

Abstract
New nonnucleoside analogs are being developed as part of a multi-drug regimen to treat hepatitis C viral infections. Particularly promising are inhibitors that bind to the surface of the thumb domain of the viral RNA-dependent RNA polymerase (NS5B). Numerous crystal structures have been solved showing small molecule non-nucleoside inhibitors bound to the hepatitis C viral polymerase, but these structures alone do not define the mechanism of inhibition. Our prior kinetic analysis showed that nonnucleoside inhibitors binding to thumb site-2 (NNI2) do not block initiation or elongation of RNA synthesis; rather, they block the transition from the initiation to elongation, which is thought to proceed with significant structural rearrangement of the enzyme-RNA complex. Here we have mapped the effect of three NNI2 inhibitors on the conformational dynamics of the enzyme using hydrogen/deuterium exchange kinetics. All three inhibitors rigidify an extensive allosteric network extending >40 Å from the binding site, thus providing a structural rationale for the observed disruption of the transition from distributive initiation to processive elongation. The two more potent inhibitors also suppress slow cooperative unfolding in the fingers extension-thumb interface and primer grip, which may contribute their stronger inhibition. These results establish that NNI2 inhibitors act through long range allosteric effects, reveal important conformational changes underlying normal polymerase function, and point the way to the design of more effective allosteric inhibitors that exploit this new information.
Keywords: allosteric regulation, hepatitis C virus (HCV), hydrogen exchange mass spectrometry, inhibition mechanism, nucleic acid enzymology
Introduction
Chronic HCV3 infections lead to life-threatening liver diseases including fibrosis, cirrhosis, and hepatocellular carcinoma, and since 2007, HCV has been responsible for more deaths in the United States per year than HIV (1). Multiple HCV proteins are targets for small molecule inhibitors, most notably the RNA-dependent RNA polymerase, termed nonstructural protein 5B (NS5B). Recently, new therapies based upon combined nucleoside analogs and nonnucleoside inhibitors acting directly against the viral replication have dramatically improved the prognosis of treatment while eliciting fewer side effects (2) compared with treatments with interferon. However, the biochemical mechanisms of action of these inhibitors are not fully understood.
Like other polymerases, NS5B consists of “fingers,” “palm,” and “thumb” subdomains (Fig. 1) whose organization can be likened to a closed right hand (3–6). Two unique features of NS5B are an extension of the fingers domain that reaches over the active site to contact the thumb domain to enclose the active site and a β-loop that projects from the thumb domain into the active site to block the binding of duplex RNA (Fig. 1). Current models suggest that the β-loop may be required for de novo initiation but then must swing out of the active site in the transition from initiation to elongation mode (7, 8). NS5B also has a C-terminal linker containing a membrane-anchoring domain (Fig. 1), and although membrane association is required for replication in vivo, it is not required for biochemical activity. In this study we use the truncated Δ21 construct lacking the membrane anchor. However, the remaining C-terminal peptide (beyond residue ∼550) occludes the active site in the apoenzyme but is missing in the structure of the initiation complex (7), so its role in initiation and elongation is unknown.
FIGURE 1.

NS5B RNA-dependent RNA polymerase in the apo form (top, 1C2P.pdb) and in the initiation complex (bottom, 4WTI.pdb). NS5B is shown in the top view (looking down into the RNA binding cleft). The architectural subdomains of the classic open right hand conformation are colored, respectively, blue, red, and green for the fingers, the palm, and the thumb. The fingers extensions and C-terminal tail are colored in cyan and gray, respectively. The β-loop is colored in yellow. Filibuvir, a NNI2 was placed at the NNI2 binding site through alignment with Filibuvir bound structure 3FRZ.pdb and represented as magenta sticks. In the initiation complex (bottom), the dinucleotide-primed RNA template is shown as spheres.
NS5B is targeted by two major classes of inhibitors; nucleoside analog inhibitors that are incorporated into the newly synthesized RNA to act as chain terminators and nonnucleoside inhibitors (NNIs) that allosterically inhibit enzyme activity, analogous to the inhibition of HIV (9). Of particular interest here are the thumb site 2 inhibitors that bind to the surface of the thumb domain (NNI2). Crystal structures of NS5B bound to NNIs have been solved (10), but the structures fail to reveal any significant changes in the enzyme active site, and little is known about their mechanisms of action or the identity of steps that are inhibited.
Delineating NNI inhibition mechanisms is complicated by the complex replication cycle of NS5B. During de novo initiation, NS5B binds to the 3′-UTR of the single-stranded RNA template and catalyzes the linkage of the first two complementary nucleotides to form a dinucleotide that will act as the primer for subsequent addition. This process is very slow and inefficient in vitro and is followed by several rounds of additional nucleotide incorporation with significant accumulation of abortive intermediates. After incorporation of ∼4–6 nucleotides the polymerase undergoes a transition to rapid processive elongation (8, 11). NNIs might in principle impact any one or more of these various stages of RNA synthesis (namely, initiation, transition, or elongation), and it is important to understand which steps are inhibited and how an allosteric effector binding to the surface of the enzyme can attenuate reactions at the active site.
In our recent work we showed that NNI2s inhibit replication by blocking the transition from initiation to the elongation mode (11), which is thought to proceed with a substantial rearrangement of the enzyme involving, at the very least, the movement of the β-loop out of the active site. Here we complement these studies using hydrogen/deuterium exchange monitored by mass spectrometry (HDX-MS). For proteins the size of NS5B, HDX-MS provides a useful approach for probing conformational dynamics and perturbation by ligands. Proteins for which HDX-MS has revealed the dynamical basis of allostery include nuclear receptors (12), G protein-coupled receptors (13), the viral polymerase HIV-1 reverse transcriptase (14), and many others (15–19).
In our initial HDX-MS studies on NS5B reported here we document the fundamental exchange dynamics observed in the apoenzyme and the changes in dynamics after binding NNI2 to the surface of the thumb domain. These data provide direct evidence for long range effects of inhibitor binding propagating from the binding site to domains throughout the protein. By correlating the solution dynamics of individual peptides observed by HDX-MS with the inhibition of specific steps of the replication cycle in kinetic studies, we provide new insights into the molecular basis of inhibition. Moreover, these studies provide some information to reveal the enzyme conformational dynamics underlying RNA replication.
