

Abstract
The class B scavenger receptors BI (SR-BI) and BII (SR-BII) are HDL receptors that recognize various pathogens, including bacteria and their products. It has been reported that SR-BI/II-null mice are more sensitive than normal mice to endotoxin-induced inflammation and sepsis. Since the SR-BI/II-knockout model demonstrates multiple immune and metabolic disorders we investigated the role of each receptor in the LPS-induced inflammatory response and tissue damage using transgenic mice with pLiv-11-directed expression of human SR-BI or SR-BII. Six hours after intraperitoneal LPS injection, transgenic hSR-BI and hSR-BII mice demonstrated markedly higher serum levels of pro-inflammatory cytokines and 2-3-fold increased expression levels of inflammatory mediators in the liver and kidney, compared to wild type (WT) mice. LPS-stimulated iNOS expression was 3-6-fold higher in the liver and kidney of both transgenic strains, although serum NO levels were similar in all mice. Despite the lower HDL plasma levels, both transgenic strains responded to LPS by a 5-fold increase of plasma corticosterone levels which were only moderately lower than in WT animals. LPS treatment resulted in MAPKs activation in tissues of all mice; however, the strongest response was detected for hepatic ERK1/2 and kidney JNK of both transgenic mice. Histological examination of hepatic and renal tissue from LPS-challenged mice revealed more injury in hSR-BII but not hSR-BI transgenic mice vs WT controls. Our findings demonstrate that hSR-BII, and to a lesser extent hSR-BI, significantly increase LPS-induced inflammation and contribute to LPS-induced tissue injury in the liver and kidney, two major organs susceptible to LPS toxicity.
Keywords: class B scavenger receptors, LPS, pro-inflammatory cytokines, iNOS, NLRP3, MAPKs, liver and kidney injury
Introduction
The class B scavenger receptors BI (SR-BI) and its splice variant BII (SR-BII) are transmembrane proteins that interact with a broad range of ligands, including native and modified lipoproteins (1–3), serum amyloid A (4, 5), beta-amyloid (6), bacteria and apoptotic cells (7–9). These receptors are known to play a role in atherosclerosis (10, 11), host defense (12–15), apoptotic cell removal (3) and hepatitis C infection (16). Both receptors are predominantly expressed in the liver and steroidogenic tissues (2, 17–19), parenchymal epithelial cells of various organs (20–22) and phagocytic cells (23, 24). We previously demonstrated that both SR-BI and SR-BII bind bacteria, facilitate their internalization and mediate bacteria-associated pro-inflammatory signaling due, in part, to their interaction with bacterial cell wall components LPS and lipoteichoic acid (LTA), and the bacterial cytosolic protein GroEL (14). LPS is a pathogen-associated molecular pattern (PAMP) bacterial cell wall phospholipid that mediates inflammation, and replicates some aspects of septic shock. LPS induces acute organ damage due to mechanisms that currently are not fully understood.
Understanding the role of SR-BI and SR-BII in SR-BI/SR-BII knockout mice during endotoxic shock is complicated by steroid insufficiency. SR-BI/SR-BII null mice had higher mortality following endotoxin injection or peritonitis, suggesting that SR-Bs are protective during endotoxemia/sepsis (12, 15, 25, 26). The protective function of SR-BI was attributed to its role in glucocorticoid (GC) production (25), reduced NO-induced cytotoxicity (26), increased hepatic LPS clearance (15) and suppressed LPS-induced TLR-4 signaling (12), resulting in reduced plasma pro-inflammatory cytokines. At the same time, our and others’ data provide evidence that the SR-B receptor family might facilitate bacterial, viral and parasitic infection and sepsis by promoting bacterial uptake, followed by cytosolic evasion resulting in incomplete phagocytosis, inflammation amplification and bacterial infection (13, 14, 16, 27).
The majority of evidence supporting a protective role of SR-BI in sepsis has been obtained using SR-BI/SR-BII KO mice as an experimental model. This loss-of-function model is characterized by the defective HDL cholesterol metabolism, impaired adrenal accumulation of cholesterol esters and female infertility (28, 29). SR-BI deficiency has also been recently shown to result in multiple other dysfunctions, including excessive lymphocyte apoptosis associated with immunosuppression (30), enhanced susceptibility to arterial thrombosis, increased reticulocytosis due to a reduced erythrocyte lifespan (31), and susceptibility to atherosclerosis due to reduced SR-BI-mediated cholesterol ester uptake. In addition, the important consequence of defective adrenal cholesterol ester uptake in SR-BI-KO mice is diminished glucocorticoid (GC) production, which has a profound effect on responses to stress, fasting and sepsis (25, 32). As a result of GC deficiency, SR-BI-KO mice are more susceptible to LPS-induced shock and cecal ligation and puncture (CLP) – induced sepsis and death (12, 13, 15). We and others reported an increased inflammatory response in SR-BI/SR-BII KO mice compared to wild-type mice following acute LPS administration (26) or CLP-induced sepsis (12, 13). However, it has been shown that the increased susceptibility of SR-BI/SR-BII KO mice to LPS could be suppressed by corticosteroid supplementation (25). Our findings further demonstrated that combined hormone (glucocorticoid (GC) + mineralocorticoid (MC)) replacement therapy introduced 24 hours in advance of the CLP-induced sepsis essentially alleviated pro-inflammatory responses and improved survival in SR-BI/SR-BII KO mice (13).
The multiple dysfunctions of SR-BI/BII-deficient mice complicate understanding the role of SR-BI and SR-BII in sepsis and infection. This prompted us to generate an alternative, gain-of-function model of hSR-BI (hSR-BI tgn) and hSR-BII (hSR-BII tgn) transgenic mice using an expression vector pLiv-11, which contains a constitutive human apoE gene promoter. Its hepatic control region results in high levels of transgene expression predominantly, but not exclusively (e.g. in the kidney), in hepatocytes (33). In this study, using hSR-BI and hSR-BII transgenic mice on a C57BL/6J background, we evaluated the individual contribution of SR-BI and SRB-II human homologues in LPS-induced inflammation, as well as hepatic and renal tissue damage. Our findings demonstrate that hSR-BII, and to a lesser extent hSR-BI, contribute to the pro-inflammatory response and organ injury induced by the acute LPS administration, and therefore, pharmacologically targeting these receptors could be a helpful tool to reduce LPS-induced liver and kidney damage.
Materials and Methods
Reagents
LPS (E. coli 0111:B4) and macrophage colony stimulating factor (M-CSF) were purchased from Sigma. All reagents used for RNA isolation, reverse transcription and real-time PCR were from Life Technologies. Enzyme-linked immunosorbent assay (ELISA) kits for quantifying mouse IL-6, IL-1β and CXCL1 and human IL-8 were from Life Technologies and for mouse MIP-2 from R&D Systems. A competitive ELISA kit for quantifying corticosterone was from Enzo Life Sciences and a kit for colorimetric assay of nitrate (NOx) was purchased from Cayman Chemical. Anti-human SR-BI/BII antibody was from BD Biosciences (cat. # 610883), rabbit anti-human SR-BI and anti-human SR-BII antibodies were custom produced against C-terminal domain specific peptides of hSR-BI (CTSAPKGSVLQEAKL, Anaspec, San Jose, CA), or hSR-BII (CLPDSPSRQPPSPTA, Sigma, St. Louis, MO). Custom antibodies were validated in Western blotting assays using cell lysates from HeLa cell line stably transfected with hSR-BI and hSR-BII (14) (suppl. Fig. 1.). Anti-mouse β-actin antibody, alkaline phosphatase secondary antibody and cholesterol quantitation kit were from Sigma Aldrich. Antibodies against phosphorylated and non-phosphorylated forms of MAP kinases (ERK1/2, SAPK/JNK and p38) were purchased from Cell Signaling Technology, Inc.
Mice
The liver-specific expression vector pLiv-11, which contains the human apoE promoter (23) was used to express hSR-BI in the liver. Full-length (1.7-kb) human SR-BI (hSR-BI) cDNA (GenBank: BC112037.1) was flanked by Not I linkers and inserted into a unique Not I site of modified pLiv-11. Clones with the correct orientation of the transgene were selected after digestion of the plasmid DNA by Sph I and Aat II. The resulting pLiv-11- hSR-BI plasmid was digested with Sal I and Spe I, and an 11.6-kb DNA fragment Liv-hSR-BI, containing the complete expression cassette was isolated, purified and used to generate hSR-BI transgenic mice on the C57BL/6J background. The Liv-hSR-BII construct was created the same way, by using the human SR-BII gene (2). In vivo studies were performed as follows: LPS (1 mg/kg) or PBS (using same volume, approximately 150 μl per mouse) was injected intraperitoneally (IP) in wild-type (WT), hSR-BI tgn and hSR-BII tgn mice. Six hours after LPS/PBS injection, mice were euthanized for blood collection and organ harvesting. 16–18 week old male mice (n=3 for PBS-treated and n=5 for LPS-treated groups) were used in this study. Mice were kept at the NIH animal facility under specific pathogen free conditions. All animal studies were approved by the NHLBI ACUC under protocols H-0050R2 and H-0100R2.
Cell culture
Human embryonic (epithelial) kidney cells (HEK293, ATCC) were stably transfected to express hSR-BI and hSR-BII (hSR-BI-HEK293 and hSR-BII-HEK293, respectively) (7). Murine wild-type (WT), hSR-BI tgn and hSR-BII tgn macrophages were isolated from mouse bone marrow cells (BMC) obtained from transgenic wild-type, hSR-BI- or hSR-BII- expressing mice, respectively. The macrophages were differentiated by culturing in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), in the presence of 10 ng/ml of mouse macrophage colony-stimulating factor (M-CSF) for 7–10 days.
Primary culture of kidney epithelial cells
Freshly harvested mouse kidneys were thoroughly separated from fat, de-capsulated, and finely dissected to obtain fragments less than 0.5mm3 in size. The resulting minced tissue from 2 kidneys was digested in 10 ml of HBSS, containing 4μM CaCl2, 10mM HEPES and 1.5 mg/ml collagenase D (Roche Diagnostics, cat. # 11088874103) at 37°C for 40 min with 120 RPM rotation. Following digestion, the cell suspension was diluted with prechilled PBS and washed three times (50g × 3min centrifugations) in ice-cold PBS. The resulting cell pellet was re-suspended in isosmotic Percoll solution (final concentration 25%) and centrifuged for 10 min at 1000 × g to separate intact cells from the cell fragments floating on the top of supernatant. Pelleted cells were re-suspended in the appropriate volume of culture media (DMEM containing 10% FBS, 10mM HEPES and 1% penicillin/streptomycin) and plated on collagen-coated plates. The average yield of kidney epithelial cells (KEC) from 2 kidneys was sufficient for plating onto ~ 200 cm2 of culture plates. The culture medium was changed after 48 hours in order to remove non-adherent cells and residual cellular fragments.
Western blot analysis of liver and kidney hSR-BI and hSR-BII protein expression
Tissue samples were weighed, resuspended in 1:10 w/v of T-Per Tissue protein Extraction Reagent (Thermo Scientific) and disrupted using a chilled glass-Teflon homogenizer. The resulting lysates were centrifuged at 10,000 × g for 10 minutes and the supernatant was collected. Supernatant aliquots mixed with 2× sample buffer (Life Technologies) and heated for 5 min at 95°C were electrophoresed by reducing SDS PAGE, using 10% Tris-Glycine gels, transferred to nitrocellulose membranes and assayed by Western blot using Alkaline Phosphatase Chromogenic Substrate (Life Technologies, cat. # WP20001).
Total RNA isolation and quantitative real-time PCR analysis
For RNA isolation, tissue samples preserved in RNAlater were homogenized in TRIzol Reagent (Precellys 24, Bertin Technologies, France). RNA was isolated with the PureLink RNA Mini Kit after DNase treatment. RNA (2 μg) was reverse-transcribed using TaqMan Reverse Transcriptase Reagent Kit. Real-time qPCR assays were performed with a StepOne Real-Time PCR System (Applied Biosystems), using 40 ng of cDNA per reaction. A list of TaqMan Gene Expression assays used in the study is shown in Table I. Relative levels of gene expression were measured by the comparative CT (ΔΔCT) method with mouse β-actin or GAPDH genes as reference genes. All gene expression results were analyzed using the 2-ΔΔCT formula and presented as normalized fold changes, compared to WT control (without LPS treatment).
Table I.
