

Abstract
Background & Aims
Small intestinal carcinoids are rare and difficult to diagnose and patients often present with advanced, incurable disease. Although the disease occurs sporadically, there have been reports of family clusters. Hereditary small intestinal carcinoid has not been recognized and genetic factors have not been identified. We performed a genetic analysis of families with small intestinal carcinoids to establish a hereditary basis and find genes that might cause this cancer.
Methods
We performed a prospective study of 33 families with at least 2 cases of small intestinal carcinoids. Affected members were characterized clinically and asymptomatic relatives were screened and underwent exploratory laparotomy for suspected tumors. Disease-associated mutations were sought using linkage analysis, whole-exome sequencing, and copy number analyses of germline and tumor DNA collected from members of a single large family. We assessed expression of mutant protein, protein activity, and regulation of apoptosis and senescence in lymphoblasts derived from the cases.
Results
Familial and sporadic carcinoids are clinically indistinguishable except for the multiple synchronous primary tumors observed in most familial cases. Nearly 34% of asymptomatic relatives older than 50 y were found to have occult tumors; the tumors were cleared surgically from 91% of these individuals (21/23). Linkage analysis and whole-exome sequencing identified a germline 4 bp deletion in the gene inositol polyphosphate multikinase (IPMK) that truncates the protein. This mutation was detected in all 11 individuals with small intestinal carcinoids and 17/35 family members whose carcinoid status was unknown. Mutant IPMK had reduced kinase activity and nuclear localization, compared with the full-length protein. This reduced activation of p53 and increased cell survival.
Conclusions
We found that small intestinal carcinoids can occur as an inherited autosomal dominant disease. The familial form is characterized by multiple synchronous primary tumors, which might account for 22%–35% of cases previously considered sporadic. Relatives of patients with familial carcinoids should be screened to detect curable early-stage disease. IPMK haplo-insufficiency promotes carcinoid tumorigenesis.
Keywords: familial, screening, tumor suppressor gene, linkage
Serotonin producing neuroendocrine tumors (NETs) arise from enterochromaffin cells (EC) of the distal small intestine (SI), appendix and proximal colon. Tumor secretion of serotonin, bioactive amines and tachykinins lead to mesenteric and cardiac fibrosis and the carcinoid syndrome (diarrhea, flushing, bronchoconstriction). SI-NETs are the most frequent tumor of the SI and their incidence has increased ~5-fold from 1973 – 20041–3. Although well differentiated and slow growing (10–20 years before detection), the term carcinoid, is a misnomer for a frequently small, micro-invasive, metastasizing tumor.4 The relative rarity, and nonspecific symptomatology (abdominal pain and diarrhea) result in either missed or delayed diagnosis (9.2 yrs)5. As a consequence, the majority of patients present with advanced disease and a poor prognosis (36% 5-year survival)3. Most cases of SI-NETs are non-familial and 22–35% of patients present with multiple synchronous primary tumors6,7. To date, no causal molecular genetic basis for non-familial carcinoid tumor has been identified8, 9. The genetic basis for other familial endocrine tumor syndromes, with clinical features similar to SI-NETs, such as multiple endocrine neoplasia (MEN), has been described leading to speculation that a discrete genetic basis exists for SI-NETs encountered in the absence of MEN mutations. Although only 6 non-MEN reported families with multiple primary SI-NETs had been described prior to 200810–15, it is plausible that SI-NETs occur on a familial basis, with their own separate genetic cause. To date, however, familial SI-NETs have not been well recognized as a distinct clinical entity and their genetic basis and pathogenesis remain unknown16.
Studies of familial cancers have enabled discovery of driver mutations often applicable to sporadic disease, such as for the genes APC and colon cancer, VHL and clear cell kidney cancer and MEN1 and Zollinger-Ellison Syndrome. We hypothesized that 1) the presence of multiple synchronous primary tumors in “sporadic disease” represented unrecognized and thus more common familial disease; 2) familial SI-NET susceptibility is transmitted in an autosomal dominant mode at least in some families; 3) screening of at-risk asymptomatic family members would detect occult tumors; 4) screening would accelerate phenotypic ascertainment and add statistical power for genetic linkage analysis in otherwise underpowered small pedigrees; and 5) earlier diagnosis and surgical resection in asymptomatic family members might cure or delay clinical onset of the disease. We initiated a prospective study in 2008 to define the clinical features of familial SI-NET, assess the impact of screening on the natural history of the disease, and elucidate the molecular genetic basis for SI-NET.
Materials & Methods
Deatails relating to study subject enrollment, clinical evaluation and genetic analysis and the evaluation of IPMK function can be found in the Supplementary Appendix.
RESULTS
Clinical Disease Characterization
One hundred eighty one members from 33 families with SI-NET, including 44 affected individuals diagnosed before entering the study (6 of whom died during the study) and 137 asymptomatic first-degree relatives were evaluated at the NIH CRC. An additional 33 of 35 affected deceased relatives were evaluated on the basis of outside medical records (Supplementary Fig. S1). Twenty-nine of the 137 asymptomatic members screened positive for suspected carcinoid tumor. The most sensitive screening tests were wireless capsule endoscopy (WCE) and 18F-DOPA PET/CT followed closely by CT. Traditional biomarkers were only useful in two patients with elevated 24 hour urine 5-HIAA (supplementary Table S2). Among these 29 individuals screening positive, 26 underwent exploratory laparotomy. Of these 26, 23 were positive (16.8% of total screened) and 3 were negative for SI-NET by surgical pathology. Another two are considering surgery and another one was lost to follow-up. Nineteen of 56 (33.9%) asymptomatic members older than 50 screened positive and were confirmed by surgical pathology. Representative diagnostic images, (CT with intravenous contrast, 18F-DOPA PET/CT and capsule endoscopy) of these multiple, small, submucosal, primary tumors and their surgical gross and microscopic appearance (typical nests of uniform, indolent tumor cells positive for serotonin, chromogranin A, synaptophysin and rare Ki-67) are shown in Figure 1.
Figure 1.

