

Summary
Pluripotent genomes are folded in a topological hierarchy that reorganizes during differentiation. The extent to which chromatin architecture is reconfigured during somatic cell reprogramming is poorly understood. Here we integrate fine-resolution architecture maps with epigenetic marks and gene expression in embryonic stem (ES) cells, neural progenitor cells (NPCs) and NPC-derived induced pluripotent stem (iPS) cells. We find that most pluripotency genes reconnect to target enhancers during reprogramming. Unexpectedly, some NPC interactions around pluripotency genes persist in our iPS clone. Pluripotency genes engaged in both ‘fully-reprogrammed-ES’ and ‘persistent-NPC’ interactions exhibit over/undershooting of target expression levels in iPS. Additionally, we identify a subset of ‘poorly-reprogrammed’ interactions that do not reconnect in iPS and display only partially recovered, ES-specific CTCF occupancy. 2i/LIF can abrogate ‘persistent-NPC’ interactions, recover ‘poorly-reprogrammed’ interactions, re-instate CTCF occupancy and restore expression levels. Our results demonstrate that iPS genomes can exhibit imperfectly rewired 3D-folding linked to inaccurately reprogrammed gene expression.
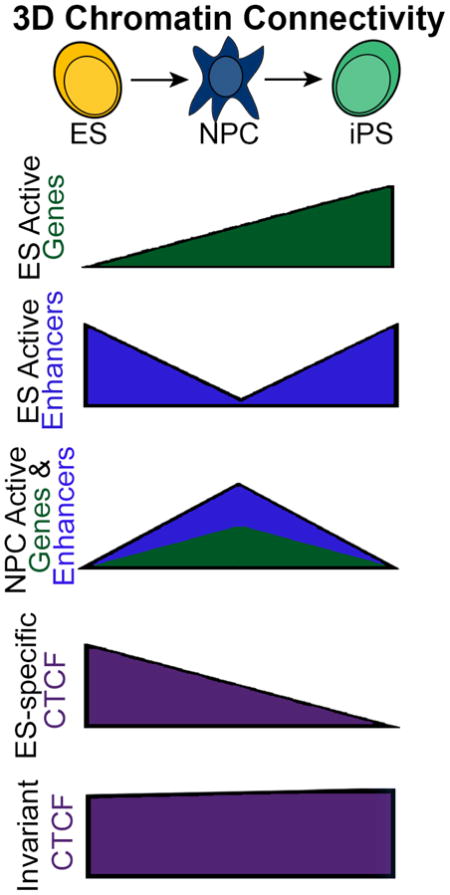
Graphical abstract
Introduction
Mammalian genomes are folded in a hierarchy of architectural configurations that are intricately linked to cellular function. Individual chromosomes are arranged in distinct territories and then are further partitioned into a nested series of Megabase (Mb)-sized topologically associating domains (TADs) (Dixon et al., 2012; Nora et al., 2012) and smaller sub-domains (sub-TADs) (Phillips-Cremins et al., 2013; Rao et al., 2014). TADs/subTADs vary widely in size (i.e. 40 kb - 3 Mb) and are characterized by highly self-associating chromatin fragments demarcated by boundaries of abruptly decreased interaction frequency. Long-range looping interactions connect distal genomic loci within and between TADs/subTADs (Jin et al., 2013; Phillips-Cremins et al., 2013; Rao et al., 2014; Sanyal et al., 2012). Single TADs, or a series of successive TAD/subTADs, in turn congregate into spatially proximal, higher-order clusters termed ‘A/B compartments’. Compartments generally fall into two classes: (i) ‘A’ compartments enriched for open chromatin, highly expressed genes and early replication timing and (ii) ‘B’ compartments enriched for closed chromatin, late replication timing and co-localization with the nuclear periphery (Dixon et al., 2015; Lieberman-Aiden et al., 2009; Pope et al., 2014; Rao et al., 2014). The organizing principles governing genome folding at each length scale remain poorly understood.
Recent high-throughput genomics studies have shed new light on the dynamic nature of chromatin folding during embryonic stem (ES) cell differentiation. Up to 25% of compartments in human ES cells switch their A/B orientation upon differentiation (Dixon et al., 2015). Compartments that switch between A and B configurations display a modest, but correlated alteration in expression of only a small number of genes, suggesting that compartmental switching does not deterministically regulate cell type-specific gene expression (Dixon et al., 2015). Similarly, lamina associated domains are dynamically altered during ES cell differentiation (Peric-Hupkes et al., 2010). For example, the Oct4, Nanog and Klf4 genes relocate to the nuclear periphery in parallel with their loss of transcriptional activity as ES cells differentiate to astrocytes. TADs are largely invariant across cell types and often maintain their boundaries irrespective of the expression of their resident genes (Dixon et al., 2012). By contrast, long-range looping interactions within and between sub-TADs are highly dynamic during ES cell differentiation (Phillips-Cremins et al., 2013; Zhang et al., 2013b). Pluripotency genes connect to their target enhancers through long-range interactions and disruption of these interactions leads to a marked decrease in gene expression (Apostolou et al., 2013; Kagey et al., 2010). Thus, data is so far consistent with a model in which chromatin interactions at the sub-Mb scale (within TADs) are key effectors in the spatiotemporal regulation of gene expression during development.
In addition to the forward progression of ES cells in development, somatic cells can also be reprogrammed in the reverse direction to induced pluripotent stem (iPS) cells via the ectopic expression of key transcription factors (Takahashi and Yamanaka, 2006). Since the initial pioneering discovery, many population-based and single cell genomics studies have explored the molecular underpinnings of transcription factor-mediated reprogramming (Hanna et al., 2009; Koche et al., 2011; Rais et al., 2013; Soufi et al., 2012). Recent efforts have uncovered changes in transcription, cell surface markers and classic epigenetic modifications during intermediate stages in the reprogramming process (Buganim et al., 2012; Lujan et al., 2015; Polo et al., 2012). Although there is some evidence of epigenetic traces from the somatic cell of origin (Bock et al., 2011; Kim et al., 2010; Polo et al., 2010), the emerging model is that ES-like epigenetic and transcriptional states can be generally reset under proper reprogramming conditions (Stadtfeld et al., 2010).