TaqMan Real-Time PCR assays used in the study.
| Species | Gene Name | Gene Symbol | Life Technologies ID number |
|---|---|---|---|
| Mouse | Interleukin 6 | Il6 | Mm00446190_m1 |
| Mouse | Chemokine (C-X-F motif) ligand 1 | Cxcl1 | Mm04207460_m1 |
| Mouse | Interleukin 1 beta | Il1b | Mm00434228_m1 |
| Mouse | Transforming growth factor, beta 1 | Tgfb1 | Mm01178820_m1 |
| Mouse | Tumor necrosis factor | Tnfa | Mm00443258_m1 |
| Mouse | NLR family, pyrin domain containing 3 | Nlrp3 | Mm00840904_m1 |
| Mouse | CD 36 antigen | Cd36 | Mm01135198_m1 |
| Mouse | Scavenger receptor, member 1 | Scarb1 | Mm00450234_m1 |
| Mouse | Glyceraldehyde-3-phosphate dehydrogenase | Gapdh | Mm03302249_g1 |
| Mouse | Actin, beta | Actb | Mm00607939_s1 |
Analysis of cytokines, corticosterone, nitric oxide and plasma total cholesterol
The IL-8 secretion by HEK293 cells was analyzed in culture supernatants after a 20h incubation period in serum-free medium with or without BSA (2 mg/ml), utilizing an ELISA kit for human IL-8. IL-6 and CXCL1 levels in culture supernatants of murine macrophages were measured following a 20h incubation in RPMI-1640 with 1% FBS, using corresponding commercial ELISA kits. Plasma levels of cytokines, corticosterone, cortisol, and nitrate (NOx) were quantified with corresponding ELISA or colorimetric kits. All samples and standards were measured in duplicate. Total cholesterol in plasma was determined as previously described (34) by enzymatic assay using commercial kit.
Histological analysis
Formalin-fixed, paraffin-embedded 4 μm thin kidney and liver sections were stained with periodic acid-Schiff reagent (PAS) (Sigma Chemical Co.). Kidney histological changes were assessed in a blind manner in 10 different randomly selected 400X fields per animal from the cortex and 10 fields from the outer stripe of the outer medulla (OSOM). Kidney tubular damage was defined as tubular epithelial swelling, loss of brush border, vacuolar degeneration, necrotic tubules, cast formation, and desquamation. Liver damage was defined as the amount of destruction of hepatic lobules, infiltration of inflammatory cells, hemorrhage, and hepatocyte necrosis. The degree of kidney and liver damage was estimated at 400× magnification by the following criteria: 1, 0 − 25% of damage; 2, 25% to 50% of damage; 3, 50% to 75% of damage; 4, 75% to 100% of damage.
Western Blot analyses of MAPKs activity
For analysis of MAPK activity, the samples of tissue lysates were prepared from livers and kidneys of WT and SR-B transgenic mice (both PBS- and LPS-treated, n=3) sacrificed at 4 h after LPS injection (1 mg/kg, intraperitoneal). This time point was selected from additional experiments where the time dependent activation of MAPKs in mouse tissues following LPS injection was assessed. The tissue lysates were mixed with 2× SDS sample buffer and loaded onto 4–20% Tris-glycine SDS/PAGE gels. After the transfer, the membranes were probed with either one of three anti-phospho-MAPK antibodies or corresponding antibodies that recognize both active and inactive forms of each subfamily of MAP kinases, according to the manufacturer’s protocols. The MAPK antibodies used in this study included anti-phospho-ERK1/2 (Thr202/Tyr204) antibody, anti-ERK1/2 antibody, anti-phospho-SAPK/JNK (Thr183/Tyr185) antibody, anti-SAPK/JNK antibody, anti-phospho-p38 MAPK (Thr180/Tyr182) antibody and anti-p38 MAPK antibody. The immunoreactive bands were detected using an alkaline phosphatase-conjugated secondary antibody (Cell Signaling Technology) and chromogenic substrate for alkaline phosphatase (Invitrogen).
Statistical Analysis
Differences between the groups were examined for statistical significance by one-way analysis of variance (ANOVA). Alternatively, a two-tailed Student’s t-test was used. All data are expressed as mean values ± standard deviation (SD) with a P value of < 0.05 considered as significant.
Results
Tissue specific expression of hSR-BI and hSR-BII in hSR-BI and hSR-BII transgenic mice
hSR-BI and hSR-BII transgenic mice had 40 and 80 copies, respectively, per homozygous genome as analyzed by the TaqMan PCR assay. hSR-BI and hSR-BII mRNA expression was highest in liver; hSR-B expression was also detected in kidney, lung and spleen, although 2–3 orders of magnitude lower than in liver (data not shown). hSR-BII protein in liver and kidney was about was 2.6-fold and 2.3-fold, respectively, more abundant than hSR-BI when using an antibody against the SR-BI/II loop domain that recognizes both human SR-B receptors (Fig. 1, upper panel). Interestingly, despite having half the number of hSR-BII transgene copies and lower liver protein levels, plasma cholesterol levels in hSR-BI tgn mice were either similar (in males) or 40% lower (in females) vs. corresponding hSR-BII transgenic mice, consistent with a higher potency of hSR-BI as a mediator of HDL cholesterol uptake (Table II). These results may reflect the higher (~80%) cell surface expression of SR-BI, essential for ligand-receptor interaction, whereas SR-BII is predominantly (80%) cytosolic (14, 35). As liver and kidney play leading roles in LPS-induced multiorgan failure; we therefore focused subsequently on these two organs.
Figure 1. Western blot analyses of liver and kidney tissue samples.
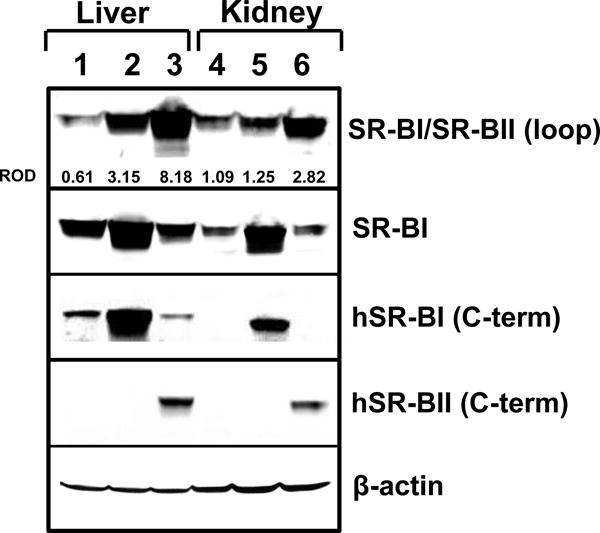
Western blot analyses for hSR-BI and hSR-BII protein expression were performed in liver and kidney tissue samples from WT (lanes 1, 4), hSR-BI tgn (lanes 2, 5) and hSR-BII tgn (lanes 3, 6) mice. hSR-BI and hSR-BII protein expression was detected by using either anti-human SR-BI and anti-human SR-BII custom antibodies (against specific peptides from C-terminal domains) or anti-human SR-BI/BII antibody (against 104–294 amino acid peptide, BD Biosciences). mSR-BI expression was detected by anti-SR-BI antibody (against C-terminal 450–509 amino acid peptide, Novus Biologicals, cat. # NB400-101). Protein expression of β-actin was measured as the loading control. ImageJ software was used to quantify the protein bands intensity, and the relative optical density (ROD) of SR-BI and SR-BII specific bands was calculated versus corresponding β-actin values (upper panel).
Table II.
Plasma levels of total cholesterol (TC) in wild type and transgenic hSR-BI and hSR-BII micea
| Mice | Males | Females | ||||
|---|---|---|---|---|---|---|
| Wild type | hSR-BI tgn | hSR-BII tgn | Wild type | hSR-BI tgn | hSR-BII tgn | |
| TC (mg/dl) | 91.0 ± 14.7 n=18 |
18.3 ± 9.6 b n=30 |
19.5 ± 5.3 b n=7 |
75.7 ± 1.4 n=15 |
17.5 ± 11.7 b n=25 |
27.9 ± 8.8 b n=16 |
Data are presented as means ± SD;
p<0.001 vs wild type.
hSR-BI tgn and hSR-BII tgn mice have an increased pro-inflammatory response to acute LPS administration compared to WT control
Our previous in vitro studies demonstrated the importance of class B scavenger receptors (SR-B), which includes SR-BI/II and CD36, for uptake of gram-negative bacteria, as well as their PAMPs such as LPS and cytosolic chaperonin 60 (GroEL), and subsequent pro-inflammatory signaling (14). In addition, our in vivo studies utilizing a steroid treated murine CLP model of sepsis demonstrated reduced systemic inflammation in SR-BI/BII-null mice, compared to their WT littermates (13). To explore each splice variant’s (SR-BI and SR-BII) influence on LPS-induced inflammation in vivo, control WT, transgenic hSR-BI and hSR-BII mice received PBS or LPS (1mg/kg, IP), and inflammatory responses were assessed by measuring serum levels of pro-inflammatory cytokines/chemokines 6 hours following injection. LPS injection resulted in a strong induction of all measured cytokines in all groups (Fig. 2). However, the LPS-induced plasma pro-inflammatory cytokine/chemokine levels were the highest in hSR-BII mice, markedly (by 2.5- to 3-fold for IL-6, MIP-2 and CXCL1) and moderately (by ~1.75-fold for IL-1β) exceeding the corresponding levels of WT animals (Fig. 2, panels A–C and Fig. 2D, respectively). The plasma levels of IL-6 and both chemokines, CXCL1 and MIP-2, in hSR-BI mice were approximately 2-fold higher than in control animals.
Figure 2. Plasma levels of inflammatory cytokines in WT, hSR-BI tgn and hSR-BII tgn mice challenged with LPS.
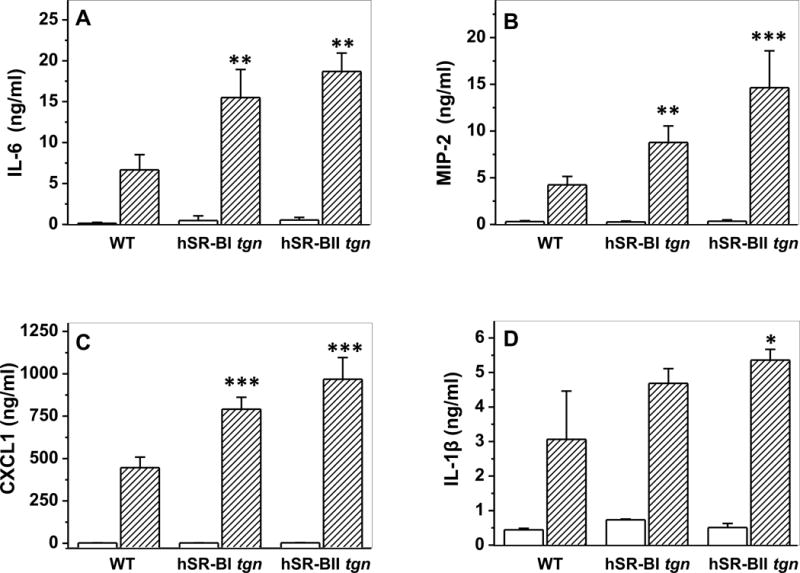
LPS (1 mg/kg, IP) or PBS was injected into WT, hSR-BI and hSR-BII tgn mice. Six hours after the LPS injection, mice were euthanized for plasma and organ collection. Plasma levels of IL-6 (A), MIP-2 (B), CXCL1 (C) and IL-1β (D) were determined by ELISA. Values are the mean ± SD (n=5). ** P<0.01, *** p<0.005, vs WT LPS-treated levels.
Liver and kidney mRNA expression of pro-inflammatory and inflammasome-associated genes in response to LPS is elevated in SR-B transgenic mice
mRNA expression levels of pro-inflammatory cytokines were assessed by quantitative PCR. Consistent with the observed difference in serum pro-inflammatory cytokines levels, LPS-treated hSR-BII tgn mice had markedly (2- to 3-fold) higher liver mRNA expression than control WT mice for all measured pro-inflammatory cytokines except CXCL1 (Fig. 3). For SR-BI tgn mice, only hepatic expression of IL-6 in response to LPS was ~2-fold higher than that of WT mice, whereas the levels of other cytokines were not statistically different between these groups. Kidney expression levels of IL-6 and CXCL1 in response to LPS in both transgenic mice were higher than those in WT mice, with a larger increase observed in the SR-BII tgn group (Fig. 4). SR-BI tgn mice also demonstrated higher kidney expression of IL-1β when compared to control mice. It has been demonstrated that LPS induces a strong hepatic expression of the NLRP3 (36, 37) inflammasome gene, suggesting a central role of the inflammasome in LPS-induced inflammation and in hepatic/renal failure. Indeed, we found that LPS strongly induced NLRP3 expression in both organs in all groups (Fig. 5). While no difference in kidney NLRP3 expression was found between the groups (Fig. 5B), hSR-BII tgn mice demonstrated a 2.5-fold higher hepatic expression of NLRP3 when compared to wild type and SR-BI tgn mice (Fig. 5A).
