

Abstract
Protein–protein interactions (PPIs) drive all biologic systems at the subcellular and extracellular level. Changes in the specificity and affinity of these interactions can lead to cellular malfunctions and disease. Consequently, the binding interfaces between interacting protein partners are important drug targets for the next generation of therapies that block such interactions. Unfortunately, protein–protein contact points have proven to be very difficult pharmacological targets because they are hidden within complex 3D interfaces. For the vast majority of characterized binary PPIs, the specific amino acid sequence of their close contact regions remains unknown. There has been an important need for an experimental technology that can rapidly reveal the functionally important contact points of native protein complexes in solution. In this review, experimental techniques employing mass spectrometry to explore protein interaction binding sites are discussed. Hydrogen–deuterium exchange, hydroxyl radical footprinting, crosslinking and the newest technology protein painting, are compared and contrasted.
Keywords: Protein-protein interactions, protein complex interface, protein painting, mass spectrometry, targeted therapeutics, proteomics
Introduction
Currently, in the realm of molecular interaction research, there are few interactions that compare to the intricacy, diversity, and rapidly expanding therapeutic interest of protein–protein interactions (PPIs) [1]. This growing interest in PPIs is demonstrated by the rapid rise in publications devoted to PPIs. From 1983 to 2012 the yearly average of PPI publications in PubMed rose from 9 to 1,362 [2]. In parallel the pharmaceutical community has placed more resources behind small molecules and monoclonal antibodies that block protein–protein interactions. Monoclonal antibody (mAb) PPI inhibitors such as Humira (adalimumbab) and Remicade (infliximab) dominated pharmaceutical sales in 2013 as the first and third highest selling biopharmaceutical products [3]. Despite this strong interest in this field, and the success of PPI inhibitors in the clinic, there have been just 60 inhibitory peptide therapeutics and 20 mAb oncology therapeutics approved by the US FDA [4]. So, with such a high demand for PPI targeted therapeutics, why is the pipeline of PPI inhibitors moving to market so small? The answer to this question is that it is extremely difficult and time consuming to elucidate the amino acid sequence of protein–protein interfaces.
The contact regions hidden within protein–protein binding interfaces are critically important targets for the next generation of therapies that disrupt PPIs. Identification of the amino acid sequences located within the contact regions of a complex between two or more disease-associated proteins is of great importance to pharmaceutical drug development [5]. These PPIs have been shown to be responsible for many common biological processes, and become deranged in disease. If left unmodulated, they can lead to malfunctions in cellular processes (such as cell growth, leading to various forms of cancer). If these regions of contact are identified, then specific therapies can be developed that would target the protein–protein interface to inhibit binding [5], signal transduction, and function.
In the past, identification of the key protein–protein interaction domains or “hot spots” has been very difficult and time consuming (6). The contact point sequences are hidden inside the highly complex binding cleft [6] that excludes solvent. PPI motifs are highly variable because their biologic function is correspondingly diverse. Some interactions are flat and featureless, while others approximate a key and binding pocket. Such biologic diversity (especially for 3 or more protein partners) makes techniques such as high-throughput chemical screening, or structure-based discovery, even more challenging [6]. Even with all the modeling and computational methods available, only 0.5% of protein–protein complexes have been revealed and made available to the public via the Protein Data Bank [5].
Despite this complexity, there has been a great deal of interest in developing peptides that act against these regions of contact between protein complexes. Peptides can span a larger area compared to a small molecule, and thereby, in theory, are better PPI inhibitors. Peptides can also exhibit high target specificity, which leads to less side interactions (or side effects) when developing a therapeutic drug [7]. As such, the peptide-based pharmaceutical market is the fastest growing share with new product success rates twice that of other small molecule pharmaceuticals [7].
An example of an emerging therapeutic peptide on the market is Exenatide (Byetta), which is an antagonist of the glucagon-like peptide-1 (GLP-1) receptor in type II diabetes patients [7]. Over its three-year clinical trial period, this peptide has shown to significantly reduce a type II diabetic patient’s hemoglobin A1c glucose levels and induce weight loss [8]. Additional examples of successful peptide-based drugs include the angiotensin-converting enzyme (ACE) inhibitor targeting hypertension named Captopril (Capoten) and the anticoagulant targeting unstable angina named Bivalirudinrub (Angiomax) [7]. The success of these targeted inhibitors demonstrates the immediate need for methods to characterize and identify the exact amino acid sequences lurking in the contact regions of PPIs.
There are some hurdles to overcome, however, when considering the use of peptides as therapeutics. Currently, there is the concern of the increased manufacturing costs to produce therapeutic peptides and the lack of a universal set of guidelines set up by regulating authorities regarding purity standards of these peptides [9]. But, as the demand for therapeutic proteins increases, production costs are being lowered and regulations regarding peptide production are being changed [9].
One major concern is the short half-life due to rapid degradation via proteases, which leads to questions concerning in vivo instability. Also, issues concerning low availability of an oral drug candidate (most peptide drug candidates require injection), hepatic and renal clearance, and the low passage of these peptides across physiological barriers (such as cellular membranes or blood–brain barriers) [10]. Poor penetration of membranes becomes a major limitation in the creation of therapeutic peptides in the field of neuroscience since crossing the blood–brain barrier is extremely difficult. But, despite these issues, having a toolset of various techniques to study PPI domains could also lead to the development of other non-peptide based drugs for targeted therapies involving the difficult to reach regions of the body (such as the brain) [11].
Currently, there are only a small number of experimental techniques that have been proven successful for the study of PPI interface sequences. In this review we will discuss the structural chemistry of PPI interfaces as a foundation for an in-depth comparison of crosslinking chemistry, hydrogen/deuterium exchange, hydroxyl radicals footprinting, and protein painting structural mass spectrometry methods.