The role for chromatin topology in the acquisition of pluripotency during reprogramming has not yet been elucidated. Recent studies have suggested that specific long-range interactions between Nanog and/or Oct4 and target enhancers can be reset during reprogramming and precede re-activation of the involved genes (Apostolou et al., 2013; de Wit et al., 2013; Denholtz et al., 2013; Wei et al., 2013; Zhang et al., 2013a). Beyond these initial locus-specific studies, it remains unknown whether the somatic cell genome unfolds/refolds at the sub-Mb scale within TADs and how chromatin topology is linked to gene expression changes during reprogramming. Here we report a detailed analysis of local chromatin folding changes during somatic cell reprogramming. We created ~4–12 kilobase (kb) resolution chromatin architecture maps in primary neural progenitor cells (NPCs), iPS cells derived from primary NPCs and pluripotent ES cells. We employed Chromosome-Conformation-Capture-Carbon-Copy (5C) to query fine-scale architectural changes in Mb-sized regions around key developmentally regulated genes. We find that chromatin folding is markedly reconfigured within TADs during the transition from primary NPCs to iPS cells. In many cases, pluripotency genes re-engage in fully reprogrammed interactions with their target ES-specific enhancers. Unexpectedly, we also observe NPC interactions around key pluripotency genes (e.g. Sox2, Klf4) that remain persistently tethered in our iPS clone. Pluripotency genes engaged in ‘persistent NPC-like’ interactions can exhibit over/under-shooting of gene expression levels in iPS, despite the fact that they may have also re-established contact with their target ES-specific enhancer(s). We also uncover a subset of ‘poorly reprogrammed’ interactions that break apart during differentiation and do not fully reconnect in our iPS clone. Many ‘poorly reprogrammed’ interactions exhibit ES-specific CTCF occupancy that is lost during differentiation and only partially recovered in iPS cells. Importantly, 2i/LIF conditions can (i) abrogate ‘persistent NPC-like’ interactions, (ii) recover ‘poorly reprogrammed’ interactions, (iii) re-instate inadequately reprogrammed CTCF occupancy and (iv) restore precise gene expression levels.
Results
Chromatin folding markedly reconfigures at the sub-Mb scale during reprogramming
To investigate changes in 3D chromatin topology during somatic cell reprogramming, we first generated ~4–12 kb-resolution chromatin architecture maps in primary NPCs, iPS cells derived from primary NPCs and ES cells (Fig. 1A). To achieve a comparable genetic background to our pluripotency model (V6.5 ES cells; 129/SvJae x C57BL/6), we selected a previously published iPS clone derived from primary NPCs isolated from neonatal brains of Sox2-green fluorescent protein (Sox2-GFP) indicator mice (mixed 129/SvJae x C57BL/6 genetic background) (Eminli et al., 2008; Stadtfeld et al., 2008). Hochedlinger and colleagues generated this iPS clone via the transduction of primary Sox2-GFP NPCs with doxycycline-inducible lentiviral vectors encoding Oct4, Klf4 and c-Myc. Importantly, this iPS clone was extensively characterized for its pluripotent properties as assessed by (i) expression of endogenous pluripotency markers (Oct4, Sox2, Nanog), (ii) demethylation of Oct4 and Nanog promoters, (iii) transgene-independent self renewal, (iv) in vivo teratoma formation of all three germ layers and (v) generation of chimeric mice (Eminli et al., 2008). Our three cellular states enable a detailed analysis of how chromatin unfolds/refolds between NPCs and iPS cells and also facilitate the comparison of genome topology between ES/iPS of comparable genetic background.
Figure 1. High-resolution architecture maps reveal marked chromatin reconfiguration during somatic cell reprogramming.

(A) Phase contrast images of the reprogramming model system. (B) Genome-wide ES cell Hi-C data (Dixon et al., 2012) at different bin sizes illustrating chromosome territories, A/B compartments and TADs. Images made with the Juicebox tool (http://www.aidenlab.org/juicebox/). The 4–12 kb resolution heatmaps from the present study query fine scale genome folding at the sub-Mb scale within TADs. (C) Relative contact frequency heatmaps are displayed for all biological replicates and regions queried. Color bars range from low (grey) to high (red/black) interaction frequencies. (D) Distance-corrected interaction score heatmaps for a select region around the Sox2 gene illustrating the presence of dynamic chromatin architecture among ES, NPC and iPS cells. Color bars range from low (blue) to high (red/black) interaction scores.
We employed 5C and high-throughput sequencing to create fine-scale chromatin architecture maps spanning > 7 Mb of the mouse genome within a set of TADs (Dostie et al., 2006). 5C combines Chromosome-Conformation-Capture (3C) with a primer-based hybrid capture step to facilitate cost-effective detection of sub-Mb scale interactions in Mb-sized loci of interest (Dekker et al., 2013). We used a tiled/alternating primer design around Nanog, Sox2, Klf4, Oct4, Nestin, and Olig1-Olig2 (described in detail (Phillips-Cremins et al., 2013)). Our 5C primer design scheme enabled the creation of ~4–12 kb resolution architecture maps for all loci combined across three cellular states with less than 30 million reads per replicate (Table S1). The power in this approach is that it focuses on elucidating fine scale architecture changes at the sub-Mb scale within TADs (Fig. 1B).
We first visualized 5C data with contact frequency heatmaps. To resolve underlying topological features, we developed an analysis pipeline to correct for known biases in 5C data and to normalize samples within and between biological replicates (described in detail Supplemental Experimental Procedures). Briefly, raw data (Fig. S1A) were quantile normalized to bring the dynamic range of all samples onto equivalent scales and to account for technical differences in sequencing depth and library complexity (Fig. S1B). To account for differences in primer efficiency that lead to non-uniformities in coverage across genomic regions, we applied our previously published primer correction algorithm to quantile-normalized data (Fig. S1C, (Phillips-Cremins et al., 2013)). We then applied a blocked binning/smoothing algorithm to attenuate spatial noise in 5C data (Fig. S1D). Our ‘Relative Contact Frequency’ heatmaps revealed striking topological patterns that are dynamic across cellular states and unique to each genomic region (Fig. 1C).
To further resolve the underlying architectural signal, we corrected for the known distance-dependence background in 5C data (Sanyal et al., 2012) (Fig. S1E–G). Consistent with recent reports (Rao et al., 2014), we found that a local distance-dependence model computed independently for each region would more precisely account for locus-specific differences in chromatin folding that are often over/under-estimated by a global background model (Fig. S1G). Our ‘Distance-Corrected Interaction Score’ heatmaps showed striking changes in topological features among NPCs, iPS and ES cells (Fig. 1D, Fig. S1E–F) with high consistency between replicates and marked differences among biological conditions (Table S2). A systematic comparative analysis at each stage in the pipeline confirmed that we have reduced known biases in 5C data (Figs. S1A–I, S2A–G).
iPS genomes can exhibit imperfectly rewired folding patterns
We next explored fine-scale chromatin folding features within TADs by visually inspecting our heatmaps. Consistent with our previous work, we observed marked changes in chromatin architecture between ES cells and NPCs. Importantly, we also noticed a striking architectural reconfiguration between NPCs and NPC-derived iPS cells (Fig. 1C–D). At many loci, iPS genome folding recapitulates the patterns seen in V6.5 ES cells. However, we also noticed several intriguing cases where iPS topology retained remnants of the folding patterns from NPCs (Fig. 1D).
