

Abstract
The glycolytic-based metabolism of cancers promotes an acidic microenvironment that is responsible for increased aggressiveness. However, the effects of acidosis on tumour metabolism have been almost unexplored. By using capillary electrophoresis with time-of-flight mass spectrometry, we observed a significant metabolic difference associated with glycolysis repression (dihydroxyacetone phosphate), increase of amino acid catabolism (phosphocreatine and glutamate) and urea cycle enhancement (arginino succinic acid) in osteosarcoma (OS) cells compared with normal fibroblasts. Noteworthy, metabolites associated with chromatin modification, like UDP-glucose and N8-acetylspermidine, decreased more in OS cells than in fibroblasts. COBRA assay and acetyl-H3 immunoblotting indicated an epigenetic stability in OS cells than in normal cells, and OS cells were more sensitive to an HDAC inhibitor under acidosis than under neutral pH. Since our data suggest that acidosis promotes a metabolic reprogramming that can contribute to the epigenetic maintenance under acidosis only in tumour cells, the acidic microenvironment should be considered for future therapies.
Keywords: Acidosis, metabolome, epigenome, osteosarcoma
Introduction
The self-organizing adaptive system to microenvironmental stress is a common hallmark of all cancer types. Among metabolic alterations, increased glycolysis is one of the most studied. Although an imbalance between enhanced oxygen consumption by cancer cells and insufficient oxygen delivery from the aberrant tumour vasculature might be the main reason for such phenomenon, glycolysis increases even in the presence of oxygen, as clearly described by Otto Warburg in 1930 [1,2]. The high-rate energetic glycolytic metabolism of cancer cells causes high lactic acid production and a high proton efflux. Notably, low pH is one of the main pathophysiological traits of solid tumours at a very early stage. However, thanks to tumour plasticity, malignant cells quickly learn how to survive and to progress under an acidic extracellular environment [3]. We recently confirmed the highly acidifying activity of sarcoma cells and their ability to survive under acidic pH; and, like in other cancers, a low extracellular pH in these tumours induces an increased invasive behaviour [4,5]. Nowadays, it is clear that extracellular acidity is a consequence of the altered metabolism of cancer cells. Several studies reported that hypoxia promotes an increase in glycolysis through the activation of the hypoxia inducible factor-1 (HIF-1), and might be one of the main causes of tumour acidosis [6-8]. An increased glycolytic rate is part of the so-called “tumour fermentation”, which includes also glutaminolysis, amino acid catabolism, and uncoupled tricarboxylic acid (TCA) cycle [9,10]. However, the effects of acidosis on tumour metabolism are almost unknown. One study in breast cancer under short-term acidosis reported an imbalance of tumour fermentation versus a glutaminolytic metabolism, involving pentose phosphate and fatty acid metabolism, associated with a strong reduction in glycolysis and increased TCA cycling [11]. The shift to glutamine metabolism was also recently demonstrated under chronic acidosis in models of cervix cancer, squamous carcinoma and colon cancer cell lines that were cultured at a low pH for several weeks [12]. However, metabolic changes of cancer cells when maintained under acidosis have not been completely elucidated, and, most importantly, the metabolic adaptation of cancer cells to acidosis has never been compared with the metabolic response of normal cells. New experimental approaches may improve our knowledge in this field and may support the identification of novel and more specific anticancer therapies. In this study, to pinpoint for the first time the different metabolic profiles between osteosarcoma (OS) cells and normal human fibroblasts (Fb) under short-term acidosis, we used capillary electrophoresis with time-of-flight mass spectrometry (CE-TOFMS) [13]. We decided to evaluate the metabolic adaptation of cancer cells in respect to the normal cells in the short term, rather than to study the consequences of the chronic exposure to acidosis for clonal evolution. Indeed, we intended to evaluate the prompt reaction of tumour cells to a hostile microenvironment. The proposed model allows the identification of the first key passages that tumour cells pass through, to survive and compete with normal cells in the tumour microenvironment. First, we identified those metabolites that significantly differ between OS and Fb cells under acidosis. Then, we confirmed that the identified metabolites were specifically associated with the tumour cells rather than with normal cells, by analyzing the variation of the concentrations of the identified metabolites under acidic condition in respect to the neutral condition, in the single group of cells, cancer or normal cell group. Finally, to make sure not to lose additional metabolic pathways associated with the metabolic reprogramming of tumour cells when cultured under acidosis, we also performed the analysis of the differences of the concentration of intracellular metabolites between the acidic condition in respect to the neutral condition only in OS cells.
The intracellular availability of specific metabolites dictates the efficacy and specificity of enzymatic reactions including for epigenetic regulation [14]. Thus, altered cancer metabolism associated with extracellular acidosis may potentially modulate the epigenetic state of cancer cells as a response to cellular stress. Indeed, intracellular pH that is affected by extracellular pH [15] may modulate histone acetylation and vice versa [16]. Epigenetic alterations of chromatin can affect gene expression profiles and contribute to tumourigenesis and tumour progression [17]. Therefore, in this study we also screened alterations of the epigenetic profiles-DNA methylation and histone acetylation-of OS cells and compared it with those of normal Fb.
Materials and methods
Cell lines
Human OS cells (MG-63, HOS) and normal fibroblasts (TIG-108, TIG-121) were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). Mesenchymal stromal cell (MSC) primary cultures (#305526, #351482; #326162) were acquired from Lonza (MD). Saos-2 human OS cells were obtained from RIKEN BioResource Center (RIKEN BRC, Tsukuba, Japan). Cells were cultured in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum, supplemented with penicillin (50 units/mL) and streptomycin (50 mg/mL) with 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid to adjust the pH at 7.4. Cells were incubated at 37°C in a humidified chamber supplemented with 5% CO2. For the different assays, cells were seeded and treated with different methods according to the specific assay, as explained as follows, and as summarized in Figure 1.
Figure 1.
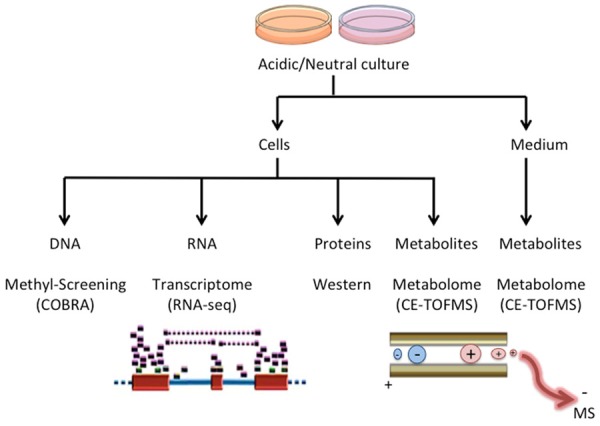
Graphical summary of the experimental protocol: The different cell models (HOS, MG-63, Saos-2, TIG-121, TIG-108) were cultured for 24 hours in the Acidic or Neutral culture medium. Samples were obtained both from the cell lysates (Cells), and by using the culture supernatant (Medium). DNA, RNA, proteins and metabolites were extracted, and analysed by COBRA, RNA-seq, Western blots, and CE-TOFMS assays. The supernatant medium was also analyzed by CE-TOFMS assay for metabolomics screening to evaluate, together with the analysis on cell lysates, the accumulation, consumption, productive increase, or decrease rates relative to each metabolite.
Metabolites extraction
Extracellular acidosis was obtained by adding to the media 10 mM piperazine-1,4-bis(2-ethanesulfonic acid) to adjust the pH at 6.5. Cells were maintained at a low pH for 24 h. To evaluate the content of intracellular metabolites, cells were cultured in 100 mm diameter dishes and washed twice with 5% mannitol solution (10 mL first and then 2 mL). Then, the cells were treated with 800 μL ice-cold methanol and left for 30 s in order to inactivate enzymes. Next, the cell extract was treated with 550 µL of Milli-Q water containing 10 μM internal standards (#H3304-1002, Human Metabolome Technologies, Inc.) and left for another 30 s. Extracellular culture medium (200 µL) was added to 500 μL ice-cold methanol containing internal standards. Once the extracts were obtained, they were centrifuged at 2,300 × g at 4°C for 5 min. The upper aqueous layer (1 mL) was centrifugally filtered through a Millipore 5-kDa cut-off filter at 9,100 × g at 4°C for 180 min to remove proteins. The filtrate was centrifugally concentrated and re-suspended in 50 µL of Milli-Q water for CE-TOFMS analysis.