Figure 3. Hepatic gene expression of inflammatory cytokines in WT, hSR-BI and hSR-BII transgenic mice challenged with LPS.
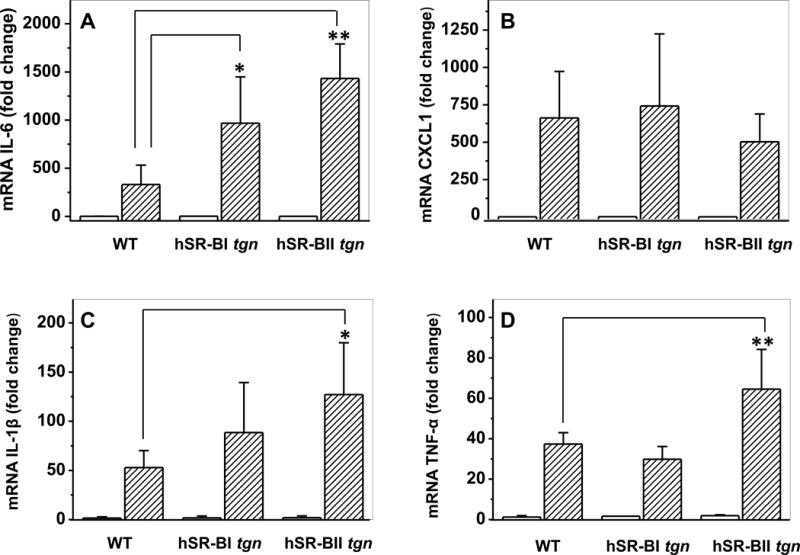
LPS (1 mg/kg, IP) or PBS was injected into WT, hSR-BI and hSR-BII tgn mice. Six hours after the LPS injection, mice were euthanized and liver tissue was collected for mRNA extraction and qRT-PCR as described in Materials and Methods. Expression levels of IL-6 (A), CXCL1 (B), IL-1β (C) and TNF-α (D) were normalized by GAPDH and presented as the fold change relative to PBS-treated control. Values shown are the mean ± SD (n=3, for PBS-treated groups, n=5 for LPS-treated groups).* P<0.05, ** P<0.01 vs WT LPS-treated mice.
Figure 4. Kidney gene expression of inflammatory cytokines in WT, hSR-BI and hSR-BII transgenic mice challenged with LPS.
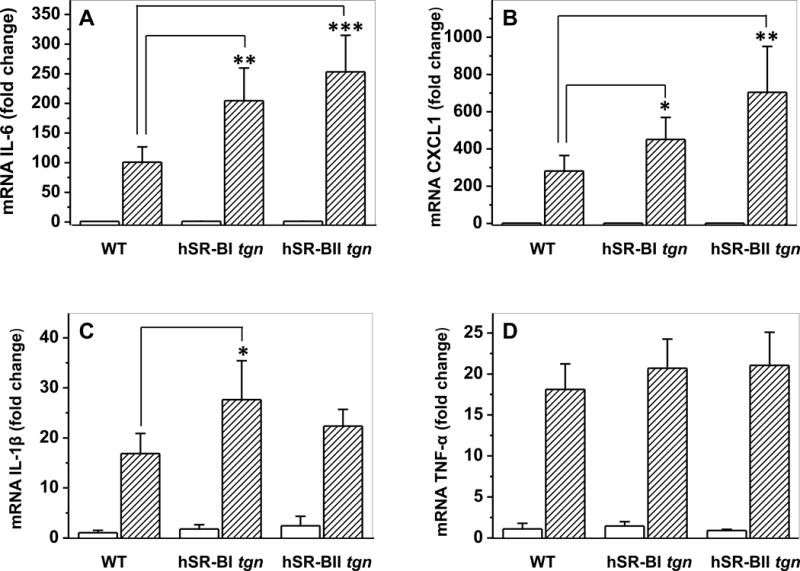
LPS (1 mg/kg, IP) or PBS was injected into WT, hSR-BI and hSR-BII tgn mice. Mice were euthanized after 6 hours; kidney samples were collected and used for mRNA extraction and qRT-PCR as described in Materials and Methods. Expression levels of IL-6 (A), CXCL1 (B), IL-1β (C) and TNF-α (D) were normalized by GAPDH and presented as the fold change relative to PBS-treated control. Values shown are the mean ± SD (n=3, for non-treated groups, n=5 for LPS-treated groups). * P<0.05, ** P<0.01 vs WT LPS-treated mice.
Figure 5. NLRP3 mRNA expression in the liver and kidney of WT, hSR-BI and hSR-BII transgenic mice challenged with LPS.
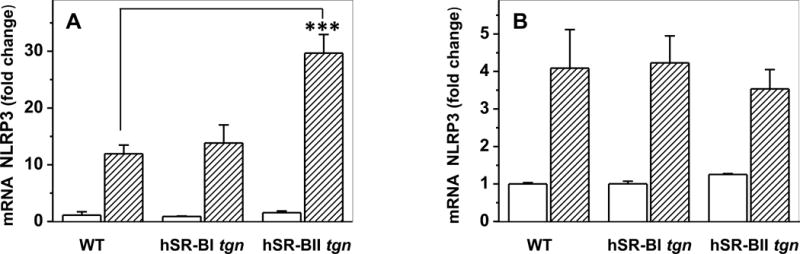
Mice were euthanized and liver and kidney samples were collected for mRNA extraction and qRT-PCR as described in Materials and Methods. Expression of liver (A) and kidney (B) NLRP3 was normalized by GAPDH and presented as the fold change relative to PBS-treated control. Values shown are the mean ± SD (n=3, for PBS-treated groups, n=5 for LPS-treated groups. *** P<0.001 vs WT LPS-treated mice.
Liver and kidney expression of iNOS in SRBI and SRBII transgenic mice is significantly elevated compared to WT mice
LPS-induced toxicity requires synergistic actions of inflammatory cytokines (TNF-α, IL-1β, IFN-gamma) and nitric oxide (NO) (38). Overproduction of NO by inducible NO synthase (iNOS), which is up-regulated following the LPS-triggered pro-inflammatory cytokine release into the circulation, has been proposed to be a major pathogenic driver of hypotension, vascular collapse and multiple organ failure during meningococcemia septic shock (39). To explore the potential impact of hSR-BI and hSR-BII expression on LPS-induced activation of iNOS, we assessed hepatic and renal mRNA levels of iNOS in mice following acute LPS injection. As expected, 6 hours following a challenge with LPS, both liver and kidney iNOS expression levels were markedly increased in all groups (Fig. 6, A, B); however, SR-BII tgn mice demonstrated a 6-fold (in the liver) and 3-fold (in the kidney) greater increase in iNOS expression over the corresponding levels of control WT mice. iNOS expression levels in both organs of SR-BI tgn mice were slightly higher than those detected in WT mice, though the difference was not statistically significant. Interestingly, despite the significantly higher hepatic and kidney levels of iNOS expression in SR-BII tgn mice, systemic serum levels of NO (nitrite) were similar in all three experimental groups (Fig. 6C), perhaps due to a saturation effect and NO metabolism, suggesting the importance of LPS-induced NO local concentrations.
Figure 6. Liver and kidney expression of iNOS and plasma nitrite/nitrate (NOx) and corticosterone levels in LPS-challenged mice.
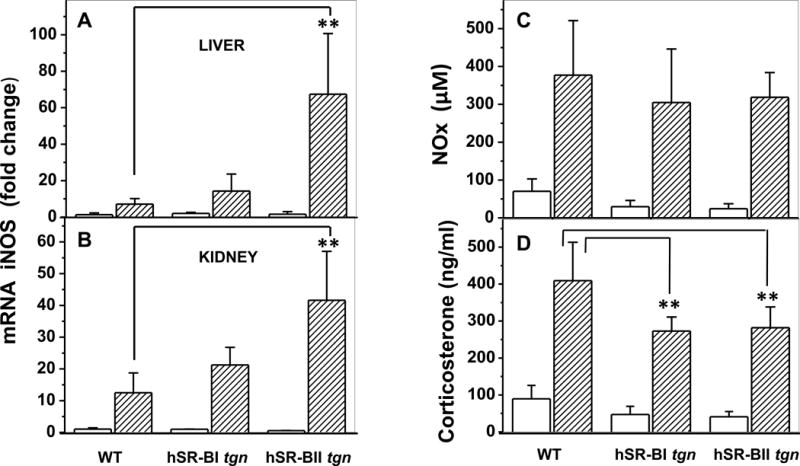
Liver (A) and kidney (B) mRNA expression of iNOS was quantified as we previously described in figure legends 3 and 4. C. Plasma NOx levels of WT, hSR-BI and hSR-BII transgenic mice were measured 6 hours following LPS injection using a colorimetric kit. Values are the mean ± SD (n=5). D. The corticosterone concentration was quantified by ELISA using plasma from hSR-BI and hSR-BII tgn mice and WT littermates 6 hours following LPS injection (n=10 for control PBS-treated groups, and n=5 for LPS-treated group, with duplicate measurements). ** P<0.01 vs WT LPS-treated mice.
Plasma from SR-B transgenic mice demonstrate mildly reduced LPS clearance and LPS-neutralization capacity compared to WT mice
Recent findings of Guo et al. (15, 40) demonstrated that in addition to LPS neutralization, HDL protects against sepsis by promoting LPS clearance, modulating leukocyte recruitment and regulating corticosterone production. Plasma HDL levels were markedly reduced in both hSR-BI (by ~70 %) and hSR-BII (by ~ 60%) tgn mice compared to WT mice as estimated by apoA-I immunoblotting assay (Suppl. Fig. 2A, insert). Using the HEK-Blue hTLR4 cells (InvivoGen), we assessed the LPS neutralization capacity of plasma from WT and hSR-B transgenic mice strictly following the protocol previously described by Guo et al. (40). Plasma samples from WT, hSR-BI and hSR-BII transgenic suppressed LPS-induced TLR4/NF-kB activation by ~ 65%, 68% and 50% respectively (Suppl. Fig. 2A), thus revealing only a mildly and not statistically significant decreased capacity of sera from SR-B transgenic mice to neutralize LPS. To test if the increased pro-inflammatory status of the LPS-challenged transgenic mice could be due to the HDL deficiency and impaired endotoxin clearance we also analyzed the endotoxicity of plasma from WT and hSR-B transgenic mice 6 hours after LPS injection. As shown in Suppl. Fig. 2B, TLR/NF-kB activation induced by plasma from LPS-treated hSR-BI and hSR-BII transgenic mice were ~1.4 times higher, than induced by the plasma from WT mice challenged with LPS. This difference was not statistically significant.
LPS-induced plasma corticosterone levels are moderately reduced in SR-BI and SR-BII tgn vs. WT mice
Because hSR-B transgenic mice have lower plasma HDL and cholesterol levels (Table II), and adrenal cholesterol uptake is essential for anti-inflammatory glucocorticoid (GC) synthesis, we assessed whether the enhanced LPS-induced pro-inflammatory responses of SR-BI and SR-BII transgenic mice might be due to impaired adrenal GC production. Prior to LPS treatment, plasma corticosterone (CS) in hSR-BI tgn and hSR-BII tgn mice was reduced by about a half compared to control WT littermates (Fig. 6D). LPS injection robustly increased plasma CS in all groups of animals, although LPS-induced corticosterone plasma levels in hSR-BI and hSR-BII tgn mice were about 70% of levels detected in control mice. Our data thus indicate that hSR-BI and hSR-BII transgenic mice, despite their markedly reduced HDL-cholesterol plasma levels, can still respond acutely to LPS challenge by increasing GC release, demonstrating only moderately decreased (vs. control mice) LPS-induced levels of CS.