Experimental Procedures
Nucleic Acids, Chemicals, and Protein
Filibuvir and GS-9669 were kindly provided by Gilead Sciences. Lomibuvir (VX222) used in HD exchange was purchased from Selleckchem Chemicals. Optima (LC/MS) grade water and acetonitrile were obtained from Fisher, and formic acid was obtained from Sigma. D2O, DCl, and NaOD were obtained from Cambridge Isotopes. N-terminal penta-His-NS5BΔ21 (con1 strain, GT1b with a 21-amino acid deletion at the C terminus) was cloned, expressed, and purified as described previously (8) and dialyzed into storage buffer (50 mm Tris-HCl, pH 7, 400 mm NaCl, 2 mm DTT, 10% glycerol).
HD Exchange Kinetics
The buffered solution containing NS5B was exchanged into elongation buffer (40 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm DTT, 2 mm MgCl2) using Amicon ultracentrifugal filters to achieve a final enzyme concentration of ∼20 μm. The coverage map for NS5B was obtained from undeuterated controls as follows: 5 μl of NS5B in elongation buffer was diluted with 45 μl of elongation buffer at room temperature followed with the addition of 200 μl of ice-cold quench (100 mm phosphate buffer, 1.5 m guanidine-HCl, pH 2.4). The samples were immediately injected into a Waters HDX nanoAcquity UPLC (Waters, Milford, MA) with in-line pepsin digestion (porozyme immobilized pepsin cartridge from Applied Biosystems). Peptic fragments were trapped on an Acquity UPLC BEH C18 peptide trap and separated on an Acquity UPLC BEH C18 column. A 7-min, 5% to 35% acetonitrile (0.1% formic acid) gradient was used to elute peptides directly into a Waters Synapt G2 mass spectrometer (Waters). MSE data were acquired with a 20–30 V ramp trap collision energy for high energy acquisition of product ions as well as continuous lock mass (Leu-enkephalin) for mass accuracy correction. Peptides were identified using the ProteinLynx Global Server 2.5.1 (PLGS) from Waters using the following settings: peptide tolerance 10 ppm, fragment tolerance 20 ppm, and a limitation on sizes of peptide lower than 4 residues used. The most stringent constraints of 0.3 fragments per residues was applied in DynamX.
NNI2s were added to the desired concentration using 100× stock in 100% DMSO. Apoenzyme control samples contained 1% DMSO (v/v). HD exchange reactions were performed as follows. 5 μl of 20 μm NS5B in elongation buffer containing 1% DMSO with or without 100 μm NNI2 was incubated in 45 μl of elongation buffer, 99.99% D2O, pD 7.4, with 1% DMSO complemented with or without 100 μm of corresponding NNI2. All reactions were performed at 25 °C. Before injection, deuteration reactions were quenched at various times (10 s, 1 min, 10 min, 1 h, and 2 h) with 200 μl of 100 mm phosphate buffer, 1.5 guanidine-HCl, pH 2.4, for Lomibuvir samples and its corresponding apo samples. Back exchange correction was performed against fully deuterated controls acquired by incubating 5 μl of NS5B in 45 μl of elongation buffer, 99.99 D2O, pD 7.4, containing 5.2 m deuterated guanidine DCl for 2 h at 25 °C before quenching (without guanidine HCl) and injection. To remedy an excessive back exchange observed with and without Lomibuvir samples, the peptide trap was removed from the trap valve and placed on the floor of HDX manager, and quenching was changed to 100 μl of 100 mm phosphate buffer, 2 m guanidine-HCl, pH 2.4, with and without Filibuvir, GS-9669 samples. As a consequence, the observed back exchange was reduced by ∼20%, and the average standard deviation over all peptic fragments and time points was reduced by ∼25%. All deuteration time points and controls were acquired in triplicates.
The deuterium uptake by the identified peptic fragments through increasing deuteration time and for the fully deuterated control was determined using Water's DynamX 2.0 software. The normalized percentage of deuterium uptake (% D) at an incubation time t for a given peptide was calculated as,
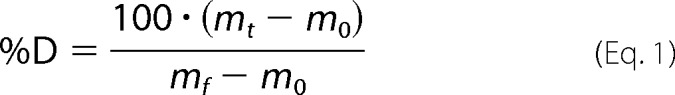 |
where mt is the centroid mass at incubation time t, and m0 is the centroid mass of the undeuterated control and the centroid mass of the fully deuterated control. The percent deuteration difference plots Δ% D (Apo-Holo) were generated using the percent deuteration calculated. Confidence intervals for the Δ% D of any individual time point were determined using the method outlined by Houde et al. (20) adjusted to percent deuteration using the fully deuterated controls. A 98% confidence interval of ±6.47% D for any single time point was determined for Lomibuvir-bound and the corresponding apoenzyme acquisitions. For GS-9669-bound, Filibuvir-bound, and their corresponding apoenzyme acquisitions, a 98% confidence interval of ± 4.91% D for any single time point. Confidence intervals (98%) were plotted on the Δ% D plots as horizontal dashed lines. EX1 type cooperative unfolding was analyzed using HX-Express2 (21).
Results
A companion paper (11) showed that NNI2s inhibit RNA replication by slowing the transition from initiation to elongation, implying long range structural effects of NNI2s on enzyme dynamics. To gain insights into the underlying molecular basis for this allosteric inhibition, we used HDX kinetics to quantify changes in enzyme dynamics in the absence and presence of three different NNI2s.
HDX Kinetics of ApoNS5B
We successfully identified 146 peptides covering 96.5% of the sequence. A representative subset of peptides (Fig. 2C) with nearly complete coverage was used to map the percent deuterium incorporation (% D) for each time point onto the NS5B structure. In Fig. 2A, the % D is color-coded from blue for highly protected to red for very labile peptic fragments. The overall HD exchange profile reveals highly protected regions of the fingers and palm. This region is characterized by protection from exchange throughout the entire deuterium incubation time range probed (10 s to 2 h) and spans buried stretches of helices N, M, C, H, K, L, G, F and E according the secondary structure naming by Bressanelli et al. (5). This highly protected scaffold parallels the overall HD exchange behavior observed for HIV-1 RT (22) pointing to potential similarities in structural and conformational dynamics design across related A-family polymerases. Another distinct yet smaller region located in the thumb is also largely protected from exchange throughout the entire deuterium incubation time range. This region is composed of buried of portions of helices Q, P, and R and parts of β-strands 17 and 18 that form the β-loop. Together, these two regions constitute the stable core of NS5B as probed by HD exchange.