CE-TOFMS analysis of intracellular metabolites and data processing
Metabolome measurements and data processing were carried out through a facility service at Human Metabolome Technology Inc. (HMT). Briefly, CE-TOFMS was carried out using an Agilent CE Capillary Electrophoresis System equipped with an Agilent 6210 Time of Flight mass spectrometer, Agilent 1100 isocratic HPLC pump, Agilent G1603A CE-MS adapter kit, and Agilent G1607A CE-ESI-MS sprayer kit (Agilent Technologies). The systems were controlled by Agilent G2201AA ChemStation software version B.03.01 for CE (Agilent Technologies). The metabolites were analysed by using a fused silica capillary (50 µm internal diameter × 80 cm total length), with commercial electrophoresis buffer (#H3301-1001 for cation analysis and #H3302-1021 for anion analysis, HMT) as the electrolyte. The sample was injected at a pressure of 50 mbar for 10 s (approximately 10 nL) in cation analysis and 25 s (approximately 25 nL) in anion analysis. The spectrometer was scanned from 50 to 1,000 m/z. Other conditions were as in the described previously [18-20]. Peaks were extracted using automatic integration software MasterHands (Keio University) in order to obtain peak information including m/z, migration time for CE-TOFMS measurement (MT) and peak area [21]. Signal peaks corresponding to isotopomers, adduct ions, and other product ions of known metabolites were excluded; remaining peaks were annotated with putative metabolites from the HMT metabolite database based on their MTs and m/z values determined by TOFMS. The tolerance range for the peak annotation was configured at ± 0.5 min for MT and ± 10 ppm for m/z. In addition, peak areas were normalized against those of the internal standards, and the resultant relative area values were further normalized according to the sample amount.
Hierarchical cluster analysis (HCA) and principal component analysis (PCA) were performed by HMT proprietary software PeakStat and SampleStat, respectively. Detected metabolites were plotted on metabolic pathway maps using the VANTED (Visualization and Analysis of Networks containing Experimental Data) software [22].
RNA extraction and RNA-seq analysis
Total RNA from each cell was extracted, prepared as the library for RNA-seq, and applied to Illumina Genome Analyzer GAIIx sequencing. Sequencing reads were aligned, mapped to Human genome build 19 (hg19) as a reference, and quantified as the expression data of transcriptome. The more detailed method was described in Supplementary information.
DNA isolation and treatment with sodium bisulfite
Cells were washed with phosphate-buffered saline (PBS) and suspended in lysis buffer (10 mM Tris-HCl and 50 mM EDTA, both at pH 8.0, 10 mM NaCl, 2% N-lauryl sarcosyl, and 200 µg/mL proteinase K). The mixture was incubated for 20 h at 55°C, followed by phenol chloroform extraction and ethanol precipitation. DNA from cell lines was extracted using QIAamp DNA Blood Mini Kits (QIAGEN). Bisulfite treatment was performed according to the method of Clark et al. [23] with variations detailed by Frevel et al. [24]. The bisulfite reaction, under mineral oil, was performed at 55°C for 16 h in a total volume of 525 μL containing 2.4 M sodium bisulfite and 123 mM hydroquinone (Sigma). Reactions were desalted using a QIAEX II gel extraction kit (QIAGEN). DNA was eluted in 50 μL of H2O, incubated with 5 μL of 3 M NaOH for 15 min at 37°C neutralized with ammonium acetate (final concentration of 3 M), and ethanol precipitated. Bisulfite-treated DNA was then resuspended in 25 μL of H2O and stored at -20°C.
Combined bisulfite restriction analysis (COBRA) for LINE1
To screen the methylation profile of genomic DNA methylation, we used COBRA for LINE1 [25,26], and the DNA extract from NEC8 testicular embryonal carcinoma cell used as a highly unmethylated control. Methylation of the LINE1 promoter was investigated as follows: PCR amplification was performed in a 25 μL volume using Ex Taq buffer (Takara) under the following conditions: 2 mM MgCl2, 200 mM each deoxynucleotide triphosphate, 0.8 mM final concentration of each primer and 0.6 unit of Ex Taq (Takara). The primer sequences related to the LINE1 promoter region were: 5’-TTGAGTTGTGGTGGGTTTTATTTAG-3’ (496-520, X58075) and 5’-TCATCTCACTAAAAAATACCAAACA-3’ (108-132, X58075). PCR cycling conditions were 95°C for 30 s, 50°C for 30 s, and 72°C for 30 s for 35 cycles. The final PCR product was digested with the HinfI restriction enzyme. The digested PCR products were separated by electrophoresis on 6% polyacrylamide gels. In COBRA analysis, the lower digested multiple bands represent methylated repetitive elements, and the upper top undigested band represents unmethylated repetitive elements or repetitive elements with a mutated restriction site. Following gel electrophoresis and ethidium bromide staining, the PCR bands were quantified through densitometric analysis to evaluate the degree of methylation determined for LINE1 elements in OS, Fb, and MSC cells. As unmethylated DNA control, DNA from NEC8, a testicular embryonal carcinoma cell line was used [26].
Western blotting
Cells were lysed in Laemmli-sodium dodecyl sulphate (SDS) buffer, subjected to SDS-polyacrylamide gel electrophoresis, and electro-transferred to membrane filters (Immuno-Blot PVDF membranes, Bio-Rad Laboratories). After blocking the filters with TBS-T (10 mM Tris-HCl (pH 7.6), 150 mM sodium chloride, 0.1% Tween 20) containing 5% bovine serum albumin (BSA), they were incubated overnight with the primary antibodies in TBS-T containing 2% BSA at 4°C. The filters were then washed in TBS-T and incubated for 1 h in horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (TrueBlotTM, eBioscience, Affymetrix Japan) or anti-goat antibodies (Santa Cruz Biotechnology) diluted 1:1,000,000 in TBS-T containing 2% BSA. After several washes with TBS-T, the immunoreaction was detected using the ECL system (GE Healthcare) with LAS4000 (Fujifilm) and quantified with Multi gauge (Fujifilm), using an anti-α tubulin (DM 1A) or anti-TATA-binding protein (TBP) antibody (Sigma) as an internal control. For the analysis of the acetylation of histone H3 in cells treated with MC1742, cells were seeded and let to adhere for 24 h in complete medium. Then, the medium was replaced with complete medium at pH 6.5 or pH 7.4, with or without HDACi MC1742 (1 or 2 μM). After 24 h, cell lysates were obtained after a washing in PBS with the hot lysis buffer. The cell lysates were prepared by direct boiling in SDS lysis buffer (1% SDS, 20 mM Tris HCl pH 7.6, 5% β-mercaptoethanol, 1 mM sodium ortho-vanadate) for 3 min followed by ultrasonication for 10-15 s. Equal amounts of protein lysates were subjected to SDS-PAGE. Antibodies against histone de-acetylase 1 (HDAC1: 10E2) and acetyl-histone H3 (Ac-H3: #06-599) were purchased from Cell Signalling Technology and Millipore, respectively. The experiments were repeated more than three times.
Viability and growth assay
Viability
Cells were seeded in 96-well plates with complete medium. After 24 h, the medium was changed with complete medium at pH 6.5 or pH 7.4, with or without HDAC inhibitor (HDACi) MC1742 (0.5, 1, or 2 μM). After additional 72 h, the cell number was indirectly evaluated by an acid phosphatase assay, as previously described [4]. Briefly, the medium was removed and each well was washed once with PBS, and the medium was replaced with 100 μL of buffer containing 0.1 M sodium acetate (pH 5.0), 0.1% Triton X-100, and 5 mM p-nitrophenil phosphate. The plate was placed at 37°C incubator for 3 h. The reaction was stopped with the addition of 10 μL of a 1 N NaOH, and colour development was assayed at 405 nm using a microplate reader (Tecan Infinite F200pro, Männedorf, Switzerland). The experiments were repeated with four replicates.
Growth assay
Cells were seeded in 6-well plates in complete medium. After 24 h, the medium was changed with complete medium at pH 6.5 or pH 7.4, with or without MC1742 (1, 2 μM). Cell growth was evaluated soon after the population doubling, at 24 h for pH 7.4 and at 48 h for pH 6.5, by the direct counting of cells using erythrosine dye (Sigma). The experiments were repeated with four replicates.
Cell cycle analysis
DNA content and bromodeoxyuridine (BrdU) incorporation were determined, by simultaneous analysis of propidium iodide (PI) and of fluorescein isothiocyanate (FITC)-conjugated anti-BrdU antibody. Cells were seeded at low density in complete medium, and after 24 h the medium was changed with complete medium at pH 6.5 or 7.4. After additional 48 h, cells were incubated with 10 mM BrdU for 60 min before harvesting. Cells were collected by trypsinization, followed by fixation in 40% ethanol for 20 min. Partial DNA denaturation was performed by incubating cells in HCl, followed by neutralization with sodium tetraborate. Samples were then exposed to a monoclonal anti-BrdU FITC antibody, washed, and finally stained with 2.5 mg/mL PI. Then, flow cytometric analysis was performed. The experiment was repeated three times.