To further explore the possibility that genome folding might be mis-wired during reprogramming, we conducted principal component analysis on our ‘Distance-Corrected Interaction Frequency’ data across all replicates and cellular states. Interestingly, we observed that genome topology in our iPS clone exhibited folding patterns that were intermediate between NPCs and the pluripotent stem cell state (Fig. 2A). To explore the functional significance of potential intermediate iPS folding patterns, we queried the transcriptome of all three cellular states using RNAseq. Consistent with our 3D observations, global gene expression profiles in our iPS clone were also parsed as intermediate between ES cells and NPCs (Fig. 2B). Together, these results support the possibility that genome architecture of some iPS clones might be imperfectly wired within TADs during reprogramming.
Figure 2. iPS genomes can exhibit intermediate folding and expression patterns between somatic and pluripotent stem cell states.

Principal component analysis of (A) distance-corrected interaction frequency data and (B) normalized RNAseq data for ES, NPC and iPS replicates. (A, B) Principal components 1 and 2 are scattered and the proportion of variance explained by each principal component is plotted below each scatterplot.
Dynamic 3-D interaction classes during cell fate transitions
To identify high-confidence, long-range interactions across all developmentally regulated loci, we fit our ‘Distance-Corrected Interaction Frequency’ data with a logistic distribution with location/scale parameters computed independently for each region (Fig. S3A, Supplemental Experimental Procedures). We then converted the p-values from our fitted models into an interaction score (− 10*log2(p-value)) that is comparable within and between experiments and allows for the robust detection of interactions that are significant above the expected background signal.
We next employed a thresholding strategy to classify 3D interactions by their dynamic contact frequencies across the three cellular states (Fig. 3A–D). To minimize false positives, we required that interaction scores cross the threshold boundaries in both replicates for a given biological condition. Moreover, we iteratively defined thresholds to achieve an empirical False Discovery Rate (eFDR) of < 10% when applied to simulated 5C replicates (Figs. 3E–H, S3B+C, Supplemental Experimental Procedures). Upon application of our classification scheme, we uncovered several dynamic interaction classes among ES, NPC and iPS cellular states (Figs. 3I–J), including: (i) 537 interactions present in ES cells, lost in NPCs and reacquired upon reprogramming (purple class) (Fig. 3K), (ii) 3004 interactions present only in ES cells and not reprogrammed (red class) (Fig. 3L), (iii) 5043 interactions absent in ES cells, acquired upon differentiation and lost in iPS cells (green class) (Fig. S3D), (iv) 1708 interactions present only in iPS cells (orange class) (Fig. S3E), (v) 148 interactions that are high in ES cells and NPCs and not present in iPS (gold class) (Fig. S3F) and (vi) 282 interactions absent in ES cells, acquired in NPCs and residually connected in iPS cells (blue class) (Fig. S3G). Noteworthy, we found that the sensitive detection of these interaction classes, particularly those that distinguish iPS from ES cells, was contingent upon the resolution and read depth afforded by the 5C approach (Figs. S3H–I).
Figure 3. Genome architecture can be classified into several distinct dynamic groups during cell fate transitions.

(A–C) Scatterplot comparison of distance-corrected interaction scores between (A) ES cells and NPCs, (B) ES and iPS cells and (C) NPCs and iPS cells. Thresholds are displayed as blue lines. For pairwise plots, cell type-specific, invariant and background interactions are represented by blue, grey and brown colored shading, respectively. (D) 3D scatterplot of distance-corrected interaction scores for cellular states in which both replicates cross the thresholds displayed in (A–C). Interaction classes are indicated by color (red, ES only; green, NPC only; orange, iPS only; gold, ES-NPC; purple, ES-iPS; blue, NPC-iPS; black, Background). Empirical false discovery rates computed from simulated data in (E–G) are reported for each classification. (E–G) Scatterplots of distance-corrected interaction scores from simulated replicates. Empirical false discovery rates were computed based on the number of interactions that cross pre-established thresholds in the simulated data versus the real data. (H) 3D scatterplot of distance-corrected interaction scores for simulated libraries that cross the thresholds displayed in (A–C, E–G). (I) Number of interactions called significant in each cell-type specific interaction class. (J) Schematic illustrating the 3D interaction behavior for each interaction class. (K–L) Zoomed-in heatmaps of distance-corrected interaction scores for specific (K) ES-iPS (purple class) and (L) ES only (red class) interactions. Classified interaction pixels are outlined in green. (M) Number of interactions called significant for each 3-D classification after clustering directly adjacent 4 kb bins. (N) Depiction of all interactions called as significant in the Sox2 region. Each interaction is outlined by the corresponding classification color.
Importantly, we note that the majority of high-count pixels were spatially adjacent each other in our ‘Distance-corrected Interaction Score’ heatmaps and appear to form larger clusters of enriched 3-D contact (Fig. 3K–L, 3N, S3D–G). To ensure that our approach was not inflating the number of significant interactions, we clustered adjacent pixels that were similarly classified, resulting in a total of only 1,248 unique interactions across three cellular states in our 5C regions (~7.5 Mb) (Fig. 3M). Our clustering approach is similar to the methodology employed by Aiden and colleagues for high-resolution Hi-C data (Rao et al., 2014). We emphasize two important points regarding the 3-D interaction classes called in this study: (i) the interactions represent both specific looping contacts and subTAD boundaries that are dynamic across three cellular states and (ii) rather than a traditional peak calling approach in just one cell type, we are reporting seven classes of long-range interactions called across three cellular states with a focus on the regions of the genome that are most likely to undergo dynamic restructuring during the reprogramming process. Overall, these results indicate that chromatin architecture is highly dynamic during cell fate transitions, with unique folding classes emerging during the reprogramming process.