FIGURE 2.

Percent deuterium incorporation of apoNS5B mapped on the structure of the NS5B polymerase (using 3FQL.pdb; Ref. 38) at each D2O incubation time point. A, the top row of structures displays the front view, the bottom row displays the top view, and the incubation time is indicated across the top. Percent deuterium incorporation is mapped according to the color gradient indicated in the legend. B, normalized deuterium uptake curves for selected peptides illustrating highly protected, unprotected, and moderately protected regions. C, representative peptides selected for the display of % D on the structures in A. In a particular peptide, overlapping regions that were omitted from representation are white. N- and C-terminal regions missing electron density in the crystal structure were also white.
In contrast, HD exchange also reveals three highly labile regions. The C-terminal tail, parts of the fingers extension loop, and parts of the NNI2 binding site show significant exchange as early as 10 s. The mostly unstructured C-terminal tail, which folds back into the RNA binding channel, is highly exchangeable, although very mild protection is observed early (10 s) at the very C-terminal end. Similarly, the Δ1 and Δ2 fingers extension loops, including the thumb-inserted αA (NNI1 and GTP binding site), are largely exchanged at 10 s with the notable exception of β-sheet-forming strands β2 and β5 that are protected from exchange for up to 10 min. Finally, a portion of αT (residues 496–511), which forms part of the NNI2 binding site, displays significant exchange at 10 s. Interestingly, this stretch spans the middle of αT, suggesting a very dynamic nature for this α-helix. With the exception of αT at the NNI2 binding site, most of these high exchange regions are somewhat unstructured and/or solvent-exposed, reflecting a possible need for conformational flexibility for efficient catalytic function.
Effects of NNI2 on Enzyme Dynamics
Fig. 3 shows the differences in % D (Δ% D) between apoNS5B or NNI2-bound NS5B (Fig. 3A, GS-9669; Fig. 3B, Lomibuvir; Fig. 3C, Filibuvir) for all peptic fragments. Individual peptides are plotted on the x axis, ordered N- to C-terminal based on the sequence number of the first residue of the peptide. The horizontal bar on top of Fig. 3A is color-coded according to subdomains as in Fig. 1, and the numbers mark the residue numbers delineating each subdomain. The Δ% D values are displayed for each peptic fragment and color-coded to indicate the time of incubation (see the Fig. 3 legend). The 98% confidence interval in Δ% D for any individual D2O incubation time point was determined based on the method outlined by Houde et al. (20), and plotted as horizontal dashed lines.
FIGURE 3.

Differences in percent deuterium incorporation of apo versus holo NS5B for NNI2 inhibitors (A, GS-9669; B, Lomibuvir; C, Filibuvir). Peptic fragments are displayed N- to C-terminal from left to right based on the residue number of the first amino acid of the peptide. The difference in % D between two states (Δ% D) is plotted for each peptic fragment for each incubation time point probed (color-coded according to incubation time). 98% confidence intervals are displayed as horizontal dashed lines. The horizontal bar on top of A corresponds to the subdomains of NS5B (blue, fingers; cyan, fingers extension; red, palm; green, thumb; yellow, β-loop; gray, C-terminal tail). The asterisks (*) denote peptides containing residues involved in inhibitor binding (His-475, Ser-476, and Tyr-477) and showing protection at the earliest time points. The plus symbol (+) denotes the peptide containing Trp-528.
Local Effects at the NNI2 Binding Site
As expected, the NNI2 binding site (Fig. 3, motif XII) displays some of the biggest Δ% D. All three NNI2s induce largely similar deuterium exchange protection patterns in the immediate vicinity of the binding site (Fig. 4), where the subdomains colored in blue show a more rigid structure and those in red show more flexibility after binding the inhibitor. As illustrated in Fig. 4, these local effects on HDX are in agreement with the binding sites defined by crystallography (23–25) or predicted by MD simulations (26). The HD exchange protection observed at the earliest incubation time (10 s, red curve in Fig. 3) is consistent with the burial and/or H-bond formation of exchangeable amide hydrogens around residues His-475, Ser-476, and Tyr-477 (asterisks in Fig. 3). Additional known inhibitor contact residues Leu-497, Arg-501, and Trp-528 are also shown in Fig. 4.
FIGURE 4.

NNI2 binding results in protection from HD exchange at the binding site. Significant differences in HD exchange upon NNI2 binding at the binding site were mapped on the structure of NS5B polymerase (3FQL.pdb). Peptic regions showing significant protection from HD exchange upon NNI2 binding are displayed in blue. Dark blue-colored regions are affected significantly by all three NNI2s. Regions showing increased HD exchange are displayed in pink. Peptic regions with effects that are not common to all three inhibitors were represented in surface rendition. Inhibitors (magenta sticks) were positioned through alignment with corresponding available structure (4EO6.pdb for GS-9669 and Lomibuvir, 3FRZ.pdb for Filibuvir).
Although our results indicate all three NNI2s induce a change in the backbone conformational flexibility around Trp-528, Filibuvir induces a markedly more pronounced effect. Indeed, the region surrounding Trp-528 is the only instance where Filibuvir resulted in a greater magnitude of Δ% D than Lomibuvir and GS-9669 (+ in Fig. 3, motif XII). Whereas Lomibuvir and GS-9669 resulted in significant Δ% D at long incubation time points for peptide 525–528, Filibuvir resulted not only in a greater magnitude Δ% D for the same stretch but also a significant decrease in exchange at 10 s in the adjacent peptide 528–534. This could be explained by the observation that Filibuvir is much more extended toward the C-terminal end of αU than Lomibuvir and GS-9669 based on a structural alignment with 4EO6 (Fig. 4). This difference in contacts may be responsible for the greater stabilization in the vicinity of the binding site seen for Filibuvir.
