


Abstract
G-quadruplexes (GQs) are 4-stranded DNA structures formed by tracts of stacked, Hoogsteen-hydrogen bonded guanosines. GQs are found in gene promoters and telomeres where they regulate gene transcription and telomere elongation. Though GQ structures are well-characterized, many aspects of their conformational dynamics are poorly understood. For example, when there are surplus guanosines in some of the tracts, they can slide with respect to one another, a process we term G-register (GR) exchange. These motions could in principle entropically stabilize the folded state, crucially benefitting GQs as their stabilities are closely tied to biological function. We have developed a method for characterizing GR exchange where each isomer in the wild-type conformational ensemble is trapped by mutation and thermal denaturation data for the set of trapped mutants and wild-type are analyzed simultaneously. This yields GR isomer populations as a function of temperature, quantifies conformational entropy and sheds light on correlated sliding motions of the G-tracts. We measured entropic stabilizations from GR exchange up to 14.3 ± 1.6 J mol−1 K−1, with melting temperature increases up to 7.3 ± 1.6°C. Furthermore, bioinformatic analysis suggests a majority of putative human GQ sequences are capable of GR exchange, pointing to the generality of this phenomenon.
INTRODUCTION
DNA G-quadruplexes (GQs) are structures adopted by deoxyguanine (dG)-rich nucleic acids. They are composed of four G-tracts of three or more consecutive dGs connected by loop sequences. The tracts come together to form G-tetrads (Figure 1A), planes of four Hoogsteen-hydrogen bonded dGs that are stacked to form the core GQ structure (1). GQs are structurally diverse and dynamic molecules; they may adopt and exchange amongst different topologies (Figure 1B and C) (2,3) and oligomeric states (4). DNA GQs are found in telomeres and gene promoters, where they regulate telomere elongation and gene transcription (5,6). In addition, several functional RNA GQs have been discovered, hinting at a general regulatory role for GQs in biology (7,8). Importantly, the thermodynamic stability of GQs has been found to correlate with their regulatory functions (9–11). Thus, a thorough understanding of GQ function is predicated on characterizing the determinants of GQ stability and dynamics. Moreover, GQs are known to interact with proteins and these dynamics may play a role in molecular recognition.
Figure 1.

Guanine quadruplex (GQ) structure. (A) The G-tetrad, a planar arrangement of four Hoogsteen-hydrogen bonded guanine residues that forms the layers of a GQ. (B) Propeller-type GQ topology with parallel G-tracts. (C) Basket-type GQ with antiparallel G-tracts.
Previous studies have investigated how topology, G-tetrad number and loop length affect GQ stability (12–14). However, one distinctive feature of GQs has attracted little attention to date. When G-tracts contain different numbers of dGs, as is the case for several known promoter GQs (15–18), several folded isomers exist for the same sequence, where each isomer contains a different subset of dGs participating in the stacked G-tetrad core. This type of GQ can potentially undergo dynamics characterized by G-tracts sliding with respect to one another. These dynamics contribute to conformational entropy of the folded state and expose different recognition motifs to potential binding partners, thereby influencing GQ stability and possibly function. We refer to these strand-shifted GQs as G-register (GR) isomers and the exchange amongst these isomers as GR exchange. For example, the c-myc Pu18 promoter GQ contains two dG3 and two dG4 tracts (19). Thus, it can form a total of four different GQ structures, with each dG4 tract contributing either the 5′ or 3′ dG to the GQ core. It has been shown using NMR and DMS foot printing experiments that all four possible GR isomers are formed at equilibrium and interconvert rapidly (6,20–22). If each GR isomer were equally populated, this would lead to a 4-fold increase in the stability of the folded state compared to a GQ with a single GR isomer, corresponding to an entropic stabilization of ΔΔS = Rln (5) where R is the ideal gas constant. However, the actual populations of the different c-myc GR isomers are not known. Individual GR isomers are challenging to characterize thermodynamically because they are potentially large in number, they interconvert on a range of timescales, and they are difficult to distinguish from one another experimentally. Therefore, the entropic contribution of GR exchange to GQ stability has not yet been quantitatively determined.
Mutations are commonly used to quench GQ conformational exchange, facilitating structural characterization and functional studies (23,24). In this approach, surplus dG residues in G-tracts are substituted with bases that are not stably incorporated into G-tetrads, such as deoxythymidine (dT) or deoxyinosine (dI), or are truncated from the sequence entirely if at the 5′ or 3′ end. This produces a unique core of 12 dG residues (in the case of four G3 G-tracts) that cannot slide with respect to one another, although exchange between different topologies is still possible (25). In principle, the thermal stabilities of the trapped mutants can be related directly to the thermal stabilities of the corresponding wild-type GR isomers. However, this assumes implicitly that the mutations cause minimal thermodynamic perturbation, beyond trapping the GQ in a single register. The validity of this assumption has not been quantitatively tested to date, which limits our ability to use this approach to unravel GR exchange dynamics.
We have developed an experimental method that yields the populations of all wild-type exchanging GR isomers as a function of temperature, while at the same time testing whether trapped GQ mutants are good thermodynamic mimics of the corresponding GR isomers. The method is based on a global analysis of thermal denaturation data, and the principle that a macromolecule populates all possible conformations in proportion to each conformation's thermodynamic stability (26). We fit data for the wild-type and trapped mutants simultaneously, in such a way that good agreement is obtained if and only if the trapped mutant GQs are thermodynamically equivalent to the corresponding wild-type GR isomers. We applied the method to study GQs found in the promoter regions of genes encoding the vascular endothelial growth factor A (VEGFA), the c-myc transcription factor (c-myc Pu18) and the Pim1 kinase (PIM1), which have 2, 4 and 12 GR isomers, respectively (Table 1 and Supplementary Figure S1). Using a combination of DSC, UV-Vis, CD, and NMR spectroscopy and our new global fitting method we showed that: (i) The trapped mutant GQs are structurally similar to the corresponding wild-type GR isomers; (ii) The trapped mutant GQs fold in an effectively two-state manner; (iii) Wild-type and trapped mutant GQ thermal melt data agree with our global fitting method; (iv) Trapping mutations do not perturb stability beyond locking the GQ in a single state, therefore, the fitted GR isomer populations are meaningful; (v) GR exchange can provide substantial stabilization the GQ folded state; and (vi) A large majority of naturally-occurring GQs can potentially undergo GR exchange.
Table 1. Wild-type and trapped mutant GQ sequences.
| Name | Sequence | Tm °Ca |
|---|---|---|
| VEGFA-WT | 5′-GGGAGGGTTGGGGTGGG-3′ | 61.2 ± 0.1 |
| VEGFA-1 | 5′-GGGAGGGTTIGGGTGGG-3′ | 58.1 ± 0.1 |
| VEGFA-2 | 5′-GGGAGGGTTGGGITGGG-3′ | 57.7 ± 0.1 |
| c-mycPu18 WT | 5′-AGGGTGGGGAGGGTGGGG-3′ | 79.9 ± 0.2b |
| c-myc Pu18 55 | 5′-AGGGTTGGGAGGGTTGGG-3′ | 66.2 ± 0.1 |
| 5′-AGGGTIGGGAGGGTIGGG-3′ | 62.00 ± 0.04 | |
| c-myc Pu18 53 | 5′-AGGGTTGGGAGGGTGGGT-3′ | 69.4 ± 0.1 |
| 5′-AGGGTIGGGAGGGTGGGI-3′ | 65.60 ± 0.04 | |
| c-myc Pu18 35 | 5′-AGGGTGGGTAGGGTTGGG-3′ | 73.6 ± 0.1 |
| 5′-AGGGTGGGIAGGGTIGGG-3′ | 74.40 ± 0.04 | |
| c-myc Pu18 33 | 5′-AGGGTGGGTAGGGTGGGT-3′ | 77.3 ± 0.1 |
| 5′-AGGGTGGGIAGGGTGGGI-3′ | 77.20 ± 0.03 | |
| PIM1-WT | 5′-GGGCGGGGCGGGGGCGGGG-3′ | 68.8 ± 1.6b |
| PIM1-1 | 5′-GGGCTGGGCTTGGGCTGGG-3′ | 54.5 ± 0.1 |
| 5′-GGGCIGGGCIIGGGCIGGG-3′ | 50.0 ± 0.1 | |
| PIM1-2 | 5′-GGGCTGGGCTTGGGCGGGT-3′ | 56.7 ± 0.2 |
| 5′-GGGCIGGGCIIGGGCGGGI-3′ | 53.4 ± 0.2 | |
| PIM1-3 | 5′-GGGCTGGGCTGGGTCTGGG-3′ | 52.5 ± 0.1 |
| 5′-GGGCIGGGCIGGGICIGGG-3′ | 49.1 ± 0.1 | |
| PIM1-4 | 5′-GGGCTGGGCTGGGTCGGGT-3′ | 55.7 ± 0.2 |
| 5′-GGGCIGGGCIGGGICGGGI-3′ | 50.2 ± 0.2 | |
| PIM1-5 | 5′-GGGCTGGGCGGGTTCTGGG-3′ | 55.9 ± 0.2 |
| 5′-GGGCIGGGCGGGIICIGGG-3′ | 49.4 ± 0.4 | |
| PIM1-6 | 5′-GGGCTGGGCGGGTTCGGGT-3′ | 56.4 ± 0.2 |
| 5′-GGGCIGGGCGGGIICGGGI-3′ | 52.9 ± 0.2 | |
| PIM1-7 | 5′-GGGCGGGTCTTGGGCTGGG-3′ | 52.4 ± 0.1 |
| 5′-GGGCGGGICIIGGGCIGGG-3′ | 53.6 ± 0.1 | |
| PIM1-8 | 5′-GGGCGGGTCTTGGGCGGGT-3′ | 61.2 ± 0.1 |
| 5′-GGGCGGGICIIGGGCGGGI-3′ | 61.8 ± 0.1 | |
| PIM1-9 | 5′-GGGCGGGTCTGGGTCTGGG-3′ | 51.4 ± 0.2 |
| 5′-GGGCGGGICIGGGICIGGG-3′ | 51.1 ± 0.1 | |
| PIM1-10 | 5′-GGGCGGGTCTGGGTCGGGT-3′ | 54.7 ± 0.2 |
| 5′-GGGCGGGICIGGGICGGGI-3′ | 55.3 ± 0.1 | |
| PIM1-11 | 5′-GGGCGGGTCGGGTTCTGGG-3′ | 51.8 ± 0.2 |
| 5′-GGGCGGGICGGGIICIGGG-3′ | 48.4 ± 0.3 | |
| PIM1-12 | 5′-GGGCGGGTCGGGTTCGGGT-3′ | 55.8 ± 0.5 |
| 5′-GGGCGGGICGGGIICGGGI-3′ | 53.3 ± 0.2 |
aDerived from UV-Vis analysis.
bAverage Tm from global fitting of dT and dI trapped mutant data sets.
