneering work of Ho and DeGrado (J Am Chem Soc 1987, 109, 6751–6758) in the late 1980s, protein design approaches have revealed many fundamental features of protein structure and stability. We are now in the era that the early work presaged – the design of new proteins with practical applications and uses. Here we briefly survey some past milestones in protein design, in addition to highlighting recent progress and future aspirations.
Keywords: protein design, computation, review, nanotechnology
INTRODUCTION
Perhaps no one was more surprised than the first protein designers themselves at how easy it was to create novel proteins that adopted the desired fold—more or less. Two complementary approaches were initially employed. DeGrado et al. sought to create four-helix bundle proteins (an apparently “simple fold” that is still a challenge today1) using the minimal number of amino acid types and a systematic approach. First helices were designed based on amino acid propensities. Helix–helix interaction interfaces were then introduced, and the four helices were linked together. At each stage the designs were checked to ensure the desired behavior (Figure 1). This strategy allowed for evaluation, and correction if needed, of each component of the design.2,3
FIGURE 1.
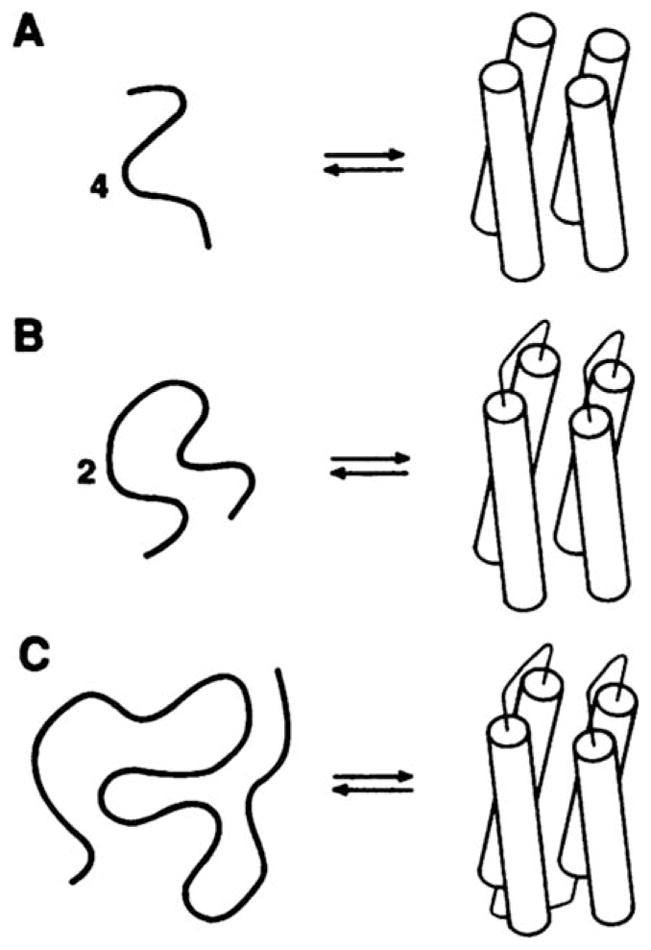
Schematic illustration of a systematic, minimal approach to the design of the four-helix bundle protein. From Regan, L.; DeGrado, W. F. Science 1988, 241, 976–978. Reprinted with permission from AAAS.
The Richardsons et al. adopted a complementary approach. They too sought to create a four-helix bundle protein, but their design goal was to maximize the number of amino acids types used so that the sequence was as “natural” as possible4 (Figure 2). Both groups created monomeric, compact helical proteins as evaluated by simple solution methods, predominantly circular dichroism (CD). It is important to note that both these approaches were considering the “protein folding problem” in reverse. They were not trying to predict what 3D structure a particular sequence would adopt, but rather identify a sequence (by no means the only sequence) that was compatible with a particular fold. In parallel with these early designs, considerable effort was also employed to delineate and experimentally quantify the thermodynamic contributions of intrinsic α-helix and β-sheet forming propensities. The results of such measurements significantly influenced future design.5–9
FIGURE 2.
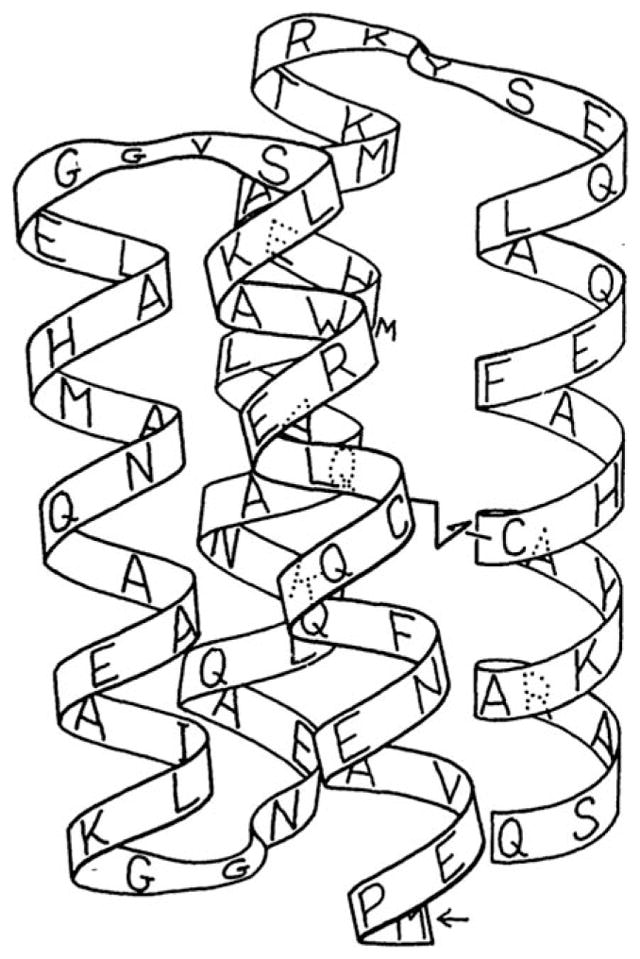
Schematic illustration showing both the sequence and proposed three-dimensional structure of the designed four-helix bundle protein, Felix. From Hecht, M. H.; Richardson, J. S.; Richardson, D. C.; Ogden, R. C. Science 1990, 249, 884–891. Reprinted with permission from AAAS.
Following these early successes, the interests of the protein design field transitioned to creating proteins with “native-like” thermodynamic properties. The zeitgeist was that although it was possible to design associating helices, they were perhaps associating in a “molten-globule like fashion,” rather than truly recapitulating the packing and associated thermodynamic properties that are characteristic of natural proteins. Subsequently, Munson et al. explicitly tracked how the thermodynamic behavior of proteins changes with hydrophobic core redesigns.10
At the same time, researchers employed coarse-grained models to computationally design protein cores, with the pervading concept being that the residues in the core must be hydrophobic and pack efficiently.11–13 Ponder and Richards promoted the concept of amino acid side-chain rotamers—that side-chains adopt a limited subset of dihedral angles. They demonstrated that rotamer and hydrophobicity constraints plus strict limits on the free volume greatly restricted the number of amino acid combinations that are compatible in the core of a small protein. Desjarlais and Handel used this type of approach with a “custom rotamer library” to redesign the core of small proteins and to subsequently make and determine the structure of repacked ubiquitin.14 Dahiyat and Mayo also used a rotamer-based approach in their Optimization of Rotamers By Iterative Techniques (ORBIT) design software. In addition, they classified every amino acid position into one of three categories: buried, surface, or boundary. Each class was given a different scoring function, which included an atomic solvation potential that favored the burial and penalized the exposure of nonpolar surface area (Figure 3 left).15
FIGURE 3.

Comparison of ORBIT and RosettaDesign design strategies. Left: In ORBIT a backbone template with known coordinates is chosen. Amino acid positions are classified into three categories: core, boundary, and surface. Dead-End Elimination is implemented to reduce the combinatorial search of energetically allowed rotamers and to obtain the global minimum energy conformation (GMEC). Lastly, Monte Carlo is used to randomly change rotamers of the GMEC sequence to sample local low-energy configurations. Right: In RosettaDesign an ensemble of de novo backbones is generated using peptide fragments that match the desired backbone. This ensemble is subjected to iterative rounds of fixed backbone sequence optimization and flexible backbone energy minimization. Amino acid sequences are optimized by sampling different residues and rotamers with a Monte Carlo search protocol. Backbones are optimized by perturbing main-chain torsion angles, cycling through rotamers for side-chains with increased energies, and minimizing backbone energy at insertion sites according to the Metropolis criterion. The table of PDB fragments in the right column is reprinted with permission from Kaufmann, K. W.; Lemmon, G. H.; Deluca, S. L.; Sheehan, J. H.; Meiler, J. Biochemistry 2010, 49(14), 2987–2998. © 2010 American Chemical Society.
Baker et al. designed and experimentally validated the first protein fold not found in nature, “Top7,” using their Rosetta-Design software.16,17 Their strategy was to construct the protein scaffold using three- and nine-residue fragments taken from the Protein Data Bank (PDB). The best combinations were then selected via Monte Carlo optimization of a number of energetic terms including hydrophobic burial, β-strand hydrogen bonding, and side-chain rotamers (Figure 3 right).