Throughout the protein there is only one instance of a statistically significant increase in deuterium exchange upon NNI2 binding, and this was observed only for thiophene derivatives Lomibuvir and GS-9669. This was revealed as the negative Δ% D in Fig. 3, motif XII, and is illustrated in Fig. 4 (pink) as the loop connecting αS and αT.
Long Range Effects of NNI2s on Enzyme Dynamics
Of greater interest are the long range effects, which we define as residues that are beyond the first or second shell of amino acids surrounding the inhibitor. Fig. 3 clearly shows that binding of each of the three NNI2s principally results in slowing the rate of deuterium exchange throughout the protein, not only at the inhibitor binding site but at distant sites as well. Indeed, peptides in the fingers, fingers extensions, palm, thumb, β-loop, and C-terminal tail display significant decreases in deuterium incorporation. In some cases the loss in % D in distant regions is comparable if not greater than at the inhibitor binding site (Fig. 3). This extended loss in deuterium uptake defines a large network of residues rigidified upon NNI2 binding.
As summarized in Fig. 5, there is an extensive overlap in the regions rigidified by the three NNI2 tested. The sequences displaying common effects across all three NNI2s have been highlighted in dark blue in Fig. 5A and mapped onto the structure of NS5B in Fig. 5B. This common allosteric network is characterized by the loss in conformational flexibility in the fingers, fingers extension loop, parts of the thumb, and the NNI2 binding site.
FIGURE 5.

Common local and long range effects on deuterium uptake induced by all three NNI2. A, differences in percent deuterium incorporation of apoversus holo NS5B for all three NNI2 inhibitors mapped on the sequence of NS5B. Peptic regions displaying significant Δ% D at any D2O incubation time between apo and each NNI2 were mapped on horizontal bars below the sequence according to the legend. Regions mapped with significant Δ% D were resolved for overlap. Effects on percent deuteration that are common to all three NNI2s, defining a common allosteric network, were highlighted on the sequence in dark blue. Effects that are restricted to one or two NNI2s only are highlighted in cyan. Subdomains were delineated above the sequence and color-coded according to Fig. 1. Notable secondary structure elements were named using the naming from Bressanelli et al. (5). B, allosteric network common to all three NNI2. Peptic fragments displaying significant Δ% D at any incubation time point for all three NNI2 were mapped on NS5B (3FQL.pdb) using a surface rendering and colored dark blue as in A. Dinucleotide primed template (orange and yellow spheres) and representative inhibitor (magenta sticks) were positioned through alignment with appropriate structure, 4WTK.pdb for the dinucleotide primed template and 3FRZ.pdb for the inhibitor (Filibuvir).
Away from the NNI2 binding site, the network extends through the fingers extensions to the fingers subdomain across the RNA binding cleft (Fig. 5B). NNI2 binding induces a loss in conformational flexibility in both Δ1 (motif I in Fig. 3) and Δ2 (motif IV in Fig. 3) fingers extension loops and a significant allosteric suppression of conformational flexibility in the fingers, including in the active site area (motif II, V, VI, and VII in Fig. 3) and at the αD-αE loop (motif III in Fig. 3) at the tip of the fingers atop the RNA binding cleft. These distant effects on deuterium incorporation are characterized by significant Δ% Ds at later deuterium incubation times (yellow, blue, and purple curves in Fig. 3), indicating that NNI2 binding results in the restriction of conformational fluctuations that lead to exposure of amide hydrogen.
Losses in Conformational Dynamics upon Binding NNI2s Is Correlated with Inhibitor Potency
Although we have observed and mapped common allosteric effects among these three NNI2s, there are also some noticeable differences between the various NNI2s tested. Initial examination of Fig. 3 clearly shows that the magnitude in Δ% Ds can be ranked in the order Lomibuvir > GS-9669 > Filibuvir. In addition, further examination reveals peptides displaying significant Δ% D specific to each NNI2 not common to all three NNI2s. These differences are mapped onto the structure in Fig. 6 with a surface rendition.
FIGURE 6.

NNI2s results in common as well as differential effects on the HD exchange protection patterns away from the binding site. Significant Δ% Ds upon NNI2 binding were mapped on the structure of NS5B polymerase (3FQL.pdb) and visualized from two different perspectives (top view and front view). Inlets show the RNA binding cleft with Δ% D mapped for each inhibitors (top, GS-9669; middle, Lomibuvir; bottom, Filibuvir). Peptic regions showing significant protection from HD exchange upon NNI2 binding are displayed in blue. Dark blue-colored regions are affected significantly by all three NNI2s. Regions showing increased HD exchange are displayed in pink. Peptic regions with effects that are not common to all three inhibitors were represented in surface rendition. Inhibitors (magenta sticks) were positioned through alignment with corresponding available structure (4EO6.pdb for GS-9669 and Lomibuvir and 3FRZ.pdb for Filibuvir) and the dinucleotide-primed (yellow spheres) template (orange spheres) was positioned through alignment with 1WTK.pdb.
Our results highlight a marked chemotype-dependent differential effect on the HD exchange behavior; Lomibuvir and GS-9669 induce significantly stronger allosteric restrictions of conformational dynamics in the front end of RNA binding cleft than Filibuvir. Most notably, the β-loop (motif XI in Fig. 3) displays losses in deuterium incorporation for Lomibuvir and GS-9669. This β-loop protrudes from the thumb into the RNA binding cleft, and its displacement from the active site is thought to be required for the transition to efficient elongation. Lomibuvir and GS9669 affect different areas of the β-loop. For Lomibuvir, it is the apex of the β-loop. For GS9669, the strands are more affected. Regardless, they stand on contrast of Filibuvir, which has no effects on the β-loop. In addition to the C-terminal tail and β-loop, helices of the fingers domain and palm and the tip of the thumb (loop αO-αP) are also differentially affected by the different NNI2 (Fig. 6). Interestingly, the distance between loop αD-αE (part of the common allosteric network) and αO-αP can characterize the open/closed state of NS5B; the distance between the side chains of residue His-95 and Pro-404 was ∼4 Å in 3FQL.pdb and ∼6 Å in the apo (1C2P.pdb). This distance increased to ∼16 Å in co-crystals with the dinucleotide primed-template, clearly showing the required displacement of αO-αP to accommodate the dinucleotide primer before the displacement of the β-loop for the elongation complex (7). For the most part, Lomibuvir and GS-9669 restricted the motions of these loops (αD-αE and αO-αP) along with the C-terminal tail and the β-loop, possibly indicating a stabilization of the closed state. In contrast, Filibuvir induces a much milder effect on this region with only the αD-αE loop and the αO-αP loops sitting atop of the β-loop displaying significant rigidification but not the β-loop/C-terminal tail motif (Fig. 6).