MATERIALS AND METHODS
Sample preparation
DNA samples were produced with a MerMade6 (Bioautomation, USA) synthesizer or were ordered from Alpha DNA (Canada) with the 5′-dimethoxytrityl group on. Syntheses utilized a high loading CPG (GlenResearch, USA) to boost yields. Oligonucleotides were removed from the CPG and deprotected using ammonium hydroxide and methylamine (1:1). Samples were purified using GlenPak columns (Glen Research, USA) and purities were determined by LC-mass spectrometry using a Bruker Maxis Impact (QTOF ESI negative mode, Bruker, USA) mass spectrometer. All samples were lyophilized, resuspended in buffers mentioned below and dialyzed against the same buffer for 24 h. Concentrations were determined by measuring the A260 and using nearest neighbor extinction coefficients (27). Samples were denatured at 90°C for 5 min and then annealed in an ice bath to promote intramolecular GQ formation prior to characterization.
Circular dichroism spectroscopy
Circular dichroism (CD) experiments were performed using a JASCO J-810 (JASCO, USA) spectropolarimeter with a cell path length of 0.1 mm. Spectra were recorded with 10 μM samples in 2.5 mM K2HPO4, 2.5 mM KH2PO4 buffer, pH 7.2. The samples were scanned six times from 330 to 230 nm at 25°C.
Nuclear magnetic resonance spectroscopy
1D 1H Nuclear magnetic resonance (NMR) spectra were collected at 25°C using an Agilent INOVA spectrometer operating at 500 MHz proton Larmor frequency. Sample concentrations were 0.2–0.3 mM in 2.5 mM K2HPO4, 2.5 mM KH2PO4 buffer, pH 7.2, with 10% D2O and referenced to 4,4-dimethyl-4-silapentane-1-sulfonic acid (DSS). Water suppression was achieved with double pulsed field gradient spin echos (DPFGSE) using a sweep width of 25 ppm. Each spectrum was recorded using 256 transients. The imino proton region of the spectrum was selected as the fingerprint for GR exchange. Spectra were processed and analyzed using MESTRENOVA NMR software.
Experimental differential scanning calorimetry
The data were collected using a NanoDSC-III microcalorimeter (TA Instruments, USA). DNA samples were 150 μM in 2.5 mM K2HPO4, 2.5 mM KH2PO4 buffer, pH 7.2. Low ionic strength was used to reduce the melting temperatures to experimentally accessible values. Samples were scanned from 5 to 90°C 15 times using a scan rate of 1°C per minute.
Experimental UV-visible spectroscopy
Spectra were recorded using a Cary Win-UV spectrophotometer (Agilent Technologies, USA). Samples were scanned 18 times at 260 and 295 nm from 15 to 95°C. Data were collected using sample concentrations of 5 or 10 μM in 2.5 mM K2HPO4, 2.5 mM KH2PO4 buffer, pH 7.2. Experiments performed at higher K+ concentrations (130 mM) showed identical trends to those performed with 7.5 mM K+, but the denaturation curves were shifted to higher and less experimentally-accessible temperatures (Supplementary Figure S2).
Thermal melt global fitting
The first step of DSC thermal melt analysis is the subtraction of data obtained for the buffer alone from the sample data (Supplementary Figure S3). The resulting raw Cp profile is then baseline-corrected, which accounts for the temperature dependence of the heat capacity, to first order. In the case of DSC data for GQs, baselines are typically polynomial (quadratic or cubic) in temperature (28–30). We employed a quadratic baseline such that the corrected heat capacity is given by:
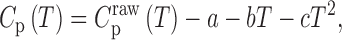 |
(1) |
where the coefficients a, b and c were optimized in the global fits (Supplementary Figure S4) as described below. The trapped mutants were assumed to fold in a two-state manner (see Results), such that the thermogram of each mutant was uniquely defined by its folding enthalpy and entropy at a reference temperature (T0) and the heat capacity difference between the folded and unfolded states (ΔCp) according to:
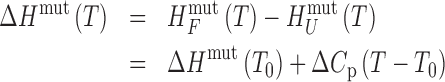 |
(2) |
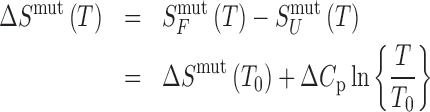 |
(3) |
It is frequently assumed that ΔCp = 0 in thermal analyses of GQs (31,32), although some studies have found that ΔCp is small and positive for GQ unfolding (29,33). We performed analyses using both ΔCp = 0 and the value of ΔCp= 1.3 kJ mol−1 K−1 that was previously measured for the human telomere GQ (29) and obtained essentially the same folding parameters and populations (Supplementary Table S1, Supplementary Figure S5). We also optimized the value of ΔCp as an adjustable parameter in the fits, yielding ΔCp ≈ 0.24 kJ mol−1 K−1. We note that our data are incompatible with ΔCp values larger than about ∼2 kJ mol−1 K−1, as at that point the quality of the fits begins to degrade substantially. Nevertheless, virtually identical folding parameters are obtained even with ΔCp fixed at 2.1 kJ mol−1 K−1. Since our results are insensitive to the choice of physically realistic ΔCp values, we performed our analysis with ΔCp = 0, for the sake of simplicity. With this assumption ΔHmut and ΔSmut are temperature-independent and define the folding equilibrium constant (Kmut) according to
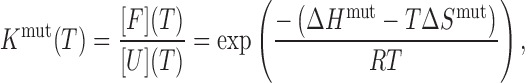 |
(4) |
which, in turn, defines the relative population of the folded state (Pmut) according to
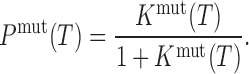 |
(5) |
Finally, the excess heat capacity thermogram for each trapped mutant is given by
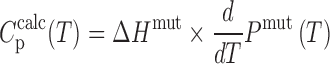 |
(6) |
In the case of a wild-type GQ undergoing GR exchange the situation is more complicated. In general, the folding enthalpy and entropy of each GR isomer can be different, such that
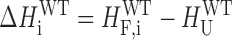 |
(7) |
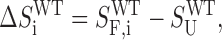 |
(8) |
where ΔHiWT and ΔSiWT are the folding enthalpy and entropy of the ith GR isomer. Note that there is only one unfolded state for a GQ that underoes GR exchange in the folded state. The folding equilibrium constant for the ith GR isomer is defined according to
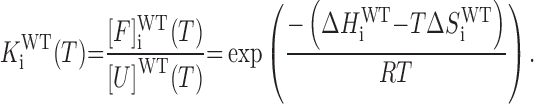 |
(9) |
The fraction of molecules populating the ith folded GR isomer is given by
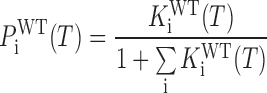 |
(10) |
where the sum runs over all GR isomers. The excess heat capacity thermogram for the wild-type GQ is then given by
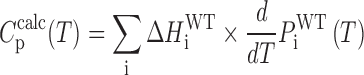 |
(11) |
Provided that the trapping mutations do not perturb stability beyond restricting the GQ to a single GR isomer, the folding thermodynamics of the wild-type and trapped mutants are related according to
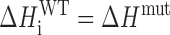 |
(12) |
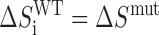 |
(13) |
where ΔHiWT and ΔSiWT are the folding enthalpy and entropy of the ith wild-type GR isomer and ΔHmut and ΔSmut are the folding enthalpy and entropy of the corresponding trapped mutant.
In the global fits, the DSC data for all trapped mutants and the wild-type GQ were analyzed simultaneously. The thermogram for each trapped mutant was calculated according to Equations (2)–(6) with ΔHmut and ΔSmut defined independently for each mutant. The thermogram for the wild-type was calculated according to Equations (9)–(11), using the thermodynamic parameters from the corresponding trapped mutants according to Equations (12) and (13). For a GQ with N GR isomers, the data set comprised N+1 DSC thermograms (N mutants and the wild-type) and the global fit contained N values of ΔH and ΔS and one second-order polynomial baseline shared between all data sets, for a total of 2N+3 adjustable parameters. The parameters were varied to minimize the residual-sum-of-squares (RSS), which is the sum of squared differences between the experimental data points and the values calculated using the global thermodynamic parameters,
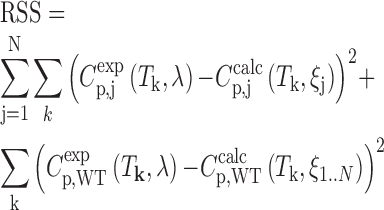 |
(14) |
where Ti is the kth experimental temperature, Cp,jexp(T) and Cp,jcalc(T) are the experimental and calculated excess heat capacity values of the jth trapped mutant, Cp,WTexp(T) and Cp,WTcalc(T) are the experimental and calculated excess heat capacity values of the wild-type, ξj = [ΔHjmut, ΔSjmut] are the folding parameters of the jth trapped mutant, and λj = [a, b, c] are the coefficients of the quadratic baseline. UV-Vis unfolding traces were modeled similarly (See Supplementary Methods).