Statistical analysis
For the CE-TOFMS, the data analysis was performed by Welch’s t-test, according to the manufacture suggestions. For all the other assays, the statistical analysis was performed with the StatViewTM 5.0.1 software (SAS Institute, Cary, NC). To evaluate the differences between the OS group of data and Fb group of data in cells cultured under acidosis, we used the Welch’s t-test. To evaluate reduced/oxidized ratio of nicotinamide adenine dinucleotide (NADH/NAD+) statistical differences among neutral OS, acidic OS, neutral Fb, and acidic Fb, one-way factorial ANOVA and multiple comparison tests accompanied by Fisher’s significance were applied. Due to the low number of observations, the other experimental data were considered as not normally distributed and non-parametric tests were used. In these cases, the Mann-Whitney U test was used for the difference between groups. Data were always expressed by mean ± standard error (SE), or standard deviation (SD), which was used only for the CE-TOFMS analysis.
Results
Osteosarcoma metabolic adaptation to acidosis
To study the metabolic profile of normal and cancer cells, we used CE-TOFMS by which we quantified the concentrations of metabolites in cell lysates and in the supernatant of both OS cells and Fb cultures. Overall, in the analysis, 243 metabolites were detected, among which the concentration of 96 were quantified also by using an internal controls as reference. To focus on a subgroup of metabolites that are specifically associated with tumour-specific adaptations to acidosis, we selected a list of metabolites that significantly differed in malignant cells (OS) in respect to normal cells under acidic conditions. We found that 14 metabolites were significantly decreased in cancer cells, whereas none was increased (Table 1). The identified decreased metabolites in OS were associated with glycolysis, amino acid catabolism and urea cycle, e.g. dihydroxyacetone phosphate (DHAP), glutamate (Glu), and argininosuccinic acid, respectively. Additionally, we found in OS cells a decrease in UDP related to the metabolic pathway of pyrimidine bases, a decrease in oxidized NAD+ related to redox status and a decrease in N8-acetylspermidine related to histone acetylation. Finally, pelargonic and decanoic acids, which are specifically related to fatty acid metabolism, were also significantly decreased. For a more comprehensive overview of the metabolic trends under acidosis, we also showed the Heat map, including additional metabolites that are related to the metabolic pathways previously described (Supplementary Table S1). All the profilings performed in the present study have been summarized in Figure 1.
Table 1.
Metabolites with lower intracellular concentration in osteosarcoma cells than in normal fibroblasts under acidosis analysed by CE-TOFMS
| Compound name | Category | OS | Fb | OS/Fb | P-value | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Mean [pmol/109 cells] | SD | Mean [pmol/109 cells] | SD | ||||
| Dihydroxyacetone phosphate | Glycolysis | 4.65 | 2.86 | 15.65 | 0.33 | 0.30 | 0.0295* |
| Phosphocreatine | Protein metabolism | 34.40 | 9.01 | 194.80 | 5.01 | 0.18 | 0.0003** |
| Glu | Glycogenic amino acid | 237.14 | 123.82 | 642.46 | 76.19 | 0.37 | 0.0424* |
| Argininosuccinic acid | Uremic toxin | 0.98 | 0.26 | 5.30 | 0.53 | 0.19 | 0.0398* |
| ATP | Purine bases | 807.41 | 217.89 | 1618.76 | 100.12 | 0.50 | 0.0218* |
| GTP | Purine bases | 34.66 | 8.57 | 100.27 | 7.71 | 0.35 | 0.0169* |
| CTP | Pyrimidine bases | 67.44 | 17.70 | 118.23 | 6.50 | 0.57 | 0.0407* |
| UTP | Pyrimidine bases | 220.87 | 152.84 | 1057.52 | 142.54 | 0.21 | 0.0379* |
| UDP-glucose, UDP-galactose | Carbohydrate metabolism | 95.97 | 48.50 | 397.60 | 27.31 | 0.24 | 0.0066** |
| CMP-N-Acetylneuraminate | Nucleotide sugars | 12.03 | 6.19 | 58.75 | 6.56 | 0.20 | 0.0308* |
| NAD+ | Electron carrier | 93.47 | 65.68 | 369.37 | 49.97 | 0.25 | 0.0373* |
| N 8-Acetylspermidine | Histone acetylation | 12.94 | 10.51 | 429.06 | 9.76 | 0.03 | 0.0006** |
| Pelargonic acid | Fatty Acids | 8.54 | 8.04 | 37.42 | 5.92 | 0.23 | 0.0489* |
| Decanoic acid | Fatty Acids | 4.10 | 2.90 | 19.12 | 0.43 | 0.21 | 0.0150* |
Welch’s t-test was used to evaluate the ratios of osteosarcomas (OS) vs. normal fibroblasts (Fb). In more details, all the data derived form OS cells (by including all the cell lines together) under acidosis were compared to all the data derived from Fb cells (by including all the different lots of cells together).
p < 0.05;
p < 0.01.
Under acidosis osteosarcoma metabolism shifts from glycolysis to OXPHOS-TCA
Among the specific metabolic pathways identified by the statistical analysis of CE-TOFMS data, we focused on glycolysis, TCA and urea cycles (Figure 2). Especially in OS cells under acidosis, intracellular intermediates of glycolysis/gluconeogenesis were increased, but only those upstream of fructose-6-phosphate (F6P) and downstream of glucose-1-phosphate (G1P), whereas intermediate metabolites that derive from fructose-1,6-biphosphate (F1,6BP) (e.g., glyceraldehyde 3-phosphate and DHAP) were all decreased (Figure 2A). In turn, pentose phosphate pathway (PPP) intermediated ribulose-5-phosphate (Ru5P) and sedoheptulose-7-phosphate (S7P) were unmodified, indicating that acidosis induces a metabolic adaptation from glycolysis to PPP metabolism. Impaired glycolysis in OS cells under acidosis was associated with reduced lactate release in cell culture supernatants (Supplementary Figure S1). Although glycolysis was strongly impaired, the content of intracellular ATP in OS cells was only mildly reduced in Saos-2 and MG-63 cells (up to 20% of inhibition), and almost unchanged in HOS cells (Supplementary Figure S2), suggesting that metabolic pathways alternative to glycolysis are active under acidosis. Under both acidic and neutral pH, TCA cycle appeared to be used in OS cells as well as in Fb. We observed an increased expression of all the complexes of OXPHOS, which was significant for complexes I and III (Supplementary Figure S3). In addition, the transcripts of enzymes related to glycolysis and TCA cycles did not change significantly under both acidic and neutral pH in both OS and Fb (Supplementary Table S2), and almost all the intermediates of the TCA cycle in metabolomics analysis showed similar levels of concentrations, with the exception of succinate in acidic OS cells that showed a trend of increase (Figure 2B). Finally, NADH/NAD+ ratio was relatively high under neutral pH in OS cells, and was drastically reduced under acidosis, suggesting that OS metabolisms shifted from glycolysis to oxidative phosphorylation (OXPHOS) and TCA cycle (Table 2).
Figure 2.