In addition to designing “native-like” proteins, protein scientists also began to introduce new functions into proteins, with much early emphasis placed on the design of metal ion binding sites. The reasons were both pragmatic and exciting: pragmatic because there are many spectroscopic methods that can be used to characterize binding geometry in solution, and exciting because many activities are associated with metal ions in proteins, including catalysis, electron transfer, and enhanced stability.18,19
The design of protein-based catalysts (retro-aldol, Kemp elimination, and stereoselective Diels-Alder reactions) followed, based on creating a binding site for the transition state of the desired reaction.20–23 The resulting designs exhibited modest activity, and most have been subsequently improved by random mutagenesis and selection.21 The power of randomization and selection to improve initial designs has been repeatedly demonstrated. For example, of 88 designs for hemagglutinin binding modules, only two displayed any binding activity, but the low binding affinity of those designs were increased substantially by affinity maturation.24 Remarkably, DeGrado et al. used chemical intuition to create a Kemp eliminase, with activity comparable to that of initial computational designs, by introducing a single Glu residue into a hydrophobic cavity of a small protein, and thus perturbing its pKa significantly.25
DeGrado and colleagues have advanced “knowledge-based” approaches for the design of constructs for diverse applications: transmembrane-binding peptides, surface-organizing peptide superstructures, and protein crystals. Their computed helical anti-membrane protein (CHAMP) protocol enabled the design of α- and β-helical peptides that bind specifically to a transmembrane (TM) helix of a target protein (Figure 4).26–28 Yin et al. leveraged known membrane-protein structures to create backbone templates and a membrane depth-dependent knowledge-based potential to sample the “relevant” sequence and rotamer space. This strategy has resulted in several designs whose binding has been experimentally verified.26,27 The association of designed CHAMP transmembrane peptides with their target integrin transmembrane peptide has been verified in micelles, using fluorescence resonance energy transfer (FRET). More importantly, the specificity and efficacy of CHAMP peptides interacting with their target integrins was also demonstrated via a biological activity assay. Designed CHAMP peptides specifically interact with, and thus stimulate the activity (plate aggregation or adhesion) of only the integrin whose transmembrane domain they were designed to interact with.
FIGURE 4.
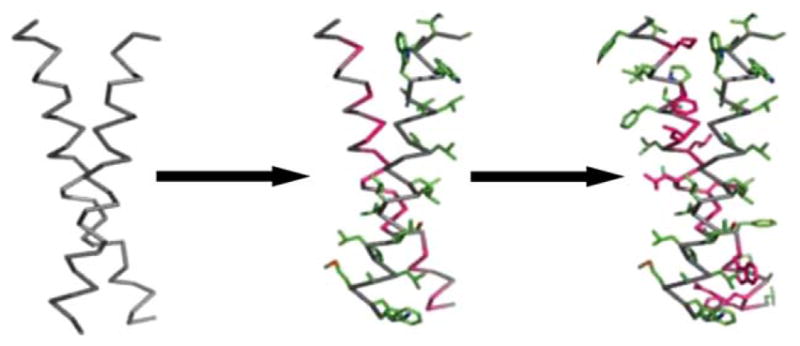
The CHAMP design process. A transmembrane helix-helix backbone pair with the desired geometry is selected from a database of membrane-protein structures (left). The original amino acid side-chains are discarded, and the helical backbones are extended to span the full length of a membrane. Next, the sequence from the target TM protein is “threaded” onto either one of the helices—in this example, the right helix with green side-chains (middle). Last, the sequence and side-chain configurations for the anti-TM peptide (represented by the left helix) is chosen via iteration over amino acids and rotamer re-packing (right). From Yin, H.; Slusky, J. S.; Berger, B. W.; Walters, R. S.; Vilaire, G; Litvinov, R.I.; Lear, J. D.; Caputo, G. A.; Bennett, J. S.; DeGrado, W. F. Science 2007, 315(5820), 1817–1822. Reprinted with permission from AAAS.
Grigoryan et al. implemented a set of rule selections to assemble a superstructure of peptides that coat single-walled nanotubes (SWNTs).29 They matched the periodicity of an α-helix to the periodic pattern surface of a SWNT via Ala Cβ methyl contacts to form a supercoil of α-helical coiled coils. In the presence of mixed types of SWNTs, the designed peptides preferentially sequestered the targeted nanotube species to produce stable aqueous suspensions. Lanci et al. implemented a similar methodology for the de novo design of a peptide that self-assembles into a P6 protein crystal.30 After determining the optimal crystalline array for a homotrimeric parallel coiled-coil and designing the sequences, they obtained a protein crystal that matched the computational design to sub-Å precision.
Current Computational Methods in Protein Design
Why has most of the computational design work employed “knowledge-based” potentials rather than potentials based solely on molecular mechanics? Classical molecular mechanics force-fields, which employ simplified interaction potentials, offer computational speed and are straightforward to implement. Typical simplifications include employing pairwise interaction potentials, treating covalent bonds as Hookean springs and using Lennard-Jones-like potentials to model van der Waals, hydrophobic, and hydrogen bonding interactions. Strengths of this approach are that it is easy to build upon and straightforward to apply in scalable computer applications. Disadvantages include the artificial separation of interactions that are deeply intertwined, including van der Waals interactions, hydrogen bonding and hydrophobic interactions. This can result in “double counting,” difficulty in calibrating the relative energetic contributions of different types of interactions and a large number of unknown parameters that must be determined. In this context, improvements or updates to widely used software packages31–33 can be classified as one of two types: (1) tweaking the relative magnitudes of different energy terms - often driven by improved experimental data, and (2) the addition of new energy terms, for example “knowledge-based” potentials that ensure that the main-chain and side-chain dihedral angles preferentially sample the observed distributions from the PDB.34
Although the global results for protein simulations and the prediction of structure from sequence are ever improving,35 their limitations have been documented, and there have been a number of suggestions for their improvement. In a recent assessment of different force-fields and their performance in predicting peptide side-chain conformations,36 the authors demonstrated that different force-fields yield significantly different predictions for χ1 side-chain dihedral angle distributions for virtually every amino acid (Figure 5). In another study, Pande et al. used 524 experimentally based metrics to assess the performance of different force-fields by running trajectories on the small protein ubiquitin. They found that different force-fields and different water models yield significantly different results37 (Figure 6). Hermans et al. compared the performance of different force-fields in reproducing the observed backbone dihedral angles of Ala and Gly.38 They concluded that none of the current classical force-fields performed satisfactorily, and suggested that quantum mechanical (QM) effects must be included to properly predict backbone conformations. Currently, such an approach is impractical for protein design. Moreover, QM-based approaches require the initial input of knowledge-based potentials (such as CMAP) for them to reliably reproduce values observed in protein structures in a reasonable time.38,39
FIGURE 5.

Comparing the performance of different force-fields in predicting side-chain dihedral angle distributions. Comparison of the relative populations of χ1 dihedral angles (where t =120° <χ1 <240°, g+=240° <χ1 <360°, and g−=0° <χ1 <120°) for different amino acids resulting from simulations of short peptides in solution, using different force-fields (CHARMM22/CMAP; FF03; FF99SB; OPLS-AA/L). The relative occupancy of each side-chain dihedral is indicated by the size and color of the associated square (see legend). Each row shows the predictions for that particular amino acid type given by the specified force-field. A good “consistent” prediction would be if each force-field gave the same result. Lys is quite good. By contrast, Val is particularly bad since its results differ with each force-field. Reprinted with permission from Vymětal, J.; Vondrášek, J. Journal of Chemical Theory and Computation 2013, 9, 441–451.© 2013 American Chemical Society.
FIGURE 6.

Comparison of force-field performance in simulations of the 78 amino acid protein, ubiquitin. 524 different parameters were used in the assessment of the score. Each column corresponds to a given force field (as indicted) and each row corresponds to a different model for explicit solvent (as indicated). For each combination of force field and water model, χ2 quantifies the agreement between simulation and experiment based on the 524 parameters, indicated by the color of the square. A smaller value of χ2 (darker blue) indicates a greater agreement between prediction and experiment. Note the differences between the different force fields with the same water models, and between different water models with the same force field. Reprinted with permission from Beauchamp, K. A.; Lin, Y. S.; Das, R.; Pande, V. S. Journal of Chemical Theory and Computation 2012, 8, 1409–1414. © 2012 American Chemical Society.
Regan et al. have taken an alternative approach to computational protein design. They have shown that simple steric-based methods, as pioneered by Ramachandran et al.,40 predict the backbone and side-chain dihedral angle combinations observed in proteins better than more elaborate techniques (Figure 7). They therefore advocate a “back to basics” approach to protein structure analysis and design. Rather than including many different terms in the molecular mechanics force-field, they argue that only a minimal set of steric interactions and stereochemical constraints should be imposed to capture the defining features of protein structure. This method has proven very effective. Not only does it provide predictions for the backbone and side-chain conformations that are observed in protein crystal structures and by NMR in solution,41–43 it also allows a mechanism to be proposed for transitions between different side-chain dihedral angle conformations.44
FIGURE 7.

Comparison between different probability distributions P(ψ,ϕ) for the backbone dihedral angles φ and ψ obtained from molecular dynamics simulations of an Ala dipeptide mimetic using different versions of the CHARMM and Amber force-fields, their associated optimized water models, and with and without the “ILDN-NMR” and “CMAP” dihedral angle potential corrections. Outlined are the Ramachandran hard sphere limits for 110°. (a) Amber99sb +TIP4P-Ew, (b) Amber99sb-ILDN-NMR +TIP4P-Ew, (c) CHARMM27 +TIP3SP, and (d) CHARMM27-CMAP +TIP3SP. Panels (e) and (f) correspond to the Alanine phi/psi distributions different subsets of the PDB. Note the different predictions in panels (a)–(d) compared with the experimental values in panels (e),(f). Reprinted with permission from Caballero, D.; Määttä, J.; Zhou, A. Q.; Sammalkorpi, M.; O’Hern, C. S.; Regan, L. Protein Science 2014, 23, 970–980.