Protein–protein Interactions
Physical Chemistry of Protein–protein Interactions
Cellular events ranging from catabolism of metabolites, cellular signaling, to the immune system recognition of a viral intruder are all possible because of protein complexes that associate rapidly and with high specificity [12]. How and why do these proteins interact in such a specific manner? What drives these PPIs to form? The answers to these questions is thought to be based on the physical chemistry of proteins, and the proximity of protein partners in the cellular or extracellular microenviroment. Protein folding and protein partner binding is strongly influenced by the hydrophobicity of amino acid residues at the interface [12]. Hydrophobic interactions are driven by the movement of non-polar amino acid residue regions from a polar to a non-polar microenvironment, resulting in a free energy gain. Van der Waals interactions within these regions allow packing of these residues and the loss of water molecules favors further stabilization of protein association and the maintenance of tertiary structure [13]. Covalent bonds, hydrogen bonds, and electrostatic interactions all influence PPI formation (Figure 1) [12].
Figure 1. Protein–protein interaction interface.
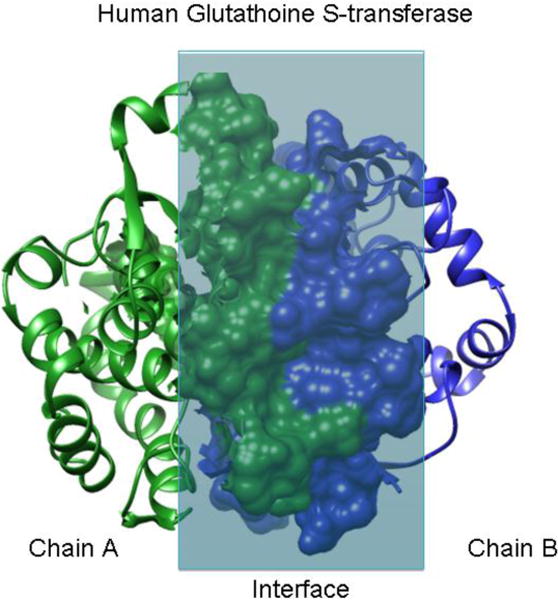
Human glutathione S-transferase. Chains A and B are shown in green and blue. The residues interacting within the interface are shown in a molecular surface representation, while the remainder of the structure is displayed in a ribbon diagram [12].
Beyond physical interactions, protein–protein interface creation is also reliant upon the complementarity of 3-D shapes at the site of interaction [13]. Sources of shape complementarity include non-polar and polar amino acid residue alignment, residue packing density, water molecule concentration within the interface, and the actual size and number of cavities at the interface itself. All these factors help determine if an interaction will occur and dictate the strength of binding [13]. Any defects in shape or residue packing can result in cavities within the interface, which are subsequentially filled with water molecules. These internal cavities are important for the formation of substrate channels, as well as general flexibility of monomer structure while binding is occurring [13].
Hot Spots
Protein complexes are very dynamic and fairly flexible, which adds to the complexity of PPI elucidation [12]. There can be different conformational states of a protein complex that depends upon its binding state. Shifts in conformational state can change the free energy landscape of a protein, which can affect the strength of binding. Shifts can also change the free energy state to promote a second or third protein to bind to the complex [12].
The energy distribution on the surface, as well as within the binding interface, changes as the protein partners undergo various conformational states [14]. Specific types of residues have greater energy contributions, compared to other residues, within the interface. In fact, even a small number of residues composing only a fraction of the PPI, contribute the vast majority of the binding free energy of the interface. This area of the PPI has now become referred to as a “hot spot” [14].
Hot spots have been shown to have a distinct composition when compared to the remaining low energy interface area. Certain residues seem to appear more frequently within hot spots, including arginine, tryptophan, and tyrosine (which have a 10% frequency within hot spot composition) [14]. Moreover, hot spots are structurally conserved with a much lower mutation rate compared to other residues within the interface [13].
Due to not only their structural conservation, but also their free energy differences and stability of coupling, hot spots have become extremely important targets for pharmaceutical drug development. Unfortunately, since these specific residues are hidden well within the cleft of the binding interface in PPIs, characterization of the hot spot can be extremely difficult [14]. Currently, other than crystallography or tomography, only a few proven methods exist to identify the sequences within PPI regions of native proteins without creating a great deal of protein modification [14].
Structural Mass Spectrometry Techniques
Crosslinking Chemistry
One prevailing method of studying PPI takes advantage of protein cross linking chemistry to covalently link regions of proteins that are in close proximity [15]. When crosslinking chemistry is combined with mass spectrometry (MS), specific residues that are crosslinked will remain linked after denaturation and protease cleavage [15] to create peptide fragments for MS analysis.
A typical protein crosslinking reaction begins with the addition of crosslinker molecule that contains a reactive group on both ends, which is then separated via a spacer (Figure 2c) [15]. Thiols, carboxylic acids, and primary amines of amino acid side chains are favored as crosslinking targets during this reaction [15]. Crosslinking can also take place between photo-activatable groups, but these linkages are generally less specific and not as well defined. The resulting products from either method, though, will now contain protein complexes with covalent bonds present between amino acids in close proximity [15].
Figure 2. Crosslinking chemistry.