Histological analyses of LPS-induced liver injury of WT, hSR-BI and hSR-BII transgenic mice
Histological changes were examined in the livers of mice 6 hours following PBS or LPS injection. As shown in Fig. 7 (panel A, B1–3), WT, hSR-BI tgn, and hSR-BII tgn mice that received PBS had liver histology scores that, although not identical, were not statistically different. In panel B, hSR-BI tgn (B2) mice had areas of the liver sections that resemble the same architecture seen in WT mice (B1), but also had hepatic areas with micro-vacuolization (blue arrow), and more infiltrating inflammatory cells. These changes were less prominent in hSR-BII tgn mice (B3). Six hours after LPS injection, all mice (B4, B5 and B6) developed statistically significant histological liver damage, which was more substantial in hSR-BII tgn mice (ANOVA with Bonferroni’s multiple comparison post-hoc test). All mice that received LPS (A, B4–6) had a pale purple/pink color on liver sections when compared with mice that received PBS (B1, B2 and B3), indicative of decreased glycogen storage after endotoxemia. After LPS injection, all mice also had more infiltrating inflammatory cells in the liver, which were especially visible in hSR-BII tgn mice that also had more prominent hepatocyte degeneration with loss of boundaries between the cells (white arrow).
Figure 7. LPS-induced histological liver damage in various mice.
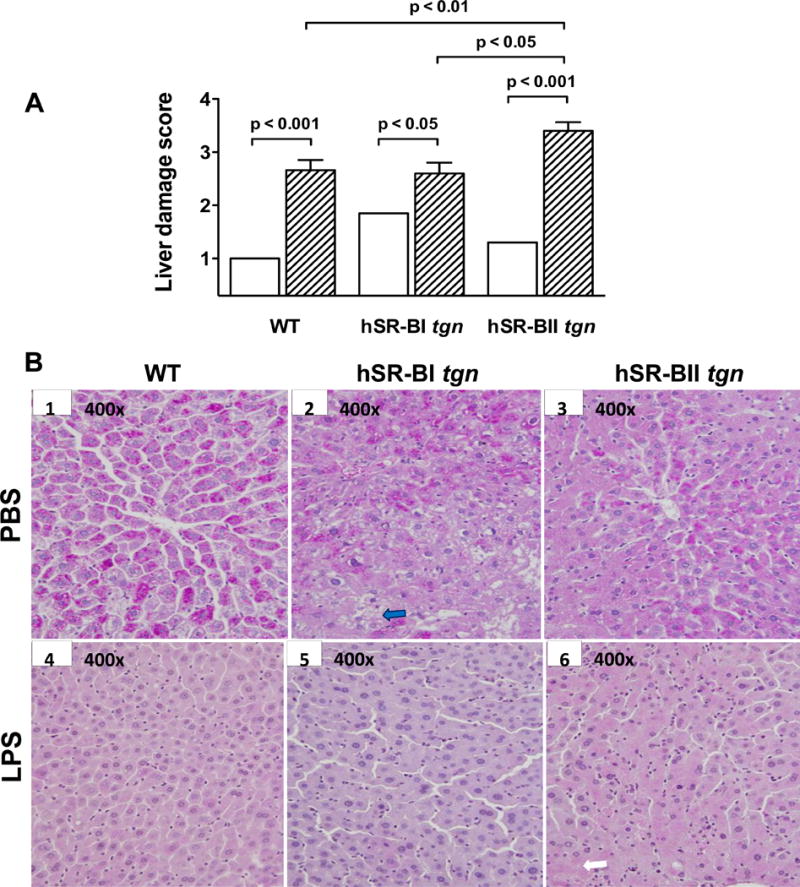
A. Semi-quantitative histological analysis of liver injury. Liver injury was defined as the amount of destruction of hepatic lobules, infiltration of inflammatory cells, hemorrhage, and hepatocyte necrosis, and scored from 1 through 4 according to % area of involvement per HPF (400×). Liver damage scores are presented for mice that received PBS (n=4–7/group, open bars) or an LPS injection (n=5/group, dashed bars). B. Representative images (400×) of liver sections stained by PAS from each group (mice that received PBS: WT – image B1, hSR-BI tgn – image B2, and hSR-BII tgn – image B3), mice that received LPS: WT – image B4, hSR-BI tgn – image B5, and hSR-BII tgn – image B6). The description of the arrows is in the Results section.
Histological analyses of LPS-induced kidney injury of WT, hSR-BI and hSR-BII transgenic mice
All mice (WT, hSR-BI tgn, and hSR-BII tgn) that received PBS had normal kidney histology with preserved tubular architecture, a preserved brush border (gray arrow, B1), and normal interstitial space (Fig 8. A, B1–3). hSR-BI tgn mice (B2) had occasional tubular vacuolization, represented by yellow arrows. Six hours after LPS injection, while neither WT nor hSR-BI tgn mice had any detectable kidney damage; hSR-BII tgn mice had statistically significant kidney damage compared to WT and hSR-BI tgn (Fig. 8, panel A; ANOVA with Bonferroni’s multiple comparison post-hoc test). No significant histological kidney damage beyond very rare tubular vacuolization (yellow arrow), and a few dilated tubules (blue arrows) was observed in either WT (B4) or hSR-BI tgn mice (B5) 6 hours following LPS injection. In contrast, hSR-BII tgn mice (B6) had pronounced kidney damage with prominent tubular vacuolization (white arrows), tubular dilation (green arrows), and increased interstitial space (tubules not adjacent) which may represent interstitial edema; however, we did not detect any glomerular changes.
Figure 8. LPS-induced kidney damage in various mice.
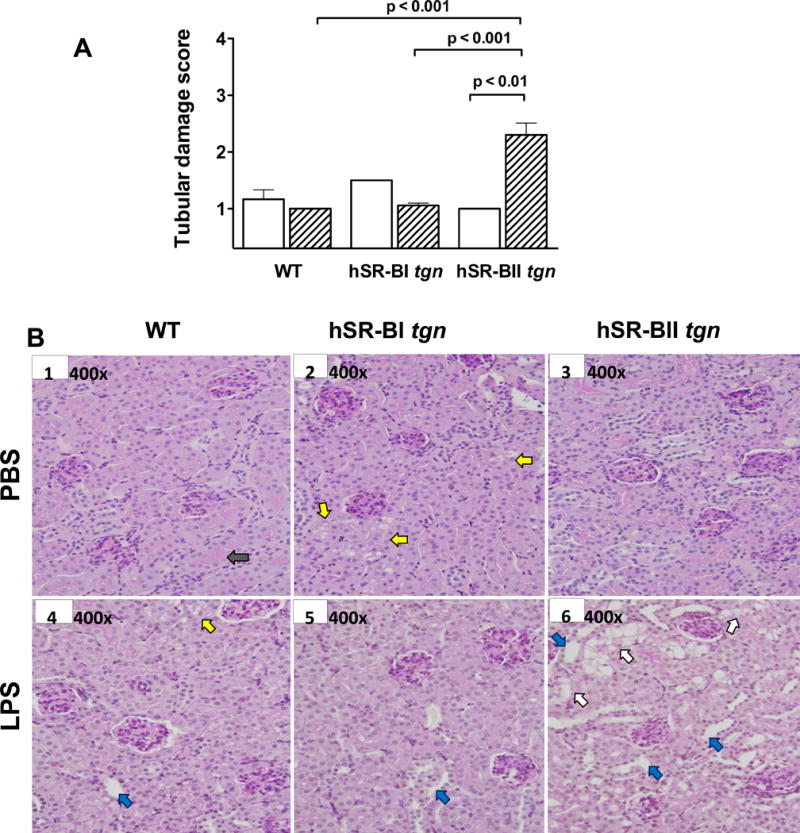
Semi-quantitative analysis of kidney injury. Kidney tubular damage was defined as tubular epithelial swelling, loss of brush border, vacuolar degeneration, necrotic tubules, cast formation, and desquamation, and scored from 1 through 4 according to % area of involvement per HPF (400×). A. Tubular damage scores of mice that received PBS (N=4–5/group, open bars), and mice that received an LPS injection (N=5/group, dashed bars). B. Representative images (400×) of kidney sections stained by PAS from each group (mice that received PBS: WT – image B1, hSR-BI tgn – image B2, and hSR-BII tgn – image B3), mice that received. LPS: WT – image B4, hSR-BI tgn – image B5, and hSR-BII tgn – image B6. The description of the arrows is in the Results section.
Effect of LPS treatment on MAPKs activation in the liver and kidney of WT, hSR-BI and hSR-BII transgenic mice
To explore potential mechanisms of the elevated LPS-induced pro-inflammatory response of hSR-B transgenic mice, we analyzed activation of several MAPK family members in the liver and kidney from LPS-challenged WT and hSR-B transgenic mice using specific antibodies against phosphorylated ERK1/2, JNK and p38 MAPKs. As shown in Fig. 9, ERK1/2 and p38 MAPKs were already activated in the liver and kidney of all control mice, with higher baseline levels observed in hSR-B transgenic animals, while the JNK activity was negligible in the tissues from all PBS-treated mice. All LPS-treated mice had an increased phosphorylation/activation of all three MAPKs in both liver and kidney tissue. However, hSR-B transgenic mice demonstrated markedly higher levels of phosphorylated MAPKs; an especially noticeable difference was observed in hepatic ERK1/2 (Fig. 9A, upper panel) and kidney JNK levels (Fig. 9B, middle panel). Among all LPS-treated mice hSR-BII transgenic mice had the highest levels of all phosphorylated MAPKs in both liver and kidney tissues compared to hSR-BI transgenic and WT mice. Total MAPKs protein levels were not affected by LPS treatment.
Figure 9. Western Blot analyses of MAPKs activity in the liver and kidney of LPS-treated WT and SR-B transgenic mice.
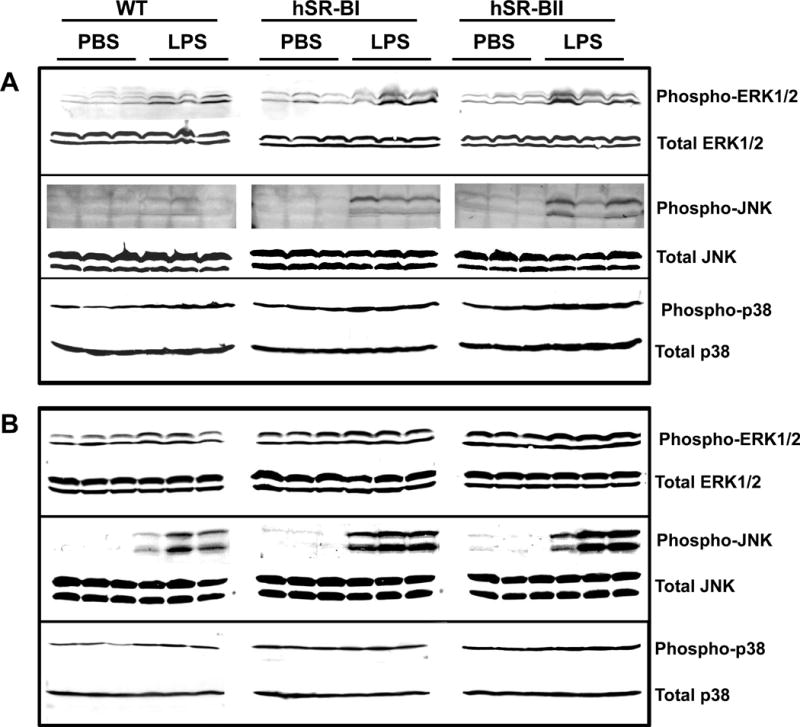
Four hours after PBS or LPS (1 mg/kg, IP) injection into WT, hSR-BI and hSR-BII tgn mice (n=3 for each group), mice were sacrificed; organs were collected and processed for assessment of MAPKs phosphorylation as described in the Materials and Methods section. A. MAPK activity of liver samples using antibodies against phospho-ERK1/2 (upper panel) or phospho-p38 (bottom panel). B. MAPK activity of kidney samples using antibodies against phospho-ERK1/2 (top panel), phospho-JNK (middle panel), or phospho-p38 (bottom panel). Equal loading of samples was ensured by using anti-ERK1/2, anti-JNK or anti-p38 MAPK antibodies against total (non-phosphorylated) MAPK protein.