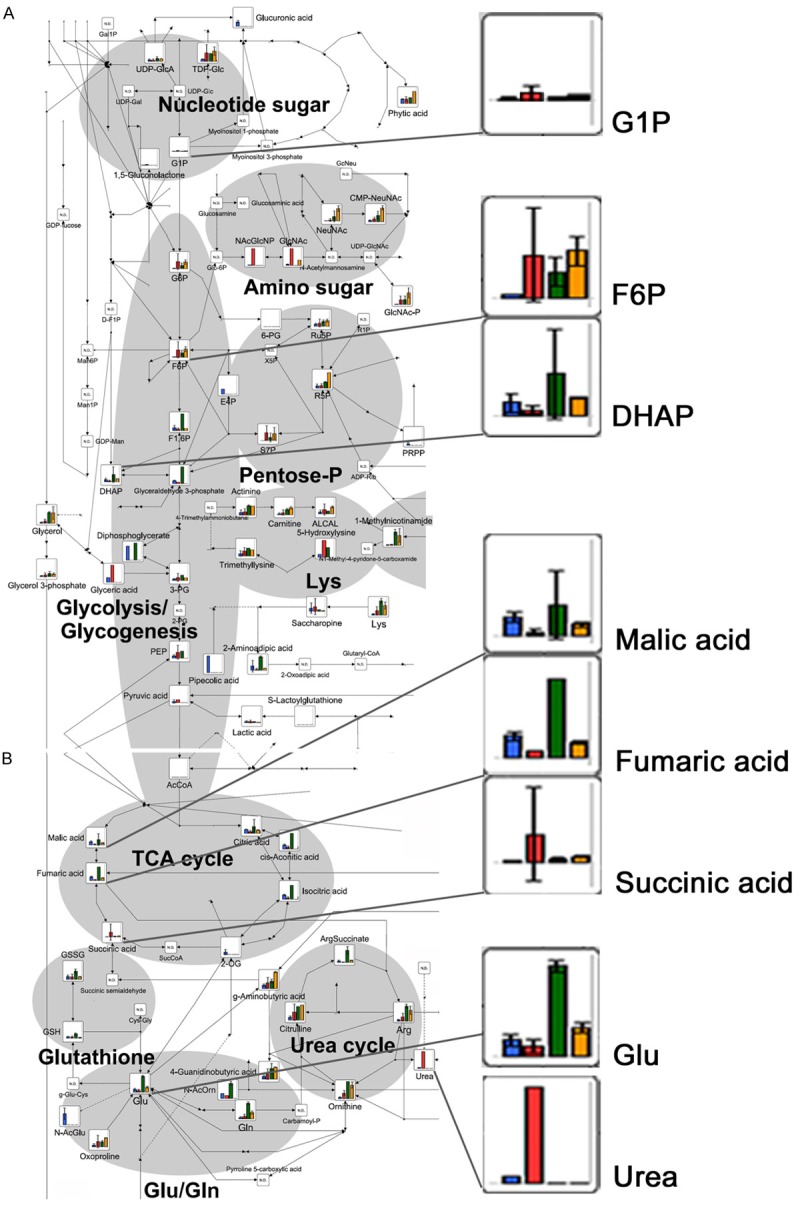
CE-TOFMS analysis of intracellular metabolites related to the main energetic metabolic pathways after 24 h of exposure to acidosis: Bar graphs show the relative intracellular amount of metabolites. The bars correspond to the mean values of the three OS cell lines included in this study, under neutral (blue) and acidic (red) conditions, and to the mean of the two normal Fb cultures under neutral (green) and acidic conditions (orange), mean ± SD. Metabolic pathway maps were first written by VANTED software at HMT, and the copyright has been provided to the authors. A. Glycolysis/glycogenesis, pentose-phosphate, amino sugar and nucleotide sugar metabolic pathways; B. TCA and urea cycles, Glu/Gln metabolic pathway.
Table 2.
NADH/NAD+ ratio of both OS and Fb cells in neutral vs. acidic conditions, by CE-TOFMS analysis
| NADH/NAD+ (x10-2) | OS | Fb | ||
|---|---|---|---|---|
|
| ||||
| Mean | SD | Mean | SD | |
| Neutral pH | 11.35* | 4.38 | 4.51 | 2.39 |
| Acidic pH | 3.36 | 3.74 | 1.00 | 0.33 |
One-way factorial ANOVA and multiple comparison tests accompanied by Fisher’s significance indicated a significant difference of NADH/NAD+ ratio in osteosarcoma (OS) cells cultured under neutral conditions than in the others; acidic OS, neutral fibroblasts (Fb) and acidic Fb.
p < 0.05.
To ensure that we included in the analysis all the significant metabolic pathways associated with the metabolic reprogramming of tumour cells as a response to extracellular acidosis, we performed a statistical analysis of the differences of the concentration of intracellular metabolites only in OS cells, between the acidic conditions in respect to the neutral conditions by the Welch’s t-test (Table 3). Under our experimental conditions, malic and fumaric acids were significantly decreased (Table 3), and succinic acid showed a trend for accumulation only in malignant OS cells under acidic versus neutral environments (Figure 2B). In our models, a decrease in glutamate (Glu) concentration suggested an increase in glutaminolysis to form Glu that is immediately used in several metabolic pathways in addition to the TCA cycle (Figure 2B), and we found a strong increase in all the intermediates of the urea cycle (citrulline, L-Arginine [Arg], ornithine, and urea) in OS cells under acidosis (Figure 2B).
Table 3.
Metabolites significantly altered in acidic vs. neutral conditions in OS cells, by CE-TOFMS analysis
| Compound name | Category | Acidic pH | Neutral pH | Acidic/Neutral | P-value | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Mean [pmol/109 cells] | SD | Mean [pmol/109 cells] | SD | ||||
| Fumaric acid | TCA cycle | 4.67 | 3.30 | 23.79 | 3.90 | 0.20 | 0.028* |
| Malic acid | TCA cycle, Pyruvate metabolism | 50.44 | 27.82 | 250.28 | 52.38 | 0.20 | 0.017* |
| Nicotinamide | Cholesterol reducer, Vitamine B | 10.86 | 2.53 | 3.70 | 0.95 | 2.94 | 0.044* |
| Glycerophospho choline | Glycerol-lipid metabolism, osmolytes | 2532.93 | 637.55 | 397.00 | 134.11 | 6.38 | 0.037* |
Welch’s t-test was used to evaluate the ratios of Acidic vs. Neutral conditions in osteosarcoma (OS) cells.
p < 0.05.
Low pH promotes the accumulation of amino sugar and nucleotide metabolites
Through the analysis of amino sugar metabolism (Figure 2A), we also observed a specific increase in β-D-N-acetylglucosamine (GlcNAc) and of its precursor N-acetylglucosamine-6-phosphate (NAcGlcNP), only in cancer cells. The increase in GlcNAc and of NAcGlcNP was associated with a reduction of pyrimidine base UTP and of purine bases ATP, CTP, and GTP; and with an accumulation of guanine, guanosine, and adenine (Figure 3A and Supplementary Table S1). Adenine and adenosine accumulation might be associated with their increased conversion to methyl-thioadenosine (MTA) and S-adenosylmethionine (SAM). We further analysed porphyrin and Gly/Ser/Cys metabolic pathways, and we noticed an increase in the level of spermine, MTA and SAM in OS cells (Figure 3B). In addition to their specific metabolic function, decreases of NAD+, UTP, and N8-acetylspermidine (Table 1) can share a common feature to chromatin regulation. Our profiling data based on the metabolomics approach suggest that metabolic adaptations under acidosis can influence tumour epigenetic alteration as a survival response to a hostile acidic microenvironment already after 24 h (Figure 4). Therefore, we decided to evaluate the methylation and acetylation profiles in our model after short-term exposure to acidosis.
Figure 3.
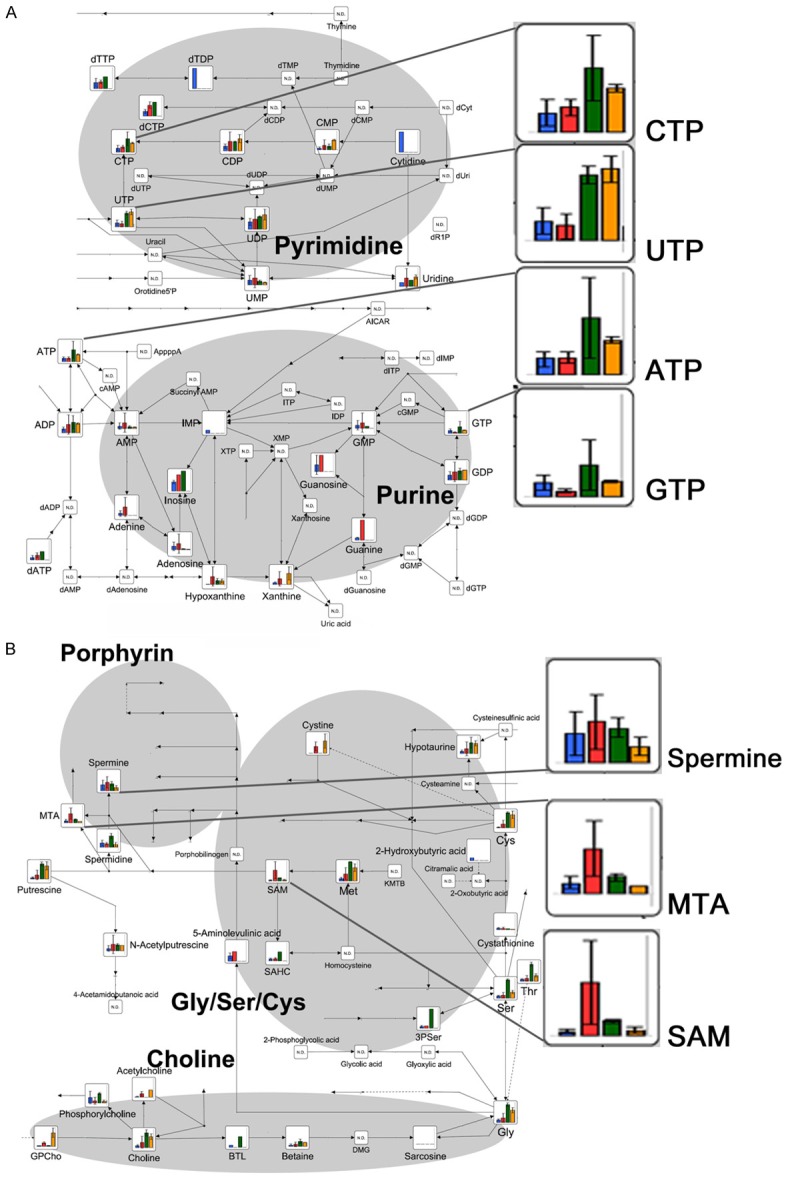
CE-TOFMS analysis of intracellular metabolites related to nucleotide and to Gly/Ser/Cys metabolic pathway after 24 h of exposure to acidosis: Bar graphs show the relative intracellular amount of metabolites. The bars correspond to the mean values of the three OS cell lines under neutral and acidic conditions (blue and red, respectively), and to the mean of the two normal Fb cultures under neutral and acidic conditions (green and orange, respectively), mean ± SD. Metabolic pathway maps were first written by VANTED software at HMT, and the copyright has been provided to the authors. A. Purine and pyrimidine metabolic pathways; B. Gly/Ser/Cys and porphyrin metabolic pathways.