Diagnostic imaging, gross and microscopic pathology of small intestinal carcinoid tumors from patients with familial carcinoid tumor. (A) Arterial enhancement of primary tumors on CT enterography. (B) 18F-DOPA PET/CT (left) and PET alone (right) of tumors in panel A. (C) WCE of a SI-NET. (D) Resected ileum demonstrating the wide distribution of tumors (identified with sutures) and a luminal view of a small umbilicated submucosal tumor before and after sectioning. (E) Insular appearance of tumoral cells and surrounding fibrosis in serial sections stained with hematoxylin and eosin (H&E), immunostained for Ki-67, serotonin (SER), and neuroendocrine marker proteins, chromogranin A (CgA) and synaptophysin (SYN).
Familial disease has a clinical presentation and course similar to sporadic disease1, 7. The 77 symptomatic relatives diagnosed before enrollment were diagnosed at an average age of 61 with approximately the same number of males and females. The most common symptoms were abdominal pain (70%), flushing (32%) and diarrhea (29%). They also presented at a late stage (stage IV, 70%) with low grade (grade I, 91%), small diameter (< 1 cm on average) primary tumors predominantly in the distal small bowel (jejunum and ileum) (Table 1). However, a higher proportion of familial patients (36 of 54, 67%) presented with multiple primary tumors (Table S1) compared to patients with no affected relatives (22–35%)1,6,7. This high proportion is likely an underestimate due to incomplete surgical exploration as suggested by the multiple tumors (average of 6.4 per patient) found in the majority of first-degree relatives (18 of 23, 78.3%) positively screened in our study (Supplementary Table S2).
Table 1.
Summary of Clinical Data for Patients with Familial SI-NET
| Parameter | Asymptomatic, Screened Positive | Symptomatic |
|---|---|---|
| Number of Patients | 26 | 77 |
| Gender (M:F) | 15:11 | 37:40 |
| Age Symptomatic (yrs) | NA | 56 (55, 16–83)(n=73) |
| Age at Diagnosis (yrs) | 58.5 (57.5, 43–75) | 61 (61, 27- |
| Diagnostic Delay (yrs)* | NA | 5 (2, 0–29)(n=73) |
| Age (LwD) (yrs) | 61 (60, 44–76) | 61 (61, 36–84) |
| Age at Death (yrs) | NA | 72 (73, 45–87) (n=38) |
| Survival (yrs) | ||
| Dead | 0 | 5.6 (3, <1–18)(n=37) |
| AwD (yrs since dx.) | 5.25 (n=2) | 6.75 (6, 2–16) (n=30) |
| NED (yrs) | 4 (4.5, 2–5.7)(n=21) | 8.5 (6, 2–26)(n=9)^ |
| Presenting Sxs. | NA | n=76 |
| Abdominal Pain | NA | 53 (70%) |
| Flushing | NA | 24 (32%) |
| Diarrhea | NA | 22 (29%) |
| Obstruction | NA | 19 (25%) |
| GI Bleeding | NA | 8 (11%) |
| Incidental | NA | 3 (4%) |
| Stage at Diagnosis | n= 23 | n=73 |
| I | 8 (32%) | 3 (4%) |
| IIA:B | 4:1 (22%) | 2:0 (3%) |
| IIIA:B | 0:8 (39%) | 1:16 (23%) |
| IV | 2 (8%) | 51 (70%) |
| Tumor Grade | ||
| Grade 1 | 22 (96%) | 50 (91%) |
| Grade 2 | 1 (4%) | 5 (9%) |
| Tumor Size (largest, cm) | 0.85 (0.8, 0.1–2.5)(n=23) | 1.9 (1.7, 0.9- |
| No. of Primary Tumors | 5.7 (4, 1–29)(n=23) | 6.8 (2, 1–50)(n=55) |
| Tumor Location | n = 23 | n = 47 |
| Jejunum | 6 | 3 |
| Jejunum-Ileum | 5 | 3 |
| Ileum | 12 | 37 |
| Appendix | _ | 2 |
| Lung | _ | 2 |
| Bowel Resection (cm)‡ | 76.5 (75, 7.5–195)(n=23) | Insufficient Data |
| Diagnostic Modalities | n=26 | n=72 |
| Imaging | 25 (96%) | 41 (57%) |
| Surgery/Biopsy † | 23 (92%) | 33 (46%) |
| Biochemistry | 2 (8%) | 6 (8%) |
| Autopsy | NA | 3 (4%) |
M, Male: F, Female: Age LwD (Living with Disease), average age of patients still living with known disease; AwD, Alive with Disease; dx., diagnosis; NED, No Evidence of Disease; Sxs., symptoms;
Age at diagnosis minus age of symptom onset; Where applicable, data are expressed as mean (median, range). n, # of patients contributing to the result.
Two patients had pulmonary carcinoid and two patients had appendiceal carcinoid, the remaining 5 patients had SI carcinoids with an average of 5.6 years with NED.
Length of bowel resected.
Only 23 of 26 positively screened patients underwent surgical exploration. Diagnostic modality percentages add to more than 100% since more than one modality was used for some patients.
While screening did not result in significantly earlier diagnosis (58 vs. 61 yo), it diagnosed significantly earlier stage disease compared to symptomatic relatives (8.7% vs. 70% stage IV, p < 0.001). Twenty of 23 patients (87%) with stage I –IIIB disease identified by screening were surgically cleared of disease and have no evidence of disease for an average follow-up of 4 years compared to 9 of 39 (23%) living symptomatic relatives with an average follow-up of 8.5 years. Thirty eight of 77 (49%) symptomatic relatives died within an average of 5.6 years of diagnosis while all of the 23 asymptomatic screened-positive patients are alive for an average of more than 4 years after their diagnosis (Table 1 and Supplementary Table S2).
Although nearly all patients had SI-NET, patient 9:III-25 had only a previously diagnosed pulmonary typical carcinoid (Figs. 2A, S1 and Supplementary Table S1). Patient 4:III-3 also had a previously diagnosed pulmonary typical carcinoid that was resected two years prior to screening positive for SI-NET in our study (Supplementary Fig. S1 and Table S2).
Figure 2.