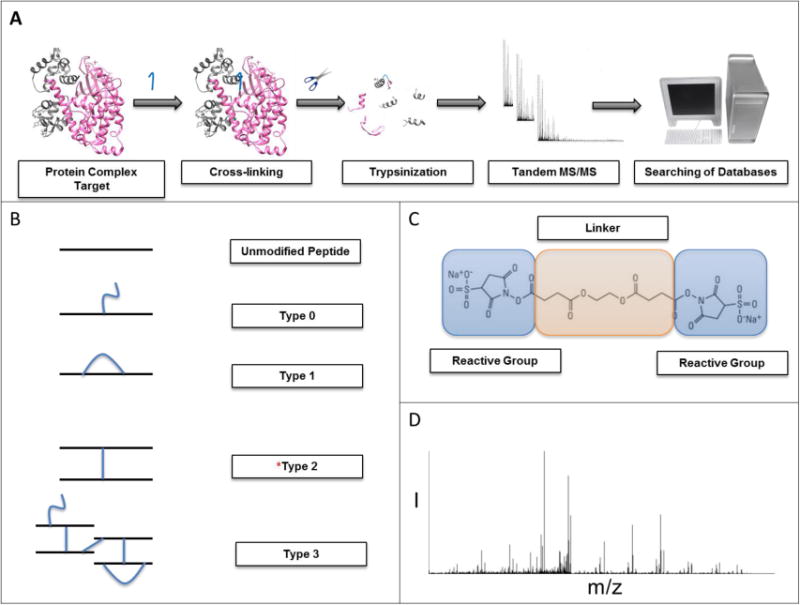
a) During crosslinking, a chemical crosslinker is added to the protein complex and after sufficient incubation (usually 30 minutes or longer) the complex undergoes trypsin digestion. The resulting fragments are separated by liquid chromatography and then run on the mass spectrometer for identification following a database search. Dedicated software builds a database of theoretical molecular species obtained from all the possible combinations of protein proteolytic fragments and the known crosslinker molecule. Statistical models are then used to match the experimental data to the prediction database. b) These are examples of types 0 to 3 crosslinked peptides. *Type 2 is the most useful during structural mass spectrometry techniques. c) Crosslinking occurs between primary amines (or reactive groups) via bifunctional molecules that possess two reactive groups that are separated by a spacer. During the crosslinking reaction, a covalent bond is formed between the amine of the protein and the leaving group portion of the crosslinker. This reaction involves the formation of a peptide bond. d) An example of a fragmentation spectrum that results from mass spectrometry analysis [15].
After completion of the crosslinking reaction, the protein complex products will fall within one of four different classifications of crosslinked peptides [17]. Type 0 peptides are considered modified peptides since they consist of a single peptide bound to a crosslinker molecule without being coupled to an adjacent peptide. Type 1 peptides occur when a peptide crosslinks internally, or between two crosslinker molecules on the same peptide, creating what is referred to as a cyclic peptide [16]. The type 2 classification consists of two independent peptides becoming crosslinked between their modified groups, resulting in a crosslinked peptide. When there is a combination of type 0, 1, or 2 present, this is considered type 3 or a higher order crosslinked peptide (Figure 2b) [16].
Following crosslink formation, the peptide complexes are then subjected to digestion with a protease (typically trypsin). This digestion creates peptide fragments with preserved crosslinkage, which can be then fractionated and analyzed by MS (Figure 2a) [15]. This analysis occurs in two steps, beginning with identification of the crosslinked peptides using the fragmentation spectrum and the mass of the fragment. The information is then elaborated with dedicated software and a list of all potential peptide crosslinkages is constructed (Figure 2d) [16].
Although this method has been successful in identifying and predicting protein crosslinks with the help of in silico protein digestion, there are also several limitations when it comes to the identification of PPIs [15]. One major issue is that the crosslinker probe molecules have a difficult time accessing the interface region of a protein–protein contact area. A second issue is false positives or covalent linkage artefacts within the fragment population, which can decrease the confidence level of the peptide crosslink match [15].
Hydrogen/deuterium Exchange
Another method for studying PPIs is Hydrogen/deuterium exchange (HDX). In this approach, deuterium is used to transiently label amides in the protein backbone [17]. To accomplish HDX, proteins are allowed to interact in buffered aqueous solutions until equilibrium is reached. Following complex formation, proteins are then diluted in a buffered D2O solution for a short time period [18]. Solvent accessible backbone amides become deuterated whereas backbone amides are protected from deuterium labeling. The exchange reaction is then stopped by diluting the sample in H2O at low pH and temperature. This process (referred to as quenching) unfolds the protein and locks the deuterium in place [18].
The samples are then subjected to immobilized pepsin digestion at low pH to prepare them for liquid chromatography (LC)/MS-MS analysis [18]. In order to evaluate the amount of deuterium that is lost, two controls are generally run along with the samples. One control is 0% deuterium labeling, and the other control has 100% deuterium labeling. In addition, an internal protein standard is run in parallel to calculate the inherent exchange rates (Figure 3) [21].
Figure 3. Hydrogen–deuterium exchange workflow.