LPS-induced pro-inflammatory response in bone marrow-derived macrophages and kidney epithelial cells from WT, hSR-BI and hSR-BII transgenic mice
Since hSR-BII tgn mice displayed profound kidney injury following LPS treatment compared to WT and hSR-BI tgn mice we attempted to identify the cell type responsible for the increased pro-inflammatory cytokine response and tissue damage. To test the potential contribution of macrophages toward elevated LPS-induced inflammatory cytokine production, we treated BMDM isolated from WT, hSR-BI and hSRBII transgenic mice with 1 μg/ml of LPS for the indicated time intervals (Suppl. Fig. 3). A lower dose of LPS (100 ng/ml) did not result in statistically different responses of BMDM IL-6 secretion between WT, SR-BI and hSR-BII transgenic mice following an 18-hour treatment (data not shown). WT and hSR-BI tgn macrophages stimulated with LPS demonstrated similar time-dependent increases of IL-6 and CXCL1 production, while secretion levels of both cytokines after 6 and 18 hours of LPS treatment were moderately (~ 30–35%) higher in macrophages from hSR-BII mice (Suppl. Fig. 3, C and D). At the same time, LPS-induced mRNA expression levels of both cytokines in BMDM from hSR-BII tgn mice exceeded those measured in macrophages from control mice by 2.1-fold (IL-6) and 1.7-fold (CXCL1). The differences in LPS-induced cytokine expression in BMDM from WT and hSR-BI tgn mice were less evident and not statistically significant (Suppl. Fig. 3, A and B). Our analysis of mRNA (data not shown) and protein (Suppl. Fig. 3D, insert) expression revealed fairly high levels of hSR-BI and hSR-BII in macrophages from hSR-B transgenic mice, thus, suggesting that the absence or low difference in LPS-induced cytokine responses of BMDM between hSR-B tgn and WT mice could be due to the predominant role of other pathogen recognition receptors in LPS-induced pro-inflammatory response of macrophages.
While macrophages and immune cells play a key role in innate immune response and inflammation, recent data also suggests that parenchymal cells in liver, kidney and intestine, are also actively involved in the initiation of the immune response and progression of inflammatory disorders (41, 42). It has been recently reported that murine kidney epithelial cells can participate in the renal clearance of bacteria and produce cytokines and chemokines in response to bacterial stimulation, via both TLR4-mediated and TLR4-independent signaling pathways (43). Western blot analysis of kidney epithelial cells (KECs) isolated from SR-BI and SR-BII tgn mice revealed increased (relative to endogenous levels) protein expression of both receptors (Fig. 10B). In order to assess the effects of SR-BI/SR-BII on LPS-induced pro-inflammatory responses of kidney epithelial cells, we compared LPS-induced IL-6 secretion in primary culture of KECs from WT, hSR-BI tgn and hSR-BII tgn mice. Following an 18hr incubation with LPS, a dose-dependent increase of IL-6 secretion was detected in culture media of all 3 cell types, however, both hSR-BI and hSR-BII expressing KECs demonstrated approximately 2-fold higher IL-6 levels compared to WT cells (Fig. 10A).
Figure 10. Dose-dependent LPS-induced IL-6 secretion in primary kidney epithelial cells (KECs) from WT, hSR-BI and hSR-BII transgenic mice.
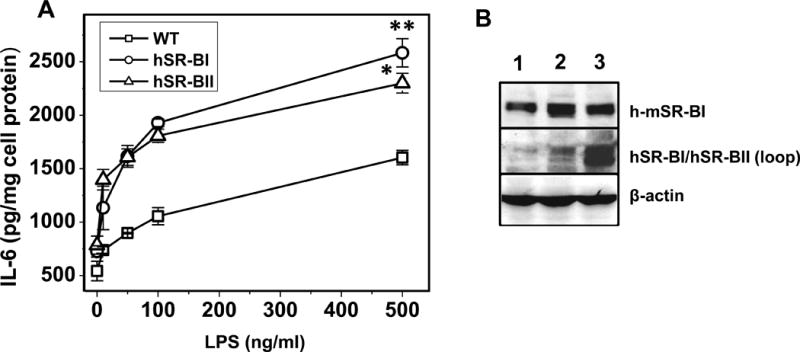
A. KECs were incubated with LPS (0, 10, 50, 100 and 500 ng/ml) in serum-free DMEM containing 2mg/ml BSA and 10mM HEPES pH 7.4 for 18 hours. Media were collected and assayed for IL-6 secretion by ELISA.
* P<0.05, ** P<0.01 vs WT LPS-treated cells. B. Western blot analysis of KECs from WT (lane 1), hSR-BI tgn (lane 2) and hSR-BII tgn (lane 3) mice. The expression of hSR-BI and mSR-BI expression was detected utilizing an anti-SR-BI antibody (against C-terminal 450–509 amino acid peptide, Novus Biologicals, cat. # NB400-101) and hSR-BI/hSR-BII protein was detected using anti-human SR-BI/BII loop antibody (against 104–294 amino acid peptide, BD Biosciences). Protein expression of β-actin was measured as the loading control.
Discussion
The innate immune response to gram-negative bacteria largely depends on the release of PAMPs such as bacterial LPS (44, 45). The mechanisms for how LPS-induced inflammation associated with cytokine and ROS overproduction and neutrophils and Kupffer cells activation causes tissue injury are not fully understood.
LPS-induced endotoxemic shock in animal models is characterized by renal inflammation, where the increased generation of pro-inflammatory cytokines/chemokines initiates and promotes acute kidney injury (AKI) (46, 47). TLR4, the major LPS receptor, is considered to be the key molecule controlling the NFkB signaling pathway (48), which in turn mediates most of inflammation and kidney damage in both LPS-induced acute kidney injury (49) and chronic kidney disease (50).
We and others previously demonstrated that gram-negative bacteria uptake is partially mediated through class B scavenger receptors, including hSR-BI/SR-BII (7, 8, 14). SR-BI/II deficiency (without steroid replacement) is associated with a hyper-inflammatory response, increased morbidity and mortality (12, 13, 25). Engagement of SR-BI and SR-BII receptors with a variety of pathogens (PAMPs) such as LPS, LTA, GroEL, and damage associated molecular patterns (DAMPs) including SAA (4, 5), induces an acute pro-inflammatory response. The role of SR-BI and SR-BII in LPS-induced inflammation and tissue damage is complex, as the SR-BI/II-KO mouse model suffers from adrenal insufficiency (25), impaired lymphocyte homeostasis and autoimmune disorders (30). To properly assess the role of SR-B receptors, models with normal adrenal and immune function are needed. Since both SR-BI and SR-BII play similar roles in lipid metabolism (2, 18, 35) with the SR-BI and SR-BII comprising 80% and 20% of scarb1 expression (2, 51), study of each receptor alone may not be complicated by adrenal insufficiency. In addition, we have found that all companies producing anti-SR-BII antibodies incorrectly used scarb2 gene as a template, which is in fact the LIMP2 (lysosomal integral membrane protein) gene sequence. As an example of this problem, the use of an actual anti-SR-BII antibody revealed a significant presence of SR-BII in the adult mink testis (52), which was not observed in other studies using LIMP2 sequence-based antibodies (53). Our specific anti hSR-BI and anti hSR-BII antibodies also demonstrated a much higher hSR-BII/SR-BI expression ratio (data not shown) than it has been previously reported. This has prompted us to study the role of hSR-BI and hSR-BII individually in transgenic mice subjected to endotoxemia.
The LPS-induced inflammatory response was assessed by measuring plasma levels of cytokines/chemokines 6 hours following LPS injection. LPS treatment induced robust pro-inflammatory responses in all three groups of experimental animals (with more than a 100-fold increase of cytokines levels); however, hSR-BI and hSR-BII tgn mice demonstrated 2- to 3-fold higher LPS-induced serum levels of pro-inflammatory cytokines than control WT littermates that received LPS. Notably, LPS increased serum levels of CXC chemokines, macrophage inflammatory protein-2 (MIP-2) and cytokine-induced neutrophil chemoattractant (KC). These are important regulators of LPS-induced transmigration and extravascular tissue accumulation of leukocytes, steps necessary for leukocytes to cause liver damage (45). Consistent with the markedly elevated CXC chemokine plasma levels, we found more profound histological changes, and in particular, a higher degree of inflammatory cell infiltration in LPS-challenged SR-BII tgn liver. Further analyses of pro-inflammatory cytokine expression profiles in liver and kidney, also revealed markedly elevated levels of mRNA expression of most cytokines measured in hSR-BII tgn mice (2000- to 3000-fold increase) and in hSR-BI tgn mice (1500- to 2000-fold increase) when compared to WT mice (1000-fold increase) following LPS injection.
Our LPS challenge model, as expected, resulted in the marked up-regulation of genes involved in the inflammatory, immune and cell adhesion responses in all three groups of mice. However, LPS administration caused histological renal injury only in SR-BII tgn mice, which was manifested by tubular vacuolization and dilation, and an enlarged interstitial space. The severity of the LPS-induced renal damage was consistent with the higher LPS-triggered pro-inflammatory response in SR-BII tgn mice, supporting the role of this SR-B splice variant as an important component of pro-inflammatory signaling pathways invoked by LPS. Data reported by others (54), as well as our own unpublished data, demonstrate prominent SR-BI expression on the surface of isolated renal proximal tubular cells, which is a major site of injury in a variety of metabolic and inflammatory diseases (55, 56). Proximal tubule cells are non-immune, parenchymal cells that can bind LPS (57), produce inflammatory mediators, such as cytokines and chemokines, and can actively participate in acute inflammatory processes by affecting leukocyte chemotaxis (58–60). Although the sites of renal expression of SR-BII are not known, our study provides new evidence implicating both SR-BI and SR-BII receptors as mediators of LPS-induced renal inflammation and injury.
The NLRP3 inflammasome, a caspase-1 activating, intracellular pathogen sensing multiprotein complex, is important in numerous inflammatory processes, including acute liver injury induced by LPS (61). LPS injection causes liver damage as indicated by elevated ALT and AST levels, and a significant upregulation of both hepatic mRNA and protein levels for all the inflammasome components including NALP3, and two cytokine targets of caspase-1, IL-1β and IL-18. While cholesterol and urate crystals stimulate NLRP3 signaling in atherosclerosis and gout, respectively (62), the role of NLRP3 is less clear in other forms of sterile inflammation or bacterial infection. In our study, we have shown that hepatic mRNA expression levels of the inflammasome NLRP3 in LPS-stimulated SRBII tgn mice exceeded those of WT mice by 2.5-fold. Recently reported data identifies the TLR4-TRIF-mediated pathway as a necessary step for inflammasome activation (63), however, the specific mechanisms of its activation and signaling remains to be determined. LPS is a ligand of TLR4, SR-BI and SR-BII, however, while signaling occurs upon its binding to all three receptors, LPS uptake is mediated only by the SR-Bs (7, 14). Thus, all these receptors could potentially contribute to the inflammasome-mediated inflammatory response via facilitating LPS translocation inside the target cell and presenting it to the inflammasome. This could be one of the mechanisms of the elevated inflammatory response observed in SR-BII mice following LPS challenge. Inflammasome activation is required to promote downstream caspase-1 cleavage and subsequent processing of pro-IL-1β into a biologically active, secreted cytokine (64). We found that following LPS challenge hSR-BII transgenic mice had moderately, but statistically significant, higher levels of plasma IL-1β, than WT littermates, potentially implicating the NLRP3/caspase-1 pathway in LPS-induced hSR-BII-dependent IL-1β release.
High levels of NO produced by the iNOS during acute inflammation and sepsis are considered to be a major cause of cytotoxicity and tissue damage (65–67). Expression of iNOS is a result of pro-inflammatory cytokine release by LPS-activated macrophages inducing iNOS gene transcription in tissue epithelial cells (68, 69). Our results demonstrate significantly (5- to 10-fold) higher mRNA expression levels of hepatic and kidney iNOS in both SR-BI and SR-BII transgenic mice compared to WT controls, which was in line with higher expression levels of pro-inflammatory cytokines responsible for iNOS activation. Interestingly, our results did not reveal any difference in systemic plasma levels of NO between the WT and hSR-BI/BII transgenic mice, perhaps reflecting a saturation effect. This may indicate that the level of NO synthesis is already limited due to the insufficient substrate or excessive arginase activity. However, higher and more rapidly rising levels of locally produced extracellular NO could exert greater damage to liver and kidneys, both being primary NO-producing organs.