Pedigree and linkage analysis for family 9. (A) Pedigree for family 9. Affected males (squares) and females (circles) with previously diagnosed disease appear as black filled symbols while asymptomatic family members who screened positive appear as red filled symbols. Subjects with unaffected and unknown disease status appear as green empty symbols, respectively. Slashes indicate deceased family members. Small Arabic numerals alongside a symbol identify individuals included in the linkage analysis (panel B) on the basis of SNP data alone (black) or SNP plus microsatellite data (red). Small Arabic numerals below symbols indicate subject age, if alive, or at death. Age in red numerals denote patients evaluated at the NIH CRC. IPMK genotypes are indicated below the patient’s age as either WT or MUT (4 bp deletion). (NA, DNA not available). (B) Genome-wide linkage analysis for family 9 pedigree shown in panel a. Shown are multi-marker LOD scores using a dominant inheritance model for all autosomes based on combined analysis of SNPs and microsatellites. A linkage peak was found within chromosome 10 with a significant LOD score of 3.566. Negative multipoint LOD scores are truncated at zero. LOD score thresholds for suggestive and genome-wide significance appear as dotted lines. (C) Sensitivity Evaluation of Linkage Analysis in Family 9 using SNP and Microsatellite Markers on Chromosome 10. LOD scores for a full penetrance and a reduced penetrance model appear as blue and cyan lines, respectively. The red line gives −log10(P-values) for a model-free linkage analysis.
Genetic Linkage Analysis
Genetic linkage analysis was performed on the largest family (pedigree #9, Fig. 2A and Supplementary Fig. S1) to identify the chromosomal location of a causal gene. DNA was obtainable from 5 of the 7 members who were recognized as affected at the initial ascertainment. However, intense screening (detailed in the online methods) of 47 asymptomatic family members identified 6 additional affected members (4, tumor pathology; 1, elevated 24 hr urinary 5-hydroxyindoleacetic acid; 1, positive CT imaging). Nine unrelated spouses and 3 related members negative for disease on the basis of either a negative surgical exploration or negative evaluation at an advanced age were considered unaffected (Fig. 2A, green filled symbols). Initial multipoint linkage analyses using SNP markers and a subset of individuals resulted in suggestive evidence of linkage in a region on chromosome 10 (logarithm of odds (LOD)=1.6). Results of model-based multi-marker linkage analysis using all available individuals and a combination of SNP and fine-mapping microsatellite markers on chromosome 10 are shown in Fig. 2B. In this analysis, the interval on chromosome 10 reached genome-wide significance with a multipoint LOD=3.57. The one-LOD-drop region extends from rs1345561 to D10s464 and contains 132 protein-coding genes within this 14 cM (25 Mb) interval. The maximum observed LOD score was similar to that seen in simulation studies to evaluate power and varying parameters had little effect on the LOD score or the interval boundaries (Fig. 2C, supplementary Fig. S2 and the Supplementary Appendix). Genetic linkage analysis of pedigrees 1 and 7 did not identify a significant or shared linkage region with pedigree 9 (Fig. S3).
Mutational Analysis
Whole exome sequencing (WES) was performed on germline DNA from 5 affected members (9:II-3, 9:III-2, 9:III-11, 9:III-12 and 9:III-22, Fig. 2A) and one unaffected member (9:III-3, 2A) (Additional information in online methods and Supplementary Appendix). WES and bioinformatics analysis identified a single novel mutation within the linkage interval consisting of a 4 bp (CAGT) deletion in exon 6 of the 76.4 Kb gene coding for a 416 a.a protein, IPMK (HGNC:20739; NM_152230). The 4 bp deletion c.990-993del results in a frame-shift with replacement of the native a.a. sequence, SQT331-333 to RLH, subsequent premature termination close to the nuclear localization signal sequence (a.a. 320-330) and elimination of the downstream ATP binding domain (Fig. 3). Sanger sequencing confirmed the 4 bp deletion in all 11 affected individuals and detected the deletion in 17 of the 35 unknown and none of the 3 unaffected family members used in the linkage analysis. No mutations outside the linkage interval shared by all 5 affected members analyzed by WES were present in all 6 other affected family members analyzed by targeted Sanger sequencing (Supplementary Table S3). This variant also had the highest two-point LOD score of all variants typed in the region (Supplementary Fig. S2). There were no mutations in IPMK cDNA in the other 32 families analyzed by Sanger sequencing.
Figure 3.
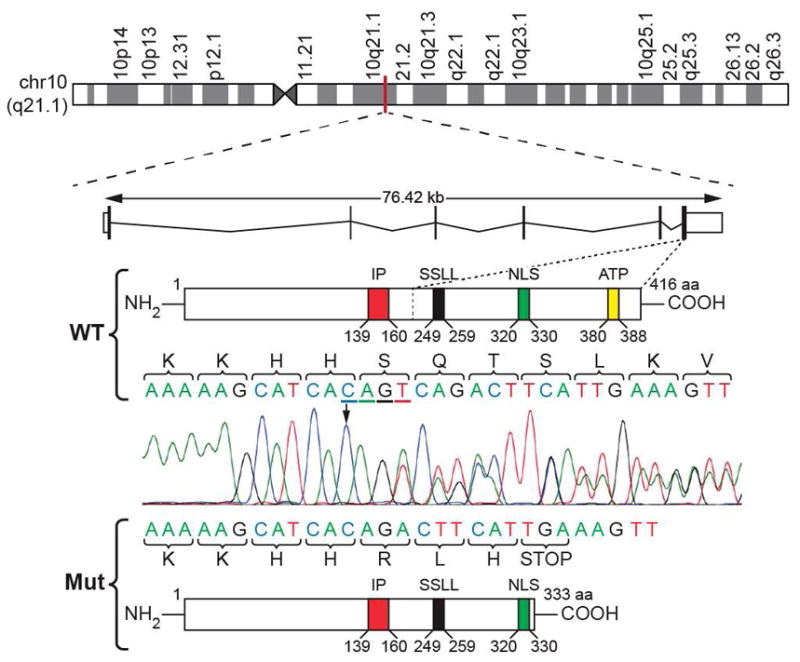
Chromosomal position, structure and sequence of WT and mutant IPMK. IPMK is positioned on the long arm of chromosome 10 at band 21.1 (ideogram, top). WT IPMK is a 76.42 kb gene consisting of 6 exons and 5 introns encoding a 416 amino acid (a.a) protein with at least 4 structural domains including an inositol phosphate (IP, red) binding site, SSLL (Ser-Ser-Leu-Leu) catalytically important domain (black), nuclear localization signal (NLS, green) and adenosine triphosphate (ATP) binding site (yellow)(middle panel). Deletion of the 4 bp, CAGT (cDNA bp 990-993; black arrow and underlined), results in a frameshift (denoted by the double lined chromatogram of Sanger sequenced germline cDNA) mutation with arginine (R), leucine (L) and histidine (H) substituted for native serine (S), glutamine (Q) and threonine (T) in positions 331-333 and subsequent premature termination (STOP).