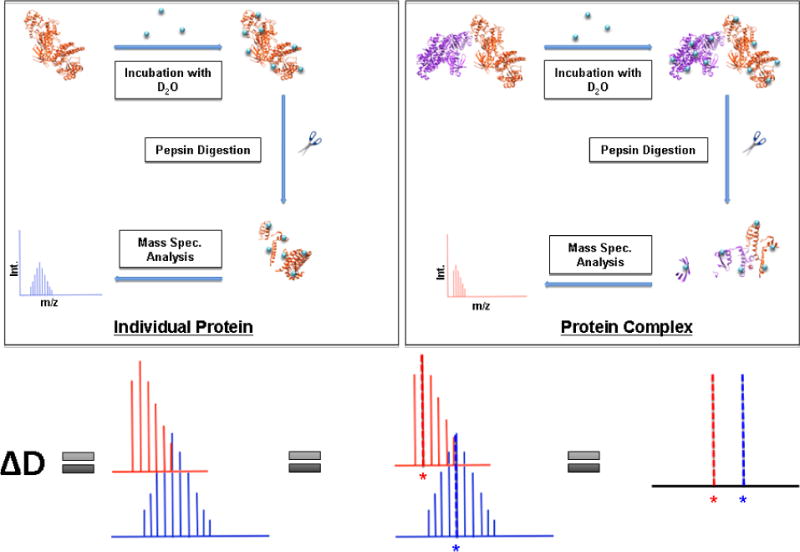
The overall overflow of a Hydrogen–deuterium exchange (HDX) experiment begins with an individual protein and its protein complex incubated separately in a buffered D2O solution and then the deuterated protein complexes are diluted (or quenched) using H2O at low pH and temperature. Following a pepsin digestion, liquid chromatography occurs to separate the peptides and then they are run on the mass spectrometer. Specific software examines the HDX mass shift to determine the amount of deuterium incorporated in the peptide fragments. These peak mass shifts are calculated as ∆D (displayed as the difference between the red and blue dotted peak lines) [20].
Protein–protein interaction sites are probed using HDX by cataloging the hydrogen/deuterium exchange rate of the amides in the test sample of complexed proteins. How fast or slow this exchange takes place is determined by the accessibility of these amides to the solvent they are dissolved in [18]. Therefore, amides in a linear external peptide should have a higher rate of exchange than those buried within a protein–protein interface since they have a higher accessibility to the solvent environment [17].
The MS analysis of the resulting HDX fragments requires specific software that compares the mass spectrum of the collection of masses represented by the deuterated protein of interest (or isotopic envelope/profile) to the undeuterated control [19]. Peptide masses are derived from calculating the centroid of its isotopic envelope, and, along with its pattern of MS/MS fragmentation, this can be used to identify the peptides as well. The software can also compare the experimental isotopic profile to theoretical isotopic profiles in order to calculate the amount of deuterium that has been incorporated by examining the HDX mass shift [19]. Those peptide fragments which contain less deuterium then their counterparts are considered to be protected, and would be part of the protein–protein interface [19].
As with crosslinking, though HDX is able to attain a wealth of information concerning individual peptides, it has several limitations when used to study PPIs. A major issue is that the MS data obtained from HDX has in the past been at the level of individual pepsin fragment peptides, which can be many amino acids in length, not single amino acid residues. This has made it difficult to confirm the exact sequence involved in the region of protein–protein interaction [21]. There have been advances in top-down HDX approaches, though, that has increased fragmentation efficiency and characterization down the single amino acid level [22]. HDX is also inefficient regarding detecting changes in the conformational state of a protein, and generally has a bias toward the average conformational state. The sensitivity of HDX may also be an issue when trying to study weaker interactions since this method requires measurable changes in deuteration exchange rates [21].
Hydroxyl Radicals Footprinting
The technique of hydroxyl radical footprinting permits mapping of protein interfaces and conformational changes by studying the solvent accessibility of the regions using hydroxyl radicals [23]. Hydroxyl radicals have the ability to react with solvent accessible protein side-chains with similar Van der Waals interactions as those of water. The resulting modifications cause a shift in mass, which can give high-resolution information concerning the accessibility of certain structural regions of a protein complex [23].
In order to begin this technique, hydroxyl radicals must first be produced. There are several methods for doing so, including hydrogen peroxide decomposition, radiolysis of water, or photolysis of hydrogen peroxide [23]. These radicals are then introduced into the unbound and bound protein complex mixtures to allow OH- labeling of the solvent accessible residues to occur. The resulting modified proteins are subjected to digestion (usually via trypsin), desalted, and prepared for MS analysis (Figure 4) [23].
Figure 4. Hydroxyl radical footprinting overview.
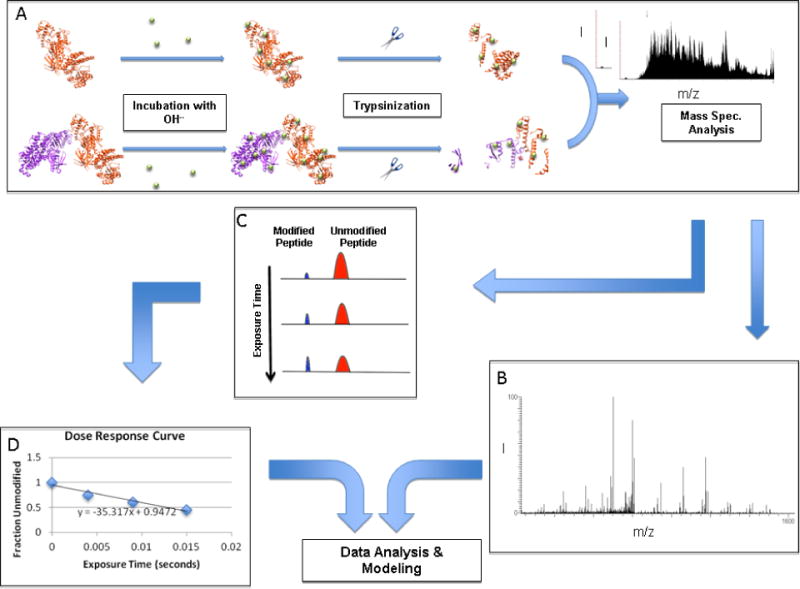
a) Following labeling with the hydroxyl radicals of the individual proteins and the subsequent protein complex, the digested/trypsinized proteins are divided up by liquid chromatography. b) Tandem mass spectrometry is performed to identify the modified peptide and examine oxidation sites. c) The rate of hydroxyl radical labeling is determined by analyzing the differences in the area of modified and unmodified peptide peaks with increasing time of radical exposure. d) These data can then be used to create a dose-response curve to calculate the rate of oxidation [23].