Protein Designs for Nanotechnology
There is great potential to harness some of the defining properties of proteins for materials applications.45 Proteins that can self-assemble into higher order structures are of particular interest and can be used to construct both amorphous materials and discrete assemblies. The unique features associated with protein-based materials make them attractive candidates for biomedical applications such as drug delivery and tissue engineering. Additionally, large, discrete protein structures that can be decorated at exact positions would facilitate several applications, including pathway engineering, sensors, and vaccines.
Protein-based hydrogels confer a number of advantages over synthetic materials for biomedical applications: (1) the features required for 3D percolation and gelation are precisely encoded by the sequence, which specifies the structure; (2) genetic engineering to create virtually any desired sequence is relatively straightforward; (3) exquisite stimuli-responsiveness can readily be controlled by appropriately engineering the interactions between protein building blocks.
Olsen et al. designed hydrogels whose formation is based on the association of α-helices into coiled-coils.46 Subsequently, Olsen et al. elaborated on these designs to create a thermosensitive hydrogel that is liquid at low temperatures (4°C) but which exhibits enhanced stiffness and durability at physiological temperature (37°C).47 There are two key components to this design: the coiled-coil based shear thinning hydrogel midblock, and endblocks comprised of the thermoresponsive polymer poly(N-isopropylacrylamide) (PNIPAM) (Figure 8). Such a shear thinning hydrogel that undergoes a transition to a more rigid, reinforced network at physiological temperatures could be used for injection, for example, in tissue repair.
FIGURE 8.
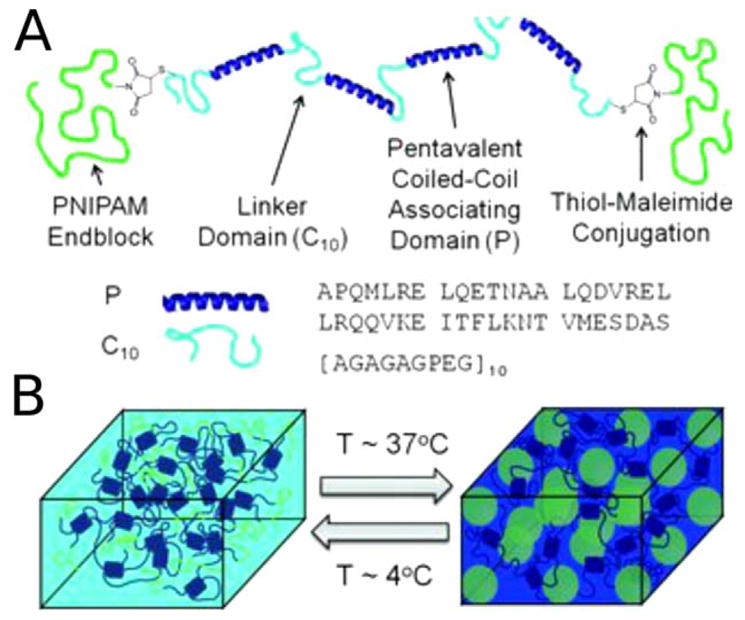
Creation of a temperature-responsive hydrogel, based on a “dual network” design. (a) Illustration of the components of the dual polymer design comprising PNIPAM ends (green), helical coiled coils (dark blue), and linker regions (pale blue). (b) Schematic representation of the temperature-dependent reinforcement of shear-thinning hydrogels by the PNIPAM triblock copolymer domains. At 4°C, the coiled coils (dark blue) fold and associate, while the PNIPAM domains (green lines) do not interact. However, at 37°C the PNIPAM blocks associate (green spheres) and reinforce the hydrogel network. Reprinted with permission from Glassman, M. J.; Chan, J.; Olsen, B. D. Advanced Functional Materials. 2013, 23, 1182–1193.
Temperature is an attractive stimulus because it is straightforward to apply. The key is to engineer stimuli-responsiveness in a regime that is compatible with physiological temperature. Woolfson and colleagues designed hydrogels using α-helical peptides with thermosensitive properties encoded by the types of interactions between entangled helical fibrils.48 The propensity for the hydrogel to become stronger or weaker after heat application is dependent on whether the fibril network is formed through hydrophobic (increase in strength with increasing temperature) or hydrogen bonding (decrease in strength with increasing temperature) interactions.
A major breakthrough in protein design that allows easy expression of branched protein building blocks was made by Howarth and colleagues. Protein expression is typically limited to linear topologies, but the development of SpyCatcher/Spy-Tag technology has changed that. SpyTag and SpyCatcher are peptide and protein components, respectively, that originated from splitting a fibronectin-binding domain of a bacterial adhesin. This fibronectin-binding domain spontaneously forms an intramolecular covalent bond in nature, and researchers were able to maintain this activity whilst dividing the protein into two separate parts49 (Figure 9, panel A). By genetically encoding SpyTag and SpyCatcher in constructs of interest, diverse topologies can be created50 (Figure 9, panel B). Arnold, Tirrell and colleagues have exploited this technology for the construction of new protein-based materials.51 A covalent hydrogel network was produced as a result of isopeptide bond formation by genetically encoding SpyTag and Spy-Catcher into elastin-like protein (ELP) constructs (Figure 10).
FIGURE 9.

A: Ribbon representation of the SpyTag-SpyCatcher complex assembly. The Lys residue (red) on the 12 kDa SpyCatcher protein spontaneously forms an isopeptide bond with the Asp (red) on the 13-reside SpyTag peptide. B: Schematic illustration of the diverse protein topologies possible by the SpyTag (red triangle)/SpyCatcher (purple crown) technology. Reprinted with permission from (A) Zakeri, B.; Fierer, J. O.; Celik, E.; Chittock, E. C.; Schwarz-Linek, U.; Moy, V. T.; Howarth, M. Proc Natl Acad Sci USA 2012, 109, E690–697, and (B) Zhang, W. B.; Sun, F.; Tirrell, D. A.; Arnold, F. H. Journal of the American Chemical Society 2013, 135, 13988–13997. © 2013 American Chemical Society.
FIGURE 10.
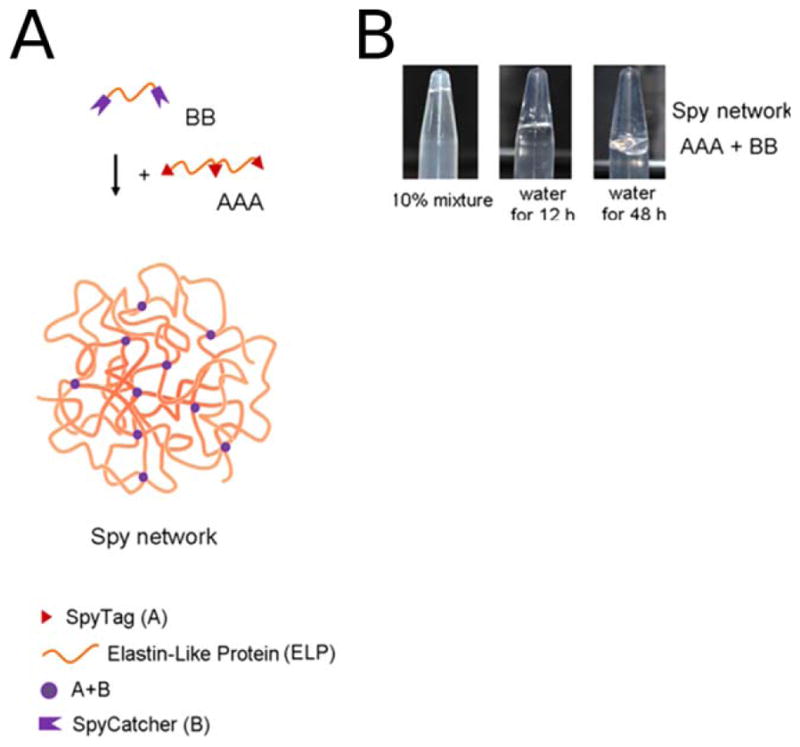
A: Cartoon of covalent hydrogel formation using SpyTag/SpyCatcher technology. Three SpyTag sequences (red triangles) are connected by an elastin-like-protein (ELP) sequence (orange strand) to make the AAA construct. Two SpyCatcher units (purple crowns) are joined by an ELP linker to form the BB construct. Mixing these two proteins results in a covalent Spy network. B: A photograph of the formed covalent Spy network. Mixing 10% wt aqueous solutions of AAA and BB in equimolar amounts of binding sites yields the hydrogel shown. Upon addition of water, the Spy network swells by 3000% after 12 h and continues to be swollen after 48 h. FROM PNAS: Sun, F.; Zhang, W. B.; Mahdavi, A.; Arnold, F. H.; Tirrell, D. A. Proc Natl Acad Sci USA 2014, 111, 11269–11274.
The development of “smart” protein-based hydrogels that can respond to various stimuli has unique potential for controlled substance release. Grove et al. demonstrated the reversible formation of self-assembling hydrogels using a protein-peptide binding interaction to encode the macroscopic properties of the gel.52,53 Concatenated arrays of tetratricopeptide repeats (TPR) were mixed with multivalent “star PEG” based arrays of cognate peptide to form noncovalently cross-linked gels (Figure 11). Because the binding interactions that form the cross-links are pH- and ionic strength-dependent, the gel dissolves and reforms in response to these stimuli. Moreover, the pH dependence of dissolution and cargo release is compatible with the decrease in pH associated with both the microenvironment of solid tumors54 and the intracellular lysosomal pathway.