Pluripotency genes form interactions that can successfully reprogram
We next set out to explore the biological relevance of our dynamic interaction classes. We utilized a series of integrative computational approaches to elucidate the underlying relationships among: (i) fine-scale chromatin folding, (ii) gene expression, (iii) histone modifications characteristic of cell type-specific regulatory elements and (iv) binding profiles of the architectural protein CTCF (Tables S1, S3, S4).
We first investigated the interactions that were present in ES cells, lost in NPCs and reconnected during reprogramming (ES-iPS; purple class) (Fig. 4A). We noticed that the Sox2 gene formed a strong 3D interaction with a pluripotent enhancer element ~120 kb downstream marked by a large domain of H3K4me1/H3K27ac in ES cells (Fig. 4B). Upon differentiation, the Sox2-pluripotent enhancer interaction disassembled in parallel with loss of H3K27ac signal and then subsequently reassembled in iPS cells (Fig. 4B,C). We also identified ES-iPS (purple class) interactions between the Oct4/Pou5f1 gene and a putative enhancer element ~20 kb upstream marked by ES-specific H3K4me1/H3K27ac (Fig. 4D). As expected given the pluripotent properties of our iPS clone, the Oct4-enhancer interaction breaks apart in NPCs and reconnects again in iPS cells (Fig. 4D,E). We next quantitatively assessed the enrichment of a wide range of genomic elements in the ES-iPS class of successfully reprogrammed 3D interactions. Consistent with previous reports (Apostolou et al., 2013) and our qualitative observations, pluripotency genes and putative ES-specific enhancers were significantly enriched at the base of ES-iPS interactions (Fig. 4F). Together, these results indicate that pluripotency genes can form long-range connections with ES-specific enhancer elements and that these interactions can reprogram in iPS cells.
Figure 4. Pluripotency gene-enhancer interactions can be re-established in iPS cells.

(A) Schematic illustrating the ES-iPS (purple) interaction class. (B,D) Relative contact frequency heatmaps (top) and zoomed-in distance-corrected interaction score heatmaps (bottom) highlighting key ES-iPS interactions (purple class) between (B) Sox2 and (D) Oct4 genes and their target enhancers. Heatmaps are overlaid on ChIPseq tracks of H3K27ac and H3K4me1 in ES cells and NPCs. (C+E) Distance-corrected interaction score changes at (C) the Sox2-enhancer interaction and (E) Oct4-enhancer interaction among ES, NPC and iPS cells. Error bars represent the standard deviation across two 5C replicates. (F) Fold enrichment of cell type-specific regulatory elements in ES-iPS (purple class) interactions compared to the enrichment expected by chance across the genome. Color bar represents fold change enrichment over background (blue, depletion; red, enrichment). P-values are computed with Fisher’s Exact test and listed in each bin. (G–H) Normalized gene expression is plotted for (G) Sox2 and (H) Oct4 genes. Error bars represent standard deviation across two RNAseq replicates.
To explore the functional significance of fully reprogrammed interactions, we next conducted genome-wide RNA-seq analysis in ES, NPCs and iPS cells. We examined Oct4 and Sox2 gene expression after normalization among libraries to account for any potential batch effects and differences in sequencing depth (Fig. S4A–D; Tables S3, S5, S6). Unexpectedly, despite reconnection with target pluripotent enhancers, Sox2 expression was markedly lower than target ES cell expression levels (Fig. 4G), whereas Oct4 expression was more than 2-fold higher than target ES cell expression levels (Fig. 4H). Our observations highlight the importance of further understanding the relationship between genome folding and expression, and led us to question if more global architectural connections around these pluripotent enhancer-promoter interactions could be linked to inaccurately reprogrammed gene expression levels in iPS cells.
Some pluripotency genes reconfigure into new NPC interactions that remain persistent in iPS
We next sought to understand larger-scale chromatin folding patterns around Sox2 (Fig. 5A). We hypothesized that chromatin architecture dynamics surrounding the short-range enhancer-promoter interaction might impact the incompletely reprogrammed Sox2 expression in our iPS clone. Unexpectedly, we observed that Sox2 is also engaged in NPC-iPS (blue class) interactions classified by (i) absence in ES cells, (ii) acquisition in NPCs and (iii) residual tethering in iPS cells (Fig. 5A–B). In NPCs, the Sox2-pluripotent enhancer interaction breaks apart and the gene forms long-range contacts with two distal NPC-specific enhancers marked by NPC-specific H3K27ac/H3K4me1. Intriguingly, although the Sox2-pluripotent enhancer interaction is reassembled (purple box), the gene also remains partially tethered to the NPC-specific enhancer in iPS cells (blue box) (Fig. 5A). We observed a similar phenomenon at the Klf4 locus, where the Klf4 gene is highly expressed in ES cells and interacts with a putative ES-specific enhancer element marked by ES-specific H3K4me1/H3K27ac ~75 kb upstream of the gene (Fig. S5A–D). In NPCs, Klf4 disconnects from its pluripotent enhancer and engages with a downstream NPC-specific enhancer (Fig. S5E–F). In iPS cells, Klf4 retains its interaction with the NPC-specific enhancer (blue box) while also partially re-tethering to its target pluripotent enhancer (purple box) (Fig. S5F).
Figure 5. Pluripotency genes can exhibit ‘persistent-NPC-like’ folding patterns in iPS cells.

(A) Relative contact frequency heatmaps (top) and zoomed-in distance-corrected interaction score heatmaps (bottom) highlighting an NPC-iPS interaction (blue class) around the Sox2 gene. Heatmaps are overlaid on ChIPseq tracks of H3K27ac and CTCF in ES cells and NPCs. (B) Schematic illustrating the NPC-iPS (blue) interaction class. (C) Distance-corrected interaction score changes at an NPC-iPS interaction around the Sox2 gene among ES, NPC, iPS, ES+2i and iPS+2i conditions. Error bars represent standard deviation across two 5C replicates. (D) Normalized expression for the Sox2 gene. Error bars represent standard deviation across two RNAseq replicates. (E, F) Fold enrichment of cell type-specific regulatory elements in NPC-iPS (blue class) interactions compared to the enrichment expected by chance across the genome. P-values are computed with Fisher’s Exact test and listed in each bin. (E) Enrichment for any given genomic annotation at the base of NPC-iPS interactions. (F) Enrichment for any given pairwise combination of genomic annotations in the two anchoring bins at the base of NPC-iPS interactions. (G) Relative ChIP-qPCR enrichment of CTCF binding at the NPC-iPS interaction (left, denoted by blue star in (A)) and ES only interaction (right, denoted by red star in (A)).