Tandem MS is performed to examine the mass shift from the hydroxyl radical modification and thereby identify a site-specific modification [23]. This analysis occurs in two steps: first being isolation of the ion of interest from a peptide precursor, followed by identification of the ions produced from fragmentation. MS spectra from both modified and unmodified proteins are compared using specific software packages, and any observable mass shifts can help locate and identify specific modified residues in a peptide’s structure [23].
From this information, conclusions can be drawn in regards to which regions of the proteins are involved in PPIs. Those residues that show less or no mass shift are thought to be unmodified, and therefore would be present in a solvent inaccessible area. When several unmodified residues are clustered together, this area could be assumed to be part of a protein–protein interface if results differ from those of the individual, unbound proteins [23].
While hydroxyl radical footprinting has shown to be a very promising technique in the world of PPI discovery, it too has some drawbacks. A major concern with this method is the length of time for the hydroxyl radical reaction to occur which is longer than the amount of time before the protein begins unfolding [24]. This would cause residues, which were once hidden in the protein–protein interface, to now be exposed to the solvent and react with the radicals. Their modification would be interpreted as a false negative. To overcome this drawback a technique has been proposed that employs radical formation by a microsecond laser pulse to reduce the time of the chemical modification [24]. A further weakness of hydroxyl radical footprinting is that certain chemicals or ionization processes involved in hydroxyl radical production can create undesirable secondary reactions, which can skew results [24].
Protein Painting
Protein Painting, a recently proposed functional approach, uses molecular “paints”, or small, organic dye molecules, to mask the external protein surfaces, to exclusively expose the amino acid sequences buried within PPI interface regions [25]. This method uses the knowledge of small molecule chemistry and the characterization power of mass spectrometry to reveal the peptide sequence hidden within the protein interaction interface without the use of crosslinking. Protein painting takes advantage of solvent exclusion to identify amino acid residues and approximate their distance from one another to determine the best targets for pharmaceutical drugs [25].
Identifying a novel series of organic dyes that bind to proteins with extremely high on rate and very low off rate was key to developing this technique [25]. These molecular “paints” also possessed the ability to block trypsin cleavage sites by binding to trypsin cleavage consensus sequences on a protein’s surface. Finally the same high affinity dyes, remain bound after denaturation, reduction and alkylation, and trypsin cleavage. Example dye chemistries effective in this method are 1-amino-9,10-dioxo-4-[3-(2-sulphonatooxyethylsulphonyl)nanilino] anthracene-2-sulphonate, sodium 4-(4-(benzyl-et-amino)-ph-azo)-2,5-di-cl-benzenesulphonate (Acid Orange 50), phenyl 4-(1-amino-4-hydroxyl-9,10-dioxo-9,10-dihydro-2-anthracenyl)oxy] benzenesulphonate (R495034), and 4-amino-3-[[4-[4-[(1-amino-4-sulphonatonaphthalen-2-yl) diazenyl] phenyl] phenyl] diazenyl] naphthanlene-1-sulphonate [25].
In order to study PPIs, a set of the protein partners must be painted individually and compared with a pre-formed complex of the same protein partners. Candidate dye solutions dissolved in PBS are introduced to the native solution phase native protein samples at room temperature and neutral pH [25]. These solutions are allowed to incubate for 5 minutes at room temperature to allow the paints to completely cover the outside of the protein complex, and then rapidly (one minute) passed through a molecular sieve Sephadex column (Figure 5a, Figure 6) [25] to separate the unbound dye from the dye-protein complexes. The eluates are then subjected to reduction and alkylation and trypsin digestion according to routine MS analysis (Figure 5b) [25].
Figure 5. Protein painting.
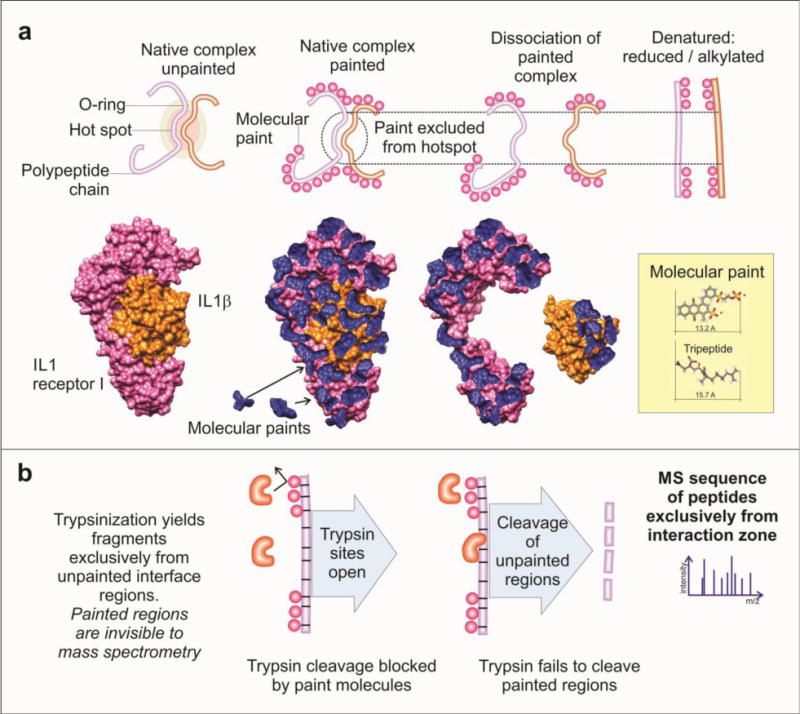
a) Protein–protein complexes are painted using surface binding molecular paints. The area of protein–protein interaction is not accessible to the solvent, so these regions remain unpainted and are available for trypsin digestion following dissociation and denaturation of the complex. A model of the IL1β complex is illustrated before and after undergoing protein painting. b) The molecular paints bind to the regions that are solvent-accessible and block trypsin cleavage sites. After digestion, the resulting peptides will be derived exclusively from the region of protein–protein interaction and can be further processed and identified by mass spectrometry [21]. Modified from Luchini et al [25].