Figure 4.
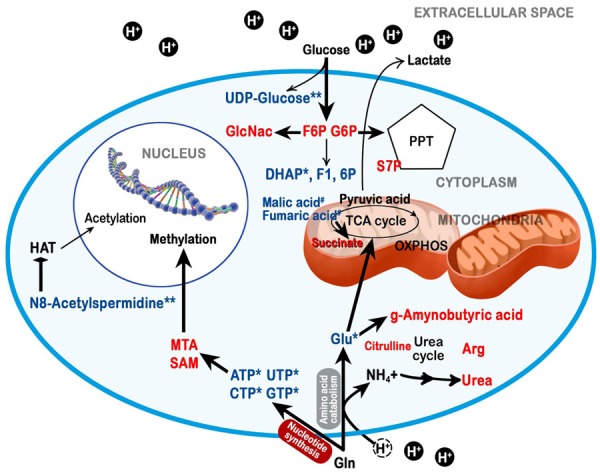
Graphical representation of the metabolic adaptations under acidosis of OS cells, as revealed by the CE-TOFMS analysis: Intracellular metabolites of tumour cells cultured in acidic medium that are reduced (in blue) or increased (in red) compared with normal cells cultured in acidic medium (0.01 ≤ *p < 0.05 and **p < 0. 01), or with tumour cells cultured under neutral conditions (#p < 0.05). The graphics are original and made by the authors.
Tumour cells that adapt to acidosis have a higher epigenetic stability
To evaluate the methylation and acetylation profiles in our model after short-term exposure to acidosis, OS cells were compared to three different primary cultures of mesenchymal stromal cells (MSCs), as an additional model of normal cells of mesenchymal lineage in addition to Fb. The profile of methylated genomic DNA was evaluated by the COBRA assay. We found that an acidic extracellular environment at pH 6.5 for 24 hrs induced demethylation of genomic DNA in normal cells of mesenchymal origin, but not in OS cells (Figure 5A). Then, we analysed the acetylation state of OS compared with the state of untransformed cells (Fb and MSCs) under acidic conditions. In particular, we analysed the status of histone deacetylase 1 (HDAC1) and of acetyl-histone H3 (Ac-H3) by Western blotting (Figure 5B). Interestingly, the acidic extracellular environment did not affect the acetylation profile of OS cells, whereas it significantly reduced HDAC1 and enhanced Ac-H3 expression (Figure 5B) in almost all primary cultures of normal cells. The reduction of HDAC1 expression specifically only in normal cells was also confirmed by transcriptional screening. The other HDAC and histone acetyltransferases (HATs) did not show any significant changes (Supplementary Table S2).
Figure 5.

Methylated DNA and histone H3 acetylation in transformed and normal cells under acidosis: (A) Methylation profile of genomic DNA was screened by COBRA for LINE1 in OS cells, Fb, and MSCs under neutral (N) and acidic (A) conditions. Representative image (upper panel), and bar graph (lower panel) of the densitometric analysis (mean ± SE, *p < 0.05, n = 3). DNA from NEC8, a testicular embryonal carcinoma, was used as an unmethylated control for LINE1. (B) HDAC1 and acetylated histone H3 (Ac-H3) were evaluated by immunoblots in OS cells, Fb, and MSCs under both neutral (N) and acidic (A) conditions using specific antibodies. The graphs (lower panel) show the densitometric analyses using α-tubulin as control (mean ± SE, *p < 0.05, n = 4).
Since we realized from our data that the maintenance of histone acetylation state is important in OS cells cultured under acidic conditions, we finally investigated the effect of HDAC inhibitors to be proposed as a targeted therapy in acidifying tumour, like OS. We treated HOS cells with the class I/IIb HDAC inhibitor (HDACi) MC1742 [27,28] under acidic and neutral conditions. First, the specific activity of the drug was confirmed to block HDAC enzymatic activity by Western blotting analysis at both pHs (pH 7.4 and 6.5) (Figure 6A). Furthermore, the treatment with MC1742 induced a higher dose-dependent inhibition of cell viability under acidosis than under normal pH conditions (Figure 6B). The growth inhibition induced by HDAC under acidosis is possibly due to a block in S phase, in addition to G2 phase, as we observed by cell cycle analysis (Figure 6C and 6D).
Figure 6.
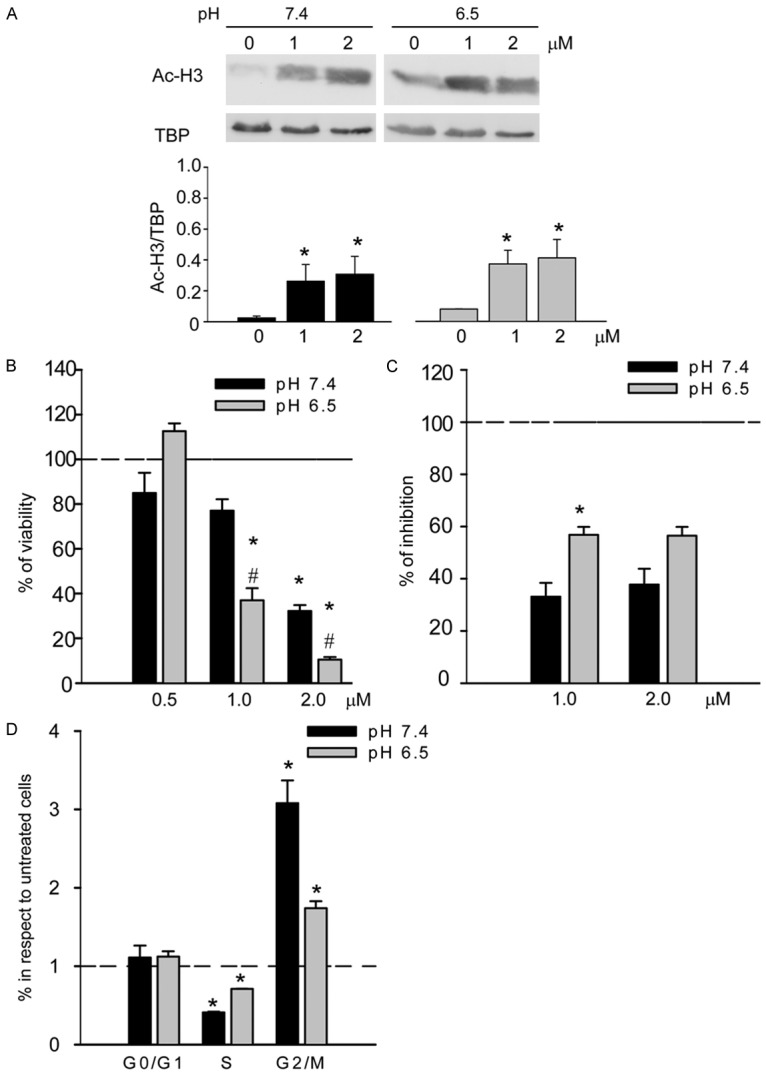
Effect of class I/IIb HDACi on HOS osteosarcoma cells under acidosis: A. Western blotting analysis of the acetylation state of histone H3 after the treatment with the HDACi MC1742 at different pHs (upper panel, representative image; lower panel, graph relative to the quantification of the bands by software analysis; *p < 0.05, n = 3); B. Analysis of cell viability by acid phosphate indirect assay after the treatment with HDACi (mean ± SE, #p vs pH 7.4 at the respective concentration, *p vs. ctr. at the respective pH; # or *p < 0.05, n = 4); C. Analysis of the HDACi effect on the number of cells, counted after 72 h of treatment with different concentrations of HDACi. Percentages of inhibitions were calculated compared with untreated cells at the respective pH (mean ± SE, *p < 0.05, n = 3); D. Cell cycle analysis of HOS cells treated with HDACi 1 μM at different pHs. Ratio of percentages of treated/untreated cells at the respective pH, mean ± SE (*p < 0.05).