Furthermore, Sanger sequencing and SNP-based copy number variation analysis of DNA isolated from laser capture micro-dissected tumor confirm the presence of the WT IPMK allele (Supplementary Fig. S4). Therefore, unlike the somatic loss of the second allele seen in some other familial cancers, there was no loss of heterozygosity in the patient’s carcinoid tumors.
Expression of WT and Mutant IPMK
The 4 bp deletion had no significant effect on IPMK transcript expression between patients and age matched controls (Fig. 4A) using semi-quantitative RT-PCR of EBV transformed B lymphoblast (shown to express IPMK, Fig. 4C) total RNA. IPMK transcripts are expressed in patient’s primary submucosal SI-NETs as detected by fluorescent in situ hybridization (FISH) using IPMK and TPH1-specific cRNA probes (Fig. 4B). Western blot of lysates of the same lymphoblasts in Fig. 4C using amino terminal epitope (a.a. 26-265) specific antibody shows expression of the truncated IPMK mutant and an approximate 50% reduction in full-length IPMK proteins. (Fig. 4C). IPMK protein was specifically expressed in carcinoid tumor relative to surrounding tissue stroma (4D). Although IPMK is widely expressed and the carcinoid tumor cell of origin, the EC cell, is present throughout multiple tissues, carcinoids are most common in the distal small intestine3. Consistent with SI-NET locations7, IPMK is most highly expressed in human SI (Fig. 5) and increases distally (Supplementary Fig. S5, mouse SI quantitative Ipmk FISH and NextBio Body Atlas microarrays, www.NextBio.com).
Figure 4.

Expression of wild type and mutant IPMK. (A) Agarose gel electrophoresis of RT-PCR exon amplicons of IPMK and β-actin (BAC) in B lymphoblasts from controls (C1–C3) and patients (P1–P3) with the heterozygous IPMK c.990-993del mutation. IPMK transcript expression did not differ between patients and controls (mean ±SEM, p=0.343). (B) Brightfield (left) and corresponding fluorescent in situ hybridization (right) images of a FFPE SI-NET from a patient with a heterozygous IPMK c.990-993del mutation. cRNA probes for IPMK and TPH-1 appear green and pink, respectively, and nuclei stained with DAPI appear blue. (C) Western blot of IPMK and β-tubulin protein expression from the same three controls and patients from panel A. Lysates of B lymphoblasts were immunoprecipitated using anti-IPMK (aa 301-337) and immunoblotted using anti-IPMK (aa 26-265). Full length IPMK (~49 kDa) was detected in both controls and patients, while faint bands of truncated IPMK were detected only in patients with heterozygous IPMK c.990-993del mutation. IPMK protein expression was reduced at least 50% in patients compared to controls (mean±SEM, p<0.001). (D) Immunohistochemical detection of IPMK expression (right, rabbit αIPMK) compared to control (left, normal rabbit serum) in FFPE sections of a SI-NET from a patient with a heterozygous IPMK c.990-993del mutation.
Figure 5.

Expression of IPMK Transcripts in Normal Human Organs. Left. Quantitative analysis of IPMK RNA in situ hybridization of normal organ tissue array. IPMK expression signal was scored as 0 (undetectable); 1 (rare); 2 (moderate) or 3 (abundant). Each bar represents the average score from three experiments on three tissue array slides (#ACI-D97, ACI-D98 and ACI-D110) performed in duplicate. Right. Representative fluorescent images of IPMK expression (red dots) from six tissues (blue, DAPI nuclear staining).
Functional Analysis of Mutant IPMK
IPMK has inositol phosphate kinase activity leading to the formation of IP417 and the subsequent rate-limiting formation of IP5 (Ins(1,3,4,5,6)P5)18 necessary for higher phosphorylated, IP6, and pyrophosphorylated InsPs, IP7 and IP8. Heterozygous loss of the ATP binding site in patients with mutant IPMK (Fig. 3A) results in a significant reduction of 32% in the formation of IP5 in patient vs. control B lymphoblasts (Fig. 6A).
Figure 6.

Effect of the heterozygous IPMK c.990-993del mutation on IPMK Function. (A) Inositol phosphate (IP) metabolism in carcinoid patients (blue) compared to WT family members (red). B Lymphoblasts were cultured with [H3]inositol and radiolabeled IPs were separated using strong anion exchange HPLC. Representative chromatograms are shown with positions of IP standards. The arrow indicates the position of IP5 analyzed in the bar graph to the right for 4 pairs of IPMK-mutant patients and WT controls (mean+/−SEM; ** p=0.01). (B) Left. Representative images of tGFP tagged-IPMK in HEK293 cells transfected with either WT (top) or mutant (bottom) form of IPMK. Cells (DAPI stained, blue) were imaged 24 hours after transfection for localization of IPMK- tGFP (green) using a confocal microscope. Right. Quantitative analysis of nuclear versus cytoplasmic localization for WT versus mutant IPMK-tGFP (mean+/−SEM; ** p<0.01). (C) Effect of mutant IPMK on Gene expression of p53 and its target genes PUMA, BAX and p21. B lymphoblasts from carcinoid patients with mutant IPMK and normal controls were treated with cisplatin (50 mM X 24 hrs.), extracted for total RNA and analyzed by qRT-PCR. Data are expressed as fold increase over untreated baseline, *p<0.05 (patients, n=4 vs. controls, n=3), **p<0.01, (patients vs. controls, n=3), mean+/−SEM. (D) Differential resistance to cellular stress of B lymphoblasts from carcinoid patients with mutant IPMK compared to normal controls. B lymphoblasts were treated with either staurosporine or cisplatin at 37° C for 24 hrs. and cell viability measured colorimetrically using WST-1. Data (n=3 experiments) are expressed as % survival, mean+/−SEM, **p</= 0.01).