Figure 6. Mass spectrometry output of protein painting is exclusive to sequences from protein–protein interaction interface area.
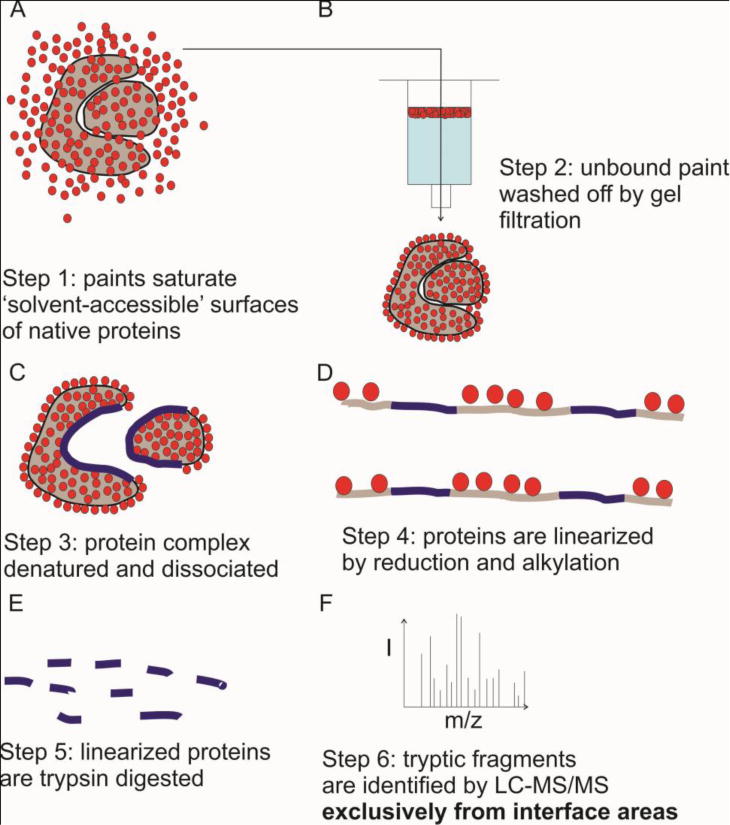
Native proteins are allowed to interact and then are briefly pulsed with excess paints. The unbound paints are removed via gel filtration columns. Proteins are dissociated and linearized. Paints remain bound to proteins during the dissociation and linearization process. The painted complexed proteins then undergo mass spectrometry analysis that identifies the unique protein–protein interaction region sequence (seen in blue). This sequence is in the solvent-inaccessible region in the points of contact between the protein and the ligand. This method involves no structural modification, which helps closer identify the region of interest in a protein’s more native form.
During the MS analysis, each sample is run through the mass spectrometer and tandem mass spectrometry is performed in a data dependent manner. Standard SEQUEST data analysis program is used to compare the fragment’s sequence to the NCBI database of the species investigated [25]. The identified fragments from the painted complexes are then compared to the MS analysis of the individual dissociated proteins with and without dye painting. The difference in the two data sets exclusively reveals the contact interface regions that were not accessible to the dye paints [25].
Protein–protein interface identification via protein painting is based on the principle that the molecular paints bind to all regions of the protein complex’s surface except those hidden within binding surfaces (due to solvent exclusion). The paints remain bound to the protein and serve to block all the trypsin sites that are not contained within the protein–protein interface [25]. The resulting fragments identified by MS, therefore, should only be the sequence of amino acids involved in the PPI of that particular protein complex. This sequence can then be used to create targeted therapeutics, such as custom peptide inhibitors, or antibodies, to block protein complex formation [25].
Comparing the four methods discussed, protein painting rapidly isolates and identifies solvent-excluded regions of a protein–protein interface with the least amount of modification to the protein’s native structure. The most important attribute of protein painting is that this method generates sequences exclusively from the interface region (true positives). Sequences outside the interface are invisible because their trypsin cleavage sites are blocked so they do not appear in the MS output. In addition the method is very rapid and requires no special software to sort peptide fragment. Nevertheless, protein painting has its limitations. One issue is the reliance upon the density and location of the trypsin cleavage sites, which could lead to reduced resolution in areas where cleavage sites are sparse [25]. In addition, the dye paints must be applied over a short pulse (5 minutes) to the pre-formed native complexes and then rapidly washed off before [25] before the protein partners spontaneously dissociate according to their intrinsic off-rate.
Conclusions
Four complementary methods are available to pharmacologists and structural biologists seeking to define the interface contact points functionally required for protein–protein interactions. A side-by-side comparison of the four protocols is summarized in Table 1.
Table 1.