Discussion
The role of intratumoral acidosis in cancer progression is increasingly appreciated especially on invasive capacity and the formation of metastasis. Also in osteosarcoma, we recently demonstrated that acidosis promotes tumour aggressiveness [4]. In this study, we wanted to further evaluate the feedback effect of this biochemical alteration associated with the tumour microenvironment and caused by the altered metabolism of glycolytic tumour cells. In particular, we focused on the rapid metabolic response to this stressing condition. Through the metabolic profile, we found that in OS cells 14 metabolites were significantly decreased. These were almost all associated with reduced glycolysis and amino acid catabolism. In addition, we found significant reductions of NAD+, UTP, and N8-acetylspermidine that are more related to epigenetic alteration (Table 1 and Figure 4). Notably, tumour and normal cells cultured under short-term acidosis are not dying cells, as we directly confirmed through the use of viability staining, and the metabolic alterations that we observed in our model define the first reaction of tumour cells to adapt to a hostile microenvironment. Our preliminary data suggested a reduced glycolytic metabolism of OS cells cultured under acidosis, as already demonstrated in other cancer [11]. The adapted metabolism to extracellular acidosis with a reduced glycolytic flux affected the proliferation rate of OS cells leading to an increase in the number of cells in G0 phase (Supplementary Figure S4). Impaired glycolysis was also associated with reduced lactate release in OS cell culture supernatants, thus confirming a repression of lactate fermentation [29]. Reductions of both glycolysis and lactate release in response to extracellular acidosis may be an efficient feedback system to avoid a further acidification of the extracellular space that would ultimately lead to an excessive acidification of the cytosol and cell death. Adversely, PPP intermediates were unmodified, indicating that acidosis induces a metabolic adaptation from glycolysis to PPP metabolism, as reported in other cancers [11,12]. Similarly, the maintained and/or increased expression of the complexes of OXPHOS, indirectly indicated that mitochondrial respiration is still used under acidosis [12]. Finally, NADH/NAD+ ratio was drastically reduced under acidosis in OS cells (Table 2), suggesting that OS metabolisms shifted from glycolysis to OXPHOS-TCA.
Accumulation of succinate is a common feature of several cancers [29] and NADH-fumarate reductase (NADH-FR) system has been involved in energy production in some cancers [30-33]. Interestingly, under the experimental acidic condition, malic and fumaric acids were significantly decreased (Table 3), and succinic acid showed a trend for accumulation only in malignant OS cells (Figure 2B). In NADH-FR system, NADH serves as the electron and proton donor, whereas fumarate serves as the ultimate electron and proton acceptor, with succinate as an end product. NADH-FR system still operates when the TCA cycle is defective and oxygen is limited; therefore, NADH-FR system should be considered as a potential source of energy to be further explored in cancer cells in hypoxic and acidic microenvironments. Several inhibitors of the NADH-FR system [34,35] or of complex II [36] at the core of this type of alternative respiration have been studied as new anticancer agents and should also be considered for the treatment of hypoxic or acidic cancers like OS. Alternatively, in the case of reduced glucose fermentation, amino acid consumption (e.g., glutaminolysis), another valuable source of energy, is notoriously associated with metabolic acidosis [37,38] and can be a main source of energy production in cancer [39-41], also in an acidic microenvironment [12]. In our models, a decrease in glutamate (Glu) concentration suggested an increase in glutaminolysis, and a strong increase in all the intermediates of the urea cycle was found in OS cells under acidosis. Under physiological conditions, a Glu/Gln cycle has been associated with increased urea cycle rate in human hepatocytes that detoxify ammonia produced from high amino acid consumption [42-44]. Therefore, the use of Glu analogues such as L-glutamic acid γ-monohydroxamate (GAH) in melanomas [45] could be explored in the future as an anticancer strategy in OS. Under acidosis, the formation of ammonia to fuel the urea cycle is also an efficient system to reduce the excess of intracellular protons.
We also observed a specific increase in GlcNAc and NAcGlcNP in cancer cells. GlcNac has been associated with O-GlcNacylation, which is important for transcriptional control [46]. Notably, we recently demonstrated an association between the accumulation of amino sugars and glucose deprivation in cancer cells [47,48]. The increase in GlcNAc and of NAcGlcNP was associated with a reduction of pyrimidine base UTP and of purine bases ATP, CTP, and GTP; and with an accumulation of guanine, guanosine, and adenine. Accumulation of guanine and guanosine might be a consequence of increased nucleotide synthesis from Glu, coupled with guanine deaminase deficiency in acidic OS, a feature that is often observed in malignancies [49,50]. Identifying the type of nucleotide necessary for cancer survival under stress conditions such as acidosis could have an impact in cancer therapy since several specific nucleotide analogues are already available and can be used in combination with conventional chemotherapy [51,52]. Adenine and adenosine accumulation might be associated with their increased conversion to MTA and SAM; and increases of spermine, MTA and SAM were noticed in OS cells, indeed. MTA and SAM are methyl donors for various substrates, such as nucleic acids, proteins, and lipids [53,54]. In addition to their specific metabolic function, decreases of NAD+, UTP, and N8-acetylspermidine can share a common feature to chromatin regulation [55,56]. On the basis of the metabolomics data, we can conclude that the availability of certain metabolic substrates is specifically changed during acidosis in OS cells, thus giving a selective advantage to tumour cells. Several metabolites have also been associated with epigenetic modifications, and metabolic adaptations to a stress condition have been linked to epigenetic regulation [57]. The ability to reprogram metabolic activities under stress conditions is responsible for tumour plasticity. Under acidosis, the metabolic reprogramming of cancer cells is associated with the prompt ability to modulate the expression of different ion/proton transporters [58,59] or to increase the lysosomal compartment [60]. On the other side, it has been reported that acidosis can strongly affect the response to hypoxia through impairment of HIF-1, inflammation and the unfolded protein response [61], thus regulating genomic transcriptional outputs. Indeed, our profiling data based on the metabolomics approach could suggest that metabolic adaptations under acidosis influence tumour epigenetic alteration as a survival response to a hostile acidic microenvironment already after 24 h (Figure 4). So far, the relationship between pH and epigenetic modifications has been barely studied in cancer. Few reports indicate that histone modification is associated with intracellular pH regulation [16,62]. Furthermore, Corbet et al. [12] demonstrated that the enzymatic activity of NAD-dependent deacetylase sirtuin-1 (SIRT1) is differently modulated in cells maintained under chronic acidosis. Similarly, we investigated the methylation and acetylation profiles in our model after short exposure to acidosis to evaluate the rapid adaptation of OS cells to this stressing condition. Interestingly, COBRA assay and acetyl-H3 immunoblotting indicated an epigenetic stability in OS cells than in normal cells. The maintenance of the epigenetic profile under stressing conditions is a key mechanism for cancer cells to survive and to promote genome integrity [63] and cloning efficiency [64]. Furthermore, when we treated OS cells under acidic and neutral conditions with an HDACi MC1742 [27,28], OS cells were more sensitive to the agent under acidosis than under neutral pH. At low pH, the growth inhibition induced by MC1742 was due to a block in G2 phase, as reported previously for this class of inhibitors [65], with an additional increase number of cells blocked also in the S phase. Our results thus suggest that the use of HDAC inhibitors or other epigenetic modulators can affect cancer cells also under an acidic microenvironment, a condition associated with chemoresistance in cancers, and should be considered as a possibly therapeutic strategy in the near future, especially for acidifying tumors like OS [66,67].
In conclusion, this study demonstrated an extensive reprogramming and adaptations of the metabolism of normal and malignant cells of mesenchymal origin after an acidic microenvironment in the short-term. In particular, we found: 1) a reduced glycolytic rate in both normal and cancer cells; 2) a shift from a Warburg glycolytic metabolism to OXPHOS and TCA cycles possibly associated with a higher consumption rate of amino acid coupled to an increase of urea cycle, especially in cancer cells; 3) the metabolic pattern observed in cancer cells cultured under acidosis could potentially support the maintenance of the epigenetic marks, and finally ensure DNA replication. These data offer novel opportunities for the identification of effective drugs that can selectively target malignant tumour cells in acidic microenvironments.