We hypothesized that the dual tethering of Sox2/Klf4 genes to their target ES-specific pluripotent enhancers and their decommissioned NPC-specific enhancers might lead to inaccurate reprogramming of proper expression levels in our iPS clone. As a first step toward testing this hypothesis, we cultured our iPS clone under 2i/LIF conditions to promote a naïve, ground state of pluripotency and ensure morphological/phenotypic uniformity across the population (Marks et al., 2012; Ying et al., 2008). Strikingly, we noticed that 2i/LIF culture of iPS cells resulted in (i) loss of the Sox2- or Klf4-NPC enhancer (blue class) interactions, (ii) a further amplification in strength of the Sox2- or Klf4- pluripotent enhancer (purple class) interactions and (iii) a fine-tuning of Sox2 or Klf4 expression to ES levels (Fig. 5A, 5C–D, S5E+F). These results indicate that 2i/LIF conditions are capable of untethering persistent somatic cell chromatin architecture in a population of iPS cells and restoring inaccurately reprogrammed gene expression to levels equivalent to those found in V6.5 ES cells. Future causative studies will be necessary to further dissect the link among architectural persistence, naïve vs. primed pluripotency and precise gene expression levels during reprogramming.
We then set out to further understand the mechanistic basis of NPC-iPS (blue class) interactions. Quantitative enrichment analysis revealed three key genomic annotations enriched at the base of NPC-iPS contacts: (i) ES-specific genes, (ii) NPC-specific CTCF and (iii) constitutive CTCF (Fig. 5E). We then computed ‘sided’ enrichments by accounting for the presence/absence of genomic annotations in both anchoring loci at the base of the NPC-iPS interactions (see schematic, Fig. 5F). Consistent with our qualitative observations, ES-specific genes most significantly contact NPC-specific enhancers when located at the base of NPC-iPS interactions (Fig. 5F). We note that Sox2 and Klf4 are classified as ES-specific genes in our study due to their markedly increased expression in ES cells vs. NPCs. However, both genes are still expressed at levels at least 8-fold higher than background in NPCs. Together, these results led us to hypothesize that genes with developmental roles in both ES cells and NPCs, but regulated by different enhancers in the two cellular states, might be particularly susceptible to inappropriate tethering to off-lineage enhancers in iPS cells.
Our quantitative enrichment analyses also indicated that ES-specific genes formed significant 3- D connections with NPC-specific and constitutively bound CTCF sites (Figs. 5E–F). Consistent with this quantitative result, we noticed a constitutively bound CTCF site at the base of the Sox2 NPC-specific enhancer (Fig. 5A) and an NPC-specific CTCF site at the base of the Klf4 NPC-specific enhancer (Fig. S5F), suggesting that CTCF might work together with enhancers to facilitate 3-D connections to the correct target gene(s). To understand how CTCF binding might be altered during reprogramming, we performed CTCF ChIP-qPCR across all five of our cellular states. We queried CTCF occupancy levels in the NPC-specific and ES-specific enhancers (Fig. 5A, blue and red stars, respectively) at the Sox2 locus. We found that the NPC-specific enhancer remains constitutively bound by CTCF in ES, NPC, iPS, ES+2i and iPS+2i conditions (Fig. 5G, left). By contrast, the ES-specific enhancer exhibited high CTCF in ES cells, loss of binding in NPCs, sustained low CTCF occupancy in iPS cells and subsequent restoration of occupancy in 2i/LIF (Fig. 5G, right).
Intriguingly, CTCF binding patterns correlate with the changes in chromatin architecture around Sox2. In ES cells, the constitutive CTCF site interacts with the ES-specific CTCF site, resulting in spatial co-localization of the ES- and NPC-specific enhancers (Fig. 5A, red box). Loss of CTCF binding at the ES-specific enhancer correlates with disconnection of the enhancer-enhancer interaction in NPCs. In parallel, the constitutive CTCF site at the NPC-specific enhancer forms a strong NPC-iPS (blue class) interaction with the Sox2 gene (Fig. 5A, blue box). We posit that the Sox2-NPC-enhancer interaction remains tethered in iPS cells because CTCF does not fully rebind to the ES-specific enhancer (Fig. 5G, right). In support of this idea, 2i/LIF leads to (i) reacquisition of CTCF binding at the ES-specific enhancer, (ii) reconnection of the interaction between both ES-specific and NPC-specific enhancers and (iii) abrogation of the Sox2-NPC-specific enhancer interaction. These observations are consistent with a working model in which ‘persistent-NPC’ interactions can remain in iPS cells when some developmentally regulated genes are tethered to NPC-specific enhancers, possibly at constitutive or NPC-specific CTCF sites.
We highlight that somatic cell-specific elements were not specifically enriched in NPC-iPS interactions (Fig. S6A–C). For example, NPC-specific genes and enhancers were primarily enriched in NPC only (green class) interactions, supporting our finding that it is ES-specific genes, particularly those that remain somewhat active in NPCs, that are redirected into NPC-iPS contacts. An example illustrating this idea can be found at the Olig1/Olig2 genes that are expressed in an NPC-specific manner and equivalently form NPC only (green class) interactions with a downstream NPC-specific enhancer (Fig. S6D–E). Expression of Olig1/2 is lost in parallel with loss of the green class 3-D interaction. Together, these results support the intriguing possibility that ES-specific genes that remain partially active in NPCs form new interactions with somatic cell-specific enhancers during differentiation and that these contacts can remain tethered as a form of architectural persistence in iPS cells. Noteworthy, because 5C is performed on a population of millions of cells, we cannot distinguish between the possibilities that (i) pluripotency genes simultaneously form both ES-iPS and NPC-iPS contacts in individual cells or (ii) pluripotency genes form two different sets of interactions in distinct ES-like subpopulations.
Pluripotent interactions that do not reprogram display dynamic CTCF occupancy
Finally, we explored the interactions that are present in ES cells and lost in NPCs, but do not reconnect in iPS cells (red group, Figs. 6A–B, S7A–B). A notable illustration of these ‘poorly reprogrammed’ interactions is found at the Zfp462 gene (highlighted in green, Fig. 6A), which interacts with a downstream putative ES-specific enhancer element in ES cells. Zfp462 expression is reduced in NPCs in parallel with loss of H3K27ac at the putative downstream enhancer and loss of the interaction. By contrast to the previously discussed ES-iPS (purple) group, this gene-enhancer interaction is not reassembled in iPS. Similarly, the genes Mis18a and Urb1 form interactions in ES cells that are not reprogrammed (highlighted in yellow and green, respectively; Fig. S7A). Together, these genomic loci reveal a class of interactions that are refractory to reprogramming in iPS cells.