FIGURE 11.

A schematic illustration of the reversible formation of a TPR-peptide based hydrogel. Consensus TPR “binding” modules (dark blue) that bind to the cognate peptide are concatenated with TPR “spacer” modules (pale blue) that do not bind the peptide so that binding sites are arrayed on different faces of the cylinder. Peptide cross-linkers were constructed by chemical attachment of the cognate peptide to functionalized 4-armed star PEG molecules (black lines with red termini). Mixing the TPR arrays with PEG-peptide cross-linkers in a stoichiometric ratio of 1:2 results in hydrogel formation, which can be reversed by increasing ionic strength or decreasing pH.
DNA origami has enabled the design of an impressive diversity of 2- and 3D structures.55 Incorporating function has proven to be more difficult. By contrast, designing structures for “protein origami” is more involved, but their functionalization is relatively straightforward. Jerala et al. took advantage of the specificity of association between coiled-coil building blocks to form a single-chain polypeptide structure that folds into a polyhedron.56 The design employed six different pairs of coiled-coils (Figure 12). A linker sequence was chosen that included residues that would enhance flexibility and disrupt helix formation—Ser-Gly-Pro-Gly. Another crucial component of the design is the orthogonality of the pairs—unintentional cross-reactivity between different coiled-coil monomers would prevent the proper assembly of the tetrahedron. The resulting 3D structure was imaged by atomic force microscopy (AFM), and the proximity of the N- and C-termini at the same vertex was confirmed by a split-fluorescent protein assay. Protein origami is attractive because such structures can be easily functionalized for use in pathway engineering, difference imaging, and novel vaccines.
FIGURE 12.

Design of a tetrahedron/trigonal pyramid using coiled-coils assembly. (a) Cartoon illustrating the pyramid components—sets of heterodimeric and homodimeric parallel and anti-parallel coiled-coils. The 12 individual peptide sequences are concatenated in the indicated order, with each sequence separated by the flexible linker Ser-Gly-Pro-Gly. Grey lines indicate the interacting pairs. (b) Schematic of the desired tetrahedron structure. Arrows indicate the direction of the helices in the coiled-coil pairs. Reprinted by permission from Macmillan Publishers Ltd: [Nature Chemical Biology] Gradisar, H.; Bozic, S.; Doles, T.; Vengust, D.; Hafner-Bratkovic, I.; Mertelj, A.; Webb, B.; Sali, A.; Klavzar, S.; Jerala, R. Nat Chem Biol 2013, 9, 362–366, © 2013.
Woolfson and colleagues created self-assembled cage-like particles (SAGEs) that form spheres of roughly 100 nm in diameter.57 Noncovalent heterodimeric and heterotrimeric coiled-coils were employed as building blocks for the design, where different coils were connected by asymmetric disulfide bonds to form hubs that assemble into a hexagonal array upon mixing (Figure 13, panel A). Interestingly, instead of forming the expected flat assembly based on the hexagonal design, the structures assembled into closed spheres (Figure 13, panels B and C). Modeling suggested that the hubs are actually wedge-shaped instead of perfect tripods with arms angled at exactly 120°. The largest reported assembly with structural validation, a 24-subunit protein cube, was designed by Yeates et al.58 Their design strategy involved making fusions between natural dimeric and trimeric proteins. The particular proteins used were chosen so that the angle of the interface would satisfy the requirements for cube formation when propagated. The trimeric protein 2-keto-3-deoxy-6-phosphogalactonate (KDPGal) aldolase and dimeric N-terminal domain of FkpA protein were connected by a flexible linker (Figure 14). When mixed, they self-assembled into a porous cube with an outer diameter of 225 Å and an inner diameter of 132 Å, as determined by X-ray crystallography. The structure was additionally validated by negative stain electron microscopy and small angle X-ray scattering (SAXS) analysis. Like the SAGE particles vide supra, the large cavities in these protein assemblies have potential applications in delivery of molecular cargo.
FIGURE 13.

Design of “spheres” using coiled coil assemblies (SAGE). A: Design of the hubs that self-assemble to form SAGE molecules. The heterotrimeric coiled-coil (CC-Tri3, green) connects to either CC-Di-A (red) and CC-Di-B (blue) via asymmetric sulfide linkages (purple lines) to form hub A (red-green) and hub B (blue-green). Mixing of hub A and hub B yields a hexagonal array by the formation of heterodimeric coiled-coils between CC-Di-A and CC-Di-B. EM (B) and LMFM (C) images of a hydrated SAGE molecule. From Fletcher, J. M.; Harniman, R. L.; Barnes, F. R.; Boyle, A. L.; Collins, A.; Mantell, J.; Sharp, T. H.; Antognozzi, M.; Booth, P. J.; Linden, N.; Miles, M. J.; Sessions, R. B.; Verkade, P.; Woolfson, D. N. Science 2013, 340, 595–599. Reprinted with permission from AAAS.
FIGURE 14.

Design of a 24-subunit protein cube. (a) The designed fusion protein with the KDPGal aldolase trimer (green) connected to the dimeric domain of FkpA protein (orange) by the helical linker (blue). The purple and cyan lines represent the two-fold and three-fold axes of symmetry, respectively. (b) A cartoon model of the 24-subunit cage design. The two-fold and three-fold axes of symmetry in the cube are shown in purple and cyan, respectively. Reprinted by permission from Macmillan Publishers Ltd: [Nature Chemistry] Lai, Y. T.; Reading, E.; Hura, G. L.; Tsai, K. L.; Laganowsky, A.; Asturias, F. J.; Tainer, J. A.; Robinson, C. V.; Yeates, T. O. Nat Chem 2014, 6, 1065–1071, © 2014.
Protein Designs That Function In Vivo
To fully understand protein function in the cellular milieu, it is desirable to be able to study and manipulate protein activity in living cells. Since Green Fluorescent Protein (GFP) was first cloned two decades ago,59 fluorescent proteins have become a powerful and omnipresent tool in biology. The potential of GFP to study protein expression and localization was recognized early on, but several important limitations had to be overcome before it could be used routinely in biological applications. Protein design has played an important role in overcoming these obstacles and in extending the in vivo potential of fluorescent proteins.
Wild-type fluorescent proteins are often oligomers, a property that could interfere with the natural activity of a protein of interest. A common design strategy to prevent oligomerization is to introduce unfavorable electrostatic interactions to disrupt the subunit interfaces. This tactic was successfully used to create the monomeric mFruit series of fluorescent proteins,60 which was expanded recently with mPapaya,61 in addition to the monomeric version of the extremely bright green vivid verde fluorescent protein (mVFP).62 New fluorescent protein colors, which allow for more proteins to be tagged and followed in the same cell, have been created by randomly mutating GFP and screening for altered fluorescence emission characteristics63 (Figure 15). A similar method was also used to identify mutations that increase brightness and shift excitation peaks,64 allow GFP to fold faster,65 and introduce a number of additional properties useful for a wide range of applications.
FIGURE 15.

Fluorescent proteins with a wide spectral range of excitation and emission. Reprinted with permission from Tsien, R. Y. Angewandte Chemie International Edition 2009, 48, 5612–5626.
Engineered versions of fluorescent proteins, such as “split GFP” and “split dsRed” have also been developed to study protein–protein interactions in vivo66–68 (Figure 16). As the name implies, these assays use versions of fluorescent proteins that have been split into N- and C-terminal halves. On their own, the two protein halves do not interact. Attaching proteins that bind to each other brings the two chains together, and the fluorescent protein is reconstituted. Libraries of potential protein–protein interacting partners can be screened by this assay. An advantage of the split GFP assay is that protein-binding partners can be examined in the context of their native cellular environment, whether it be E. coli, yeast, worm, or mammalian cells.
FIGURE 16.
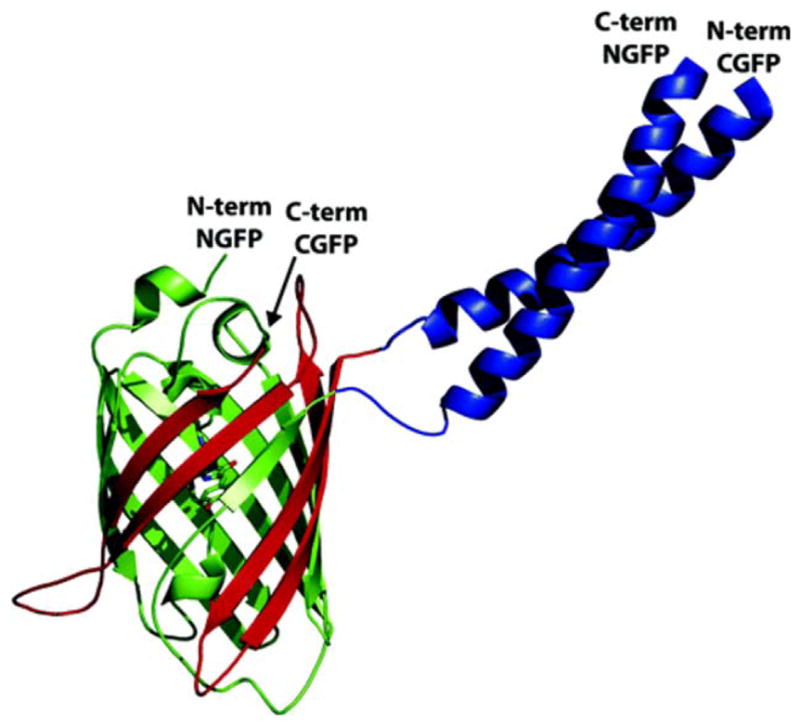
Schematic illustration of the split GFP system used to identify protein-protein interactions. GFP is split into N-terminal (green) and C-terminal (red) halves, which do not associate on their own. Attaching two interacting proteins (depicted here are a designed pair of coiled-coil dimers) forces the two halves to associate, producing the native fold and fluorophore. Reprinted with permission from Magliery, T. J.; Wilson, C. G.; Pan, W.; Mishler, D.; Ghosh, I.; Hamilton, A. D.; Regan, L. Journal of the American Chemical Society 2005, 127, 146–157. © 2005 American Chemical Society.