Adrenal expression of SR-BI is required for cortical cholesterol accumulation and acute adrenal steroidogenesis during stress or inflammation (25, 32, 70, 71). SR-BI/II KO mice are unable to increase steroid production in response to LPS, and consequently, manifest increased susceptibility to toxic LPS effects and an uncontrolled hyper-inflammatory response (25, 71). To examine whether the reduced cholesterol levels associated with SR-B overexpression could attenuate the acute adrenal production of corticosteroids in hSRBI tgn and hSRBII tgn mice, we compared LPS-induced plasma corticosterone levels of WT and SR-B transgenic mice. In contrast to SR-BI/II KO mice, WT, hSR-BI and hSR-BII transgenic mice were capable of responding to LPS challenge by 6, 9- and 18-fold increases (vs baseline levels) of plasma corticosterone, respectively. However, the absolute values of CS plasma levels in LPS-treated WT animals were 30% higher (p < 0.05) than in hSR-BI and hSR-BII tgn mice. Our data (not shown here) demonstrated that although a SAA acute injection did not induce substantial glucocorticoid increase, there was still a marked increase in inflammatory response in both hSR-BI and hSR-BII transgenic mice when compared to WT mice, further confirming that the higher pro-inflammatory status of SR-BI/SR-BII tgn mice is unlikely due to the insufficient GCs production by the adrenals.
Our results revealing higher systemic and local pro-inflammatory responses of hSR-B transgenic mice relative to wild type mice in response to LPS do not support the previously reported beneficial role of SR-BI due to its anti-inflammatory activity during endotoxemia/sepsis. Previous studies demonstrated higher susceptibility of SR-BI-null mice to endotoxin-induced inflammation leading to a higher death rate, thus suggesting a protective role of SR-BI in sepsis (12, 15, 25). Most of these studies employed the model of SR-BI-null mice characterized by multiple immune and metabolic disorders including impaired homeostasis of lymphocytes and splenic macrophages (30), defective lipoprotein metabolism (29) and adrenal insufficiency (13, 25), resulting in an abnormally high inflammatory cytokine response to LPS (32). Consequently, without an appropriate compensation for adrenal corticosteroid deӿciency, the true role of SR-BI during LPS-induced inӾammation could be misinterpreted. Indeed, our previous ӿndings demonstrated that glucocorticoid/mineralocorticoid supplementation could signiӿcantly normalize the acute inӾammation response in mice with SR-BI/II deӿciency (13). SRBI/BII-null mice did not survive CLP sepsis in the absence of steroids. In contrast, these SRBI/BII-deficient mice had 48% survival rate and markedly reduced plasma IL-6 and TNF-α levels compared to a wild type mice, as a result of steroid replacement therapy. A recent study of Gilibert et al. (72) utilizing transgenic mice with tissue-specific SR-BI expression further highlighted the critical importance of the intact adrenal SR-BI-dependent glucocorticoid production for the host response to LPS. Their data demonstrated that mice lacking SR-BI in the adrenal cortex exposed to an endotoxin challenge exhibited an exacerbated systemic and local inflammatory response and higher lethality rate compared to normal mice, whereas mice deficient for SR-BI in hepatocytes, endothelial cells or the myeloid cell lineage (including monocytes, macrophages, and granulocytes) were not more susceptible to LPS-induced death. In addition, they also found that SR-BI ablation in hepatocytes led to a moderate increase in systemic inflammatory markers, and SR-BI deficiency in myeloid cells was associated with an anti-inflammatory effect.
As was suggested by the earlier report of Guo et al. (12) the protective role of SR-BI could be attributed to its ability to modulate the inflammatory response to LPS in macrophages as their results demonstrated moderately increased pro-inflammatory cytokine production to LPS by SR-BI/BII-deficient macrophages relative to cells from normal mice. Another study (30) demonstrated a significant increase in the number of splenic monocytes/macrophages, having markedly elevated expression of pro-inflammatory cytokines in SR-BI-null mice, thus revealing impaired homeostasis of myeloid cells due to SR-BI deficiency. In our study, on the contrary, we did not observe a statistically significant difference in cytokine response to LPS between the bone marrow-derived macrophages from WT and SR-BI transgenic mice and only modestly (30–35%) higher levels of IL6 and CXCL1 were produced by LPS-treated cells from SR-BII tgn mice when compared to normal cells. We suggest that the higher pro-inflammatory response to LPS found by Guo et al. (12) in macrophages from SR-BI-null mice could be attributed to the abnormally heightened activation of these cells. In addition, the results of Gilibert et al. (72) revealing a trend for increased survival of CLP-treated mice with SR-BI deletion in myeloid cells would also not support a major protective function for SR-BI expressed in macrophages.
In addition to the well-known role of HDL in LPS neutralization, recently reported data (40) demonstrated its ability to protect against CLP-induced sepsis through promoting LPS clearance and leucocyte recruitment. Our data revealed a ~ 70% and ~ 60% reduction of plasma HDL levels (assessed by an apoA-I immunoblotting assay) in hSR-BI and hSR-BII transgenic mice, respectively, when compared to WT mice. Nonetheless, despite the markedly lower HDL levels in plasma of hSR-B transgenic mice the endotoxicity of their plasma following LPS challenge was only moderately higher (by ~ 40%) when compared to WT mice, perhaps due to the increased LPS clearance mediated by the hepatic hSR-BI (15, 25) and hSR-BII.
Involvement of MAPK signaling pathways is implicated in LPS-induced release of pro-inflammatory cytokines such as IL6, IL-1β and TNF-a (73, 74). In this study we examined the potential contribution of ERK1/2, JNK and p38 MAPKs in the enhanced inflammatory response of LPS-challenged hSR-B transgenic mice. We have found that LPS treatment results in higher phosphorylation levels of all three MAP kinases in liver and kidney tissues of both transgenic mice strains compared to WT mice with the most pronounced difference observed for the hepatic ERK1/2 and kidney JNK activity. Our results indicate that MAPKs provide differential and tissue-specific contributions to the hSR-BI- and hSR-BII-dependent pro-inflammatory LPS response.
Previous reports have implicated mouse SR-BI as a signal transducing receptor that interacts with a PDZ domain-containing adaptor protein PDZK1 (75), and binds HDL to stimulate eNOS activity and cell migration through the Src family kinase/MAPK-mediated signaling pathway (76). Our earlier studies demonstrated that SAA activates p44/p42 and p38 MAP kinases in epithelial cells in a CLA-1 (hSR-BI)-dependent manner (4). Additional studies in bovine aortic endothelial cells have revealed that Src family kinase(s) (SFKs) participate in the proximal signaling events activated by HDL–SR-BI in the endothelium (77) and co-immunoprecipitation experiments have further confirmed that c-Src can interact with SR-BI, although the specific domain participating in the interaction has not been identified. There have been no reports regarding the signaling capacity of the SR-BII, however, several sequences, including six proline-rich motifs, capable to bind SH3 (Src homology 3) domains, and one SH2 (Src homology 2)-domain recognition site, have been identified within the C-terminal region of human SR-BII (78). We suggest that SFKs could be important upstream regulators of the hSR-B-mediated LPS-induced MAPK signaling pathway due to an interaction of the SR-BII cytoplasmic tail with SH2 and/or SH3 domains of the SFKs, initiated by ligand-receptor binding. Additional studies to analyze the activation/phosphorylation of SFKs as well as co-immunoprecipitation assays utilizing SR-B expressing cells and tissues exposed to LPS are required to further investigate the above proposed mechanism.
To investigate which cell type within the kidney could be responsible for hSR-BI/II-mediated effects, LPS-induced pro-inflammatory responses of bone marrow derived macrophages (BMDM) or kidney epithelial cells from hSR-BI/II tgn mice were compared to WT mice. Our data demonstrating only moderate difference in LPS-induced cytokine response of BMDM from WT and hSR-B transgenic mice suggests that other LPS receptors, e.g. TLR4, CD36 and SR-A could play much more prominent roles as mediators of LPS-induced inflammatory responses in macrophages, while the contribution of SR-Bs in these cells is limited.
It has been demonstrated that Kupffer cells and kidney interstitial macrophages are important sources of hepato- and nephrotoxicity during LPS-induced acute organ injury (79–82). However, recent findings also reveal that parenchymal cells, such as hepatocytes, intestinal and kidney epithelial cells are important in the innate immune response, contributing to cytokine/chemokine production (43, 83, 84) as well as being the principal source of high systemic NO (41) levels during septic shock.
SR-BI is abundantly expressed in hepatocytes and kidney epithelial cells, both being important LPS target cells during sepsis (42, 85, 86). The expression of SR-BII has been also reported in mouse and human liver (51, 87). Since BMDM from SR-B transgenic mice demonstrated only a moderately enhanced pro-inflammatory phenotype compared to WT (Suppl. Fig. 1), epithelial cells could represent another important SR-BI/II-related target of LPS. Indeed, our data demonstrating a 2-fold increase of IL-6 response in primary culture of LPS-treated KEC from both hSR-BI and hSR-BII transgenic mice, compared to the WT control cells, support this hypothesis.
In conclusion, we found that both SR-BI and SR-BII transgenic mice had significantly higher expression of pro-inflammatory mediators and more prominent hepatic and renal injury following LPS challenge vs. control mice. These results indicate, that in addition to the well-established LPS effects mediated by TLR4, the ability of SR-BI and SR-BII to recognize LPS, and more generally, gram-negative bacteria, enable these receptors to substantially contribute to the LPS/PAMP-induced pro-inflammatory responses and tissue damage during endotoxemia/sepsis.
Supplementary Material
Acknowledgments
Grant support
This research was supported by the NIH Intramural Research Programs at the CC, NIDDK, NIAID and NHLBI.
Abbreviations
- BMDM
bone marrow-derived macrophages
- CLP
cecal ligation and puncture
- CXCL1
chemokine (C-X-C motif) ligand 1
- CS
corticosterone
- ERK1/2
extracellular signal-regulated protein kinase 1 and 2
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GCs
glucocorticoids
- HPF
high-power field
- iNOS
inducible NO synthase
- JNK
c-Jun N-terminal kinase
- KECs
kidney epithelial cells
- MAPK
mitogen-activated protein kinases
- NLRP3
NLR family pyrin domain containing 3
- LPS
lipopolysaccharide
- LTA
lipoteichoic acid
- ROS
reactive oxygen species
- SAA
serum amyloid A
- SR-BI-KO
scavenger receptor BI knockout mice
- hSR-BI tgn
human scavenger receptor BI transgenic mice
- hSR-BII tgn
human scavenger receptor BII transgenic mice
- SFKs
Src-family kinases
- WT
wild type mice.