IPMK is a regulated nucleocytoplasmic protein with predominant nuclear localization19,20,21. The 4 bp deletion in mutant IPMK affects amino acids occurring immediately following the nuclear localization signal (NLS) with alteration of the subsequent 3 a.a. followed by premature truncation (Fig. 3A). This significantly affects IPMK localization. Expression of transfected, amino-terminally fused tGFP to the IPMK-4bp del. mutant in the human embryonic kidney cell line (HEK293) results in nearly an 80% reduction (p<0.01) in nuclear localization vs. tGFP-WT IPMK (Fig. 6B).
Independent of its catalytic activity, the amino terminus of IPMK binds and co-activates p53 in the nucleus to enhance target gene transcription of pro-apoptotic and cell cycle arrest target genes22. Reduction in the nuclear localization of mutant IPMK has no significant effect on p53 transcript expression in patient vs. control B lymphoblasts under genotoxic stress from cisplatin (Fig. 6C). However, under these conditions, p53 pro-apoptotic and cell cycle arrest target gene transcripts, PUMA (p53 up-regulated modulator of apoptosis), BAX (B cell lymphoma 2-associated X) and p21 (cyclin dependent kinase inhibitor 1, CDKN1A), respectively, are significantly reduced by 40–45% in patients with mutant IPMK vs. control B lymphoblasts (Fig. 6C). The IPMK mutant-related reduction in p53 target gene transcription is associated with resistance to apoptosis due to DNA damage as evidenced by equivalent induction of γH2AX, reduction in cleavage of poly(ADP-ribose) polymerase (PARP) (Fig. S6 A and B) and increase in survival of patient vs. control B lymphoblasts treated with cisplatin (Fig. 6D). The IPMK mutant effect on p53-mediated apoptosis was also assessed in the HEK293 cell line treated with staurosporine. Overexpression of the IPMK mutant vs. WT IPMK results in less caspase-3 cleavage and greater survival as measured by propidium iodide (PI) exclusion (Supplementary Fig. S6 C and D).
DISCUSSION
This prospective study shows that the familial occurrence of SI-NET is similar to sporadic disease except for multiple synchronous primary tumors, hinting at a germline predisposition. While inspection of the 33 pedigrees (Supplementary Fig. S1) suggests autosomal dominant transmission with late onset and variable penetrance23,24, other causes for familial occurrence cannot be excluded. Analysis of the only family with a sufficient number of affected individuals to detect significant linkage established for the first time a genetic basis for familial carcinoid consistent with autosomal dominant inheritance. Autosomal dominant transmission and slow tumor growth allowed not only a high rate of detection, especially in older family members, but also diagnosis of early-stage disease unlike their symptomatic relatives and patients with non-familial disease1,4.
The 3-year trend in earlier diagnosis of screened patients versus their relatives with symptomatic late-stage disease suggests that there is a critical period prior to metastasis when primary tumors are smaller (less likely to metastasize7) but detectable and thus surgically curable. WCE and 18F-DOPA/PET CT followed by CT were the most sensitive pre-operative screening tests, however, no one test was uniformly positive nor sufficiently sensitive to detect all tumors palpated at surgery. Biomarkers such as serotonin, chromogranin A and 24 hour urine 5-HIAA considered useful for symptomatic sporadic SI-NET were rarely positive for detection of early disease in asymptomatic patients. Despite the relatively short follow up for this slow growing tumor, positive screening of asymptomatic relatives and earlier surgery results in a disease-free and survival advantage that positively impacts the natural history of the disease (Table 1). The ability to screen for asymptomatic carriers by gene sequencing not only reduces the need for close clinical follow-up for at risk carriers but also ensures the earliest detection of disease.
The reported 22–35% occurrence6,7 of multiple synchronous tumors on presentation of putatively non-familial disease likely represents germline origin and suggests that the familial form is much more common than previously appreciated. A recent Scandinavian population study estimated a 10–30 fold increased incidence among first degree relatives of patients with SI carcinoid25.
Carcinoid tumors occur throughout the body as a result of the diffuse distribution of the precursor neuroendocrine derived EC cell. After the small intestine, the lung is the second most common site for sporadic carcinoid tumors and accounts for approximately 25% of carcinoids2. Pulmonary carcinoids originate from the embryonic foregut unlike SI-NET tumors that originate from the midgut. In addition to this difference in embryonic origin, the tumor development, secretory products, genetic mutations and molecular pathogenesis are also different26. There has been a single report of two families with pulmonary carcinoids27 and a single report of both pulmonary and SI-NET in a family with one member with a pulmonary carcinoid and another person with both a pulmonary and SI carcinoid as reported here for family 416. While the genetic basis has not been established for the family of patient 4:III-3 (Supplementary Fig. S1) who developed both a pulmonary and SI-NET, the presence of a single mutation in the IPMK gene giving rise to a pulmonary carcinoid (IPMK is well expressed in lung, Fig. 5) in patient 9:III-25 (Fig. 2 and Supplementary Fig. S1) and SI-NETs in his affected relatives indicates that a single gene mutation can initiate both types of tumors. Therefore, the pathogenesis of both types of tumors may be more similar than previously appreciated. The occurrence of pulmonary carcinoids also indicates the need for screening of pulmonary tumors in addition to SI-NET in families with carcinoid.
WES identified a single novel mutation within the linkage interval causing a 4 bp deletion in exon 6 of IPMK. The absence of other novel gene coding sequence mutations shared by all affected family members and the coincident high-level expression of IPMK consistent with tumor distribution along the SI lends additional support for IPMK as the causal gene.