The binding of Lomibuvir or GS-9669, but not Filibuvir, resulted in significantly decreased deuterium uptake in residues 550–564 of the C-terminal tail (motif XIV in Figs. 3 and 6). This region includes Trp-550 and Phe-551 (Fig. 6), that are known to interact with the β-loop (27), and Asp-559, which is known to interact with the tip of the fingers (28). This Lomibuvir/GS-9669-induced Δ% D was observable at early incubation times (10 s, red curve motif XIV in Fig. 3), suggesting a small yet significant increase in secondary or tertiary structure stability upon inhibitor binding. This structural difference is accompanied by decreases in conformational flexibility in the vicinity of this C-terminal tail stretch for Lomibuvir and GS-9669 that are largely absent for Filibuvir.
Compared with Filibuvir, the additional restrictions of the conformational dynamics induced by thiophene-based Lomibuvir/GS-9669 outline a more extensive allosteric network that entirely encircles the RNA binding cleft. The order Lomibuvir > GS-9669 > Filibuvir, observed in ranking the magnitude of the effects on Δ% D, correlates with the potency of inhibition seen in kinetic studies (11).
Cooperative Unfolding of Δ2 Loop/Primer Grip
Detailed examination of the exchange kinetics also revealed the slow cooperative unfolding of the Δ2 loop/primer grip motif. This cooperative unfolding is denoted by the characteristic EX1 kinetics behavior displaying double envelope isotopic peak distributions (29) with the progressive disappearance of an exchange-protected envelope concomitant with appearance of an unprotected envelope. Fig. 7 shows two representative peptides, illustrating this cooperatively unfolding. Fig. 7, D and E, shows stacked spectra with the overall distribution along with each individual envelope. Fig. 7B localizes the peptic fragments involved in the cooperative unfolding on the structure. These regions span β4 and β5 of the Δ2 loop and C-terminal end of β16 into the N-terminal half of αO, which is the primer grip motif. Along with loop αS-αT and αA helix from the Δ1 fingers extension loop, they form part of the interface between the fingers extensions and the thumb with residues of β5 making contact with αO (Fig. 7B). The slow cooperative unfolding indicates that residues in the region undergo correlated motions that expose multiple amide hydrogens. The difference in the center of each isotopic envelope suggests that these motions involve ∼4–5 amide hydrogens in each peptide. Such a large conformational change may point to motions that lead to the unraveling of β5 and the N-terminal portion of αO. Fig. 7, D and E, show that, in the apo form, ∼50% sampling of unfolded state occurs after ∼10 min of incubation in deuterium for fragment 137–145 (Δ2 loop) and ∼1–2 h for fragment 384–397. These slow exchange kinetics indicate that the exchange-competent conformation in these regions is a rarely sampled state.
FIGURE 7.

Cooperative unfolding of Δ2 fingers extension loop and primer grip motif was decreased by thiophene-based Lomibuvir and GS-9669. Cooperative unfolding of peptic fragment 137–145 (right) and 384–397 (left) revealed by the characteristic EX1 double envelope isotopic distribution. Representative stacked spectra (D, for peptide 384–397; E, for peptide 137–145) at 0 s, 10 min, 1 h, 2 h, and fully deuterated (TD) are displayed for apo and holo NS5B (GS-9669, Lomibuvir and Filibuvir). The relative proportion of the unfolded envelope is plotted versus D2O incubation time for each peptic fragment (A, for peptide 384–397; C, for peptide 137–145) in the presence and absence of NNI2s. Peptic fragments are localized on the structure of NS5B (4WTC.pdb) containing an RNA duplex in the RNA binding channel (B) and colored according to the colors assigned in A and C. Divalent cations are displayed as black spheres and conserved arginines are displayed as sticks. The αS-αT loop is represented as a surface rendition.
The observed Δ2 loop/primer grip cooperative unfolding described in apoNS5B is also suppressed in a chemotype-dependent manner. Fig. 7, A and C, show the relative unfolding of Δ2 loop and primer grip motifs in the absence and presence of all three inhibitors. Lomibuvir greatly reduces the unfolding of both the Δ2 loop and the primer grip motif. Similarly, GS-9669 reduces the unfolding of both structural elements, albeit to a lesser extent for the primer grip motif. This behavior stands in contrast with Filibuvir, which does not seem to have an effect on these motifs.
Discussion
Recent advances in combination therapies have greatly improved the prognosis for treatment of HCV infections (2). However, as one component of the combination therapy, the mechanism of action of nonnucleoside inhibitors is poorly understood. Here we provide the first evidence to relate structure to the observed kinetic effects of thumb site 2 inhibitors using hydrogen/deuterium exchange monitored by mass spectral analysis of individual peptides. The NNI2 binding site is located on the surface of the enzyme away from the RNA binding channel, active site, and other functionally relevant motifs (β-loop, C-terminal tail, and fingers extension). Although there are numerous NNI-bound structures, a molecular mechanism of inhibition cannot be inferred from structure alone. Multiple studies have shown that binding of these NNI2s to NS5B is accompanied by a significant increase in Tm (27, 29, 30); however, an increase in Tm could be simply attributed to the stabilization of the folded state by the inhibitor binding energy and need not require long range conformational changes.