Figure 6. Interactions that do not reprogram display poorly reprogrammed CTCF occupancy.

(A) Relative contact frequency heatmaps (top) and zoomed-in distance-corrected interaction score heatmaps (bottom) highlighting an ES only (red class) interaction at ES-specific CTCF binding sites at the Zfp462 gene (indicated in green). Heatmaps are overlaid on ChIPseq tracks of H3K27ac and CTCF in ES cells and NPCs. (B) Schematic illustrating the ES only (red class) interactions. (C) Fraction of ES only (red class) interactions enriched with distinct cell type-specific regulatory elements compared to the expected enrichment in background. P-values are computed with Fisher’s Exact test and listed in each bin. (D) Bar plot displaying the fraction of each interaction class containing ES-specific CTCF binding sites compared to the expected background fraction. Fisher’s Exact test: *, P= 2.06016e-21; **, P= 0.000541696. (E) Distance-corrected interaction score changes at an ES only interaction around the Zfp462 gene among ES, NPC, iPS, ES+2i and iPS+2i conditions. Error bars represent standard deviation across two 5C replicates. (F) Zfp462 gene expression among ES, NPC, iPS, ES+2i and iPS+2i conditions. Error bars represent standard deviation across two RNAseq replicates. (G) Aggregate distance-corrected interaction score changes among ES, NPC, iPS, ES+2i and iPS+2i conditions for genes anchoring red class. (H) Relative ChIP-qPCR enrichment of CTCF binding at the ES only interaction (denoted by blue star in (A)).
To investigate the mechanistic basis for poorly reprogrammed (red class) interactions, we again looked for possible dynamic CTCF binding. We noticed that genomic loci where CTCF is bound in ES cells, but severely depleted in NPCs, were preferentially located at the base of poorly reprogrammed interactions (green boxes; Figs. 6A, S7A). Consistent with this observation, ES-specific CTCF sites were significantly enriched in ES only (red class) interactions (Fig. 6C+D). ChIP-qPCR analysis of CTCF occupancy revealed consistent depletion of CTCF in our iPS clone compared to ES cells (Fig. 5G, 6H, S7G). Importantly, culture of our iPS clone in 2i/LIF media resulted in (i) reacquisition of the red group interactions, (ii) re-establishment of CTCF occupancy and (iii) restoration of gene expression levels in iPS (Figs. 6E–H, S7C–G). Corroborating locus-specific observations, a global analysis of red class interactions demonstrated a marked increase in interaction score upon addition of 2i/LIF media to iPS cells (Fig. 6G). On the basis of these results, we posit that the loss of CTCF binding at critical developmentally regulated loci can be inefficiently restored during a cell-fate transition like somatic cell reprogramming.
Somatic elements are disconnected and pluripotent genes hyperconnected in our iPS clone
We hypothesized that distinct types of regulatory elements exhibit differential connectivity patterns as ES cells transition to NPCs and back to iPS cells. To address this hypothesis, we computed a ‘connectivity’ metric for each class of genomic element in each of the three cellular states. ES-specific enhancers lose their connectivity in NPCs and then reconnect in iPS cells (Fig. 7A). Intriguingly, ES-specific genes become increasingly more connected upon differentiation and subsequent reprogramming (Fig. 7B). By contrast, NPC-specific genes/enhancers increase connectivity in NPCs, but then resume ground state ES-like connectivity in iPS (Fig. 7C–D). Poised enhancers and invariant CTCF sites display minor differences in connectivity across the three cellular states (Figs. 7E+F), whereas ES- specific CTCF sites lose their interactions upon differentiation and only partially gain back connectivity in iPS (Fig. 7G). NPC-specific CTCF sites increase in connectivity in NPCs and then partially resume their disconnected state in iPS cells (Fig. 7H).
Figure 7. Pluripotency genes can be hyperconnected in iPS cells.

Connectivity of distinct regulatory elements in ES cells, ES-derived NPCs and NPC-derived iPS cells. (A) ES-specific enhancers; (B) ES-specific genes; (C) NPC-specific enhancers; (D) NPC-specific genes; (E) Poised enhancers; (F) Invariant CTCF; (G) ES-specific CTCF; (H) NPC-specific CTCF. (I) Schematic illustrating a model of the ‘hyper-connectivity’ of certain pluripotency genes in our NPC-derived iPS clone. Key ES-specific genes (denoted by colored arrows) display the ability to reprogram their connections with ES-specific enhancers (denoted by green/blue ‘transcription factor’ binding sites) and retain remnants of their somatic connections. This intermediate architectural state correlates with inaccurate reprogramming of gene expression levels (represented by colored +/−) and can be fully restored upon culture in 2i/LIF media.
Overall, our results support a model in which somatic cell regulatory elements reconfigure to a ground connectivity state during reprogramming, whereas pluripotency genes (particularly those that retain a low level of activity in NPCs) can be ‘hyperconnected’ in our iPS clone due to persistent cell-of-origin interactions (Fig. 7I). We hypothesize that ‘persistent-NPC’ and ‘poorly reprogrammed’ interactions contribute to inaccurate reprogramming of gene expression levels. Consistent with this idea, 2i/LIF can erase ‘persistent-NPC’ interactions, restore ‘poorly reprogrammed’ interactions and re-establish precise ES-like expression levels in our iPS clone.
Discussion
Understanding the molecular mechanism(s) governing somatic cell reprogramming is of paramount importance to our knowledge of cell fate commitment and the use of iPS cells for regenerative medicine applications. Mechanistic studies have primarily focused on profiling gene expression and classic epigenetic modifications at intermediate stages in the reprogramming process (Koche et al., 2011; Polo et al., 2012; Soufi et al., 2012; Stadtfeld et al., 2008). However, the molecular roadblocks that impede the efficiency and timing of epigenome resetting in iPS cells are just beginning to emerge. Here we examine a unique aspect of reprogramming: the higher-order folding of chromatin in the 3D nucleus. We demonstrate that iPS genome architecture at the sub-Mb scale within TADs can be imperfectly rewired during transcription factor-mediated reprogramming.