Tumors expressed WT IPMK and IPMK LOH was not observed in tumors by sequencing and SNP based CNV analysis. The heterozygous IPMK in patient’s lymphoblasts and transfected HEK293 cells caused only partial loss of protein expression and function (Figs. 4 and 6). Together, these findings indicate IPMK haplo-insufficiency as a likely basis for IPMK diminished function. The 4 bp deletion leading to the loss of a functional NLS and early protein truncation with loss of the ATP binding site decreased IPMK nuclear localization and kinase activity (respectively), diminished p53 activation/apoptosis and increased survival under genotoxic stress conditions (Fig. 6). The decrease in nuclear localization (Fig. 6) is most likely the result of the mutation/truncation near the NLS20, 28 that would diminish p53 transcriptional co-activation of downstream effectors of apoptosis and cell cycle arrest22. This interaction between IPMK and p53 is consistent with the identification of IPMK in a loss of function screen for genes required for p53–dependent oncogene-induced senescence.29 It is also in line with the number of sporadic carcinoid tumors with either copy number variations or mutations observed along the p53 apoptotic pathway8 as well as the identification of recurrent haplo-insufficient cyclin-dependent kinase inhibitor 1B (CDKN1B) somatic mutations9 and germline mutations in several cyclin-dependent kinase inhibitors responsible for MEN4.30 Mutant p53 can behave as a haplo-insufficient tumor suppressor gene resulting in delayed onset of cancers31. This effect may be mimicked by the decreased interaction of haplo-insufficient IPMK with WT p53 resulting in the late onset of carcinoid tumors.
IPMK has additional non-catalytic activity stabilizing the mTORC1 complex32 and catalytic activity acting as a soluble inositol phosphate and lipid kinase33. Inositol polyphosphates regulate chromatin remodeling, 34,35 transcription,36,37 and mRNA export38 as does IPMK nuclear PI3 kinase activity19, 39,40,41. These varied catalytic and non-catalytic functions have been shown in vitro to regulate both proliferation32, 33, 42 and apoptosis43–46. Further investigation is needed to understand how these opposing actions are perturbed by the mutation in IPMK in this family and whether the balance of these actions contributes to carcinoid tumor initiation beyond the demonstrated effect of the mutation on p53 activation. (An expanded Discussion is in the Supplementary Appendix.)
In vivo mouse studies show that homozygous deletion of either exon 4 or exon 6 of Ipmk is embryonic lethal31,47. The truncated proteins from the exon-4 (~107 a.a.) and exon-6 (~197 a.a.) Ipmk hemizygous deleted mice do not have any catalytic activity, and would not be expected to have non-catalytic/physical interaction activity reported for WT Ipmk 22, 32,41. Mice with a hemizygous deletion of exon 4 have a normal phenotype for at least 1.5 years of study45. The phenotype of the longer Ipmk protein expected in heterozygous exon-6-deleted mice has not been reported33. The 4 bp deletion leading to a 333 a.a. truncated IPMK reported here for family 9 would be expected to retain some non-catalytic activity22, 32 and therefore, the lack of a disease phenotype for the Ipmk hemizygous mice would not be applicable. Furthermore, the late onset slow growth characteristics of carcinoid tumor make the adequacy of a 1.5 year observation in a mouse model questionable 47. It is conceivable that the reduced nuclear and increased cytosolic localization of the truncated 333 a.a. IPMK protein results not only in the demonstrated reduction in activation of p53 but also increased stabilization of mTOR/Raptor in the TORC1 complex32 leading to decreased apoptosis and increased proliferation, respectively. While this hypothesis needs experimental validation, sporadic carcinoid tumors demonstrate increased PI3K/AKT/mTOR signaling and are sensitive to mTOR inhibitory therapy with everolimus48–50.
The absence of IPMK mutations in 32 unrelated families underscores the genetic heterogeneity of this disease and is consistent with recent studies that found either no recurrent mutations among 48 sporadic carcinoid tumors analyzed by WES8, or only one gene (CDKN1B) with multiple somatic mutations in 8% of 180 patients analyzed by exome and genome-sequence analysis9. No somatic mutations have been reported in IPMK in sporadic carcinoid tumors. Interestingly, a search of the Catalog of Somatic Mutations in Cancer (COSMIC, http://www.sanger.ac.uk/cosmic) revealed 54 unique somatic variants in a variety of tumor types, including 6 indel or missense mutations in the part of IMPK near and immediately preceding the germline deletion we report (Figure S7). Among these somatic mutations, IPMK deletion mutations, 920_921delAG and 986delA, result in strikingly similar truncations of 337 and 335 a.a. compared to the 333 a.a. truncation reported here.
Hopefully, future exome sequencing and comparative germline variant analysis among remaining families with fewer affected members will be a successful strategy for the identification of additional susceptibility genes for familial SI-NET, so that screening can be limited to at-risk family members and new insights into the pathogenesis and treatment can be realized.
Supplementary Material
Acknowledgments
Grant Support:
This work was supported by the Intramural Research Programs of the National Institute of Diabetes, Digestive and Kidney Diseases, National Human Genome Research Institute, Clinical Research Center, National Cancer Institute and National Library of Medicine, National Institutes of Health.
We thank Elaine F. Remmers and Colleen Satorius for SNP genotyping; Terri Wakefield for protocol support; Richard Warner for family referral; and the subjects and their families for their cooperation and participation in this study.
Abbreviations used in this paper
- SI
small intestine
- IPMK
inositol polyphosphate multikinase
- WES
whole exome sequence
- NET
neuroendocrine tumor
- EC
enterochromaffin cell
- MEN1
multiple endocrine neoplasia type 1
- APC
adenomatous polyposis coli
- VHL
von Hippel-Lindau
- LOD
logarithm of the odds
- SNP
single nucleotide polymorphism
- GFP
green fluorescent protein
- TPH-1
tryptophan hydroxylase type 1
- RT-PCR
reverse- transcription polymerase chain reaction
Footnotes
Disclosures: The authors disclose no conflicts of interest.
Authorship:
Study concept and design: R.A, A.A.S., J.E.B-W., S.A.W.
Acquisition of data: Y.S., X.Z., J.F., A.T., M.V., G.J., J.F., M.W., M.S.J., U.L.H., M.S.H. and S.A.W.
Analysis and interpretation of data: Y.S., X.Z., J.F., J.F., S.J.M., A.M.V., S.C.C., M.R., M.M.Q., A.L., C.C.C., R.M.L., R.A., A.A.S., S.S., Q.L., M.S.H., J.E.B-W. and S.A.W.
Manuscript drafting: Y.S., X.Z., R.A., A.A.S., S.S., J.E.B-W and S.A.W.