Identification of GR exchange in GQ sequences from the Eukaryotic Promoter Database
GQs are formed by sequences containing four Gn-tracts, of n contiguous dG residues per tract, separated by loop sequences Nm of any composition, where m is the number of loop bases. Biological GQs are thought to form according to the rule devised by Balasubramanian et al. which identifies 5′-G≥3N1-7G≥3N1-7G≥3N1-7G≥3-3′ as the consensus sequence for stable folding (34). GQ stability decreases with increasing loop length and stable biological GQs tend to contain short loops, thus the folding rule loop length is defined with 1 ≤ m ≤ 7. GQs formed from G-tract lengths n = 1–2 are known, yet these are typically unstable relative to other conformational states (35) and so are not likely to be well populated under biological conditions. When n = 3 for all four Gn-tracts and the loops are free of dG residues, three G-tetrads are formed and every dG participates in the GQ core. Interestingly, Fox et al. demonstrated GQ sequences with all G4 or G5 tracts did not form more than three G-tetrads (12), suggesting multiple GR isomers are possible for these sequences with different subsets of dG residues participating in the formation of three G-tetrads. GQs with all G6 or G7 tracts did form >3 G-tetrads. However, such GQs are biologically rare, their dynamics are likely complicated, and they were not included in our analysis, i.e. we only considered sequences with n ≤ 5. We therefore used a query sequence, 5′-G3-5N1-7G3-5N1-7G3-5N1-7G3-5-3′, where N = dA, dC, dT, or dG to search the Eukaryotic Promoter Database (36) for GQ sequences that could potentially undergo GR exchange. No more than two consecutive dG residues were allowed in any loop sequence, nor were dG residues allowed immediately adjacent to a flanking G-tract. We examined both coding and complementary strands from the −499 to +100 position of 23 322 human promoter sequences. All sequences matching the query were analyzed to determine the number of possible GR isomers (see Supplementary Methods).
RESULTS
Systematically trapping GR isomers with mutations
We studied three promoter GQ sequences, referred to here as VEGFA, c-myc Pu18 and PIM1, which contain 1, 2 and 4 surplus dG residues, respectively. We note that it could be possible for surplus dG residues to be accommodated in the form of a G-pentad or G-hexad, rather than leading to GR exchange. However, the pentad and hexad arrangements observed to date contain four dG residues and either one or two dA residues. Furthermore, these unusual structures exist only in the context of dimers (37,38). We selected monomeric GQs (31) in which it is likely that surplus dG residues lead to GR exchange rather than G-pentad or G-hexad formation. Therefore, the VEGFA, c-myc Pu18 and PIM1, GQs populate 2, 4 and 12 GR isomers, respectively. The sequences were systematically trapped in structures mimicking each of their GR isomers by substituting all surplus dG residues with dT or dI (Table 1 and Supplementary Figure S1). In the following we refer to these sequence variants as trapped mutants, where we use the term mutant to indicate alterations of naturally occurring DNA sequences. We note that the difference between dI and dG is the presence of a hydrogen atom versus an amino group at the 2-position, respectively. Replacing dG with dI in a G-tetrad therefore comes at the cost of one hydrogen bond, estimated at several kcal mol−1 (39,40). This translates into a greater than 100:1 preference for dG relative to dI in the core of the GQ. In the case of dT, the preference for dG is even stronger. Therefore we consider both dI- and dT-containing mutants to be effectively trapped in a single GR isomer with dGs located in the core and dI or dT residues in the loops or flanking regions. For instance in VEGFA (5′-G1G2G3-A4-G5G6G7-T8T9-G10G11G12G13-T14-G15G16G17-3′), the mutation 10dG>dX locks the third G-tract in a 5′ shifted position, while 13dG>dX locks it in a 3′ shifted position, relative to the core of 3 G-tetrads (dX = dT or dI). For the c-myc Pu18 GQ (5′-A1-G2G3G4-T5-G6G7G8G9-A10-G11G12G13-T14-G15G16G17G18-3′), the double mutations 6dG,15dG>dX, 6dG,18dG>dX, 9dG,15dG>dX and 9dG,18dG>dX lock the second and fourth G-tracts in the 5′5′, 5′3′, 3′5′ and 3′3′ registers relative to the core, respectively. In what follows, we will refer to the wild-type GR isomers corresponding to these trapped mutants as 55, 53, 35 and 33. The PIM1 GQ 5′-G1G2G3-C4-G5G6G7G8-C9-G10G11G12G13G14-C15-G16G17G18G19-3′ has 12 possible GR isomers. To mutationally trap this sequence in a single register isomer, four substitutions per mutant sequence are required: one in the second G-tract, two in the third G-tract, and one in the fourth G-tract. For example, in the third G-tract, we employed the following pairs of mutations to force adoption of a single register with respect to a GQ core of three G-tetrads: 5′-…X10X11G12G13G14…-3′, 5′-…X10G11G12G13X14…-3′ and 5′-…G10G11G12X13X14…-3′, where (…) denotes the rest of the PIM1 sequence in the 5′ and 3′ directions.
Structural analyses
We used a combination of CD and 1H NMR spectroscopy to structurally characterize the wild-type GQs and their trapped mutants. In the case of both c-myc Pu18 and VEGFA, the CD spectra of all trapped mutants closely overlay those of the wild-type GQs (Figure 2A and B and Supplementary Figure S6A–C). The sharp maxima at 265 and minima at 240 nm imply that the wild-type GQs and all mutants adopt parallel topologies (41), as expected. Furthermore, the 1H NMR spectra of the wild-type GQs closely correspond to a superposition of the trapped mutant spectra (Supplementary Figures S7A and B, S8A). This strongly supports the idea that the wild-type GQs populate a mixture of conformational isomers, each of which resembles one of the trapped mutants, with exchange occurring relatively slowly on the NMR chemical shift timescale (≥seconds). In other words, the trapped mutants are good structural mimics of the wild-type GR isomers, at this level of resolution.
Figure 2.

Guanine quadruplex circular dichroism (CD) spectra. CD spectra for wild type (black) and trapped mutant (colored) (A) VEGFA dG>dI, (B) c-myc Pu18 dG>dI and (C) PIM1 dG>dI GQs. In (C) and (D), the solid black line corresponds to the corrected wild-type PIM1 CD spectrum. In (D) the population-weighted average spectrum of the dG>dT and dG>dI trapped mutants are shown with filled and empty symbols, respectively. (E) Comparison of ΔCD265–290 (CD (265 nm)–CD (290 nm)) signals for dT and dI trapped mutants of the PIM1 GQ. The empty circle indicates data for the outlying PIM1-10 trapped mutant (Table 1). The line indicates the best linear fit to the 11 filled data points (R = 0.90).
Although the CD spectrum of the PIM1 wild-type GQ (Supplementary Figure S9) is consistent with a predominantly parallel topology, it exhibits a small shoulder at 290 nm (Figure 2D and Supplementary Figure S6F), indicating that some members of the conformational ensemble contain antiparallel strands (3). Interestingly, while the CD spectra of many mutants closely match that of the wild-type GQ, several others are quite different, with maxima near 290 nm and shoulders at 265, indicating that these trapped mutants adopt predominantly antiparallel topologies. The corresponding dG>dT and dG>dI spectra closely superimpose, with only one exception (Supplementary Figure S10). In order to quantify this agreement, we have compared the difference in ellipticities at 265 and 290 nm (ΔCD265–290 = CD (265 nm) – CD (290 nm)) for dG>dT and dG>dI mutants. Positive, zero and negative ΔCD265–290 values are consistent with predominantly parallel, mixed and predominantly antiparallel topologies respectively. The ΔCD265–290 values obtained for the dG>dT and dG>dI trapped mutants agree closely, yielding a correlation coefficient of R = 0.90 (Figure 2E). This implies that the same topology is obtained regardless of whether dT or dI residues are present at the mutated loop positions. It seems likely that the wild-type GR isomer, which contains dG at these positions, would prefer the same topology favored by the corresponding dT and dI trapped mutants as well. This idea is supported by the excellent agreement between the CD spectrum of the wild-type GQ and the averaged spectra of the dG>dT and dG>dI trapped mutants (Figure 2D). In other words, GR exchange is linked to topological dynamics with some GR isomers favoring parallel topologies and other GR isomers favoring antiparallel topologies.
1H NMR spectra of the wild-type PIM1 GQ and many of the trapped mutants are highly broadened, indicative of conformational dynamics on the microsecond to millisecond timescales (Supplementary Figures S7C, S8B) (42). Similarly broadened 1H NMR spectra have been previously observed for GQs that interconvert between different GR isomers (17) and/or topologies (43). In the case of the PIM1 trapped mutants, the broadening is likely due to interconversion between parallel and various antiparallel topologies, as discussed above. For the wild-type PIM1 GQ, the extensive spectral broadening is likely due to dynamics among different topologies within a given GR isomer (as observed for the trapped mutants), as well as interconversion between different GR isomers. The population-weighted average NMR spectrum of the trapped mutants is in good agreement with that of the wild-type, consistent with the notion that the trapped mutants are good structural mimics of the corresponding wild-type GR isomers (using globally-fit populations).
Trapped mutant folding
The thermodynamic stabilities of the wild-type GQs and trapped mutants were characterized by DSC and UV-Vis thermal melts, shown in Figures 3 and 4. In what follows, the data for the trapped mutants were analyzed assuming a two-state model comprising only fully folded and fully unfolded states. While two-state models have previously been applied to GQ folding (31,44–45), some GQs are known to populate folding intermediates (29,45–47), therefore the two-state assumption warrants closer examination. The DSC and UV-Vis data show simple monophasic melting transitions, which provides some indication that folding is two-state, since GQs that populate folding intermediates such as G-triplexes often yield DSC thermograms and UV unfolding traces with distinctive shoulders or multiple distinct transitions (29,48–50). Nevertheless, more complicated folding processes can be present despite the absence of multiple or broad transitions (46,51–53), and further tests are required. A classic approach for establishing two-state folding is to separately analyze the shape of a DSC thermogram, which yields the van ‘t Hoff enthalpy (ΔHVH) and entropy (ΔSVH), and the overall magnitude of the thermogram, i.e. the areas under the Cp and Cp/T traces, which yield the calorimetric enthalpy (ΔHcal) and entropy (ΔScal) (28,54–55), respectively. When folding is two-state, ΔHVH = ΔHcal and ΔSVH = ΔScal. We performed this test on c-myc Pu18 GQ trapped mutant DSC data and obtained excellent agreement (Supplementary Table S2). We further applied the DSC deconvolution method of Freire and Biltonen (45,56) and found again that these thermograms are consistent with two-state unfolding (Supplementary Figure S11). Additionally, we compared c-myc Pu18 UV-Vis thermal melt data collected at 260 and 295 nm. When folding is two-state these sets of data are expected to coincide, while they are likely to diverge when intermediates are present (46,52). The 260 and 295 melting profiles are nearly superimposable (Supplementary Figure S12) and yield identical folding parameters (Supplementary Figure S13A and B). Together this gives us confidence that the c-myc Pu18 GQ trapped mutants fold in a two-state manner.