Acknowledgements
The authors thank Hiroko Kita (Shiga University of Medical Science) and Silvia Lemma (Istituto Ortopedico Rizzoli) for experimental assistance in immunoblot analysis and epigenomic screening. The study was partly supported by KAKENHI (Grant-in-Aid for Scientific Research, n. 25293130 to T.C.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, from AIRC (Associazione Italiana per la Ricerca sul Cancro, n. 11426 to N.B.), from IIT-Sapienza Project (to A.M.), and from FP7 Projects BLUEPRINT/282510 and A-PARADDISE/602080 (to A.M.).
Disclosure of conflict of interest
None.
Authors’ contribution
T.C., S.A., K.K., P.S., A.M. and N.B. designed the research and wrote the paper. T.C. did all the metabolomics experiments and the acetylation, methylation, and data analyses. D.R. prepared the HDACi MC1742. S.A. and G.B. performed cellular analysis. S.A. also analysed the data.
Supporting Information
References
- 1.Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 4.Avnet S, Di Pompo G, Lemma S, Salerno M, Perut F, Bonuccelli G, Granchi D, Zini N, Baldini N. V-ATPase is a candidate therapeutic target for Ewing sarcoma. Biochim Biophys Acta. 2013;1832:1105–1116. doi: 10.1016/j.bbadis.2013.04.003. [DOI] [PubMed] [Google Scholar]
- 5.Perut F, Avnet S, Fotia C, Baglio SR, Salerno M, Hosogi S, Kusuzaki K, Baldini N. V-ATPase as an effective therapeutic target for sarcomas. Exp Cell Res. 2014;320:21–32. [PubMed] [Google Scholar]
- 6.Goldblatt H, Friedman L, Cechner RL. On the malignant transformation of cells during prolonged culture under hypoxic conditions in vitro. Biochem Med. 1973;7:241–252. doi: 10.1016/0006-2944(73)90079-3. [DOI] [PubMed] [Google Scholar]
- 7.Harguindey S, Pedraz JL, Garcia Canero R, Perez de Diego J, Cragoe EJ Jr. Hydrogen ion-dependent oncogenesis and parallel new avenues to cancer prevention and treatment using a H(+)-mediated unifying approach: pHrelated and pH-unrelated mechanisms. Crit Rev Oncog. 1995;6:1–33. doi: 10.1615/critrevoncog.v6.i1.20. [DOI] [PubMed] [Google Scholar]
- 8.Chiche J, Brahimi-Horn MC, Pouyssegur J. Tumour hypoxia induces a metabolic shift causing acidosis: a common feature in cancer. J Cell Mol Med. 2010;14:771–794. doi: 10.1111/j.1582-4934.2009.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seyfried TN, Flores RE, Poff AM, D’Agostino DP. Cancer as a metabolic disease: implications for novel therapeutics. Carcinogenesis. 2014;35:515–527. doi: 10.1093/carcin/bgt480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dang CV. Glutaminolysis: supplying carbon or nitrogen or both for cancer cells? Cell Cycle. 2010;9:3884–3886. doi: 10.4161/cc.9.19.13302. [DOI] [PubMed] [Google Scholar]
- 11.LaMonte G, Tang X, Chen JL, Wu J, Ding CK, Keenan MM, Sangokoya C, Kung HN, Ilkayeva O, Boros LG, Newgard CB, Chi JT. Acidosis induces reprogramming of cellular metabolism to mitigate oxidative stress. Cancer Metab. 2013;1:23. doi: 10.1186/2049-3002-1-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Corbet C, Draoui N, Polet F, Pinto A, Drozak X, Riant O, Feron O. The SIRT1/HIF2alpha axis drives reductive glutamine metabolism under chronic acidosis and alters tumor response to therapy. Cancer Res. 2014;74:5507–5519. doi: 10.1158/0008-5472.CAN-14-0705. [DOI] [PubMed] [Google Scholar]
- 13.Monton MR, Soga T. Metabolome analysis by capillary electrophoresis-mass spectrometry. J Chromatogr A. 2007;1168:237–246. doi: 10.1016/j.chroma.2007.02.065. discussion 236. [DOI] [PubMed] [Google Scholar]
- 14.Sassone-Corsi P. Physiology. When metabolism and epigenetics converge. Science. 2013;339:148–150. doi: 10.1126/science.1233423. [DOI] [PubMed] [Google Scholar]
- 15.Riemann A, Ihling A, Schneider B, Gekle M, Thews O. Impact of extracellular acidosis on intracellular pH control and cell signaling in tumor cells. Adv Exp Med Biol. 2013;789:221–228. doi: 10.1007/978-1-4614-7411-1_30. [DOI] [PubMed] [Google Scholar]
- 16.McBrian MA, Behbahan IS, Ferrari R, Su T, Huang TW, Li K, Hong CS, Christofk HR, Vogelauer M, Seligson DB, Kurdistani SK. Histone acetylation regulates intracellular pH. Mol Cell. 2013;49:310–321. doi: 10.1016/j.molcel.2012.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen H, Laird PW. Interplay between the cancer genome and epigenome. Cell. 2013;153:38–55. doi: 10.1016/j.cell.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soga T, Heiger DN. Amino acid analysis by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem. 2000;72:1236–1241. doi: 10.1021/ac990976y. [DOI] [PubMed] [Google Scholar]
- 19.Soga T, Ueno Y, Naraoka H, Ohashi Y, Tomita M, Nishioka T. Simultaneous determination of anionic intermediates for Bacillus subtilis metabolic pathways by capillary electrophoresis electrospray ionization mass spectrometry. Anal Chem. 2002;74:2233–2239. doi: 10.1021/ac020064n. [DOI] [PubMed] [Google Scholar]
- 20.Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J Proteome Res. 2003;2:488–494. doi: 10.1021/pr034020m. [DOI] [PubMed] [Google Scholar]
- 21.Sugimoto M, Wong DT, Hirayama A, Soga T, Tomita M. Capillary electrophoresis mass spectrometry-based saliva metabolomics identified oral, breast and pancreatic cancer-specific profiles. Metabolomics. 2010;6:78–95. doi: 10.1007/s11306-009-0178-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junker BH, Klukas C, Schreiber F. VANTED: a system for advanced data analysis and visualization in the context of biological networks. BMC Bioinformatics. 2006;7:109. doi: 10.1186/1471-2105-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Frevel MA, Sowerby SJ, Petersen GB, Reeve AE. Methylation sequencing analysis refines the region of H19 epimutation in Wilms tumor. J Biol Chem. 1999;274:29331–29340. doi: 10.1074/jbc.274.41.29331. [DOI] [PubMed] [Google Scholar]
- 25.Yang AS, Estecio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32:e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ushida H, Kawakami T, Minami K, Chano T, Okabe H, Okada Y, Okamoto K. Methylation profile of DNA repetitive elements in human testicular germ cell tumor. Mol Carcinog. 2012;51:711–722. doi: 10.1002/mc.20831. [DOI] [PubMed] [Google Scholar]
- 27.Mai A, Massa S, Rotili D, Simeoni S, Ragno R, Botta G, Nebbioso A, Miceli M, Altucci L, Brosch G. Synthesis and biological properties of novel, uracil-containing histone deacetylase inhibitors. J Med Chem. 2006;49:6046–6056. doi: 10.1021/jm0605536. [DOI] [PubMed] [Google Scholar]
- 28.Di Pompo G, Salerno M, Rotili D, Valente S, Zwergel C, Avnet S, Lattanzi G, Baldini N, Mai A. Novel Histone Deacetylase Inhibitors Induce Growth Arrest, Apoptosis, and Differentiation in Sarcoma Cancer Stem Cells. J Med Chem. 2015;58:4073–9. doi: 10.1021/acs.jmedchem.5b00126. [DOI] [PubMed] [Google Scholar]
- 29.Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, Casimiro MC, Aquila S, Ando S, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget. 2012;3:798–810. doi: 10.18632/oncotarget.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, Kinoshita T, Saito N, Ochiai A, Tomita M, Esumi H, Soga T. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time-of-flight mass spectrometry. Cancer Res. 2009;69:4918–4925. doi: 10.1158/0008-5472.CAN-08-4806. [DOI] [PubMed] [Google Scholar]
- 31.Chen Z, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007;39:267–274. doi: 10.1007/s10863-007-9086-x. [DOI] [PubMed] [Google Scholar]
- 32.Tomitsuka E, Kita K, Esumi H. The NADHfumarate reductase system, a novel mitochondrial energy metabolism, is a new target for anticancer therapy in tumor microenvironments. Ann N Y Acad Sci. 2010;1201:44–49. doi: 10.1111/j.1749-6632.2010.05620.x. [DOI] [PubMed] [Google Scholar]