Accessible via the online version of Gastroenterology at www.gastrojournal.org.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Strosberg J. Neuroendocrine tumours of the small intestine. Best Pract Res Clin Gastroenterol. 2012;26:755–73. doi: 10.1016/j.bpg.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Yao JC, Hassan M, Phan A, et al. One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol. 2008;26:3063–72. doi: 10.1200/JCO.2007.15.4377. [DOI] [PubMed] [Google Scholar]
- 3.Kulke MH, Mayer RJ. Carcinoid tumors. N Engl J Med. 1999;340:858–68. doi: 10.1056/NEJM199903183401107. [DOI] [PubMed] [Google Scholar]
- 4.Soga J. The term “carcinoid” is a misnomer: the evidence based on local invasion. J Exp Clin Cancer Res. 2009;28:15. doi: 10.1186/1756-9966-28-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vinik AI, Silva MP, Woltering EA, et al. Biochemical testing for neuroendocrine tumors. Pancreas. 2009;38:876–89. doi: 10.1097/MPA.0b013e3181bc0e77. [DOI] [PubMed] [Google Scholar]
- 6.Strosberg JR, Weber JM, Feldman M, et al. Prognostic validity of the american joint committee on cancer staging classification for midgut neuroendocrine tumors. J Clin Oncol. 2013;31:420–5. doi: 10.1200/JCO.2012.44.5924. [DOI] [PubMed] [Google Scholar]
- 7.Moertel CG. Karnofsky memorial lecture. An odyssey in the land of small tumors. J Clin Oncol. 1987;5:1502–22. doi: 10.1200/JCO.1987.5.10.1502. [DOI] [PubMed] [Google Scholar]
- 8.Banck MS, Kanwar R, Kulkarni AA, et al. The genomic landscape of small intestine neuroendocrine tumors. J Clin Invest. 2013;123:2502–8. doi: 10.1172/JCI67963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Francis JM, Kiezun A, Ramos AH, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013;45:1483–6. doi: 10.1038/ng.2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wale RJ, Williams JA, Beeley AH, et al. Familial occurrence in carcinoid tumours. Aust N Z J Surg. 1983;53:325–8. doi: 10.1111/j.1445-2197.1983.tb02456.x. [DOI] [PubMed] [Google Scholar]
- 11.Eschbach JWaR, Joseph A., Jr Metastatic Carcinoid, A Familial Occurrence. Annals of Internal Medicine. 1962;57:647–650. [Google Scholar]
- 12.Moertel CG, Dockerty MB. Familial occurrence of metastasizing carcinoid tumors. Ann Intern Med. 1973;78:389–90. doi: 10.7326/0003-4819-78-3-389. [DOI] [PubMed] [Google Scholar]
- 13.Pal T, Liede A, Mitchell M, et al. Intestinal carcinoid tumours in a father and daughter. Can J Gastroenterol. 2001;15:405–9. doi: 10.1155/2001/908056. [DOI] [PubMed] [Google Scholar]
- 14.Lengyel G, Karteszi M, Vallent K, et al. Familial occurrence of carcinoid of the small intestine. Orv Hetil. 1989;130:573–5. [PubMed] [Google Scholar]
- 15.Kinova S, Duris I, Kovacova E, et al. Malignant carcinoid in two brothers. Bratisl Lek Listy. 2001;102:231–4. [PubMed] [Google Scholar]
- 16.Hemminki K, Li X. Familial carcinoid tumors and subsequent cancers: a nation-wide epidemiologic study from Sweden. Int J Cancer. 2001;94:444–8. doi: 10.1002/ijc.1473. [DOI] [PubMed] [Google Scholar]
- 17.Saiardi A, Nagata E, Luo HR, et al. Mammalian inositol polyphosphate multikinase synthesizes inositol 1,4,5-trisphosphate and an inositol pyrophosphate. Proc Natl Acad Sci U S A. 2001;98:2306–11. doi: 10.1073/pnas.041614598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chang SC, Miller AL, Feng Y, et al. The human homolog of the rat inositol phosphate multikinase is an inositol 1,3,4,6-tetrakisphosphate 5-kinase. J Biol Chem. 2002;277:43836–43. doi: 10.1074/jbc.M206134200. [DOI] [PubMed] [Google Scholar]
- 19.Resnick AC, Snowman AM, Kang BN, et al. Inositol polyphosphate multikinase is a nuclear PI3-kinase with transcriptional regulatory activity. Proc Natl Acad Sci U S A. 2005;102:12783–8. doi: 10.1073/pnas.0506184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nalaskowski MM, Deschermeier C, Fanick W, et al. The human homologue of yeast ArgRIII protein is an inositol phosphate multikinase with predominantly nuclear localization. Biochem J. 2002;366:549–56. doi: 10.1042/BJ20020327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kublun I, Ehm P, Brehm MA, et al. Efficacious inhibition of Importin alpha/beta-mediated nuclear import of human inositol phosphate multikinase. Biochimie. 2014;102:117–23. doi: 10.1016/j.biochi.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Xu R, Sen N, Paul BD, et al. Inositol Polyphosphate Multikinase Is a Coactivator of p53-Mediated Transcription and Cell Death. Sci Signal. 2013;6:ra22. doi: 10.1126/scisignal.2003405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cunningham JL, Diaz de Stahl T, Sjoblom T, et al. Common pathogenetic mechanism involving human chromosome 18 in familial and sporadic ileal carcinoid tumors. Genes Chromosomes Cancer. 2011;50:82–94. doi: 10.1002/gcc.20834. [DOI] [PubMed] [Google Scholar]
- 24.Jarhult J, Landerholm K, Falkmer S, et al. First report on metastasizing small bowel carcinoids in first-degree relatives in three generations. Neuroendocrinology. 2010;91:318–23. doi: 10.1159/000299790. [DOI] [PubMed] [Google Scholar]
- 25.Kharazmi E, Pukkala E, Sundquist K, et al. Familial risk of small intestinal carcinoid and adenocarcinoma. Clin Gastroenterol Hepatol. 2013;11:944–9. doi: 10.1016/j.cgh.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 26.Leotlela PD, Jauch A, Holtgreve-Grez H, et al. Genetics of neuroendocrine and carcinoid tumours. Endocr Relat Cancer. 2003;10:437–50. doi: 10.1677/erc.0.0100437. [DOI] [PubMed] [Google Scholar]