In signaling networks a single protein may interact with multiple targets, and teasing apart the different activities can be a challenge. Several protein design strategies have been developed to address this issue. The simplest approach is to treat the binding and activating domains of a signaling protein as independent modules that can be mixed and matched. By fusing domains from different proteins, it is possible to create novel networks that possess different input–output combinations. Park et al. transplanted new protein binding domains onto the MAP kinase scaffolding domain Ste5 to create a strain of yeast that responds to stimulation by mating hormone with an osmolarity response.69 Similarly, Howard et al. created a novel adaptor protein by fusing protein binding domains from different signaling networks to produce a new strain of cells that underwent apoptosis in response to factors that normally trigger cell growth and survival.70 These fusion proteins allow scientists to separate different activities of a signaling protein in order to systematically investigate each function.
Another approach to dissecting protein-protein interaction networks and pathways is to block the interaction of a protein with a particular binding partner. This can be accomplished without directly mutating the target proteins themselves by expressing a second protein that binds to a specific surface on the protein of interest. “Native” antibodies are not appropriate for such intracellular applications because they contain disulfide bonds that are not stable in the reducing environment of the cell. As a result, other protein scaffolds that lack disulfide bonds have been used to create functional intracellular binding modules. Amstutz et al. used site-specific randomization in combination with an in vitro binding screen to isolate an ankyrin repeat protein that binds the kinase aminoglycoside phosphotransferase (APH) with high affinity. This ankyrin module binds and inhibits APH both in vitro and in vivo.71 Cortajarena et al. randomized the substrate-binding residues of a TPR domain and sorted variants using a split GFP assay and FACS sorting in E. coli to isolate TPR variants that bind the human protein DSS1. Overexpressing the DSS1-binding modules in yeast phenocopied a Sem1 deletion mutant (Sem1 is the yeast homologue of Dss1). Because Sem1/DSS1 has been proposed to interact with a number of different partners, and its function in yeast is still unknown, having a TPR module that can inhibit a particular region of interaction is a powerful new tool.72
An elegant example of a protein design approach to delineate protein function in vivo is seen in the work of Shokat et al.73 By designing a protein kinase with altered ATP-binding specificity they were able to identify substrates of that kinase. The strategy was to first introduce mutations in the kinase that enlarged the substrate-binding pocket such that bulky ATP derivatives could be bound. The binding pocket on the wild-type kinase is too small to bind the bulky ATP derivative. Thus, by supplying radioactively labeled ATP derivatives to cells expressing the mutant kinase, only substrates of that particular kinase would be labeled (Figure 17).
FIGURE 17.
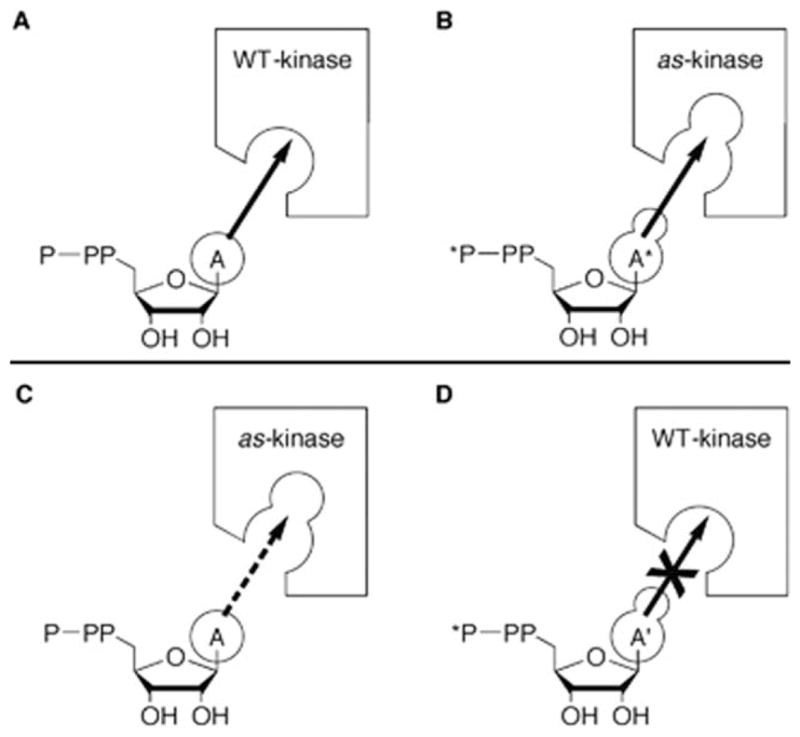
Schematic illustration of the method developed by Shokat et al. to identify kinase substrates. Analogue sensitive (As)-kinase contains a mutation in the ATP binding domain that enables it to function with both ATP (A) and a bulky ATP derivative (A*), while wild-type (WT) kinase can only functional with A. Hodgson, D. R.; Schröder, M. Chemical Society Reviews 2011, 40, 1211–1223. Reproduced with permission from The Royal Society of Chemistry.
A current goal in protein design is to create new methods to control protein activity in vivo. These methods would be a useful tool for studying processes that occur on fast timescales, as well as for rewiring existing pathways. Ideally, the stimulus would be fast, induce a high relative change in activity, and affect only the desired target. A creative strategy is to use small molecules to control protein localization. One way to achieve this is by attaching the ligand-binding domain of a nuclear hormone receptor to the protein of interest. Hormone receptors contain several conserved domains, among which are a ligand-binding domain (LBD) and a DNA-binding domain (DBD). In the absence of hormone, the LBD binds the chaperone Hsp90 and prevents entry of the receptor into the nucleus. Binding of hormone to the LBD induces a conformational change that releases the receptor, allowing nuclear entry and binding of the DBD to its cognate sequence.74 Hybrid receptors have been created that recognize a variety of hormones and DNA sequences.75,76 Protein localization can also be controlled by taking advantage of proteins that interact only in the presence of a small molecule. Many of these strategies are based on rapamycin, a small molecule that can simultaneously bind the FK506 binding protein (FKBP) as well as the rapamycin binding domain of mTOR (FKBP-rapamycin binding domain or FRB) to form a ternary complex. By attaching either FRB or FKBP to the protein of interest and the other to a transmembrane protein, it is possible to induce protein localization to a specific subcellular compartment by adding rapamycin. This strategy can either be used to induce the activity of a protein whose substrate lies at the plasma membrane77 or to sequester proteins away from cytosolic substrates.78
Small molecule-based methods are effective, but they are restricted by molecular diffusion through the plasma membrane and cell walls. In principle, light would be an ideal stimulus because illumination can be rapidly switched on or off, resulting in essentially instantaneous addition or removal of signal. Moreover, it can reach any part of the cell, a property that is not necessarily true for small molecules. Most strategies modify natural plant photoreceptors to create light sensitive proteins. One popular choice is the light oxygen voltage domain from phototropin (LOV2), which consists of a core flavin mononucleotide-binding domain followed by a C-terminal Jα helix. Illumination with blue light triggers formation of a covalent bond between the excited flavin and a cysteine residue in the core domain, which induces a conformational rearrangement that results in unfolding of the Jα helix. Renicke et al. fused a short, synthetic, destabilizing domain from murine ornithine decarboxylase (cODC1) to LOV2 to create a photosensitive degron.79 cODC1 is degraded through an ubiquitin independent mechanism, one of the requirements for which is exposure of a short unstructured region. Attaching cODC1 immediately after the Jα helix produced a protein that is only degraded when illuminated with blue light (Figure 18). By varying the length of cODC1 and its attachment point to LOV2, Renicke et al. were ultimately able to isolate a module that rapidly and extensively reduced protein target concentrations upon illumination.
FIGURE 18.

Cartoon depiction of the LOV2-based photosensitive degron (psd). Psd consists of a fusion between the LOV2 photosensitive domain and the proteasome binding peptide cODC. In the dark state, the Jα helix on the LOV2 domain is folded and associates with cODC, preventing it from interacting with the proteasome. Absorption of blue light triggers unfolding of Jα, releasing the cODC peptide to bind the proteasome and induce degradation of the psd module, along with any protein fused to it. Reprinted from Chemistry & Biology, Vol 20, Renicke, C.; Schuster, D.; Usherenko, S.; Essen, L. O.; Taxis, C., “A LOV2 Domain-Based Optogenetic Tool to Control Protein Degradation and Cellular function,” 619–626, © 2013, with permission from Elsevier.