Figure 3.

Global fits of guanine quadruplex DSC thermal denaturation data. Thermograms (points) and global best-fit lines (lines) obtained for the complete set of c-myc Pu18 wild-type, (A) dT-containing, (C) dI-containing trapped mutant GQs. Black and colored lines correspond to data for the wild-type and trapped mutant GQs, respectively. The GR isomer populations (i.e. the fraction of wild-type DNA chains adopting each conformation) were extracted from the global fits (Equation 10) and are plotted in the panels immediately to the right, for (B) dT-containing and (D) dI-containing trapped mutants. Colored dashed lines indicate the populations of each folded GR isomer, while black and grey lines correspond to the total population of the folded state and the population of the unfolded state, respectively. (E) Cartoon of the c-myc Pu18 GQ undergoing exchange between four folded GR isomers and the unfolded state. KX-Y= PX/PY is the equilibrium constant for isomers X and Y, while the values indicated are for the dG>dI trapped mutants. Equilibrium constants have been expressed as >1 for ease of comparison.
Figure 4.

Global fits of guanine quadruplex UV-Visible thermal denaturation data. Thermograms (points) and global best-fit (lines) for complete sets of wild-type and dI-containing trapped mutants of (A) VEGFA, (C) c-myc Pu18, and (E) PIM1 GQs. Black and colored lines correspond to data for the wild-type and trapped mutant GQs, respectively. The GR isomer populations (i.e. the fraction of wild-type DNA chains adopting each conformation) were extracted from the global fits (Equation 10) and are plotted in the panels immediately to the right for (B) VEGFA, (D) c-myc Pu18, and (F) PIM1 GQs. Colored dashed lines indicate the populations of each folded GR isomer, while black and grey lines correspond to the total population of the folded state and the population of the unfolded state, respectively.
The situation is more complicated for the PIM1 trapped mutants, as the CD and NMR data show that some of them populate multiple folded topologies, and thus there are formally more than two thermodynamic states accessible to these molecules. Nevertheless, if the stabilities of the different topologies are similar, then folding can still be considered a pseudo-two-state process in which the population of the unfolded state increases and the populations of all folded sub-states decrease in concert as the temperature is raised. In order to test whether this approximation holds, we compared UV-Vis data of PIM1 trapped mutants obtained at 260 and 295 nm (Supplementary Figure S14). Data at the two wavelengths exhibit quite different baseline slopes, perhaps due in part to topology exchange (25,57. Nevertheless, the melting transition regions coincide closely, and stability analyses performed on either the 260 or 295 nm data yield virtually the same results (Supplementary Figure S13C and D). We have therefore treated folding of the PIM1 trapped mutants as pseudo-two-state. The melt is well-defined at all temperatures by effective two-state folding parameters, in which the ‘fraction folded’, plotted in Figure 4E refers to the fraction of chains adopting any folded topology.
Globally fitting GQ thermal denaturation data
We developed a global fitting method for characterizing GR exchange that can be applied to any set of thermal denaturation data (in this study DSC and UV-Vis). In a standard thermodynamic analysis, unfolding data for a single sample are fitted independently of all other samples to yield a single set of thermodynamic parameters. In our method, data for a complete set of trapped mutants and the wild-type GQ are analyzed simultaneously to yield a combined set of thermodynamic parameters describing the stabilities of all mutants, as well as the populations of all wild-type GR isomers (see Materials and Methods section). Our method is based on the assumption that the free energy difference between the folded and unfolded states of a trapped mutant is equal to the free energy difference between the corresponding folded GR isomer and the unfolded state of the wild-type GQ. To illustrate, data for a wild-type GQ with two GR isomers (such as VEGFA) would be analyzed together with those of the two corresponding trapped mutants. If at a given temperature, the folded:unfolded ratio for mutant A is 1:1, and that of mutant B is 2:1, then the assumption is that for the wild-type GQ, the (isomer A):(isomer B):(unfolded) ratio is 1:2:1. This relationship between the stabilities of trapped mutants and those of the corresponding wild-type GR isomers is applied across the full temperature range. Violations of this assumption lead to poor agreement across the data set in the global analysis (see below). Notably, we observe excellent agreement between the experimental data and global fits suggesting that the trapped mutants are good thermodynamic mimics of the corresponding wild-type GR isomers. This implies that our description of wild-type GQ conformational sampling is accurate. For example, Figure 3A and C shows complete sets of DSC data for the c-myc Pu18 GQ, comprising thermograms for the wild-type and four trapped mutants employing dG>dT (Figure 3A) and dG>dI (Figure 3C) substitutions. Interestingly, the wild-type GQ is more thermally stable than any of the trapped mutants, with thermal upshifts of the melting point of as much as 18°C relative to the least stable of the trapped mutants. This increase in stability is caused by entropic stabilization of the wild-type GQ folded state due to the presence of multiple GR isomers (19,20). The entropically driven thermal up-shift indicates that the wild-type populates multiple GR isomers at equilibrium and that the interconversion rate is relatively rapid (≤minutes). If the wild-type populated just one GR isomer, then we would expect its DSC thermogram to overlay that of the corresponding trapped mutant (i.e. no shift in melting temperature). If the wild-type exchanged very slowly (≥minutes) among the four GR isomers, the wild-type DSC trace would be the population-weighted average of the four traces of the trapped mutants, which would appear as a slight thermal down-shift from the most stable mutant (Supplementary Figure S15). Figure 3B and D shows the extracted populations of the four wild-type GR isomers as a function of temperature. Notably, very similar values are obtained with either dG>dT or dG>dI mutations, suggesting that this method is relatively insensitive to the precise chemistry of the substitute residues. The results show that the wild-type conformational ensemble is highly skewed with dominant populations of the 33 and 53 GR isomers. Interestingly, the proportions of 53, 35 and 55 GR isomers increase with increasing temperature, however they never represent more than ∼30–40, 10 and 5% respectively.
UV-Vis absorbance spectroscopy is a more high-throughput method for thermal analysis than DSC, requires approximately 100-fold lower sample concentrations, and is equally amenable to global analyses of GR exchange. Due to these advantages, it was possible to apply UV-Vis spectroscopy to a range of different GQs and trapped mutants (see Supplementary Methods and Supplementary Figures S16–S17 for sets of raw and globally analyzed melting profiles of dG>dT and dG>dI trapped mutants, respectively). Figure 4A shows absorbance melting profiles of the wild-type VEGFA GQ and a complete set of two dG>dI trapped mutants. Figure 4C shows melting profiles for the wild-type c-myc Pu18 GQ and complete sets of four trapped mutants employing dG>dI substitutions. Figure 4E shows melting profiles for the wild-type PIM1 GQ and complete set of 12 trapped mutants employing dG>dI substitutions. The extracted populations of the GR isomers comprising the wild-type ensembles are plotted in the panels to the right (Figure 4B, D and F). Importantly the UV-Vis results for the c-myc Pu18 GQ (Figure 4D) closely match those obtained by DSC (Figure 3B and D), indicating that the global fitting approach can be robustly applied to different experimental modalities. In addition this confirms that the c-myc Pu18 GQ is monomeric, as the same stabilities were obtained at very different concentrations, 10 versus 150 μM for UV-Vis and DSC, respectively. In all cases, the conformational ensemble is skewed. In the case of VEGFA, the two GR isomers are populated with a 3:2 ratio. For c-myc Pu18, one of the four GR isomers represents more than 60% of the ensemble, while for PIM1, just one of the twelve GR isomers is populated to 50%. This has implications for the entropy contribution of GR exchange, as discussed below.
Trapped mutants as thermodynamic mimics of GR isomers
The key assumption of the global analysis is that thermal stability of each trapped mutant relative to its unfolded state is identical to that of the corresponding wild-type GR isomer relative to the wild-type unfolded state. However, the trapped mutants and the GR isomers differ; the wild-type contains one or more dG residues in loop positions whereas trapped mutants contain dT or dI substitutions. Therefore, it is of paramount importance to determine if the trapped mutants are good thermodynamic mimics of the wild-type GR isomers. Fortunately, the global fitting approach is well-suited to evaluating whether or not this is the case. If the mutations lead to thermodynamic perturbations, then the complete set of thermal denaturation data for the wild-type and trapped mutants will be mutually inconsistent. This will be reflected in poor agreement between experimental and calculated heat capacities or spectroscopic absorbances. We used Monte Carlo computer simulations to evaluate the ability of the global fitting approach to detect such inconsistencies (Supplementary Methods). Using the thermodynamic parameters of the c-myc Pu18 GQ as a starting point, we generated synthetic sets of thermal denaturation data in which the thermodynamic folding parameters of the trapped mutants differed from those of the corresponding GR isomers, i.e. the synthetic data sets were in violation of the key assumption stated above. We then subjected the synthetic data sets to global analysis. The simulations indicate the differences between the folding parameters of the trapped mutants and the corresponding wild-type GR isomers are no more than a maximum of ±1.5 kJ mol−1 for c-myc Pu18 (Figure 5A and B and Supplementary Figure S18). Using the same analysis, we find the maximum difference to be ∼±2.5 kJ mol−1 for PIM1 (Figure 5C and Supplementary Figure S19).
Figure 5.