Recent studies focusing on a single locus (e.g. Nanog, Oct4) reported that pluripotency genes can re-establish long-range connections with their target enhancers in iPS cells (Apostolou et al., 2013; de Wit et al., 2013; Denholtz et al., 2013; Wei et al., 2013; Zhang et al., 2013a). Motivated by the need to understand how chromatin unfolds/refolds more generally in iPS, we created high-resolution maps of chromatin architecture in Mb-sized regions around developmentally regulated genes. Consistent with previous reports, we observe that many pluripotency genes interact with ES-specific enhancers in ES cells; these interactions break apart in NPCs and then reassemble in iPS cells. Additionally, we find that somatic cell interactions between NPC-specific genes and NPC-specific enhancers generally disconnect in iPS cells. Thus, our data confirm and extend several known locus-specific principles of genome folding during reprogramming.
We also uncover new classes of chromatin interactions that do not behave in the expected manner. We identified a small subset of NPC-iPS (blue class) interactions representing persistent chromatin folding patterns from the somatic cell of origin in iPS cells. Unexpectedly, we find that some key pluripotency genes can form new 3-D connections in NPCs that remain tethered in our iPS clone. For example, Klf4 and Sox2 are dually tethered to their target ES-specific enhancers and their decommissioned NPC-specific enhancers in iPS cells. We posit that this rare, but intriguing form of ‘architectural persistence’ might be causally linked to inaccurate reprogramming of target gene expression levels in certain iPS clones. In support of this working model, we find that 2i/LIF conditions are capable of untethering persistent somatic cell chromatin architecture and restoring the inaccurately reprogrammed expression to levels equivalent to those found in a genetically comparable ES cell line. Noteworthy, NPC-specific genes/enhancers form contacts in NPCs that subsequently disassemble in iPS, suggesting that somatic genes are not driving the architectural persistence in iPS cells. These results agree with previous studies suggesting that somatic cell gene expression is downregulated during the initiation phase of reprogramming and precedes the re-activation of the pluripotency network (Polo et al., 2012). We favor a model in which reconfiguration of higher-order chromatin topology could be a potential rate-limiting step in the reprogramming process and that architectural persistence or incomplete architectural reprogramming (discussed below) can block the formation of fully reprogrammed iPS cells (Buganim et al., 2012; Tanabe et al., 2013).
CTCF is a key player in the organization of the 3D genome and anchors the base of a large number of long-range interactions in ES cells (Dixon et al., 2012; Rao et al., 2014; Handoko et al., 2011; Phillips-Cremins et al., 2013). Here we provide a new link between CTCF and reprogramming. We identify a new class of chromatin interactions that are high in ES cells, break apart in NPCs and are not fully reconfigured in iPS cells. Importantly, we find that these ‘poorly reprogrammed’ interactions often contain ES-specific CTCF binding sites that lose occupancy in NPCs and do not re-acquire full binding in our iPS clone. CTCF has largely stable occupancy patterns during development, with 60–90% of sites remaining bound to the genome between cell types (Kim et al., 2007). Thus, we speculate a model in which CTCF binding is difficult to lose during differentiation, but once occupancy is abolished it is inefficiently re-established during reprogramming. Importantly, DNA methylation is refractory to CTCF binding (Bell and Felsenfeld, 2000), suggesting a possible link between poorly reprogrammed chromatin contacts and previously reported sources of cell of origin epigenetic persistence (Kim et al., 2010; Polo et al., 2010). Indeed, because ES cells cultured in 2i/LIF display global hypomethylation (Ficz et al., 2013; Habibi et al., 2013), we speculate that the interplay between CTCF and dynamic DNA methylation might serve as a mechanism underlying our observation that 2i/LIF media can fully restore CTCF occupancy and ‘poorly reprogrammed’ interactions.
Epigenetic and transcriptional signatures are generally reset in fully reprogrammed iPS cells cultured under optimal conditions (Cahan et al., 2014; Stadtfeld et al., 2010). However, variations in epigenetic profiles among iPS clones have been attributed to reprogramming method, passage number, genetic background or lab-to-lab procedural discrepancies (Bock et al., 2011; Polo et al., 2010). Therefore, we sought to confirm that our observations were truly linked to inefficiencies in the reprogramming of our iPS clone, and not experimental artifacts due to (i) residual somatic cells in our iPS population or (ii) lab-specific culture conditions. Importantly, Hochedlinger and colleagues have extensively characterized the iPS clone used in this manuscript for its pluripotent properties (Eminli et al., 2008). Additionally, our iPS clone was cultured to > 15 passages in serum+LIF-containing growth conditions not amenable to NPC proliferation/survival. Finally, known NPC markers are not upregulated in our iPS population vs. ES cells (Fig. S6E–G). Thus, we see no evidence of contaminating NPCs in our iPS cells. Although somatic cells are absent, we cannot rule out the possibility that there could be a gradient of pluripotent properties (e.g. a continuum between naïve and primed pluripotency) across single cells within our fully reprogrammed iPS clonal population. Because we are conducting population-based assays, we would detect all interactions that exist across the different pluripotent states. Consistent with this possibility, we see that conversion of the population to a uniform, naïve pluripotent state with 2i/LIF media abrogates “architectural persistence” interactions and re-instates “poorly reprogrammed” interactions. Additionally, although we subjected our iPS cells with or without 2i/LIF to the same number of passages (p > 15), we cannot rule out the possibility that further long-term passaging might also resolve any mis-wired chromatin interactions. Noteworthy, these results raise the interesting possibility that an iPS clone capable of creating transgenic mice might still exhibit some level of architectural heterogeneity that can be fully resolved with 2i/LIF media. Exciting lines of future inquiry will query genome folding in higher passages, alternative reprogramming conditions, tetraploid-complementation verified iPS cells and a range of iPS clones derived from multiple somatic cell lineages.
While Beagan et al. was under review, de Laat, Graf and colleagues published a genome-wide analysis of chromatin architecture in iPS cells derived from four independent somatic cell lineages (Krijger et al., 2016). The authors take a top-down approach in which they generate genome-wide, albeit low resolution, Hi-C maps suited to query higher-order levels of genome organization (i.e. A/B compartments, TADs, nuclear positioning of TADs). Importantly, they demonstrate that A/B compartments are largely reset during reprogramming. Moreover, consistent with the leading idea that TADs are largely invariant among cell types (Dixon et al., 2012), TAD boundaries remained for the most part consistent among iPS clones and ES cells. At the level of sub-Mb scale genome folding, however, the design of the two studies is such that different findings arise. Here we take a bottom-up approach in which we create high-resolution, high-complexity maps focused on fine-scale chromatin folding dynamics within TADs around developmentally regulated genes. Given the sensitivity and statistical power afforded by the 5C assay, it is not surprising that we detect a larger number of dynamic looping interactions and subTAD boundaries than reported in Krijger et al. during the transition among ES, iPS and NPC cellular states. Noteworthy, when we increase our bin size from 4 kb up to 300 kb (Fig. S3H), we can recapitulate the author’s high level of correlation between the ES and iPS cells (Fig. S3I). Krijger et al. and Beagan et al. offer complementary viewpoints into genome architecture dynamics across a wide range of length scales and resolutions during reprogramming. Together, the findings from these studies are consistent with our working hypothesis that architectural changes causally linked to developmentally relevant alterations in gene expression occur within TADs at the sub-Mb scale.