Strickland et al. also took advantage of the LOV2 domain to create TULIPs (tunable, light-controlled interacting protein tags).80 A short peptide recognized by Erbin PDZ (ePDZ, an engineered protein binding domain) was placed downstream of the Jα helix and designed so that a portion of the peptide would be incorporated in the α-helix under dark conditions. In the dark state, the peptide ligand is partially in a helical conformation, and binding to ePDZ is blocked. Illumination with blue light triggers Jα unfolding, and frees the peptide to interact with ePDZ. By fusing ePDZ to a transmembrane protein, it was possible to induce protein localization to the plasma membrane by illuminating cells with blue light. A number of peptide mutations were identified during the design process that varied in affinity for ePDZ. When combined with the ePDZ variants that bind peptides with different strengths, the authors were able to create a series of interaction pairs that covered a wide spectrum of binding affinities.
Future Directions for Protein Design
We still do not have the theoretical or computational tools to design any protein structure or any protein-protein interaction interface on demand. Although different approaches have had some successes, it is not uncommon for a randomization plus screen or selection step to be required after the initial design to achieve the desired activity.23,24 Currently, it is often easier to skip the design process entirely, and obtain the activity of interest solely by randomization and selection.72,81 There are a number of factors underlying these struggles, the most significant of which are outlined below.
In computational protein design, a common approach is to add knowledge-based terms to existing force-fields [e.g., the cross-term energy correction map (CMAP) correction for the backbone dihedral angle distributions in CHARMM] so that they can recapitulate the experimentally observed backbone and side-chain dihedral angle distributions.34 However, this strategy has disadvantages. For example the PDB includes many more α-helices than β-sheets, so the CMAP correction necessarily overemphasizes α-helical structures. To solve this issue, we need unbiased experimental measurements of intrinsic backbone and side-chain conformational propensities.82–85
In a recent community-wide assessment of the current challenges in designing protein–protein interfaces, the inability to accurately model certain types of interactions, such as electrostatics and hydrogen bonding, was mentioned as a major limitation.86 The issue of how to appropriately balance electrostatic and solvation effects has recently been discussed in detail.87 A related question is whether or not quantum mechanical effects—such as the polarizability of electron clouds or solvation energies of compounds—are necessary to calibrate classical potentials.
How to best model the interplay between local steric interactions and backbone motion remains an unsolved problem. Consider the example of correctly balancing the energetics of introducing a Val residue versus an Ala residue at a certain position. Accommodating a Val necessitates a small movement of the backbone, but its side chain can interact favorably with a nearby hydrophobic patch. On the other hand, insertion of an Ala requires no backbone movement but abolishes the hydrophobic interaction. The correct balancing of such energetics continues to be a challenge. This scenario was encountered in state-of-the-art design work by Fleishman et al.,24 in which initial low-affinity designs for hemagglutinin-binding proteins were improved by randomization and selection. One of the mutations that resulted in a 25-fold increase in affinity was an Ala to Val mutation, in a situation similar to that described above. Other similar examples have been reported,12,88,89 and several strategies are being developed to address this issue.88,90–94
Correctly ranking computational designs remains a challenge for several reasons. First, when both physics- and knowledge-based terms are included in the force-field to generate and evaluate the designs, it is difficult to calibrate the relative strengths of the terms and determine the energy of the design in physical units. Thus, it would be preferable to rank designs using several metrics rather than using the same force-field that guided the design process. In addition, when using approaches that mix physics- and knowledge-based terms, it is difficult to ensure that all protein–protein and protein–water enthalpic contributions are properly accounted for and that the protein and solvent entropic contributions are included. Ideally, one would need to calculate the free energies of the designs to rank them. Privett et al.23 addressed some of these issues by using all-atom MD simulations in addition to their standard design protocol (Figure 3 left), to assess iterative designs of a Kemp eliminase, resulting in a functional enzyme after three rounds.
Despite these problems, there has been significant progress in developing computational techniques for predicting and designing protein structures de novo. This is in part because by examining “static” protein structures, designers can delineate design goals in a relatively straightforward manner. By contrast, when designing functional proteins, the goal is by no means clear. Certain elements can be designed for, such as “binding of the transition state” but the dynamics that accompany—and may be essential for—activity are not nearly so obvious. The connection between structure, dynamic protein–protein interactions and catalysis is not well understood.
The future of designed materials and assemblies lies in the creation of more diverse and robust protein building blocks. Orthogonality and specificity will be very important in creating new materials. For example, an expanded toolkit of coiled-coil interactions, in particular heterodimers, with minimal cross-reactivity would greatly benefit the construction of new self-assembling complexes.95–99 Using building blocks that interact only with the desired ligands will allow for functionalization at discrete locations. The development of new types of specific protein–protein interaction modules with minimal cross-reactivity with each other or with cellular components would also be useful.100 Additionally, new and improved “smart” materials will focus on enhanced responsiveness to molecular stimuli. By continuing to design protein building blocks that are more sensitive to stimuli such as pH, light, ionic strength, and temperature, we will expand the functionality of designed materials in the future.
For future in vivo applications, it would be useful to develop fluorescent proteins that are brighter and which mature faster than existing variants. In addition, proteins that emit at longer wavelengths would be beneficial to allow for deeper tissue penetration necessary for imaging in live multicellular organisms. Some progress towards this goal has been made with the discovery of several fluorescent proteins that fluoresce in the near IR region, although these variants suffer from low brightness,101,102 increased toxicity,103 and are limited by slow and/or incomplete fluorophore maturation.104 Future efforts will also focus on developing new fluorescent protein-based biosensors that function in vivo,105 as well as identifying new variants that can be used for super-resolution microscopy.106 Often when introducing new properties into fluorescent proteins, scientists select for a single attribute at the expense of other spectroscopic properties. For example, the mutations introduced to produce a monomeric VFP also decreased fluorescent brightness.62 Future design efforts will also focus on improving the fluorescent properties of novel proteins. This will be achieved through iterative rounds of design, solving crystal structures of the new protein, and then using these structures as a guide for further improvement. This approach has been used to improve the quantum yield of Cyan Fluorescent Protein (CFP) from 0.21 to most recently 0.93, the highest value to date for a monomeric protein.107 Improved selection procedures, as well as past experience, will expedite this process.
To better understand signaling networks, tools will need to be developed that can distinguish between proteins with different post-translational modifications. Progress has been made with the design of protein domains that recognize phosphorylated substrates,108 and future efforts will be made easier by the recent creation of bacterial strains that can incorporate unnatural amino acids at specific positions.109
New small molecule-inducible domains that respond to novel stimuli would also be useful. To create more intricate synthetic pathways it will be necessary to develop additional switches that can be used to control the activity of multiple proteins independently while minimizing interference with native cellular processes. It will also be useful to develop molecular switches that can respond to biologically relevant stimuli. These would greatly aid the creation of synthetic circuits for therapeutic, industrial, and detection applications. Another major focus will be to create new methods for controlling proteins with light. Light-sensitive domains will be developed that can control a wider range of protein activities, respond to light stimuli faster, and be induced by longer wavelengths than existing designs.
Efforts to design new proteins were first undertaken with the intent to increase our knowledge of structure and activity but also with the promise of creating new practical protein tools. Through the years, our basic understanding of proteins has increased greatly, and we have begun to enter the era when we will be able to produce functional proteins that will revolutionize medicine and technology.