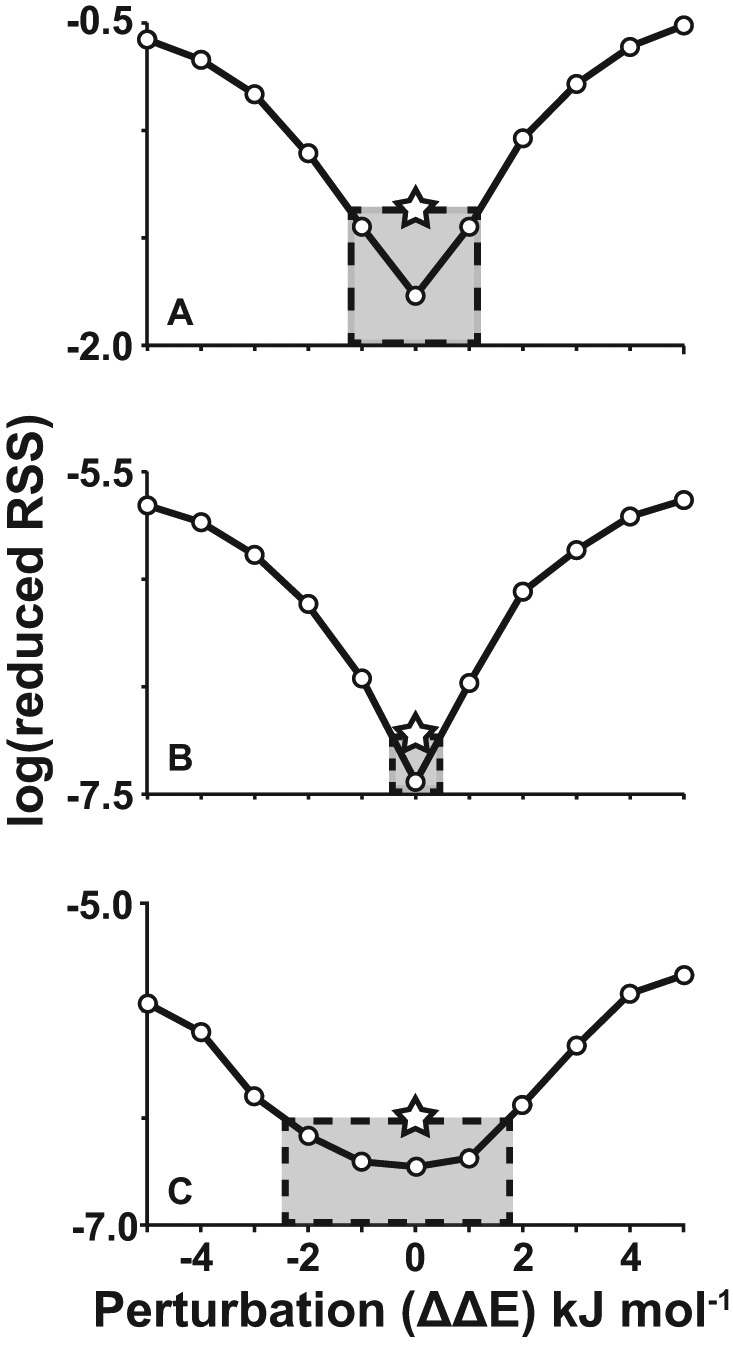
Sensitivity of the global fit to thermodynamic perturbations. Increasing thermodynamic mismatch between trapped mutants and their corresponding GR isomers (ΔΔE) leads to larger values of ‘reduced RSS’ in global fits, i.e. poorer overall agreement. The reduced residual-sum-of-squares (RSS) is the residual-sum-of-squared differences between the simulated data points and the best global fit to the simulated data points, divided by the degrees of freedom of the fit (#simulated data points – #adjusted parameters). Simulations are shown for c-myc Pu18 GQ (A) DSC and (B) UV-Vis data, and (C) PIM1 UV-Vis data using dG>dI mutations (See Supplementary Figures S5 and S6 for dG>dT simulations). Simulated data sets (1000 and 25 iterations for c-myc and PIM1 data sets, respectively) were prepared in which the folding ΔH and T0ΔS values of the trapped mutants differed from those of the corresponding wild-type GR isomers by random perturbations with a mean of zero and standard deviation of ΔΔE kJ mol−1. Simulated data sets were analyzed according to the global fitting method (Equations 1–14). Larger thermodynamic perturbations led to poorer fits, as indicated by larger RSS fitting residuals (Equation 14). The stars correspond to the ‘reduced RSS’ values obtained for the true experimental data sets. These define the maximum average mismatch between the folding thermodynamics of trapped mutants and corresponding wild-type GR isomers (gray shaded boxes). For thermodynamic perturbations larger than 1.1–1.5 kJ mol−1 (DSC) or 0.2–0.5 kJ mol−1 (UV-Vis), the RSS is greater than what we observe experimentally. In other words, the root-mean-squared (RMS) difference between the folding parameters of the trapped mutants and the corresponding wild-type GR isomers is not much more than this tolerance. For points of reference, the ceiling for perturbations is about 0.5% of the total folding enthalpies (ΔHF) of the GQs and 3% of the ΔΔHF between the most and least stable trapped mutants.
The thermodynamic parameters extracted from dG>dT and dG>dI trapped mutants are quite similar (Supplementary Tables S3 and S4), indicating that the stabilities of the GQs are not very sensitive to the identities of residues in the targeted loop positions, whether these be dT, dI, or presumably dG. In the case of c-myc Pu18, the extracted folding enthalpies, populations of the GR isomers and the melting temperatures based on dG>dT and dG>dI mutations correlate very well with correlation coefficients (R) varying between 0.80 and 0.99 with a mean of 0.94 ± 0.09, for both DSC and UV-Vis measurements (Supplementary Figure S20A–F). For PIM1, the Tms of the individual trapped mutants and the populations of the GR isomers also agree well. The ΔH values of GR isomer folding are less well reproduced by dG>dT and dG>dI substitutions (Supplementary Figure S20G–I). This is likely because the PIM1 trapped mutants harbor more substitutions, giving larger thermodynamic perturbations (Figure 5C). Nevertheless, the fact that we extracted similar GR isomer populations with dG>dT or dG>dI substitutions for both c-myc Pu18 and PIM1 gives us confidence that we can use these values to better understand GQ function.
Entropy effects and correlated motions in GR exchange
One strength of our approach is that we are able to directly compute a portion of the entropic stabilization of the folded state due to internal dynamics. The entropy of a folded GQ exchanging among N GR isomers is given by
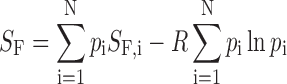 |
(15) |
where pi is the fraction of folded chains adopting the ith GR isomer, and SF,i is the molar entropy of ith folded isomer. The first term in Equation (15) is simply the population-weighted average entropy over all the isomers and the second is the entropy due to interconversion between multiple states. This can be calculated directly, as the set of pi is obtained in our global analysis. It is typically difficult to separate solvent and conformational contributions in experimental measurements of entropy changes, for biomolecules in aqueous solution (58). In contrast, our global fitting approach gives us direct access to a well-defined component of conformational entropy, i.e. that due to GR exchange. The resulting contributions to the entropy of the folded state, increases in the folding equilibrium constant and thermal upshifts in Tm (ΔGF = 0) are listed in Table 2 for the three GQs studied here. Interestingly, due to the skewed populations of the GR isomers, the actual entropic stabilization is less than the maximum that would be obtained with equal populations. Not surprisingly, the largest stabilization is seen for wild-type PIM1, which has 12 GR isomers and whose melting temperature is increased by 7.3±1.6°C compared to that of the most stable GR isomer. Even the VEGFA GQ with only 2 GR isomers exhibits a 1.8 ± 0.1-fold increase in the population of the folded state and a 3.1 ± 0.3°C upshift in melting temperature.
Table 2. Entropic stabilization of folded guanine quadruplexes by G-register exchange.
| VEGFA | c-myc Pu18 | PIM1 | |
|---|---|---|---|
| Number of GR isomers (N) | 2 | 4 | 12 |
| ΔΔStheo= Rln(N) J mol−1 K−1a | 5.8 | 11.5 | 20.7 |
| ΔΔSexp J mol−1 K−1b | 5.20 ± 0.01 | 6.2 ± 0.9 | 14.3 ± 1.6 |
| KF,exch/KF,indivc | 1.8 ± 0.1 | 2.1 ± 0.5 | 4.5 ± 1.9 |
| ΔTm°Cd | 3.1 ± 0.3 | 3.4 ± 1.2 | 7.3 ± 1.6 |
Parameters given here are the average of the results from dT and dI trapped mutant data sets where applicable (DSC and UV-Vis for c-myc Pu18, UV-Vis for PIM1).
aMaximum conformational entropy contribution for exchange among N isomers.
bActual conformation entropy contribution calculated using trapped mutant populations at 25°C.
cRatios of wild-type and most-populated trapped mutant folding equilibrium constants evaluated at the wild-type Tm.
dWild-type thermal upshifts relative to the most-populated GR isomer. Errors were calculated as the standard deviations of the values extracted from global fits of DSC and UV-Vis data according to variance/co-variance method (See Supplementary Methods).
In addition, our results give a direct measure of concerted dynamics via a comparison of the populations of the different GR isomers. We find that the position of one G-tract influences the sliding motions of the adjacent G-tracts. For instance, in c-myc Pu18, the 4th G-tract populates the 3′-shifted register over the 5′-shifted register with a ∼10:1 ratio when the 2nd G-tract is in the 5′-shifted register (P53/P55 = 12.8, Figure 3E, Supplementary Table S5). When the 2nd G-tract is 3′-shifted, this ratio becomes ∼30:1 (P33/P35 = 33.5). Cooperative motions are more pronounced in PIM1 (∼50-fold differences in shifting, Supplementary Figure S21, Supplementary Table S6).
GR exchange in the human genome
In order to better understand the relevance of GR exchange to biological GQs, we conducted a bioinformatic analysis of promoter regions of the human genome (36), as GQ formation in these regions has been linked to biological function (59–61). We found 28 377 potential GQ-forming sequences following the pattern 5′-G3-5N1-7G3-5N1-7G3-5N1-7G3-5-3′ (N = dA, dC, dT or dG), in both strands of the 23 322 human promoter regions from the EPD (see Materials and Methods). The number of retrieved sequences implies that, on average, each 600 base-pair promoter region contained one or two putative GQ-forming sequences. The number of possible GR isomers was calculated for each putative GQ sequence (Supplementary Methods), and the resulting histogram is shown in Figure 6. Notably, 89% of GQ sequences following this pattern can potentially adopt 2 or more folded GR isomers, with 19% potentially exchanging among 12 or more. A sizeable fraction of potential GQ sequences have 20 or more different GR isomers which could stabilize the folded state by as much as ∼17°C (See Supplementary Methods). Within the selected sequences, 52, 35 and 13% of G-tracts contain 3, 4 and 5 residues, respectively. Longer G-tracts were not included in the analysis, as these can lead to more complicated dynamics that are beyond the scope of this work. Interestingly the distribution of the number of GR isomers closely matches that obtained if the lengths of adjacent G-tracts are uncorrelated. For example, the probability that a given G-tract contains 4 dG residues is independent of the lengths of the other three G-tracts in the sequence. It should be noted that since G-tracts longer than 5 dG residues were excluded, certain numbers of GR isomers do not appear in the analysis, explaining the gaps at 5, 7, 10, etc. in Figure 6. Furthermore, it has been recently shown that small-molecule guanine derivatives can ‘fill in’ G-tetrads in GQs with G-vacancies, i.e. those lacking a full complement of dG residues (62). This raises the interesting possibility that even GQs with the canonical four G3 tracts might be able to undergo GR exchange, with guanine derivatives filling in the vacancies left by the shifted strands. Similar arguments apply to GQs with G-tracts of unequal length, further increasing the availability of potential GR isomers.