References
- 1.Krieger M. Scavenger receptor class B type I is a multiligand HDL receptor that influences diverse physiologic systems. J Clin Invest. 2001;108:793–797. doi: 10.1172/JCI14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webb NR, Connell PM, Graf GA, Smart EJ, de Villiers WJ, de Beer FC, van der Westhuyzen DR. SR-BII, an isoform of the scavenger receptor BI containing an alternate cytoplasmic tail, mediates lipid transfer between high density lipoprotein and cells. J Biol Chem. 1998;273:15241–15248. doi: 10.1074/jbc.273.24.15241. [DOI] [PubMed] [Google Scholar]
- 3.Murao K, Terpstra V, Green SR, Kondratenko N, Steinberg D, Quehenberger O. Characterization of CLA-1, a human homologue of rodent scavenger receptor BI, as a receptor for high density lipoprotein and apoptotic thymocytes. J Biol Chem. 1997;272:17551–17557. doi: 10.1074/jbc.272.28.17551. [DOI] [PubMed] [Google Scholar]
- 4.Baranova IN, Vishnyakova TG, Bocharov AV, Kurlander R, Chen Z, Kimelman ML, Remaley AT, Csako G, Thomas F, Eggerman TL, Patterson AP. Serum amyloid A binding to CLA-1 (CD36 and LIMPII analogous-1) mediates serum amyloid A protein-induced activation of ERK1/2 and p38 mitogen-activated protein kinases. J Biol Chem. 2005;280:8031–8040. doi: 10.1074/jbc.M405009200. [DOI] [PubMed] [Google Scholar]
- 5.Cai L, de Beer MC, de Beer FC, van der Westhuyzen DR. Serum amyloid A is a ligand for scavenger receptor class B type I and inhibits high density lipoprotein binding and selective lipid uptake. J Biol Chem. 2005;280:2954–2961. doi: 10.1074/jbc.M411555200. [DOI] [PubMed] [Google Scholar]
- 6.Husemann J, Loike JD, Kodama T, Silverstein SC. Scavenger receptor class B type I (SR-BI) mediates adhesion of neonatal murine microglia to fibrillar beta-amyloid. J Neuroimmunol. 2001;114:142–150. doi: 10.1016/s0165-5728(01)00239-9. [DOI] [PubMed] [Google Scholar]
- 7.Vishnyakova TG, Kurlander R, Bocharov AV, Baranova IN, Chen Z, Abu-Asab MS, Tsokos M, Malide D, Basso F, Remaley A, Csako G, Eggerman TL, Patterson AP. CLA-1 and its splicing variant CLA-2 mediate bacterial adhesion and cytosolic bacterial invasion in mammalian cells. Proc Natl Acad Sci USA. 2006;103:16888–16893. doi: 10.1073/pnas.0602126103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Philips JA, Rubin EJ, Perrimon N. Drosophila RNAi screen reveals CD36 family member required for mycobacterial infection. Science. 2005;309:1251–1253. doi: 10.1126/science.1116006. [DOI] [PubMed] [Google Scholar]
- 9.Bird DA, Gillotte KL, Horkko S, Friedman P, Dennis EA, Witztum JL, Steinberg D. Receptors for oxidized low-density lipoprotein on elicited mouse peritoneal macrophages can recognize both the modified lipid moieties and the modified protein moieties: implications with respect to macrophage recognition of apoptotic cells. Proc Natl Acad Sci USA. 1999;96:6347–6352. doi: 10.1073/pnas.96.11.6347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang W, Yancey PG, Su YR, Babaev VR, Zhang Y, Fazio S, Linton MF. Inactivation of macrophage scavenger receptor class B type I promotes atherosclerotic lesion development in apolipoprotein E-deficient mice. Circulation. 2003;108:2258–2263. doi: 10.1161/01.CIR.0000093189.97429.9D. [DOI] [PubMed] [Google Scholar]
- 11.Pei Y, Chen X, Aboutouk D, Fuller MT, Dadoo O, Yu P, White EJ, Igdoura SA, Trigatti BL. SR-BI in bone marrow derived cells protects mice from diet induced coronary artery atherosclerosis and myocardial infarction. PloS one. 2013;8:e72492. doi: 10.1371/journal.pone.0072492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo L, Song Z, Li M, Wu Q, Wang D, Feng H, Bernard P, Daugherty A, Huang B, Li XA. Scavenger Receptor BI Protects against Septic Death through Its Role in Modulating Inflammatory Response. J Biol Chem. 2009;284:19826–19834. doi: 10.1074/jbc.M109.020933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leelahavanichkul A, Bocharov AV, Kurlander R, Baranova IN, Vishnyakova TG, Souza AC, Hu X, Doi K, Vaisman B, Amar M, Sviridov D, Chen Z, Remaley AT, Csako G, Patterson AP, Yuen PS, Star RA, Eggerman TL. Class B scavenger receptor types I and II and CD36 targeting improves sepsis survival and acute outcomes in mice. J Immunol. 2012;188:2749–2758. doi: 10.4049/jimmunol.1003445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baranova IN, Vishnyakova TG, Bocharov AV, Leelahavanichkul A, Kurlander R, Chen Z, Souza AC, Yuen PS, Star RA, Csako G, Patterson AP, Eggerman TL. Class B scavenger receptor types I and II and CD36 mediate bacterial recognition and proinflammatory signaling induced by Escherichia coli, lipopolysaccharide, and cytosolic chaperonin 60. J Immunol. 2012;188:1371–1380. doi: 10.4049/jimmunol.1100350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo L, Zheng Z, Ai J, Huang B, Li XA. Hepatic scavenger receptor BI protects against polymicrobial-induced sepsis through promoting LPS clearance in mice. J Biol Chem. 2014;280:14666–14673. doi: 10.1074/jbc.M113.537258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Catanese MT, Ansuini H, Graziani R, Huby T, Moreau M, Ball JK, Paonessa G, Rice CM, Cortese R, Vitelli A, Nicosia A. Role of scavenger receptor class B type I in hepatitis C virus entry: kinetics and molecular determinants. Journal Virol. 2010;84:34–43. doi: 10.1128/JVI.02199-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Acton SL, Scherer PE, Lodish HF, Krieger M. Expression cloning of SR-BI, a CD36-related class B scavenger receptor. The Journal of biological chemistry. 1994;269:21003–21009. [PubMed] [Google Scholar]
- 18.Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, Krieger M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science. 1996;271:518–520. doi: 10.1126/science.271.5248.518. [DOI] [PubMed] [Google Scholar]
- 19.Azhar S, Reaven E. Scavenger receptor class BI and selective cholesteryl ester uptake: partners in the regulation of steroidogenesis. Mol Cell Endocrinol. 2002;195:1–26. doi: 10.1016/s0303-7207(02)00222-8. [DOI] [PubMed] [Google Scholar]
- 20.Brundert M, Ewert A, Heeren J, Behrendt B, Ramakrishnan R, Greten H, Merkel M, Rinninger F. Scavenger receptor class B type I mediates the selective uptake of high-density lipoprotein-associated cholesteryl ester by the liver in mice. Arterioscler Thromb Vasc Biol. 2005;25:143–148. doi: 10.1161/01.ATV.0000149381.16166.c6. [DOI] [PubMed] [Google Scholar]
- 21.Duncan KG, Bailey KR, Kane JP, Schwartz DM. Human retinal pigment epithelial cells express scavenger receptors BI and BII. Biochem Biophys Res Commun. 2002;292:1017–1022. doi: 10.1006/bbrc.2002.6756. [DOI] [PubMed] [Google Scholar]
- 22.Miquel JF, Moreno M, Amigo L, Molina H, Mardones P, Wistuba, Rigotti A. Expression and regulation of scavenger receptor class B type I (SR-BI) in gall bladder epithelium. Gut. 2003;52:1017–1024. doi: 10.1136/gut.52.7.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chinetti G, Gbaguidi FG, Griglio S, Mallat Z, Antonucci M, Poulain P, Chapman J, Fruchart JC, Tedgui A, Najib-Fruchart J, Staels B. CLA-1/SR-BI is expressed in atherosclerotic lesion macrophages and regulated by activators of peroxisome proliferator-activated receptors. Circulation. 2000;101:2411–2417. doi: 10.1161/01.cir.101.20.2411. [DOI] [PubMed] [Google Scholar]
- 25.Cai L, Ji A, de Beer FC, Tannock LR, van der Westhuyzen DR. SR-BI protects against endotoxemia in mice through its roles in glucocorticoid production and hepatic clearance. J Clin Invest. 2008;118:364–375. doi: 10.1172/JCI31539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li XA, Guo L, Asmis R, Nikolova-Karakashian M, Smart EJ. Scavenger receptor BI prevents nitric oxide-induced cytotoxicity and endotoxin-induced death. Circ Res. 2006;98:e60–65. doi: 10.1161/01.RES.0000219310.00308.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gowda NM, Wu X, Kumar S, Febbraio M, Gowda DC. CD36 contributes to malaria parasite-induced pro-inflammatory cytokine production and NK and T cell activation by dendritic cells. PloS one. 2013;8:e77604. doi: 10.1371/journal.pone.0077604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci USA. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yesilaltay A, Morales MG, Amigo L, Zanlungo S, Rigotti A, Karackattu SL, Donahee MH, Kozarsky KF, Krieger M. Effects of hepatic expression of the high-density lipoprotein receptor SR-BI on lipoprotein metabolism and female fertility. Endocrinology. 2006;147:1577–1588. doi: 10.1210/en.2005-1286. [DOI] [PubMed] [Google Scholar]
- 30.Feng H, Guo L, Wang D, Gao H, Hou G, Zheng Z, Ai J, Foreman O, Daugherty A, Li XA. Deficiency of scavenger receptor BI leads to impaired lymphocyte homeostasis and autoimmune disorders in mice. Arterioscler Thromb Vasc Biol. 2011;31:2543–2551. doi: 10.1161/ATVBAHA.111.234716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meurs I, Hoekstra M, van Wanrooij EJ, Hildebrand RB, Kuiper J, Kuipers F, Hardeman MR, Van Berkel TJ, Van Eck M. HDL cholesterol levels are an important factor for determining the lifespan of erythrocytes. Exp Hematol. 2005;33:1309–1319. doi: 10.1016/j.exphem.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 32.Hoekstra M, Meurs I, Koenders M, Out R, Hildebrand RB, Kruijt JK, Van Eck M, Van Berkel TJ. Absence of HDL cholesteryl ester uptake in mice via SR-BI impairs an adequate adrenal glucocorticoid-mediated stress response to fasting. J Lipid Res. 2008;49:738–745. doi: 10.1194/jlr.M700475-JLR200. [DOI] [PubMed] [Google Scholar]
- 33.Simonet WS, Bucay N, Lauer SJ, Taylor JM. A far-downstream hepatocyte-specific control region directs expression of the linked human apolipoprotein E and C-I genes in transgenic mice. J Biol Chem. 1993;268:8221–8229. [PubMed] [Google Scholar]
- 34.Vaisman BL, Klein HG, Rouis M, Berard AM, Kindt MR, Talley GD, Meyn SM, Hoyt RF, Jr, Marcovina SM, Albers JJ, et al. Overexpression of human lecithin cholesterol acyltransferase leads to hyperalphalipoproteinemia in transgenic mice. J Biol Chem. 1995;270:12269–12275. doi: 10.1074/jbc.270.20.12269. [DOI] [PubMed] [Google Scholar]
- 35.Eckhardt ER, Cai L, Sun B, Webb NR, van der Westhuyzen DR. High density lipoprotein uptake by scavenger receptor SR-BII. J Biol Chem. 2004;279:14372–14381. doi: 10.1074/jbc.M313793200. [DOI] [PubMed] [Google Scholar]
- 36.Boaru SG, Borkham-Kamphorst E, Tihaa L, Haas U, Weiskirchen R. Expression analysis of inflammasomes in experimental models of inflammatory and fibrotic liver disease. J Inflam. 2012;9:49. doi: 10.1186/1476-9255-9-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim SJ, Lee SM. NLRP3 inflammasome activation in D-galactosamine and lipopolysaccharide-induced acute liver failure: role of heme oxygenase-1. Free Radic Biol Med. 2013;65:997–1004. doi: 10.1016/j.freeradbiomed.2013.08.178. [DOI] [PubMed] [Google Scholar]
- 38.Novogrodsky A, Vanichkin A, Patya M, Gazit A, Osherov N, Levitzki A. Prevention of lipopolysaccharide-induced lethal toxicity by tyrosine kinase inhibitors. Science. 1994;264:1319–1322. doi: 10.1126/science.8191285. [DOI] [PubMed] [Google Scholar]
- 39.Brandtzaeg P, Kierulf P, Gaustad P, Dobloug J, Mollnes TE, Sirnes K. Systemic meningococcal disease: a model infection to study acute endotoxinemia in man. Prog Clin Biol Res. 1988;272:263–271. [PubMed] [Google Scholar]
- 40.Guo L, Ai J, Zheng Z, Howatt DA, Daugherty A, Huang B, Li XA. High density lipoprotein protects against polymicrobe-induced sepsis in mice. J Biol Chem. 2013;288:17947–17953. doi: 10.1074/jbc.M112.442699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bultinck J, Sips P, Vakaet L, Brouckaert P, Cauwels A. Systemic NO production during (septic) shock depends on parenchymal and not on hematopoietic cells: in vivo iNOS expression pattern in (septic) shock. FASEB J. 2006;20:2363–2365. doi: 10.1096/fj.06-5798fje. [DOI] [PubMed] [Google Scholar]