Acknowledgments
Contract grant sponsors: Raymond and Beverly Sackler Institute for Biological, Physical, and Engineering Sciences; National Science Foundation; the National Institutes of Health; and the Ford Foundation
References
- 1.Murphy GS, Sathyamoorthy B, Der BS, Machius MC, Pulavarti SV, Szyperski T, Kuhlman B. Protein Sci. 2014;4:434–445. doi: 10.1002/pro.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ho SP, DeGrado WF. J Am Chem Soc. 1987;109:6751–6758. [Google Scholar]
- 3.Regan L, DeGrado WF. Science. 1988;241:976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 4.Hecht MH, Richardson JS, Richardson DC, Ogden RC. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 5.Padmanabhan S, Marqusee S, Ridgeway T, Laue TM, Baldwin RL. Nature. 1990;344:268–270. doi: 10.1038/344268a0. [DOI] [PubMed] [Google Scholar]
- 6.Kim CA, Berg JM. Nature. 1993;362:267–270. doi: 10.1038/362267a0. [DOI] [PubMed] [Google Scholar]
- 7.Smith CK, Withka JM, Regan L. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 8.Minor DL, Jr, Kim PS. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 9.Smith CK, Regan L. Science. 1995;270:980–982. doi: 10.1126/science.270.5238.980. [DOI] [PubMed] [Google Scholar]
- 10.Munson M, Balasubramanian S, Fleming KG, Nagi AD, O’Brien R, Sturtevant JM, Regan L. Protein Sci. 1996;5:1584–1593. doi: 10.1002/pro.5560050813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ponder JW, Richards FM. J Mol Biol. 1987;193:775–791. doi: 10.1016/0022-2836(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 12.Lim WA, Sauer RT. Nature. 1989;339:31–36. doi: 10.1038/339031a0. [DOI] [PubMed] [Google Scholar]
- 13.Yue K, Dill KA. Proc Natl Acad Sci USA. 1992;89:4163–4167. doi: 10.1073/pnas.89.9.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Desjarlais JR, Handel TM. Protein Sci. 1995;4:2006–2018. doi: 10.1002/pro.5560041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahiyat BI, Mayo SL. Protein Sci. 1996;5:895–903. doi: 10.1002/pro.5560050511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuhlman B, Dantas G, Ireton GC, Varani G, Stoddard BL, Baker D. Science. 2003;302:1364–1368. doi: 10.1126/science.1089427. [DOI] [PubMed] [Google Scholar]
- 17.Butterfoss GL, Kuhlman B. Annu Rev Biophys Biomol Struct. 2006;35:49–65. doi: 10.1146/annurev.biophys.35.040405.102046. [DOI] [PubMed] [Google Scholar]
- 18.Regan L, Clarke ND. Biochemistry. 1990;29:10878–10883. doi: 10.1021/bi00501a003. [DOI] [PubMed] [Google Scholar]
- 19.Regan L. Annu Rev Biophys Biomol Struct. 1993;22:257–287. doi: 10.1146/annurev.bb.22.060193.001353. [DOI] [PubMed] [Google Scholar]
- 20.Jiang L, Althoff EA, Clemente FR, Doyle L, Rothlisberger D, Zanghellini A, Gallaher JL, Betker JL, Tanaka F, Barbas CF, III, Hilvert D, Houk KN, Stoddard BL, Baker D. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rothlisberger D, Khersonsky O, Wollacott AM, Jiang L, DeChancie J, Betker J, Gallaher JL, Althoff EA, Zanghellini A, Dym O, Albeck S, Houk KN, Tawfik DS, Baker D. Nature. 2008;453:190–195. doi: 10.1038/nature06879. [DOI] [PubMed] [Google Scholar]
- 22.Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St Clair JL, Gallaher JL, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D. Science. 2010;329:309–313. doi: 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Privett HK, Kiss G, Lee TM, Blomberg R, Chica RA, Thomas LM, Hilvert D, Houk KN, Mayo SL. Proc Natl Acad Sci USA. 2012;109:3790–3795. doi: 10.1073/pnas.1118082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fleishman SJ, Whitehead TA, Ekiert DC, Dreyfus C, Corn JE, Strauch EM, Wilson IA, Baker D. Science. 2011;332:816–821. doi: 10.1126/science.1202617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Korendovych IV, Kulp DW, Wu Y, Cheng H, Roder H, DeGrado WF. Proc Natl Acad Sci USA. 2011;108:6823–6827. doi: 10.1073/pnas.1018191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin H, Slusky JS, Berger BW, Walters RS, Vilaire G, Litvinov RI, Lear JD, Caputo GA, Bennett JS, DeGrado WF. Science. 2007;315:1817–1822. doi: 10.1126/science.1136782. [DOI] [PubMed] [Google Scholar]
- 27.Shandler SJ, Korendovych IV, Moore DT, Smith-Dupont KB, Streu CN, Litvinov RI, Billings PC, Gai F, Bennett JS, DeGrado WF. J Am Chem Soc. 2011;133:12378–12381. doi: 10.1021/ja204215f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walters R, DeGrado W. Proc Natl Acad Sci USA. 2006;103:13658–13663. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lanci CJ, MacDermaid CM, Kang SG, Acharya R, North B, Yang X, Qiu XJ, DeGrado WF, Saven JG. Proc Natl Acad Sci USA. 2012;109:7304–7309. doi: 10.1073/pnas.1112595109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jorgensen WL, Tirado-Rives J. J Am Chem Soc. 1988;110:1657–1666. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 32.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FT, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Case DA, Babin V, Berryman JT, Betz RM, Cai Q, Cerutti DS, TEC, Darden TA, Duke RE, Gohlke H, Goetz AW, Gusarov S, Homeyer N, Janowski P, Kaus J, Kolossváry I, Kovalenko A, Lee TS, LeGrand S, Luchko T, Luo R, Madej B, Merz KM, Paesani F, Roe DR, Roitberg A, Sagui C, Salomon-Ferrer R, Seabra G, Simmerling CL, Smith W, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Kollman PA. University of California; San Francisco: 2014. [Google Scholar]
- 34.MacKerell AD, Jr, Feig M, Brooks CL., III J Am Chem Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- 35.Kryshtafovych A, Moult J, Bales P, Bazan JF, Biasini M, Burgin A, Chen C, Cochran FV, Craig TK, Das R, Fass D, Garcia-Doval C, Herzberg O, Lorimer D, Luecke H, Ma X, Nelson DC, van Raaij MJ, Rohwer F, Segall A, Seguritan V, Zeth K, Schwede T. Proteins. 2014;82:26–42. doi: 10.1002/prot.24489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vymětal J, Vondrášek J. J Chem Theory Comput. 2012;9:441–451. doi: 10.1021/ct300794a. [DOI] [PubMed] [Google Scholar]
- 37.Beauchamp KA, Lin YS, Das R, Pande VS. J Chem Theory Comput. 2012;8:1409–1414. doi: 10.1021/ct2007814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hu H, Elstner M, Hermans J. Proteins. 2003;50:451–463. doi: 10.1002/prot.10279. [DOI] [PubMed] [Google Scholar]
- 39.Zhu X, Lopes PEM, Shim J, MacKerell AD. J Chem Inform Modeling. 2012;52:1559–1572. doi: 10.1021/ci300079j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramakrishnan C, Ramachandran GN. Biophys J. 1965;5:909–933. doi: 10.1016/S0006-3495(65)86759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou AQ, O’Hern CS, Regan L. Biophys J. 2012;102:2345–2352. doi: 10.1016/j.bpj.2012.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou AQ, Caballero D, O’Hern CS, Regan L. Biophys J. 2013;105:2403–2411. doi: 10.1016/j.bpj.2013.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou AQ, O’Hern CS, Regan L. Proteins. 2014;82:2574–2584. doi: 10.1002/prot.24621. [DOI] [PubMed] [Google Scholar]
- 44.Caballero D, Maatta J, Zhou AQ, Sammalkorpi M, O’Hern CS, Regan L. Protein Sci. 2014;23:970–980. doi: 10.1002/pro.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grove TZ, Cortajarena AL, Regan L. Curr Opin Struct Biol. 2008;18:507–515. doi: 10.1016/j.sbi.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olsen BD, Kornfield JA, Tirrell DA. Macromolecules. 2010;43:9094–9099. doi: 10.1021/ma101434a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glassman MJ, Chan J, Olsen BD. Adv Funct Mater. 2013;23:11. doi: 10.1002/adfm.201202034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banwell EF, Abelardo ES, Adams DJ, Birchall MA, Corrigan A, Donald AM, Kirkland M, Serpell LC, Butler MF, Woolfson DN. Nat Mater. 2009;8:596–600. doi: 10.1038/nmat2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zakeri B, Fierer JO, Celik E, Chittock EC, Schwarz-Linek U, Moy VT, Howarth M. Proc Natl Acad Sci USA. 2012;109:E690–E697. doi: 10.1073/pnas.1115485109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang WB, Sun F, Tirrell DA, Arnold FH. J Am Chem Soc. 2013;135:13988–13997. doi: 10.1021/ja4076452. [DOI] [PubMed] [Google Scholar]
- 51.Sun F, Zhang WB, Mahdavi A, Arnold FH, Tirrell DA. Proc Natl Acad Sci USA. 2014;111:11269–11274. doi: 10.1073/pnas.1401291111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grove TZ, Osuji CO, Forster JD, Dufresne ER, Regan L. J Am Chem Soc. 2010;132:14024–14026. doi: 10.1021/ja106619w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grove TZ, Forster J, Pimienta G, Dufresne E, Regan L. Biopolymers. 2012;97:508–517. doi: 10.1002/bip.22033. [DOI] [PubMed] [Google Scholar]
- 54.Warburg O. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 55.Sacca B, Niemeyer CM. Angew Chem Int Ed Engl. 2012;51:58–66. doi: 10.1002/anie.201105846. [DOI] [PubMed] [Google Scholar]