Figure 6.

Occurrence of G-register exchange in predicted human guanine quadruplex sequences. Black bars show the number of putative GQ sequences identified in a database of human promoter regions (36) (28 377 putative GQ sequences in total) with a given GR isomer multiplicity. White bars show the number of sequences that would be expected to have a given GR isomer multiplicity if G-tracts containing 3, 4, and 5 consecutive dG residues are distributed randomly within GQ sequences. The inset shows the number G-tracts from the putative GQ sequences that contain 3, 4, and 5 consecutive dG residues.
DISCUSSION
Our results clearly show that GQs with G-tracts of different lengths adopt multiple folded isomers with different core dG residues. Previous NMR spectroscopy and DMS modification studies on the c-myc Pu18 GQ have demonstrated that all possible G-register isomers are populated at equilibrium and interconvert rapidly (19–21). Our NMR results are in agreement. The spectra of the wild-type GQs correspond closely to the population-weighted average spectra of the trapped mutants, strongly suggesting that all three wild-type GQs populate multiple GR isomers at equilibrium. Furthermore, DSC and UV-Vis thermal analyses indicated that all three wild-type GQs are entropically stabilized with respect to the most stable of the trapped mutants. This is consistent with the wild-type GQs undergoing relatively rapid exchange among multiple GR isomers. These dynamics likely impact GQ function. GR exchange stabilizes the folded state, and there is a well-established link between GQ stability and gene expression (20,63). In addition we find that GR exchange is coupled to topological rearrangements and can alter binding motifs presented by GQs. The relationship between GR isomerism and topology switching has been previously noted in a GQ from the human telomerase promoter region which has two well-populated GR isomers, one of which favors a parallel configuration, while the other is mixed (3 + 1) parallel/antiparallel (25). The differing topological preferences were explained in terms of the different loop interactions in the GR isomers. For instance, in the mixed topology GR isomer, strands 2 and 3 are anti-parallel and the intervening loop is two nucleotides long, stabilized by hydrogen bonding with the third loop. In the parallel GR isomer, strands 2 and 3 are parallel and the intervening loop contains three nucleotides which pack against the GQ core (25). For PIM1, the situation is more complicated as it exchanges among 12 GR isomers, each with distinct topological preferences, likely governed by the drive to simultaneously optimize hydrogen bonding, base stacking and the formation of stable features such as single-residue double chain reversal loops (64,65). Nevertheless, these results suggest that coupling between GR exchange and topological interconversion may be a general feature of GQ dynamics. The fact that motions on different sides of the GQ are correlated is significant, as coupled conformational changes at distal sites in biological macromolecules has been identified as a prerequisite for the existence of allostery (66). Many synthetic ligands that target GQs bind with stoichiometries greater than one, and GQs are known to interact with each other and with proteins (67–69). Furthermore, it has been shown that different conformational states of the same GQ can have different protein binding competencies (70). Coupled motions of the G-tracts could thus modulate the molecular recognition of GQs in vivo and influence their interactions with drugs.
It must be noted that we performed in vitro measurements on large populations of molecules. In a living cell, a given GQ-forming sequence is present only once, or at most in a few copies. To extrapolate our results to the cellular level, one may invoke the ergodic hypothesis (71) which states that the population of a given conformational state in a large ensemble is proportional to the length of time spent by a single molecule in that state, evaluated over a long period. In other words, our finding that GR exchange increases the stability of the folded state by factors of up to 4.5 implies that an isolated G-rich DNA sequence would spend 4.5-fold more of its time in the folded rather than the unfolded state, as a direct result of GR exchange. Since folded GQs can block polymerase read-through (72), this could directly modulate transcriptional control by an individual G-rich sequence. Furthermore, the stabilities that we have determined for the different GR isomers are directly related to the total amounts of time that an individual GQ-forming sequence spends in each state, which modulates the probability that an encounter with a GQ-binding protein (68) will result in complex formation.
The internal dynamics of nucleic acids are increasingly recognized as being essential to their biological activity. Conformational transitions in RNA molecules are involved in metabolite and temperature sensing by riboswitches (73,74), catalysis by ribozymes (75), and the assembly of RNA–protein complexes (76), among other processes. DNA duplex dynamics have been linked to recognition by transcription factors (77), DNA damage (78), and ligand binding (79). DNA GQs are well known to be flexible in the native state (80) and can populate equilibrium folding intermediates (29,81). Some G-rich sequences assemble into multimeric structures (4) while others contain more than four G-tracts, with the ability to substitute the extra "spare tire" tracts into the four-stranded core (82). Thus the GQ dynamical repertoire is complex and varied. Studies have characterized nucleic acid motion mechanically, defining the ranges of motion of RNA hinges and relating these to molecular recognition (83). Others have determined the thermodynamic and kinetic parameters governing excursions to weakly-populated excited states (84). What sets the current work apart from these studies is that we have determined quantitative folding parameters for each member of large conformational ensembles containing as many as 12 discrete isomers. To our knowledge, these are the largest biomolecular conformational ensembles to be characterized at this level of detail. Our data provide a rare opportunity to precisely define the highly complex internal motions of nucleic acids.
We have identified the sliding of G-tracts with respect to each other as a prevalent form of internal dynamics in GQs. GR exchange likely extends to guanine-vacant, ‘spare-tire’, multimeric, and RNA GQs (4,11,62,82,85), where the GQ core could undergo similar conformational rearrangements. This reinforces the idea that GQs must be considered as dynamic ensembles with differentially populated structural forms. We have shown GR exchange contributes directly to thermodynamic stability and it could potentially modulate higher-order interactions and biological function. We demonstrate that mutant GQs harboring dG>dT and dG>dI substitutions that quench these sliding motions are good structural and thermodynamic mimics of the corresponding wild-type GR isomers. Globally fitting thermal denaturation data for complete sets of wild-type GQs and trapped mutants thus yields the populations of all GR isomers as a function of temperature. We observed melting point elevations of up to 7.3 ± 1.6°C for a GQ undergoing 12-state exchange relative to the single most stable GR isomer. A bioinformatic analysis of human promoter sequences revealed that great majority of naturally-occurring GQ sequences can potentially undergo GR exchange with many having 20 or more different GR isomers. Thus a full description of GR exchange is critical for understanding GQ stability and function. The global fitting method we introduce here is inexpensive, rapid (particularly for UV-Vis data) and yields exquisitely detailed information on the conformational sub-states comprising the native structural ensemble of GQs. We believe it represents a new and powerful tool for elucidating structure-dynamics-function relationships for this important class of molecule.
Supplementary Material
Acknowledgments
The authors would like to thank Dr Hanadi Sleiman, Dr Masad Damha and Dr Graham Hamblin for help with nucleic acid synthesis and characterization. We thank Amani Hariri for insightful manuscript comments. R. W. H. V. was supported by the Canadian Institutes of Health Research Drug Development Training Program, the Groupe de Recherche Axé sur la Structure des Protéines, The National Science and Engineering Research Council Bionanomachines CREATE training program, and Dr Julian Adams (Infinity Pharmaceuticals, Boston, USA).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Natural Sciences and Engineering Research Council [NSERC, Canada, 327028-09]. Funding for open access charge: Natural Sciences and Engineering Research Council [NSERC, Canada, 327028-09].
Conflict of interest statement. None declared.