We performed these studies to characterize hydrogen/deuterium exchange dynamics using enzyme in the absence of RNA. Because of the initiation reaction is only ∼50% efficient (8, 11), HDX studies using a reaction mixture used in kinetic studies would be uninterruptable. Moreover, there are multiple states (initiation, transition, and elongation) that one would like to study individually. Our kinetic studies argue that RNA is stably bound only in the elongation complex, but perhaps of most interest would be the complex representing the transition from initiation to elongation, which may be formed only transiently. Indeed, to date there is only one crystal structure mimicking the initiation complex (7) and none of the elongation complex with an intact β-loop. In future studies we will work to isolate elongation complex in sufficient quantities and purity to perform HDX measurements, but that represents a significant technical challenge. Our current work addresses the fundamental properties of the apoenzyme and reveals significant long range effects of NNI2 on enzyme dynamics. Moreover, the magnitude of the effect of different NNI2s on exchange dynamics is correlated with their potency seen in kinetic analysis of inhibition (11), supporting our conclusion that studies on the free enzyme reflect fundamental dynamics that persist in the enzyme-RNA complex.
NNI2 binding induces significant reductions in deuterium uptake at both short and long labeling times. Reductions in exchange at the shortest labeling times (10 s) are generally attributed to the formation of new secondary structure or the burial of exchangeable amide hydrogens in an interface, whereas reductions in exchange at longer incubation times, such as minutes or hours, is attributed to reductions in the frequency of small or large scale “breathing motions” that transiently expose amide hydrogens for exchange (31). Persson and Halle (32) recently analyzed a millisecond-length molecular dynamics simulation of the protein BPTI and concluded that the locally “open” exchange competent states have a mean residence time of 100 ps. Thus, at least for exchange in the EX2 regime, even HDX monitored on the minutes-to-hours timescale reports on the frequency of fast timescale dynamics.
Our work provides the first experimental evidence to show that NNI2s induce long range perturbation of the conformational dynamics of NS5B. These results are especially suggestive when combined with data showing that NNI2s block the transition from initiation to elongation, resulting in the accumulation of abortive products, three-five nucleotides in length (11). The magnitude of this effect follows the order Lomibuvir > GS-9669 > Filibuvir. In Fig. 6 it is clear that the extent of rigidification of NS5B by NNI2s follows this same trend, with the most extensive effects seen for Lomibuvir followed by GS-9669 and then Filibuvir. The parallel between enzymatic assays and HDX suggests that suppression of conformational dynamics provides a rationale for the long range effects leading to inhibition. Our results suggest that NNI2 stabilizes the closed initiation-mode state and thereby inhibits the transition from the closed, de novo-initiated primed-template bound state into the more open, processive elongation-competent state.
Biochemical and structural studies have pointed to several structural motifs whose conformational dynamics may be involved in this transition from the initiation-competent closed state to the elongation-competent open state. For instance, the restriction of motions of the Δ1 fingers extension loop has been implicated in the initiation stages of RNA synthesis by NS5B by limited proteolysis (33). Moreover, the displacement of the finger extensions away from the thumb subdomain through mutagenesis were also shown to result in an initiation-incompetent yet elongation-enhanced construct exhibiting increased double-stranded RNA binding (34). On the frontal side of the protein, the presence of the β-loop in the RNA binding channel was shown to be critical for de novo initiation (35), yet attempts to bind duplex RNA into the active site indicate that the β-loop must be displaced (or removed) to form a stable elongation complex.
The requirement for the progressive removal of the C-terminal tail and β-loop during transition into elongation state was first suggested by Harrus et al. (36). Recently reported co-crystal structures of NS5B with various RNA constructs including a dinucleotide primed template mimicking a primed-initiation complex and a symmetrical 8-mer RNA duplex approximating the elongation complex (with a β-loop deletion) showed the progressive opening of the RNA binding channel in the transition from initiation to elongation (7). This transition appears to proceed with an increased distance between fingers and thumb domains as illustrated by the increasing distance between loops αD-αE and αO-αP, displacement of the β-loop. It appears that the C-terminal tail may be displaced upon binding template RNA (7). Finally, MD simulation studies have linked NNI2 binding with changes in conformational sampling of NS5B between a closed, initiation-competent state and a more open, elongation-competent state (37). Overall, there is ample evidence to suggest that the transition from a closed, initiation-competent state into an open, elongation-competent state would involve significant motions both in the fingers/fingers extension as well as the C-terminal tail/β-loop regions.
The NNI2-induced rigidification revealed in this study is consistent with the overall mechanism of inhibition revealed by nucleotide incorporation kinetic assays (11) but also provides molecular details underlying a conformational basis for differences in potency among the three NNI2s studied.
We have shown that all three NNI2s significantly decrease the conformational fluctuations sampled in a common network of residues that extends from the binding site through the fingers extension into the fingers domain. This allosterically induced rigidification of the fingers and fingers extension is consistent with a more stable closed state that would hinder the sampling of open conformations that have been suggested to be required for elongation (34, 35). In addition, thiophene-based Lomibuvir and GS-9669 confer additional loss in conformational flexibility in the C-terminal tail/β-loop motif, critical regions that may be linked to the transition into the open elongation-competent state (7, 36). The regions rigidified by Lomibuvir/GS-9669 binding completely encircle the RNA binding cleft and are suggestive of an increased difficulty in transitioning into an open elongation. The chemotype-specific suppression of dynamics underlying EX1 exchange in the RNA binding cleft by the two thiophene-based NNI2s may also contribute to their more pronounced inhibition of the transition to elongation, and moreover, the trends follow the inhibitor potency observed in kinetic studies with Lomibuvir > GS-9669 > Filibuvir. Taken together, the HDX and nucleotide incorporation data provide evidence that the primary mode of action of NNI2 is to inhibit the transition from an initiation-competent closed state into an elongation-competent open state leading to the accumulation of abortive intermediates. The differences in potency observed is attributable to differences in intensity and extent of these allosteric effects that would hinder this transition.
A detailed examination of these differences in conformational dynamics as it relates to differences in potency can potentially provide valuable insight for medicinal chemistry and further development of more effective NNI2. For instance, it is worth asking how thiophene-based inhibitors Lomibuvir and GS-9669 suppress conformational dynamics in the β-loop/C-terminal motif but Filibuvir does not. Relating chemotype-dependent contacts in the binding site to long range chemotype-dependent effects on dynamics may allow inferences to be made on how local variations in inhibitor-protein interactions may lead to differences in potency. Such analysis could illuminate the path to more potent inhibitors.