- 42.Vodovotz Y, Liu S, McCloskey C, Shapiro R, Green A, Billiar TR. The hepatocyte as a microbial product-responsive cell. J Endotoxin Res. 2001;7:365–373. [PubMed] [Google Scholar]
- 43.Chassin C, Goujon JM, Darche S, du Merle L, Bens M, Cluzeaud F, Werts C, Ogier-Denis E, Le Bouguenec C, Buzoni-Gatel D, Vandewalle A. Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J Immunol. 2006;177:4773–4784. doi: 10.4049/jimmunol.177.7.4773. [DOI] [PubMed] [Google Scholar]
- 44.Rietschel ET, Kirikae T, Schade FU, Mamat U, Schmidt G, Loppnow H, Ulmer AJ, Zahringer U, Seydel U, Di Padova F, et al. Bacterial endotoxin: molecular relationships of structure to activity and function. FASEB J. 1994;8:217–225. doi: 10.1096/fasebj.8.2.8119492. [DOI] [PubMed] [Google Scholar]
- 45.Li X, Klintman D, Liu Q, Sato T, Jeppsson B, Thorlacius H. Critical role of CXC chemokines in endotoxemic liver injury in mice. J Leukoc Biol. 2004;75:443–452. doi: 10.1189/jlb.0603297. [DOI] [PubMed] [Google Scholar]
- 46.Wang W, Bansal S, Falk S, Ljubanovic D, Schrier R. Ghrelin protects mice against endotoxemia-induced acute kidney injury. Am J Physiol Renal Physiol. 2009;297:F1032–1037. doi: 10.1152/ajprenal.00044.2009. [DOI] [PubMed] [Google Scholar]
- 47.Zahedi K, Barone S, Kramer DL, Amlal H, Alhonen L, Janne J, Porter CW, Soleimani M. The role of spermidine/spermine N1-acetyltransferase in endotoxin-induced acute kidney injury. Am J Physiol Cell Physiol. 2010;299:C164–174. doi: 10.1152/ajpcell.00512.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ. TLR4 activation mediates kidney ischemia/reperfusion injury. J Clin Invest. 2007;117:2847–2859. doi: 10.1172/JCI31008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172:2629–2635. doi: 10.4049/jimmunol.172.4.2629. [DOI] [PubMed] [Google Scholar]
- 50.Campbell MT, Hile KL, Zhang H, Asanuma H, Vanderbrink BA, Rink RR, Meldrum KK. Toll-like receptor 4: a novel signaling pathway during renal fibrogenesis. The J Surg Res. 2011;168:e61–69. doi: 10.1016/j.jss.2009.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chiba-Falek O, Nichols M, Suchindran S, Guyton J, Ginsburg GS, Barrett-Connor E, McCarthy JJ. Impact of gene variants on sex-specific regulation of human Scavenger receptor class B type 1 (SR-BI) expression in liver and association with lipid levels in a population-based study. BMC Med Genet. 2010;11:9. doi: 10.1186/1471-2350-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Akpovi CD, Yoon SR, Vitale ML, Pelletier RM. The predominance of one of the SR-BI isoforms is associated with increased esterified cholesterol levels not apoptosis in mink testis. J Lipid Res. 2006;47:2233–2247. doi: 10.1194/jlr.M600162-JLR200. [DOI] [PubMed] [Google Scholar]
- 53.Cai SF, Kirby RJ, Howles PN, Hui DY. Differentiation-dependent expression and localization of the class B type I scavenger receptor in intestine. J Lipid Res. 2001;42:902–909. [PubMed] [Google Scholar]
- 54.Tsun JG, Yung S, Chau MK, Shiu SW, Chan TM, Tan KC. Cellular cholesterol transport proteins in diabetic nephropathy. PloS one. 2014;9:e105787. doi: 10.1371/journal.pone.0105787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Kooten C, Daha MR, van Es LA. Tubular epithelial cells: A critical cell type in the regulation of renal inflammatory processes. Exp Nephrol. 1999;7:429–437. doi: 10.1159/000020622. [DOI] [PubMed] [Google Scholar]
- 56.Tang SC, Leung JC, Chan LY, Tsang AW, Lai KN. Activation of tubular epithelial cells in diabetic nephropathy and the role of the peroxisome proliferator-activated receptor-gamma agonist. Journal of the American Society of Nephrology: JASN. 2006;17:1633–1643. doi: 10.1681/ASN.2005101113. [DOI] [PubMed] [Google Scholar]
- 57.Kalakeche R, Hato T, Rhodes G, Dunn KW, El-Achkar TM, Plotkin Z, Sandoval RM, Dagher PC. Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J Am Soc Nephrol. 2011;22:1505–1516. doi: 10.1681/ASN.2011020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.van Kooten C, Daha MR. Cytokine cross-talk between tubular epithelial cells and interstitial immunocompetent cells. Curr Opin Nephrol Hypertens. 2001;10:55–59. doi: 10.1097/00041552-200101000-00009. [DOI] [PubMed] [Google Scholar]
- 59.Schmouder RL, Strieter RM, Wiggins RC, Chensue SW, Kunkel SL. In vitro and in vivo interleukin-8 production in human renal cortical epithelia. Kidney Int. 1992;41:191–198. doi: 10.1038/ki.1992.26. [DOI] [PubMed] [Google Scholar]
- 60.Franquesa M, Riera M, Herrero-Fresneda I, Sola A, Hotter G, Lloberas N, Cruzado JM, Torras J, Grinyo JM. Tubular epithelial cells transfected with hHGF counteracts monocyte chemotactic protein-1 up-regulation after hypoxia/reoxygenation insult. Transplant Proc. 2009;41:2069–2072. doi: 10.1016/j.transproceed.2009.05.023. [DOI] [PubMed] [Google Scholar]
- 61.Ganz M, Csak T, Nath B, Szabo G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J Gastroenterol. 2011;17:4772–4778. doi: 10.3748/wjg.v17.i43.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 63.Tsutsui H, Imamura M, Fujimoto J, Nakanishi K. The TLR4/TRIF-Mediated Activation of NLRP3 Inflammasome Underlies Endotoxin-Induced Liver Injury in Mice. Gastroenterol Res Pract. 2010;2010:641865. doi: 10.1155/2010/641865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 65.Tunctan B, Uludag O, Altug S, Abacioglu N. Effects of nitric oxide synthase inhibition in lipopolysaccharide-induced sepsis in mice. Pharmacol Res. 1998;38:405–411. doi: 10.1006/phrs.1998.0381. [DOI] [PubMed] [Google Scholar]
- 66.Numata M, Suzuki S, Miyazawa N, Miyashita A, Nagashima Y, Inoue S, Kaneko T, Okubo T. Inhibition of inducible nitric oxide synthase prevents LPS-induced acute lung injury in dogs. J Immunol. 1998;160:3031–3037. [PubMed] [Google Scholar]
- 67.Ahmad N, Chen LC, Gordon MA, Laskin JD, Laskin DL. Regulation of cyclooxygenase-2 by nitric oxide in activated hepatic macrophages during acute endotoxemia. J Leukoc Biol. 2002;71:1005–1011. [PubMed] [Google Scholar]
- 68.Wong JM, Billiar TR. Regulation and function of inducible nitric oxide synthase during sepsis and acute inflammation. Adv Pharmacol. 1995;34:155–170. doi: 10.1016/s1054-3589(08)61084-4. [DOI] [PubMed] [Google Scholar]
- 69.Bogdan C. Nitric oxide and the immune response. Nat Immunol. 2001;2:907–916. doi: 10.1038/ni1001-907. [DOI] [PubMed] [Google Scholar]
- 70.Temel RE, Trigatti B, DeMattos RB, Azhar S, Krieger M, Williams DL. Scavenger receptor class B, type I (SR-BI) is the major route for the delivery of high density lipoprotein cholesterol to the steroidogenic pathway in cultured mouse adrenocortical cells. Proc Natl Acad Sci USA. 1997;94:13600–13605. doi: 10.1073/pnas.94.25.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hoekstra M, Ye D, Hildebrand RB, Zhao Y, Lammers B, Stitzinger M, Kuiper J, Van Berkel TJ, Van Eck M. Scavenger receptor class B type I-mediated uptake of serum cholesterol is essential for optimal adrenal glucocorticoid production. J Lipid Res. 2009;50:1039–1046. doi: 10.1194/jlr.M800410-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gilibert S, Galle-Treger L, Moreau M, Saint-Charles F, Costa S, Ballaire R, Couvert P, Carrie A, Lesnik P, Huby T. Adrenocortical scavenger receptor class B type I deficiency exacerbates endotoxic shock and precipitates sepsis-induced mortality in mice. J Immunol. 2014;193:817–826. doi: 10.4049/jimmunol.1303164. [DOI] [PubMed] [Google Scholar]
- 73.Kim SH, Kim J, Sharma RP. Inhibition of p38 and ERK MAP kinases blocks endotoxin-induced nitric oxide production and differentially modulates cytokine expression. Pharmacol Res. 2004;49:433–439. doi: 10.1016/j.phrs.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 74.Lee SJ, Lim KT. Inhibitory effect of ZPDC glycoprotein on the expression of inflammation-related cytokines through p38 MAP kinase and JNK in lipopolysaccharide-stimulated RAW 264.7 cells. Inflamm Res. 2009;58:184–191. doi: 10.1007/s00011-008-8118-2. [DOI] [PubMed] [Google Scholar]
- 75.Ikemoto M, Arai H, Feng D, Tanaka K, Aoki J, Dohmae N, Takio K, Adachi H, Tsujimoto M, Inoue K. Identification of a PDZ-domain-containing protein that interacts with the scavenger receptor class B type I. Proc Natl Acad Sci USA. 2000;97:6538–6543. doi: 10.1073/pnas.100114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mineo C, I, Yuhanna S, Quon MJ, Shaul PW. High density lipoprotein-induced endothelial nitric-oxide synthase activation is mediated by Akt and MAP kinases. J Biol Chem. 2003;278:9142–9149. doi: 10.1074/jbc.M211394200. [DOI] [PubMed] [Google Scholar]
- 77.Zhu W, Saddar S, Seetharam D, Chambliss KL, Longoria C, Silver DL, Yuhanna IS, Shaul PW, Mineo C. The scavenger receptor class B type I adaptor protein PDZK1 maintains endothelial monolayer integrity. Circ Res. 2008;102:480–487. doi: 10.1161/CIRCRESAHA.107.159079. [DOI] [PubMed] [Google Scholar]
- 78.Mulcahy JV, Riddell DR, Owen JS. Human scavenger receptor class B type II (SR-BII) and cellular cholesterol efflux. Biochem J. 2004;377:741–747. doi: 10.1042/BJ20030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fukuda M, Yokoyama H, Mizukami T, Ohgo H, Okamura Y, Kamegaya Y, Horie Y, Kato S, Ishii H. Kupffer cell depletion attenuates superoxide anion release into the hepatic sinusoids after lipopolysaccharide treatment. J Gastroenterol Hepatol. 2004;19:1155–1162. doi: 10.1111/j.1440-1746.2004.03408.x. [DOI] [PubMed] [Google Scholar]
- 80.El Kasmi KC, Anderson AL, Devereaux MW, Fillon SA, Harris JK, Lovell MA, Finegold MJ, Sokol RJ. Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology. 2012;55:1518–1528. doi: 10.1002/hep.25500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Juskewitch JE, Platt JL, Knudsen BE, Knutson KL, Brunn GJ, Grande JP. Disparate roles of marrow- and parenchymal cell-derived TLR4 signaling in murine LPS-induced systemic inflammation. Sci Rep. 2012;2:918. doi: 10.1038/srep00918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rogers NM, Ferenbach DA, Isenberg JS, Thomson AW, Hughes J. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol. 2014;10:625–643. doi: 10.1038/nrneph.2014.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Liu S, Gallo DJ, Green AM, Williams DL, Gong X, Shapiro RA, Gambotto AA, Humphris EL, Vodovotz Y, Billiar TR. Role of toll-like receptors in changes in gene expression and NF-kappa B activation in mouse hepatocytes stimulated with lipopolysaccharide. Infect Immun. 2002;70:3433–3442. doi: 10.1128/IAI.70.7.3433-3442.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Y, Rangan GK, Goodwin B, Tay YC, Harris DC. Lipopolysaccharide-induced MCP-1 gene expression in rat tubular epithelial cells is nuclear factor-kappaB dependent. Kidney Int. 2000;57:2011–2022. doi: 10.1046/j.1523-1755.2000.00051.x. [DOI] [PubMed] [Google Scholar]
- 85.Tiwari MM, Messer KJ, Mayeux PR. Inducible nitric oxide synthase and apoptosis in murine proximal tubule epithelial cells. Toxicol Sci. 2006;91:493–500. doi: 10.1093/toxsci/kfj168. [DOI] [PubMed] [Google Scholar]
- 86.Scott MJ, Liu S, Shapiro RA, Vodovotz Y, Billiar TR. Endotoxin uptake in mouse liver is blocked by endotoxin pretreatment through a suppressor of cytokine signaling-1-dependent mechanism. Hepatology. 2009;49:1695–1708. doi: 10.1002/hep.22839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Graf GA, Roswell KL, Smart EJ. 17beta-Estradiol promotes the up-regulation of SR-BII in HepG2 cells and in rat livers. J Lipid Res. 2001;42:1444–1449. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.