Summary of crosslinking, hydrogen–deuterium exchange, hydroxyl footprinting and protein painting structural mass spectrometry techniques [15].
| Crosslinking | Hydrogen–Deuterium Exchange | Hydroxyl Footprinting | Protein Painting | |
|---|---|---|---|---|
| Experimental Configuration | Standard | Optimized for deuterium retention | Optimized for UV pulse shorter than 1 microsecond | Standard |
| pH | Neutral-basic | Strongly acidic (pH=2) | Neutral-slightly basic | Neutral |
| Temperature | Room temperature, −20°C for delayed MS analysis | Room temperature, 4 °C, and −80 °C | Room temperature, −20°C for delayed MS analysis | Room temperature, −20°C for delayed MS analysis |
| Treatment Duration | 0.5–2 hours | Short (few minutes) | 1 microsecond and shorter | Short (few minutes) |
| Software Analysis | Dedicated software | Dedicated software | Manually search in the MS spectra for oxidized products | No special requirements: standard MS workflow and software |
| Results | Restricted to trypsin fragments that contain primary amine, carboxyl, sulfhydryl, or carbonyl groups depending on the crosslinker of choice | Pepsin fragment length, average 10 amino acids | Half of trypsin fragment with caveat that oxidized arginine might not be cleaved by trypsin | Half of any trypsin fragment (for two interacting partners, average 4.5 amino acids, resolution of paint molecules 3 amino acids) |
| Positive Output | Binding partners are identified with low specificity for interface solvent excluded binding regions | Interaction regions are identified by a small 1.0073 Dalton shift in peptic fragment peptide mass | Interaction regions are identified by absence of oxidization | Interaction regions are identified by presence of tryptic peptides exclusively derived from both sides of the interface |
| Protein Conformation | Pre-formed complex covalently crosslinked | Pre-formed complex deuterated | Pre-formed complex oxidized | Pre-formed complex coated non covalently with small dye molecules |
| Coverage Type | Protease fragments that contain primary amine, carboxyl, sulfhydryl, or carbonyl groups depending on the crosslinker of choice. Crosslinked lysine will not be cleaved by trypsin. | Pepsin cleavage peptides | Trypsin cleavage sites with possible exception of oxidized Arginine | Known distribution of trypsin cleavage sites preferred for MS |
| Negative Outputs or Side Reaction Products | Internal crosslinks, modified peptide (type 0) and cyclic peptide (type 1) are identified as side reaction products | Within-protein interactions are not identified as false positive because the method is differential (unbound – bound state) | OH radical reaction can cause proteins to unfold. Oxidized residues can pre-exist prior to treatment (e.g methionine) | Within-protein interactions are not identified as false positive because the method is differential (unbound – bound state) |
In terms of positive outputs, each method has its advantages and disadvantages. The resulting outputs range from examining shifts in fragment mass in HDX to tryptic peptides from unpainted, solvent excluded regions in protein painting. Methods such as crosslinking and hydroxyl footprinting create false positives compared to the other methods due to internal crosslinking and the generation of false positives due to pre-emptive protein unfolding. Protein painting offers a method that is less time consuming then other methods, has the highest rate of true positives [25] and requires no modification to the actual structure of the proteins involved in the PPIs. (Figure 6).
Despite the limitless applications of these technologies, all of these methods are currently limited to studying known protein interacting partners. In order to study unknown proteins and their interacting partners, a de novo screening tool would need to be implemented prior to using one of the structural MS techniques [26]. Immunoprecipitation of unknown proteins, followed by SDS-PAGE and LC-MS/MS, has shown to be an effective technique to identify previously unknown interacting partners, as well as assist in building protein interaction networks [26].
Expert commentary & five-year view
Future directions in the field of structural mass spectrometry include using one or more of the reviewed experimental techniques together to obtain the most complete picture of the protein–protein interface structure [27]. Integration of these techniques with in silico modeling and crystallography data, if available, can be the starting point for delineating candidate regions within the complex interface that have the highest likelihood of being effective drug targets. We expect that the next 5 years will see a series of inhibitors emerge from the application these technologies to important disease associated protein–protein interactions. Ion mobility (IM) MS has already been used to elucidate the exact structure of large multi-protein complexes, such as the tryptophan RNA binding attenuation protein (TRAP) [28]. Moreover, protein painting in conjunction with in silico computational models, has produced a completely new class of inhibitor that will block the three-way interaction of IL-1β, IL1R1, and IL1RacP, to abolish downstream inflammatory signaling [25]. This inhibitor has applications to arthritis and other diseases associated with aberrant or persistent inflammation.
Key Issues.
The contact regions hidden within protein–protein binding interfaces are critically important targets for the next generation of therapies that disrupt protein–protein interactions (PPIs).
Currently, other then using such methods as crystallography or tomography to attempt structure definition, it is difficult to identify the sequences within the PPI regions of native proteins without a great deal of protein modification.
There are a small number of techniques that have been proven successful in studying and identifying PPIs using structural mass spectrometry methods: crosslinking chemistry, hydrogen/deuterium exchange, hydroxyl radical footprinting, and protein painting.
Methods such as crosslinking and hydroxyl footprinting may have more of a reduced specificity and a lower number of true positive hits when compared to the other methods due to internal crosslinking leading side products and chemical modification of unfolded domains.
Protein painting offers a method that is less time consuming compared to other methods, is associated with a high specificity of hits, and requires no covalent modification to the region of interaction on proteins in the native complex.
Future directions in the field of structural mass spectrometry include integrating two or more of these methods in combination with in silico modeling. The methods are complementary since they use different chemical principles.
Acknowledgments
LA Liotta and A Luchini received NIH funding for this project (grants NIH NCI 1R21CA177535-01, NIH NCI 1R33CA173359-01 and NIH NIAMS 1R21AR061075-01).