Author Contributions
D. D. performed the experiments, prepared the figures, and wrote the first draft of the paper. P. L. W. guided in the design of the measurements and in interpreting the results and assisted in writing the paper. J. L. prepared the protein and assisted in interpreting the results and writing the paper. K. A. J. assisted in the design of the experiments, interpretation of the results, and writing the paper. All four authors reviewed and approved the final version of the manuscript.
Acknowledgment
Nonnucleoside inhibitors were a gift from Gilead Sciences, Inc.
This work was supported, in whole or in part, by National Institutes of Health Grant 1R01AI110577 (NIAID; to K. A. J.). This work was also supported by the University of Maryland Baltimore, School of Pharmacy Mass Spectrometry Center (SOP1841-IQB2014) and by the Welch Foundation (F-1604; to K. A. J.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HCV
- hepatitis C virus
- NS5B
- nonstructural protein 5B
- NNI
- nonnucleoside inhibitor
- HDX
- hydrogen/deuterium exchange.
References
- 1. Ly K. N., Xing J., Klevens R. M., Jiles R. B., Ward J. W., and Holmberg S. D. (2012) The increasing burden of mortality from viral hepatitis in the United States between 1999 and 2007. Ann. Intern. Med. 156, 271–278 [DOI] [PubMed] [Google Scholar]
- 2. Muir A. J., and Naggie S. (2015) Hepatitis C virus treatment: is it possible to cure all hepatitis C virus patients? Clin. Gastroenterol. Hepatol. 13, 2166–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lesburg C. A., Cable M. B., Ferrari E., Hong Z., Mannarino A. F., and Weber P. C. (1999) Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6, 937–943 [DOI] [PubMed] [Google Scholar]
- 4. Ago H., Adachi T., Yoshida A., Yamamoto M., Habuka N., Yatsunami K., and Miyano M. (1999) Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 7, 1417–1426 [DOI] [PubMed] [Google Scholar]
- 5. Bressanelli S., Tomei L., Roussel A., Incitti I., Vitale R. L., Mathieu M., De Francesco R., and Rey F. A. (1999) Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 96, 13034–13039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mosley R. T., Edwards T. E., Murakami E., Lam A. M., Grice R. L., Du J., Sofia M. J., Furman P. A., and Otto M. J. (2012) Structure of hepatitis C virus polymerase in complex with primer-template RNA. J. Virol. 86, 6503–6511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Appleby T. C., Perry J. K., Murakami E., Barauskas O., Feng J., Cho A., Fox D. 3rd, Wetmore D. R., McGrath M. E., Ray A. S., Sofia M. J., Swaminathan S., and Edwards T. E. (2015) Viral replication: structural basis for RNA replication by the hepatitis C virus polymerase. Science 347, 771–775 [DOI] [PubMed] [Google Scholar]
- 8. Jin Z., Leveque V., Ma H., Johnson K. A., and Klumpp K. (2012) Assembly, purification, and pre-steady-state kinetic analysis of active RNA-dependent RNA polymerase elongation complex. J. Biol. Chem. 287, 10674–10683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Spence R. A., Kati W. M., Anderson K. S., and Johnson K. A. (1995) Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science 267, 988–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Caillet-Saguy C., Simister P. C., and Bressanelli S. (2011) An objective assessment of conformational variability in complexes of hepatitis C virus polymerase with non-nucleoside inhibitors. J. Mol. Biol. 414, 370–384 [DOI] [PubMed] [Google Scholar]
- 11. Li J., and Johnson K. A. (2016) Thumb Site 2 Inhibitors of Hepatitis C viral RNA-dependent RNA polymerase allosterically block the transition from initiation to elongation. J. Biol. Chem. 291, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang J., Chalmers M. J., Stayrook K. R., Burris L. L., Garcia-Ordonez R. D., Pascal B. D., Burris T. P., Dodge J. A., and Griffin P. R. (2010) Hydrogen/deuterium exchange reveals distinct agonist/partial agonist receptor dynamics within vitamin D receptor/retinoid X receptor heterodimer. Structure 18, 1332–1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. West G. M., Chien E. Y., Katritch V., Gatchalian J., Chalmers M. J., Stevens R. C., and Griffin P. R. (2011) Ligand-dependent perturbation of the conformational ensemble for the GPCR β2 adrenergic receptor revealed by HDX. Structure 19, 1424–1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Seckler J. M., Barkley M. D., and Wintrode P. L. (2011) Allosteric suppression of HIV-1 reverse transcriptase structural dynamics upon inhibitor binding. Biophys. J. 100, 144–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rand K. D., Jørgensen T. J., Olsen O. H., Persson E., Jensen O. N., Stennicke H. R., and Andersen M. D. (2006) Allosteric activation of coagulation factor VIIa visualized by hydrogen exchange. J. Biol. Chem. 281, 23018–23024 [DOI] [PubMed] [Google Scholar]
- 16. Shi Z., Resing K. A., and Ahn N. G. (2006) Networks for the allosteric control of protein kinases. Curr. Opin. Struct. Biol. 16, 686–692 [DOI] [PubMed] [Google Scholar]