- 56.Gradisar H, Bozic S, Doles T, Vengust D, Hafner-Bratkovic I, Mertelj A, Webb B, Sali A, Klavzar S, Jerala R. Nat Chem Biol. 2013;9:362–366. doi: 10.1038/nchembio.1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fletcher JM, Harniman RL, Barnes FR, Boyle AL, Collins A, Mantell J, Sharp TH, Antognozzi M, Booth PJ, Linden N, Miles MJ, Sessions RB, Verkade P, Woolfson DN. Science. 2013;340:595–599. doi: 10.1126/science.1233936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lai YT, Reading E, Hura GL, Tsai KL, Laganowsky A, Asturias FJ, Tainer JA, Robinson CV, Yeates TO. Nat Chem. 2014;6:1065–1071. doi: 10.1038/nchem.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Prasher DC, Eckenrode VK, Ward WW, Prendergast FG, Cormier MJ. Gene. 1992;111:229–233. doi: 10.1016/0378-1119(92)90691-h. [DOI] [PubMed] [Google Scholar]
- 60.Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- 61.Hoi H, Howe ES, Ding Y, Zhang W, Baird MA, Sell BR, Allen JR, Davidson MW, Campbell RE. Chem Biol. 2013;20:1296–1304. doi: 10.1016/j.chembiol.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 62.Ilagan RP, Rhoades E, Gruber DF, Kao HT, Pieribone VA, Regan L. FEBS J. 2010;277:1967–1978. doi: 10.1111/j.1742-4658.2010.07618.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heim R, Prasher DC, Tsien RY. Proc Natl Acad Sci. 1994;91:12501–12504. doi: 10.1073/pnas.91.26.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Heim R, Cubitt AB, Tsien RY. Nature. 1995;373:663–664. doi: 10.1038/373663b0. [DOI] [PubMed] [Google Scholar]
- 65.Pédelacq J-D, Cabantous S, Tran T, Terwilliger TC, Waldo GS. Nat Biotechnol. 2005;24:79–88. doi: 10.1038/nbt1172. [DOI] [PubMed] [Google Scholar]
- 66.Ghosh I, Hamilton AD, Regan L. J Am Chem Soc. 2000;122:5658–5659. [Google Scholar]
- 67.Hu C-D, Chinenov Y, Kerppola TK. Mol Cell. 2002;9:789–798. doi: 10.1016/s1097-2765(02)00496-3. [DOI] [PubMed] [Google Scholar]
- 68.Magliery TJ, Wilson CG, Pan W, Mishler D, Ghosh I, Hamilton AD, Regan L. J Am Chem Soc. 2005;127:146–157. doi: 10.1021/ja046699g. [DOI] [PubMed] [Google Scholar]
- 69.Park S-H, Zarrinpar A, Lim WA. Science. 2003;299:1061–1064. doi: 10.1126/science.1076979. [DOI] [PubMed] [Google Scholar]
- 70.Howard PL, Chia MC, Del Rizzo S, Liu F-F, Pawson T. Proc Natl Acad Sci. 2003;100:11267–11272. doi: 10.1073/pnas.1934711100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Amstutz P, Binz HK, Parizek P, Stumpp MT, Kohl A, Grütter MG, Forrer P, Plückthun A. J Biol Chem. 2005;280:24715–24722. doi: 10.1074/jbc.M501746200. [DOI] [PubMed] [Google Scholar]
- 72.Cortajarena AL, Liu TY, Hochstrasser M, Regan L. ACS Chem Biol. 2010;5:545–552. doi: 10.1021/cb9002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Shah K, Liu Y, Deirmengian C, Shokat KM. Proc Natl Acad Sci. 1997;94:3565–3570. doi: 10.1073/pnas.94.8.3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Aranda A, Pascual A. Physiol Rev. 2001;81:1269–1304. doi: 10.1152/physrev.2001.81.3.1269. [DOI] [PubMed] [Google Scholar]
- 75.Godowski PJ, Picard D, Yamamoto KR. Science. 1988;241:812–816. doi: 10.1126/science.3043662. [DOI] [PubMed] [Google Scholar]
- 76.Picard D. Methods Enzymol. 1999;327:385–401. doi: 10.1016/s0076-6879(00)27291-1. [DOI] [PubMed] [Google Scholar]
- 77.Spencer DM, Graef I, Austin DJ, Schreiber SL, Crabtree GR. Proc Natl Acad Sci USA. 1995;92:9805–9809. doi: 10.1073/pnas.92.21.9805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson MS, Sahlender DA, Foster SD. Dev Cell. 2010;18:324–331. doi: 10.1016/j.devcel.2009.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Renicke C, Schuster D, Usherenko S, Essen L-O, Taxis C. Chem Biol. 2013;20:619–626. doi: 10.1016/j.chembiol.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 80.Strickland D, Lin Y, Wagner E, Hope CM, Zayner J, Antoniou C, Sosnick TR, Weiss EL, Glotzer M. Nat Methods. 2012;9:379–384. doi: 10.1038/nmeth.1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dreier B, Pluckthun A. Methods Mol Biol. 2012;805:261–286. doi: 10.1007/978-1-61779-379-0_15. [DOI] [PubMed] [Google Scholar]
- 82.Butterfoss GL, Richardson JS, Hermans J. Acta Crystallogr D Biol Crystallogr. 2005;61:88–98. doi: 10.1107/S0907444904027325. [DOI] [PubMed] [Google Scholar]
- 83.Hagarman A, Measey TJ, Mathieu D, Schwalbe H, Schweitzer-Stenner R. J Am Chem Soc. 2010;132:540–551. doi: 10.1021/ja9058052. [DOI] [PubMed] [Google Scholar]
- 84.Hansen DF, Neudecker P, Kay LE. J Am Chem Soc. 2010;132:7589–7591. doi: 10.1021/ja102090z. [DOI] [PubMed] [Google Scholar]
- 85.Hansen DF, Neudecker P, Vallurupalli P, Mulder FA, Kay LE. J Am Chem Soc. 2010;132:42–43. doi: 10.1021/ja909294n. [DOI] [PubMed] [Google Scholar]
- 86.Fleishman SJ, Whitehead TA, Strauch EM, Corn JE, Qin S, Zhou HX, Mitchell JC, Demerdash ON, Takeda-Shitaka M, Terashi G, Moal IH, Li X, Bates PA, Zacharias M, Park H, Ko JS, Lee H, Seok C, Bourquard T, Bernauer J, Poupon A, Aze J, Soner S, Ovali SK, Ozbek P, Tal NB, Haliloglu T, Hwang H, Vreven T, Pierce BG, Weng Z, Perez-Cano L, Pons C, Fernandez-Recio J, Jiang F, Yang F, Gong X, Cao L, Xu X, Liu B, Wang P, Li C, Wang C, Robert CH, Guharoy M, Liu S, Huang Y, Li L, Guo D, Chen Y, Xiao Y, London N, Itzhaki Z, Schueler-Furman O, Inbar Y, Potapov V, Cohen M, Schreiber G, Tsuchiya Y, Kanamori E, Standley DM, Nakamura H, Kinoshita K, Driggers CM, Hall RG, Morgan JL, Hsu VL, Zhan J, Yang Y, Zhou Y, Kastritis PL, Bonvin AM, Zhang W, Camacho CJ, Kilambi KP, Sircar A, Gray JJ, Ohue M, Uchikoga N, Matsuzaki Y, Ishida T, Akiyama Y, Khashan R, Bush S, Fouches D, Tropsha A, Esquivel-Rodriguez J, Kihara D, Stranges PB, Jacak R, Kuhlman B, Huang SY, Zou X, Wodak SJ, Janin J, Baker D. J Mol Biol. 2011;414:289–302. doi: 10.1016/j.jmb.2011.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stranges PB, Kuhlman B. Protein Sci. 2013;22:74–82. doi: 10.1002/pro.2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Davis IW, Arendall WB, 3rd, Richardson DC, Richardson JS. Structure. 2006;14:265–274. doi: 10.1016/j.str.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 89.Grove TZ, Hands M, Regan L. Protein Eng Des Sel. 2010;23:449–455. doi: 10.1093/protein/gzq015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Su A, Mayo SL. Protein Sci. 1997;6:1701–1707. doi: 10.1002/pro.5560060810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Farinas E, Regan L. Protein Sci. 1998;7:1939–1946. doi: 10.1002/pro.5560070909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ross SA, Sarisky CA, Su A, Mayo SL. Protein Sci. 2001;10:450–454. doi: 10.1110/ps.32501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Friedland GD, Linares AJ, Smith CA, Kortemme T. J Mol Biol. 2008;380:757–774. doi: 10.1016/j.jmb.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Georgiev I, Keedy D, Richardson JS, Richardson DC, Donald BR. Bioinformatics. 2008;24:i196–i204. doi: 10.1093/bioinformatics/btn169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.O’Shea EK, Lumb KJ, Kim PS. Curr Biol. 1993;3:658–667. doi: 10.1016/0960-9822(93)90063-t. [DOI] [PubMed] [Google Scholar]
- 96.Gurnon DG, Whitaker JA, Oakley MG. J Am Chem Soc. 2003;125:7518–7519. doi: 10.1021/ja0357590. [DOI] [PubMed] [Google Scholar]
- 97.Woolfson DN. Adv Protein Chem. 2005;70:79–112. doi: 10.1016/S0065-3233(05)70004-8. [DOI] [PubMed] [Google Scholar]
- 98.Grigoryan G, Reinke AW, Keating AE. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Thompson KE, Bashor CJ, Lim WA, Keating AE. ACS Synth Biol. 2012;1:118–129. doi: 10.1021/sb200015u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Speltz EB, Nathan A, Regan L. ACS Chem Biol. 2015 doi: 10.1021/acschembio.5b00415. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 101.Shcherbo D, Shemiakina II, Ryabova AV, Luker KE, Schmidt BT, Souslova EA, Gorodnicheva TV, Strukova L, Shidlovskiy KM, Britanova OV. Nat Methods. 2010;7:827–829. doi: 10.1038/nmeth.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Filonov GS, Piatkevich KD, Ting L-M, Zhang J, Kim K, Verkhusha VV. Nat Biotechnol. 2011;29:757–761. doi: 10.1038/nbt.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Davidson MW, Campbell RE. Nat Methods. 2009;6:713–717. doi: 10.1038/nmeth1009-713. [DOI] [PubMed] [Google Scholar]
- 104.Subach FV, Verkhusha VV. Chem Rev. 2012;112:4308–4327. doi: 10.1021/cr2001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ibraheem A, Campbell RE. Current Opin Chem Biol. 2010;14:30–36. doi: 10.1016/j.cbpa.2009.09.033. [DOI] [PubMed] [Google Scholar]
- 106.Nienhaus K, Nienhaus GU. Chem Soc Rev. 2014;43:1088–1106. doi: 10.1039/c3cs60171d. [DOI] [PubMed] [Google Scholar]
- 107.Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, van Weeren L, Gadella TW, Jr, Royant A. Nat Commun. 2012;3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sawyer N, Gassaway BM, Haimovich AD, Isaacs FJ, Rinehart J, Regan L. ACS Chem Biol. 2014;9:2502–2507. doi: 10.1021/cb500658w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, Soll D. Science. 2011;333:1151–1154. doi: 10.1126/science.1207203. [DOI] [PMC free article] [PubMed] [Google Scholar]