- 27.Oliveira AM, Tazelaar HD, Wentzlaff KA, et al. Familial pulmonary carcinoid tumors. Cancer. 2001;91:2104–9. doi: 10.1002/1097-0142(20010601)91:11<2104::aid-cncr1238>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 28.Meyer R, Nalaskowski MM, Ehm P, et al. Nucleocytoplasmic shuttling of human inositol phosphate multikinase is influenced by CK2 phosphorylation. Biol Chem. 2012;393:149–60. doi: 10.1515/hsz-2011-0209. [DOI] [PubMed] [Google Scholar]
- 29.Drost J, Mantovani F, Tocco F, et al. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol. 2010;12:380–9. doi: 10.1038/ncb2038. [DOI] [PubMed] [Google Scholar]
- 30.Agarwal SK, Mateo CM, Marx SJ. Rare germline mutations in cyclin-dependent kinase inhibitor genes in multiple endocrine neoplasia type 1 and related states. J Clin Endocrinol Metab. 2009;94:1826–34. doi: 10.1210/jc.2008-2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger AH, Pandolfi PP. Haplo-insufficiency: a driving force in cancer. J Pathol. 2011;223:137–46. doi: 10.1002/path.2800. [DOI] [PubMed] [Google Scholar]
- 32.Kim S, Kim SF, Maag D, et al. Amino acid signaling to mTOR mediated by inositol polyphosphate multikinase. Cell Metab. 2011;13:215–21. doi: 10.1016/j.cmet.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maag D, Maxwell MJ, Hardesty DA, et al. Inositol polyphosphate multikinase is a physiologic PI3-kinase that activates Akt/PKB. Proc Natl Acad Sci U S A. 2011;108:1391–6. doi: 10.1073/pnas.1017831108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Steger DJ, Haswell ES, Miller AL, et al. Regulation of chromatin remodeling by inositol polyphosphates. Science. 2003;299:114–6. doi: 10.1126/science.1078062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen X, Xiao H, Ranallo R, et al. Modulation of ATP-dependent chromatin-remodeling complexes by inositol polyphosphates. Science. 2003;299:112–4. doi: 10.1126/science.1078068. [DOI] [PubMed] [Google Scholar]
- 36.Odom AR, Stahlberg A, Wente SR, et al. A role for nuclear inositol 1,4,5-trisphosphate kinase in transcriptional control. Science. 2000;287:2026–9. doi: 10.1126/science.287.5460.2026. [DOI] [PubMed] [Google Scholar]
- 37.Watson PJ, Fairall L, Santos GM, et al. Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature. 2012;481:335–40. doi: 10.1038/nature10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.York JD, Odom AR, Murphy R, et al. A phospholipase C-dependent inositol polyphosphate kinase pathway required for efficient messenger RNA export. Science. 1999;285:96–100. doi: 10.1126/science.285.5424.96. [DOI] [PubMed] [Google Scholar]
- 39.Blind RD, Suzawa M, Ingraham HA. Direct modification and activation of a nuclear receptor-PIP(2) complex by the inositol lipid kinase IPMK. Sci Signal. 2012;5:ra44. doi: 10.1126/scisignal.2003111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bashamboo A, Ferraz-de-Souza B, Lourenco D, et al. Human male infertility associated with mutations in NR5A1 encoding steroidogenic factor 1. Am J Hum Genet. 2010;87:505–12. doi: 10.1016/j.ajhg.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wickramasinghe VO, Savill JM, Chavali S, et al. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol Cell. 2013;51:737–50. doi: 10.1016/j.molcel.2013.08.031. [DOI] [PubMed] [Google Scholar]
- 42.Wang Y, Wang HY. Dvl3 translocates IPMK to the cell membrane in response to Wnt. Cell Signal. 2012;24:2389–95. doi: 10.1016/j.cellsig.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu R, Snyder SH. Gene transcription by p53 requires inositol polyphosphate multikinase as a co-activator. Cell Cycle. 2013;12:1819–20. doi: 10.4161/cc.25119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koldobskiy MA, Chakraborty A, Werner JK, Jr, et al. p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A. 2010;107:20947–51. doi: 10.1073/pnas.1015671107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maffucci T, Piccolo E, Cumashi A, et al. Inhibition of the phosphatidylinositol 3-kinase/Akt pathway by inositol pentakisphosphate results in antiangiogenic and antitumor effects. Cancer Res. 2005;65:8339–49. doi: 10.1158/0008-5472.CAN-05-0121. [DOI] [PubMed] [Google Scholar]
- 46.Piccolo E, Vignati S, Maffucci T, et al. Inositol pentakisphosphate promotes apoptosis through the PI 3-K/Akt pathway. Oncogene. 2004;23:1754–65. doi: 10.1038/sj.onc.1207296. [DOI] [PubMed] [Google Scholar]
- 47.Frederick JP, Mattiske D, Wofford JA, et al. An essential role for an inositol polyphosphate multikinase, Ipk2, in mouse embryogenesis and second messenger production. Proc Natl Acad Sci U S A. 2005;102:8454–9. doi: 10.1073/pnas.0503706102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shah T, Hochhauser D, Frow R, et al. Epidermal growth factor receptor expression and activation in neuroendocrine tumours. J Neuroendocrinol. 2006;18:355–60. doi: 10.1111/j.1365-2826.2006.01425.x. [DOI] [PubMed] [Google Scholar]
- 49.Yao JC, Phan AT, Chang DZ, et al. Efficacy of RAD001 (everolimus) and octreotide LAR in advanced low- to intermediate-grade neuroendocrine tumors: results of a phase II study. J Clin Oncol. 2008;26:4311–8. doi: 10.1200/JCO.2008.16.7858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pavel ME, Hainsworth JD, Baudin E, et al. Everolimus plus octreotide long-acting repeatable for the treatment of advanced neuroendocrine tumours associated with carcinoid syndrome (RADIANT-2): a randomised, placebo-controlled, phase 3 study. Lancet. 2011;378:2005–12. doi: 10.1016/S0140-6736(11)61742-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.