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
- 1.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.Ivanov AA, Khuri FR, Fu H. Targeting protein-protein interactions as an anticancer strategy. Trends in Pharmacological Science. 2013;34(7):393–400. doi: 10.1016/j.tips.2013.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Walsh G. Biopharmaceutical benchmarks 2014. Nature Biotechnology. 2014;32(10):992–1000. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 4.Miller MJ, Foy KC, Kaumaya PTP. Cancer immunotherapy: present status, future perspective, and a new paradigm of peptide immunotherapeutics. Discovery Medicine. 2013;15(82):166–176. [PubMed] [Google Scholar]
- 5.Nero TL, Morton CJ, Holien JK, Wielens J, Parker MW. Oncogenic protein interfaces: small molecules, big challenges. Nat Rev Cancer. 2014;14:248–262. doi: 10.1038/nrc3690. [DOI] [PubMed] [Google Scholar]
- 6.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 7.Craik DJ, Fairlie DP, Liras S, Price D. The future of peptide-based drugs. Chem Biol Drug Des. 2013;81:136–147. doi: 10.1111/cbdd.12055. [DOI] [PubMed] [Google Scholar]
- 8.LaRue S, DeYoung MB, Blickensderfer A, Chen S. Once-weekly glucose-lowering therapy for type 2 diabetes. Clinical Diabetes. 2012;30(3):95–100. [Google Scholar]
- 9.Lax R. The future of peptide development in the pharmaceutical industry. Pharmanufacturing: The International Peptide Review. 2010:10–15. [Google Scholar]
- 10.Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discovery Today. 2010;15(1/2):40–56. doi: 10.1016/j.drudis.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Gozes I. Neuroprotective peptide drug delivery and development: potential new therapeutics. TRENDS in Neurosciences. 2001;24(12):700–705. doi: 10.1016/s0166-2236(00)01931-7. [DOI] [PubMed] [Google Scholar]
- 12.Keskin O, Gursoy A, Ma B, Nussinov R. Principles of protein-protein interactions: what are the preferred ways for proteins to interact? Chem Rev. 2008;108:1225–1244. doi: 10.1021/cr040409x. [DOI] [PubMed] [Google Scholar]
- 13.Sharma SK, Ramsey TM, Bair KW. Protein-protein interaction: lessons learned. Curr Med Chem. 2002;2:311–330. doi: 10.2174/1568011023354191. [DOI] [PubMed] [Google Scholar]
- 14.Morrow JK, Zhang S. Computational prediction of hot spot residues. Curr Pharm Des. 2012;18(9):1255–1265. doi: 10.2174/138161212799436412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rappsilber J. The beginning of a beautiful friendship: cross-linking/mass spectrometry and modeling of proteins and multi-protein complexes. J Struct Biol. 2011;173:530–540. doi: 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shilling B, Row RH, Gibson BW, Guo X, Young MM. MS2Assign, automated assignment and nomenclature of tandem mass specra of chemically crosslinked peptides. J Am Soc Mass Spectrom. 2013;14:834–850. doi: 10.1016/S1044-0305(03)00327-1. [DOI] [PubMed] [Google Scholar]
- 17.Katta V, Chait BT. Hydrogen/deuterium exchange electrospray ionization mass spectrometry: a method for probing protein conformational changes in solution. J Am Chem Soc. 1993;115:6317–6321. [Google Scholar]
- 18.Mandell JG, Falick AM, Komives EA. Identification of protein-protein interfaces by decreased amide proton solvent accessibility. Proc Natl Acad Sci. 1998;95:14750–14710. doi: 10.1073/pnas.95.25.14705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Z, Zhang A, Xiao G. Improved protein hydrogen/deuterium exchange mass spectrometry platform with fully automated data processing. Anal Chem. 2012;84:4942–4949. doi: 10.1021/ac300535r. [DOI] [PubMed] [Google Scholar]
- 20.Hydrogen deuterium exchange mass spectrometry. Washington University in St. Louis St. Louis, MO: School of Medicine; 2014. Available at: http://msr.dom.wustl.edu/hydrogen-deuterium-exchange-mass-spectrometry-intro/ [Last accessed 244 2015] [Google Scholar]
- 21.Percy AJ, Rey M, Burns KM, Schriemer DC. Probing protein interactions with hydrogen/deuterium exchange and mass spectrometry – a review. Analytica Chimica Acta. 2012;721:7–21. doi: 10.1016/j.aca.2012.01.037. [DOI] [PubMed] [Google Scholar]
- 22.Kaltashov I, Bobst CE, Abzalimov RR. H/D exchange and mass spectrometry in the studies of protein conformation and dynamics: is there a need for a top-down approach? Anal Chem. 2009;81(19):7892–7899. doi: 10.1021/ac901366n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L, Chance MR. Structural mass spectrometry of proteins using hydroxyl radical based protein footprinting. Anal Chem. 2011;83:7234–7241. doi: 10.1021/ac200567u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidze protein solvent-accessible residues on the microsecond timescale. Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Luchini A, Espina V, Liotta L. Protein painting reveals solvent-excluded drug targets hidden within native protein-protein interfaces. Nat Commun. 2014;5:4413. doi: 10.1038/ncomms5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malovannaya A, Li Y, Bulynko Y, Jung YJ, Wang Y, Lanz RB, O’Malley BW, Qin J. Streamlined analysis schema for high-throughput identification of endogenous protein complexes. PNAS. 2010;107(6):2431–2436. doi: 10.1073/pnas.0912599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mo J, Tymiak AA, Chen G. Structural mass spectrometry in biologics discovery: advances and future trends. Drug Discovery Today. 2012;17:1323–1330. doi: 10.1016/j.drudis.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 28.Hyung S, Ruotolo BT. Integrating mass spectrometry of intact protein complexes into structural proteomics. Proteomics. 2012;12:1547–1564. doi: 10.1002/pmic.201100520. [DOI] [PMC free article] [PubMed] [Google Scholar]