- 33.Sakai C, Tomitsuka E, Miyagishi M, Harada S, Kita K. Type II Fp of human mitochondrial respiratory complex II and its role in adaptation to hypoxia and nutrition-deprived conditions. Mitochondrion. 2013;13:602–609. doi: 10.1016/j.mito.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Tomitsuka E, Kita K, Esumi H. An anticancer agent, pyrvinium pamoate inhibits the NADH-fumarate reductase system--a unique mitochondrial energy metabolism in tumour microenvironments. J Biochem. 2012;152:171–183. doi: 10.1093/jb/mvs041. [DOI] [PubMed] [Google Scholar]
- 35.Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: Treating cancer like an infectious disease. Oncotarget. 2015;6:4569–4584. doi: 10.18632/oncotarget.3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sakai C, Tomitsuka E, Esumi H, Harada S, Kita K. Mitochondrial fumarate reductase as a target of chemotherapy: from parasites to cancer cells. Biochim Biophys Acta. 2012;1820:643–651. doi: 10.1016/j.bbagen.2011.12.013. [DOI] [PubMed] [Google Scholar]
- 37.Epler MJ, Souba WW, Meng Q, Lin C, Karinch AM, Vary TC, Pan M. Metabolic acidosis stimulates intestinal glutamine absorption. J Gastrointest Surg. 2003;7:1045–1052. doi: 10.1016/j.gassur.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 38.Adam W, Simpson DP. Glutamine transport in rat kidney mitochondria in metabolic acidosis. J Clin Invest. 1974;54:165–174. doi: 10.1172/JCI107738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McKeehan WL. Glycolysis, glutaminolysis and cell proliferation. Cell Biol Int Rep. 1982;6:635–650. doi: 10.1016/0309-1651(82)90125-4. [DOI] [PubMed] [Google Scholar]
- 40.Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- 41.Mazurek S. Tumor cell energetic metabolome. Weinheim, Germany: Wiley-VCH; 2007. [Google Scholar]
- 42.Haussinger D, Lamers WH, Moorman AF. Hepatocyte heterogeneity in the metabolism of amino acids and ammonia. Enzyme. 1992;46:72–93. doi: 10.1159/000468779. [DOI] [PubMed] [Google Scholar]
- 43.Haussinger D. Hepatic glutamine transport and metabolism. Adv Enzymol Relat Areas Mol Biol. 1998;72:43–86. doi: 10.1002/9780470123188.ch3. [DOI] [PubMed] [Google Scholar]
- 44.Haussinger D. Nitrogen metabolism in liver: structural and functional organization and physiological relevance. Biochem J. 1990;267:281–290. doi: 10.1042/bj2670281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomasset N, Chabanas A, Tournaire R, Malley S, Navarro C, Lawrence JJ, Dore JF, Vila J. Selective cytotoxicity of L-glutamic acid gamma-monohydroxamate (GAH) for melanoma tumor cells. Anticancer Res. 1993;13:1393–1398. [PubMed] [Google Scholar]
- 46.Slawson C, Hart GW. O-GlcNAc signalling: implications for cancer cell biology. Nat Rev Cancer. 2011;11:678–684. doi: 10.1038/nrc3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Isono T, Chano T, Okabe H, Suzaki M. Study of global transcriptional changes of N-GlcNAc2 proteins-producing T24 bladder carcinoma cells under glucose deprivation. PLoS One. 2013;8:e60397. doi: 10.1371/journal.pone.0060397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Isono T, Chano T, Kitamura A, Yuasa T. Glucose Deprivation Induces G2/M Transition-Arrest and Cell Death in N-GlcNAc2-Modified Protein-Producing Renal Carcinoma Cells. PLoS One. 2014;9:e96168. doi: 10.1371/journal.pone.0096168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Durak I, Cetin R, Canbolat O, Cetin D, Yurtarslani Z, Unal A. Adenosine deaminase, 5’-nucleotidase, guanase and cytidine deaminase activities in gastric tissues from patients with gastric cancer. Cancer Lett. 1994;84:199–202. doi: 10.1016/0304-3835(94)90376-x. [DOI] [PubMed] [Google Scholar]
- 50.Snyder FF, Cruikshank MK, Seegmiller JE. A comparison of purine metabolism and nucleotide pools in normal and hypoxanthine-guanine phosphoribosyltransferase-deficient neuroblastoma cells. Biochim Biophys Acta. 1978;543:556–569. doi: 10.1016/0304-4165(78)90310-0. [DOI] [PubMed] [Google Scholar]
- 51.Bertino JR, Waud WR, Parker WB, Lubin M. Targeting tumors that lack methylthioadenosine phosphorylase (MTAP) activity: current strategies. Cancer Biol Ther. 2011;11:627–632. doi: 10.4161/cbt.11.7.14948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Skipper HE, Robins RK, Thomson JR, Cheng CC, Brockman RW, Schabel FM Jr. Structure-activity relationships observed on screening a series of pyrazolopyrimidines against experimental neoplasms. Cancer Res. 1957;17:579–596. [PubMed] [Google Scholar]
- 53.Huang S. Histone methyltransferases, diet nutrients and tumour suppressors. Nat Rev Cancer. 2002;2:469–476. doi: 10.1038/nrc819. [DOI] [PubMed] [Google Scholar]
- 54.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–597. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gut P, Verdin E. The nexus of chromatin regulation and intermediary metabolism. Nature. 2013;502:489–498. doi: 10.1038/nature12752. [DOI] [PubMed] [Google Scholar]
- 56.Brett-Morris A, Wright BM, Seo Y, Pasupuleti V, Zhang J, Lu J, Spina R, Bar EE, Gujrati M, Schur R, Lu ZR, Welford SM. The polyamine catabolic enzyme SAT1 modulates tumorigenesis and radiation response in GBM. Cancer Res. 2014;74:6925–6934. doi: 10.1158/0008-5472.CAN-14-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Johnson C, Warmoes MO, Shen X, Locasale JW. Epigenetics and cancer metabolism. Cancer Lett. 2015;356:309–314. doi: 10.1016/j.canlet.2013.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Parks SK, Mazure NM, Counillon L, Pouyssegur J. Hypoxia promotes tumor cell survival in acidic conditions by preserving ATP levels. J Cell Physiol. 2013;228:1854–1862. doi: 10.1002/jcp.24346. [DOI] [PubMed] [Google Scholar]
- 59.Xu K, Mao X, Mehta M, Cui J, Zhang C, Mao F, Xu Y. Elucidation of how cancer cells avoid acidosis through comparative transcriptomic data analysis. PLoS One. 2013;8:e71177. doi: 10.1371/journal.pone.0071177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salerno M, Avnet S, Bonuccelli G, Hosogi S, Granchi D, Baldini N. Impairment of lysosomal activity as a therapeutic modality targeting cancer stem cells of embryonal rhabdomyosarcoma cell line RD. PLoS One. 2014;9:e110340. doi: 10.1371/journal.pone.0110340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tang X, Lucas JE, Chen JL, LaMonte G, Wu J, Wang MC, Koumenis C, Chi JT. Functional interaction between responses to lactic acidosis and hypoxia regulates genomic transcriptional outputs. Cancer Res. 2012;72:491–502. doi: 10.1158/0008-5472.CAN-11-2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurdistani SK. Histone modifications in cancer biology and prognosis. Prog Drug Res. 2011;67:91–106. doi: 10.1007/978-3-7643-8989-5_5. [DOI] [PubMed] [Google Scholar]
- 63.Bosch-Presegue L, Vaquero A. Sirtuindependent epigenetic regulation in the maintenance of genome integrity. FEBS J. 2015;282:1745–1767. doi: 10.1111/febs.13053. [DOI] [PubMed] [Google Scholar]
- 64.Morita T, Nagaki T, Fukuda I, Okumura K. Clastogenicity of low pH to various cultured mammalian cells. Mutat Res. 1992;268:297–305. doi: 10.1016/0027-5107(92)90235-t. [DOI] [PubMed] [Google Scholar]
- 65.Li Y, Li X, Guo B. Chemopreventive agent 3,3’-diindolylmethane selectively induces proteasomal degradation of class I histone deacetylases. Cancer Res. 2010;70:646–654. doi: 10.1158/0008-5472.CAN-09-1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Thews O, Riemann A, Nowak M, Gekle M. Impact of hypoxia-related tumor acidosis on cytotoxicity of different chemotherapeutic drugs in vitro and in vivo. Adv Exp Med Biol. 2014;812:51–58. doi: 10.1007/978-1-4939-0620-8_7. [DOI] [PubMed] [Google Scholar]
- 67.Raghunand N, Martinez-Zaguilan R, Wright SH, Gillies RJ. pH and drug resistance. II. Turnover of acidic vesicles and resistance to weakly basic chemotherapeutic drugs. Biochem Pharmacol. 1999;57:1047–1058. doi: 10.1016/s0006-2952(99)00021-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.