- 17. Hawse W. F., Champion M. M., Joyce M. V., Hellman L. M., Hossain M., Ryan V., Pierce B. G., Weng Z., and Baker B. M. (2012) Cutting edge: evidence for a dynamically driven T cell signaling mechanism. J. Immunol. 188, 5819–5823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wales T. E., Hochrein J. M., Morgan C. R., Emert-Sedlak L. A., Smithgall T. E., and Engen J. R. (2015) Subtle dynamic changes accompany hck activation by HIV-1 Nef and are reversed by an antiretroviral kinase inhibitor. Biochemistry 54, 6382–6391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chen H., Ricklin D., Hammel M., Garcia B. L., McWhorter W. J., Sfyroera G., Wu Y. Q., Tzekou A., Li S., Geisbrecht B. V., Woods V. L. Jr., and Lambris J. D. (2010) Allosteric inhibition of complement function by a staphylococcal immune evasion protein. Proc. Natl. Acad. Sci. U.S.A. 107, 17621–17626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Houde D., Berkowitz S. A., and Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guttman M., Weis D. D., Engen J. R., and Lee K. K. (2013) Analysis of overlapped and noisy hydrogen/deuterium exchange mass spectra. J. Am. Soc. Mass Spectrom. 24, 1906–1912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seckler J. M., Howard K. J., Barkley M. D., and Wintrode P. L. (2009) Solution structural dynamics of HIV-1 reverse transcriptase heterodimer. Biochemistry 48, 7646–7655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li H., Tatlock J., Linton A., Gonzalez J., Jewell T., Patel L., Ludlum S., Drowns M., Rahavendran S. V., Skor H., Hunter R., Shi S. T., Herlihy K. J., Parge H., Hickey M., Yu X., Chau F., Nonomiya J., and Lewis C. (2009) Discovery of (R)-6-cyclopentyl-6-(2-(2,6-diethylpyridin-4-yl)ethyl)-3-((5,7-dimethyl-[1,2,4]tr iazolo[1,5-a]pyrimidin-2-yl)methyl)-4-hydroxy-5,6-dihydropyran-2-one (PF-00868554) as a potent and orally available hepatitis C virus polymerase inhibitor. J. Med. Chem. 52, 1255–1258 [DOI] [PubMed] [Google Scholar]
- 24. Canales E., Carlson J. S., Appleby T., Fenaux M., Lee J., Tian Y., Tirunagari N., Wong M., and Watkins W. J. (2012) Tri-substituted acylhydrazines as tertiary amide bioisosteres: HCV NS5B polymerase inhibitors. Bioorg. Med. Chem. Lett. 22, 4288–4292 [DOI] [PubMed] [Google Scholar]
- 25. Lazerwith S. E., Lew W., Zhang J., Morganelli P., Liu Q., Canales E., Clarke M. O., Doerffler E., Byun D., Mertzman M., Ye H., Chong L., Xu L., Appleby T., Chen X., et al. (2014) Discovery of GS-9669, a thumb site II non-nucleoside inhibitor of NS5B for the treatment of genotype 1 chronic hepatitis C infection. J. Med. Chem. 57, 1893–1901 [DOI] [PubMed] [Google Scholar]
- 26. Xue W., Jiao P., Liu H., and Yao X. (2014) Molecular modeling and residue interaction network studies on the mechanism of binding and resistance of the HCV NS5B polymerase mutants to VX-222 and ANA598. Antiviral Res. 104, 40–51 [DOI] [PubMed] [Google Scholar]
- 27. Boyce S. E., Tirunagari N., Niedziela-Majka A., Perry J., Wong M., Kan E., Lagpacan L., Barauskas O., Hung M., Fenaux M., Appleby T., Watkins W. J., Schmitz U., and Sakowicz R. (2014) Structural and regulatory elements of HCV NS5B polymerase – β-loop and C-terminal tail – are required for activity of allosteric thumb site II inhibitors. PloS ONE 9, e84808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cherry A. L., Dennis C. A., Baron A., Eisele L. E., Thommes P. A., and Jaeger J. (2015) Hydrophobic and charged residues in the C-terminal arm of hepatitis C virus RNA-dependent RNA polymerase regulate initiation and elongation. J. Virol. 89, 2052–2063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Engen J. R., Smithgall T. E., Gmeiner W. H., and Smith D. L. (1997) Identification and localization of slow, natural, cooperative unfolding in the hematopoietic cell kinase SH3 domain by amide hydrogen exchange and mass spectrometry. Biochemistry 36, 14384–14391 [DOI] [PubMed] [Google Scholar]
- 30. Winquist J., Abdurakhmanov E., Baraznenok V., Henderson I., Vrang L., and Danielson U. H. (2013) Resolution of the interaction mechanisms and characteristics of non-nucleoside inhibitors of hepatitis C virus polymerase. Antiviral Res. 97, 356–368 [DOI] [PubMed] [Google Scholar]
- 31. Morgan C. R., and Engen J. R. (2009) Investigating solution-phase protein structure and dynamics by hydrogen exchange mass spectrometry. Curr. Protoc. Protein Sci. 10.1002/0471140864.ps1706s58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Persson F., and Halle B. (2015) How amide hydrogens exchange in native proteins. Proc. Natl. Acad. Sci. U.S.A. 112, 10383–10388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rigat K., Wang Y., Hudyma T. W., Ding M., Zheng X., Gentles R. G., Beno B. R., Gao M., and Roberts S. B. (2010) Ligand-induced changes in hepatitis C virus NS5B polymerase structure. Antiviral Res. 88, 197–206 [DOI] [PubMed] [Google Scholar]
- 34. Chinnaswamy S., Yarbrough I., Palaninathan S., Kumar C. T., Vijayaraghavan V., Demeler B., Lemon S. M., Sacchettini J. C., and Kao C. C. (2008) A locking mechanism regulates RNA synthesis and host protein interaction by the hepatitis C virus polymerase. J. Biol. Chem. 283, 20535–20546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hong Z., Cameron C. E., Walker M. P., Castro C., Yao N., Lau J. Y., and Zhong W. (2001) A novel mechanism to ensure terminal initiation by hepatitis C virus NS5B polymerase. Virology 285, 6–11 [DOI] [PubMed] [Google Scholar]
- 36. Harrus D., Ahmed-El-Sayed N., Simister P. C., Miller S., Triconnet M., Hagedorn C. H., Mahias K., Rey F. A., Astier-Gin T., and Bressanelli S. (2010) Further insights into the roles of GTP and the C terminus of the hepatitis C virus polymerase in the initiation of RNA synthesis. J. Biol. Chem. 285, 32906–32918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Davis B. C., and Thorpe I. F. (2013) Thumb inhibitor binding eliminates functionally important dynamics in the hepatitis C virus RNA polymerase. Proteins 81, 40–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hang J. Q., Yang Y., Harris S. F., Leveque V., Whittington H. J., Rajyaguru S., Ao-Ieong G., McCown M. F., Wong A., Giannetti A. M., Le Pogam S., Talamás F., Cammack N., Nájera I., and Klumpp K. (2009) Slow binding inhibition and mechanism of resistance of non-nucleoside polymerase inhibitors of hepatitis C virus. J. Biol. Chem. 284, 15517–15529 [DOI] [PMC free article] [PubMed] [Google Scholar]