REFERENCES
- 1.Bochman M.L., Paeschke K., Zakian V.A. DNA secondary structures: stability and function of G-quadruplex structures. Nat. Rev. Genet. 2012;13:770–780. doi: 10.1038/nrg3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hu L., Lim K.W., Bouaziz S., Phan A.T. Giardia telomeric sequence d(TAGGG)4 forms two intramolecular G-quadruplexes in K+ solution: effect of loop length and sequence on the folding topology. J. Am. Chem. Soc. 2009;131:16824–16831. doi: 10.1021/ja905611c. [DOI] [PubMed] [Google Scholar]
- 3.Burge S., Parkinson G.N., Hazel P., Todd A.K., Neidle S. Quadruplex DNA: sequence, topology and structure. Nucleic Acids Res. 2006;34:5402–5415. doi: 10.1093/nar/gkl655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chaires J.B., Trent J.O., Gray R.D., Dean W.L., Buscaglia R., Thomas S.D., Miller D.M. An improved model for the hTERT promoter quadruplex. PLoS ONE. 2014;9:e115580. doi: 10.1371/journal.pone.0115580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Paeschke K., Juranek S., Simonsson T., Hempel A., Rhodes D., Lipps H.J. Telomerase recruitment by the telomere end binding protein-beta facilitates G-quadruplex DNA unfolding in ciliates. Nat. Struct. Mol. Biol. 2008;15:598–604. doi: 10.1038/nsmb.1422. [DOI] [PubMed] [Google Scholar]
- 6.Siddiqui-Jain A., Grand C.L., Bearss D.J., Hurley L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11593–11598. doi: 10.1073/pnas.182256799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Murat P., Zhong J., Lekieffre L., Cowieson N.P., Clancy J.L., Preiss T., Balasubramanian S., Khanna R., Tellam J. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol. 2014;10:358–364. doi: 10.1038/nchembio.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arora A., Suess B. An RNA G-quadruplex in the 3′ UTR of the proto-oncogene PIM1 represses translation. RNA Biol. 2011;8:802–805. doi: 10.4161/rna.8.5.16038. [DOI] [PubMed] [Google Scholar]
- 9.Pennarun G., Granotier C., Gauthier L.R., Gomez D., Hoffschir F., Mandine E., Riou J.F., Mergny J.L., Mailliet P., Boussin F.D. Apoptosis related to telomere instability and cell cycle alterations in human glioma cells treated by new highly selective G-quadruplex ligands. Oncogene. 2005;24:2917–2928. doi: 10.1038/sj.onc.1208468. [DOI] [PubMed] [Google Scholar]
- 10.Endoh T., Kawasaki Y., Sugimoto N. Suppression of gene expression by G-quadruplexes in open reading frames depends on G-quadruplex stability. Angew. Chem. Int. Ed. Engl. 2013;52:5522–5526. doi: 10.1002/anie.201300058. [DOI] [PubMed] [Google Scholar]
- 11.Endoh T., Kawasaki Y., Sugimoto N. Stability of RNA quadruplex in open reading frame determines proteolysis of human estrogen receptor alpha. Nucleic Acids Res. 2013;41:6222–6231. doi: 10.1093/nar/gkt286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rachwal P.A., Brown T., Fox K.R. Effect of G-tract length on the topology and stability of intramolecular DNA quadruplexes. Biochemistry. 2007;46:3036–3044. doi: 10.1021/bi062118j. [DOI] [PubMed] [Google Scholar]
- 13.Pandey S., Agarwala P., Maiti S. Effect of loops and G-quartets on the stability of RNA G-quadruplexes. J. Phys. Chem. B. 2013;117:6896–6905. doi: 10.1021/jp401739m. [DOI] [PubMed] [Google Scholar]
- 14.Guedin A., Gros J., Alberti P., Mergny J.L. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010;38:7858–7868. doi: 10.1093/nar/gkq639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phan A.T., Kuryavyi V., Burge S., Neidle S., Patel D.J. Structure of an unprecedented G-quadruplex scaffold in the human c-kit promoter. J. Am. Chem. Soc. 2007;129:4386–4392. doi: 10.1021/ja068739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agrawal P., Hatzakis E., Guo K., Carver M., Yang D. Solution structure of the major G-quadruplex formed in the human VEGF promoter in K+: insights into loop interactions of the parallel G-quadruplexes. Nucleic Acids Res. 2013;41:10584–10592. doi: 10.1093/nar/gkt784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang D., Hurley L.H. Structure of the biologically relevant G-quadruplex in the c-MYC promoter. Nucleosides Nucleotides Nucleic Acids. 2006;25:951–968. doi: 10.1080/15257770600809913. [DOI] [PubMed] [Google Scholar]
- 18.Agrawal P., Lin C., Mathad R.I., Carver M., Yang D. The major G-quadruplex formed in the human BCL-2 proximal promoter adopts a parallel structure with a 13-nt loop in K+ solution. J. Am. Chem. Soc. 2014;136:1750–1753. doi: 10.1021/ja4118945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Phan A.T., Modi Y.S., Patel D.J. Propeller-type parallel-stranded G-quadruplexes in the human c-myc promoter. J. Am. Chem. Soc. 2004;126:8710–8716. doi: 10.1021/ja048805k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seenisamy J., Rezler E.M., Powell T.J., Tye D., Gokhale V., Joshi C.S., Siddiqui-Jain A., Hurley L.H. The dynamic character of the G-quadruplex element in the c-MYC promoter and modification by TMPyP4. J. Am. Chem. Soc. 2004;126:8702–8709. doi: 10.1021/ja040022b. [DOI] [PubMed] [Google Scholar]
- 21.Ambrus A., Chen D., Dai J., Jones R.A., Yang D. Solution structure of the biologically relevant G-quadruplex element in the human c-MYC promoter. Implications for G-quadruplex stabilization. Biochemistry. 2005;44:2048–2058. doi: 10.1021/bi048242p. [DOI] [PubMed] [Google Scholar]
- 22.Hatzakis E., Okamoto K., Yang D. Thermodynamic stability and folding kinetics of the major G-quadruplex and its loop isomers formed in the nuclease hypersensitive element in the human c-Myc promoter: effect of loops and flanking segments on the stability of parallel-stranded intramolecular G-quadruplexes. Biochemistry. 2010;49:9152–9160. doi: 10.1021/bi100946g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith F.W., Feigon J. Strand orientation in the DNA quadruplex formed from the Oxytricha telomere repeat oligonucleotide d(G4T4G4) in solution. Biochemistry. 1993;32:8682–8692. doi: 10.1021/bi00084a040. [DOI] [PubMed] [Google Scholar]
- 24.Adrian M., Heddi B., Phan A.T. NMR spectroscopy of G-quadruplexes. Methods. 2012;57:11–24. doi: 10.1016/j.ymeth.2012.05.003. [DOI] [PubMed] [Google Scholar]
- 25.Lim K.W., Lacroix L., Yue D.J., Lim J.K., Lim J.M., Phan A.T. Coexistence of two distinct G-quadruplex conformations in the hTERT promoter. J. Am. Chem. Soc. 2010;132:12331–12342. doi: 10.1021/ja101252n. [DOI] [PubMed] [Google Scholar]
- 26.Zuckerman D.M. Statistical Physics of Biomolecules: An Introduction. Boca Raton: CRC Press; 2010. [Google Scholar]
- 27.Kallansrud G., Ward B. A comparison of measured and calculated single- and double-stranded oligodeoxynucleotide extinction coefficients. Anal. Biochem. 1996;236:134–138. doi: 10.1006/abio.1996.0141. [DOI] [PubMed] [Google Scholar]
- 28.Spink C.H. Differential scanning calorimetry. Methods Cell. Biol. 2008;84:115–141. doi: 10.1016/S0091-679X(07)84005-2. [DOI] [PubMed] [Google Scholar]
- 29.Boncina M., Lah J., Prislan I., Vesnaver G. Energetic basis of human telomeric DNA folding into G-quadruplex structures. J. Am. Chem. Soc. 2012;134:9657–9663. doi: 10.1021/ja300605n. [DOI] [PubMed] [Google Scholar]
- 30.Pagano B., Randazzo A., Fotticchia I., Novellino E., Petraccone L., Giancola C. Differential scanning calorimetry to investigate G-quadruplexes structural stability. Methods. 2013;64:43–51. doi: 10.1016/j.ymeth.2013.02.018. [DOI] [PubMed] [Google Scholar]
- 31.Kumar N., Maiti S. A thermodynamic overview of naturally occurring intramolecular DNA quadruplexes. Nucleic Acids Res. 2008;36:5610–5622. doi: 10.1093/nar/gkn543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dettler J.M., Buscaglia R., Le V.H., Lewis E.A. DSC deconvolution of the structural complexity of c-MYC P1 promoter G-quadruplexes. Biophys. J. 2011;100:1517–1525. doi: 10.1016/j.bpj.2011.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Majhi P.R., Qi J., Tang C.F., Shafer R.H. Heat capacity changes associated with guanine quadruplex formation: an isothermal titration calorimetry study. Biopolymers. 2008;89:302–309. doi: 10.1002/bip.20918. [DOI] [PubMed] [Google Scholar]
- 34.Huppert J.L., Balasubramanian S. G-quadruplexes in promoters throughout the human genome. Nucleic Acids Res. 2007;35:406–413. doi: 10.1093/nar/gkl1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Z., Dai J., Veliath E., Jones R.A., Yang D. Structure of a two-G-tetrad intramolecular G-quadruplex formed by a variant human telomeric sequence in K+ solution: insights into the interconversion of human telomeric G-quadruplex structures. Nucleic Acids Res. 2010;38:1009–1021. doi: 10.1093/nar/gkp1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cavin Perier R., Junier T., Bucher P. The eukaryotic promoter database EPD. Nucleic Acids Res. 1998;26:353–357. doi: 10.1093/nar/26.1.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kettani A., Gorin A., Majumdar A., Hermann T., Skripkin E., Zhao H., Jones R., Patel D.J. A dimeric DNA interface stabilized by stacked A.(G.G.G.G).A hexads and coordinated monovalent cations. J. Mol. Biol. 2000;297:627–644. doi: 10.1006/jmbi.2000.3524. [DOI] [PubMed] [Google Scholar]
- 38.Zhang N., Gorin A., Majumdar A., Kettani A., Chernichenko N., Skripkin E., Patel D.J. V-shaped scaffold: a new architectural motif identified in an A x (G x G x G x G) pentad-containing dimeric DNA quadruplex involving stacked G(anti) x G(anti) x G(anti) x G(syn) tetrads. J. Mol. Biol. 2001;311:1063–1079. doi: 10.1006/jmbi.2001.4916. [DOI] [PubMed] [Google Scholar]
- 39.Risitano A., Fox K.R. Inosine substitutions demonstrate that intramolecular DNA quadruplexes adopt different conformations in the presence of sodium and potassium. Bioorg. Med. Chem. Lett. 2005;15:2047–2050. doi: 10.1016/j.bmcl.2005.02.050. [DOI] [PubMed] [Google Scholar]
- 40.Szatylowicz H., Sadlej-Sosnowska N. Characterizing the strength of individual hydrogen bonds in DNA base pairs. J. Chem. Inf. Model. 2010;50:2151–2161. doi: 10.1021/ci100288h. [DOI] [PubMed] [Google Scholar]
- 41.Karsisiotis A.I., Hessari N.M., Novellino E., Spada G.P., Randazzo A., Webba da Silva M. Topological characterization of nucleic acid G-quadruplexes by UV absorption and circular dichroism. Angew. Chem. Int. Ed. Engl. 2011;50:10645–10648. doi: 10.1002/anie.201105193. [DOI] [PubMed] [Google Scholar]
- 42.Hsu S.T., Varnai P., Bugaut A., Reszka A.P., Neidle S., Balasubramanian S. A G-rich sequence within the c-kit oncogene promoter forms a parallel G-quadruplex having asymmetric G-tetrad dynamics. J. Am. Chem. Soc. 2009;131:13399–13409. doi: 10.1021/ja904007p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ambrus A., Chen D., Dai J., Bialis T., Jones R.A., Yang D. Human telomeric sequence forms a hybrid-type intramolecular G-quadruplex structure with mixed parallel/antiparallel strands in potassium solution. Nucleic Acids Res. 2006;34:2723–2735. doi: 10.1093/nar/gkl348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mergny J.L., Lacroix L. Curr. Protoc. Nucleic Acid Chem. 2009. UV Melting of G-Quadruplexes. Chapter 17, Unit 17.11. [DOI] [PubMed] [Google Scholar]
- 45.Spink C.H. The deconvolution of differential scanning calorimetry unfolding transitions. Methods. 2015;76:78–86. doi: 10.1016/j.ymeth.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 46.Gray R.D., Buscaglia R., Chaires J.B. Populated intermediates in the thermal unfolding of the human telomeric quadruplex. J. Am. Chem. Soc. 2012;134:16834–16844. doi: 10.1021/ja307543z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gray R.D., Trent J.O., Chaires J.B. Folding and unfolding pathways of the human telomeric G-quadruplex. J. Mol. Biol. 2014;426:1629–1650. doi: 10.1016/j.jmb.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Limongelli V., De Tito S., Cerofolini L., Fragai M., Pagano B., Trotta R., Cosconati S., Marinelli L., Novellino E., Bertini I., et al. The G-triplex DNA. Angew. Chem. Int. Ed. Engl. 2013;52:2269–2273. doi: 10.1002/anie.201206522. [DOI] [PubMed] [Google Scholar]
- 49.Mergny J.L., Lacroix L. Analysis of thermal melting curves. Oligonucleotides. 2003;13:515–537. doi: 10.1089/154545703322860825. [DOI] [PubMed] [Google Scholar]
- 50.Payet L., Huppert J.L. Stability and structure of long intramolecular G-quadruplexes. Biochemistry. 2012;51:3154–3161. doi: 10.1021/bi201750g. [DOI] [PubMed] [Google Scholar]
- 51.Buscaglia R., Gray R.D., Chaires J.B. Thermodynamic characterization of human telomere quadruplex unfolding. Biopolymers. 2013;99:1006–1018. doi: 10.1002/bip.22247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray R.D., Chaires J.B. Curr. Protoc. Nucleic Acid Chem. 2011. Analysis of multidimensional G-quadruplex melting curves. Chapter 17, Unit 17.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gray R.D., Chaires J.B. Kinetics and mechanism of K+- and Na+-induced folding of models of human telomeric DNA into G-quadruplex structures. Nucleic Acids Res. 2008;36:4191–4203. doi: 10.1093/nar/gkn379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haq I., Chowdhry B.Z., Jenkins T.C. Calorimetric techniques in the study of high-order DNA-drug interactions. Methods Enzymol. 2001;340:109–149. doi: 10.1016/s0076-6879(01)40420-4. [DOI] [PubMed] [Google Scholar]
- 55.Wintrode P.L., Griko Y.V., Privalov P.L. Structural energetics of barstar studied by differential scanning microcalorimetry. Protein Sci. 1995;4:1528–1534. doi: 10.1002/pro.5560040810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freire E., Biltonen R. L. Statistical mechanical deconvolution of thermal transitions in macromolecules. I. Theory and application to homogeneous systems. Biopolymers. 1978;17:463–479. [Google Scholar]
- 57.Phan A.T., Patel D.J. Two-repeat human telomeric d(TAGGGTTAGGGT) sequence forms interconverting parallel and antiparallel G-quadruplexes in solution: distinct topologies, thermodynamic properties, and folding/unfolding kinetics. J. Am. Chem. Soc. 2003;125:15021–15027. doi: 10.1021/ja037616j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fitter J. A measure of conformational entropy change during thermal protein unfolding using neutron spectroscopy. Biophys. J. 2003;84:3924–3930. doi: 10.1016/S0006-3495(03)75120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brooks T.A., Hurley L.H. Targeting MYC expression through G-quadruplexes. Genes Cancer. 2010;1:641–649. doi: 10.1177/1947601910377493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou W., Suntharalingam K., Brand N.J., Barton P.J., Vilar R., Ying L. Possible regulatory roles of promoter g-quadruplexes in cardiac function-related genes - human TnIc as a model. PLoS One. 2013;8:e53137. doi: 10.1371/journal.pone.0053137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balasubramanian S., Hurley L.H., Neidle S. Targeting G-quadruplexes in gene promoters: a novel anticancer strategy? Nat. Rev. Drug Discov. 2011;10:261–275. doi: 10.1038/nrd3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li X.M., Zheng K.W., Zhang J.Y., Liu H.H., He Y.D., Yuan B.F., Hao Y.H., Tan Z. Guanine-vacancy-bearing G-quadruplexes responsive to guanine derivatives. Proc. Natl. Acad. Sci. U.S.A. 2015;112:14581–14586. doi: 10.1073/pnas.1516925112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ou T.M., Lu Y.J., Tan J.H., Huang Z.S., Wong K.Y., Gu L.Q. G-quadruplexes: targets in anticancer drug design. ChemMedChem. 2008;3:690–713. doi: 10.1002/cmdc.200700300. [DOI] [PubMed] [Google Scholar]
- 64.Olsen C.M., Gmeiner W.H., Marky L.A. Unfolding of G-quadruplexes: energetic, and ion and water contributions of G-quartet stacking. J. Phys. Chem. B. 2006;110:6962–6969. doi: 10.1021/jp0574697. [DOI] [PubMed] [Google Scholar]
- 65.Lim K.W., Amrane S., Bouaziz S., Xu W., Mu Y., Patel D.J., Luu K.N., Phan A.T. Structure of the human telomere in K+ solution: a stable basket-type G-quadruplex with only two G-tetrad layers. J. Am. Chem. Soc. 2009;131:4301–4309. doi: 10.1021/ja807503g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Motlagh H.N., Wrabl J.O., Li J., Hilser V.J. The ensemble nature of allostery. Nature. 2014;508:331–339. doi: 10.1038/nature13001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Miyoshi D., Nakao A., Sugimoto N. Structural transition from antiparallel to parallel G-quadruplex of d(G4T4G4) induced by Ca2+ Nucleic Acids Res. 2003;31:1156–1163. doi: 10.1093/nar/gkg211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Brazda V., Haronikova L., Liao J.C., Fojta M. DNA and RNA quadruplex-binding proteins. Int. J. Mol. Sci. 2014;15:17493–17517. doi: 10.3390/ijms151017493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dai J., Carver M., Hurley L.H., Yang D. Solution structure of a 2:1 quindoline-c-MYC G-quadruplex: insights into G-quadruplex-interactive small molecule drug design. J. Am. Chem. Soc. 2011;133:17673–17680. doi: 10.1021/ja205646q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moye A.L., Porter K.C., Cohen S.B., Phan T., Zyner K.G., Sasaki N., Lovrecz G.O., Beck J.L., Bryan T.M. Telomeric G-quadruplexes are a substrate and site of localization for human telomerase. Nat. Commun. 2015;6:7643. doi: 10.1038/ncomms8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moore C.C. Ergodic theorem, ergodic theory, and statistical mechanics. Proc. Natl. Acad. Sci. U.S.A. 2015;112:1907–1911. doi: 10.1073/pnas.1421798112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Broxson C., Beckett J., Tornaletti S. Transcription arrest by a G quadruplex forming-trinucleotide repeat sequence from the human c-myb gene. Biochemistry. 2011;50:4162–4172. doi: 10.1021/bi2002136. [DOI] [PubMed] [Google Scholar]
- 73.Nudler E. Flipping riboswitches. Cell. 2006;126:19–22. doi: 10.1016/j.cell.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 74.Narayan S., Kombrabail M.H., Das S., Singh H., Chary K.V., Rao B.J., Krishnamoorthy G. Site-specific fluorescence dynamics in an RNA ‘thermometer’ reveals the role of ribosome binding in its temperature-sensitive switch function. Nucleic Acids Res. 2015;43:493–503. doi: 10.1093/nar/gku1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhuang X., Bartley L.E., Babcock H.P., Russell R., Ha T., Herschlag D., Chu S. A single-molecule study of RNA catalysis and folding. Science. 2000;288:2048–2051. doi: 10.1126/science.288.5473.2048. [DOI] [PubMed] [Google Scholar]
- 76.Stone M.D., Mihalusova M., O'Connor C.M., Prathapam R., Collins K., Zhuang X. Stepwise protein-mediated RNA folding directs assembly of telomerase ribonucleoprotein. Nature. 2007;446:458–461. doi: 10.1038/nature05600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hogan M.E., Austin R.H. Importance of DNA stiffness in protein-DNA binding specificity. Nature. 1987;329:263–266. doi: 10.1038/329263a0. [DOI] [PubMed] [Google Scholar]
- 78.Bohnuud T., Beglov D., Ngan C.H., Zerbe B., Hall D.R., Brenke R., Vajda S., Frank-Kamenetskii M.D., Kozakov D. Computational mapping reveals dramatic effect of Hoogsteen breathing on duplex DNA reactivity with formaldehyde. Nucleic Acids Res. 2012;40:7644–7652. doi: 10.1093/nar/gks519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harris S.A., Gavathiotis E., Searle M.S., Orozco M., Laughton C.A. Cooperativity in drug-DNA recognition: a molecular dynamics study. J. Am. Chem. Soc. 2001;123:12658–12663. doi: 10.1021/ja016233n. [DOI] [PubMed] [Google Scholar]
- 80.Tippana R., Xiao W., Myong S. G-quadruplex conformation and dynamics are determined by loop length and sequence. Nucleic Acids Res. 2014;42:8106–8114. doi: 10.1093/nar/gku464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Stadlbauer P., Krepl M., Cheatham T.E. 3rd, Koca J., Sponer J. Structural dynamics of possible late-stage intermediates in folding of quadruplex DNA studied by molecular simulations. Nucleic Acids Res. 2013;41:7128–7143. doi: 10.1093/nar/gkt412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fleming A.M., Zhou J., Wallace S.S., Burrows C.J. A role for the fifth G-track in G-quadruplex forming oncogene promoter sequences during oxidative stress: do these ‘Spare Tires’ have an evolved function. ACS Cent. Sci. 2015;1:226–233. doi: 10.1021/acscentsci.5b00202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Reblova K., Sponer J., Lankas F. Structure and mechanical properties of the ribosomal L1 stalk three-way junction. Nucleic Acids Res. 2012;40:6290–6303. doi: 10.1093/nar/gks258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nikolova E.N., Kim E., Wise A.A., O'Brien P.J., Andricioaei I., Al-Hashimi H.M. Transient Hoogsteen base pairs in canonical duplex DNA. Nature. 2011;470:498–502. doi: 10.1038/nature09775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heddi B., Martin-Pintado N., Serimbetov Z., Kari T.M., Phan A.T. G-quadruplexes with (4n - 1) guanines in the G-tetrad core: formation of a G-triad.water complex and implication for small-molecule binding. Nucleic Acids Res. 2016;44:910–916. doi: 10.1093/nar/gkv1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.