Overall, we present high-coverage, fine scale maps of chromatin folding within TADs in iPS cells and use our maps to uncover several new organizing principles for genome folding during reprogramming. We find that different cell type-specific regulatory elements exhibit contrasting 3-D connectivity patterns as cells switch fates in forward and reverse directions. A deeper understanding of the role for chromatin folding at each step in the reprogramming process is of critical importance toward the use of iPS cells for disease modeling and regenerative medicine purposes. Future work combining high- and low-resolution mapping approaches will provide a comprehensive view of genome folding across length scales and cellular states to create a catalogue of “hotspots” of incomplete architectural reprogramming and address whether specific somatic cell types are more or less resistant to topological changes.
Experimental Procedures
Cell culture, differentiation and reprogramming
V6.5 ES cells, primary NPCs and NPC-derived iPS cells were cultured as described in the Supplemental Experimental Procedures. Briefly, ES cells were expanded on Mitomycin-C inactivated MEFs under standard pluripotent conditions and passaged onto feeder-free gelatin-coated plates before fixation. Primary NPCs were isolated from whole brains of P1 129SvJae x C57/BL6, Sox2-eGFP mice and cultured as neurospheres for two passages before adherent culture and fixation. The iPS clone used in this paper was derived and characterized in (Eminli et al., 2008) and expanded/cultured for use in this study to >15 passages with or without 2i/LIF media as described in the Supplemental Experimental Procedures.
Generation and analysis of 5C libraries
5C libraries were generated according to standard procedures described in the Supplemental Experimental Procedures. Paired-end reads were aligned to a pseudo-genome consisting of all 5C primers using Bowtie. Interactions were counted when both paired-end reads were uniquely mapped to the 5C primer pseudo-genome. Counts were converted to contact matrices for each genomic region queried and processed, normalized and modeled as described in the Supplemental Experimental Procedures. Customized algorithms for classification of 5C interactions and the downstream integration of interaction classes with ChIP-seq and RNA-seq data are detailed in the Supplemental Experimental Procedures.
RNA-seq library preparation and analysis
Cells were lysed with Trizol and total RNA was extracted as detailed in the Supplementary Experimental Procedures. Samples were prepared for sequencing using the Illumina TruSeq Stranded Total RNA Library Prep kit with RiboZero (Illumina RS-122–2202) following the supplier’s protocol and sequenced on the Illumina NextSeq500. Libraries were analyzed and corrected for any sequencing depth or batch effect differences with methods described in the Supplemental Experimental Procedures.
ChIP-seq analysis and ChIP-qPCR
A summary of published ChIP-seq libraries re-analyzed in this study is provided in Table S4. Reads were aligned to mm9 with Bowtie using default parameters. Only uniquely mapped reads were used for downstream analyses. ChIP-seq peak calling and ChIP-qPCR experiments are detailed the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
3D genome architecture is markedly reconfigured during reprogramming
Some pluripotency genes engage in persistent, NPC-like interactions reversed in 2i
Poorly reprogrammed interactions exhibit dynamic CTCF binding reinstated in 2i
Imperfect iPS genome topology linked to inaccurately reprogrammed expression
Acknowledgments
We thank members of the Cremins lab for helpful discussions and Michael Duong for cell culture assistance. J.E.P.C. is a New York Stem Cell Foundation Robertson Investigator and an Alfred P. Sloan Foundation Fellow. This work was funded by The New York Stem Cell Foundation (J.E.P.C), the Alfred P. Sloan Foundation (J.E.P.C), the NIH Director’s New Innovator Award from the National Institute of Mental Health (1DP2MH11024701; J.E.P.C), 4D Nucleome Common Fund grants (1U01HL12999801 to J.E.P.C; U54DK107980 to J.D.) and an R01 from the National Human Genome Research Institute (R01 HG003143; J.D.). J.D. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Accession Numbers
5C data has been deposited in GEO under accession number GSE68582. http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token=enefisykjrwdtuh&acc=GSE68582
Author Contributions
T.G.G., J.K. and Z.P. contributed equally to this work. J.E.P.C, V.G.C and J.D. conceived the project. J.A.B, G.H. and J.E.P.C designed and performed 3C/5C experiments. K.H. and E.A. provided iPS cells and Sox2- GFP neonates. H.K.N. performed RNA-seq experiments. J.A.B., S.C.H., and X.L. performed ChIP experiments. T.G.G., J.K., Z.P., J.A.B. and J.E.P.C. developed code and analyzed data. J.A.B. and J.E.P.C wrote the paper with input from E.A., K.H., V.G.C. and J.D.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apostolou E, Ferrari F, Walsh RM, Bar-Nur O, Stadtfeld M, Cheloufi S, Stuart HT, Polo JM, Ohsumi TK, Borowsky ML, et al. Genome-wide chromatin interactions of the Nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell. 2013;12:699–712. doi: 10.1016/j.stem.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482–485. doi: 10.1038/35013100. [DOI] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. Reference Maps of human ES and iPS cell variation enable high-throughput characterization of pluripotent cell lines. Cell. 2011;144:439–452. doi: 10.1016/j.cell.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buganim Y, Faddah DA, Cheng AW, Itskovich E, Markoulaki S, Ganz K, Klemm SL, van Oudenaarden A, Jaenisch R. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahan P, Li H, Morris SA, Lummertz da Rocha E, Daley GQ, Collins JJ. CellNet: network biology applied to stem cell engineering. Cell. 2014;158:903–915. doi: 10.1016/j.cell.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit E, Bouwman BA, Zhu Y, Klous P, Splinter E, Verstegen MJ, Krijger PH, Festuccia N, Nora EP, Welling M, et al. The pluripotent genome in three dimensions is shaped around pluripotency factors. Nature. 2013;501:227–231. doi: 10.1038/nature12420. [DOI] [PubMed] [Google Scholar]
- Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denholtz M, Bonora G, Chronis C, Splinter E, de Laat W, Ernst J, Pellegrini M, Plath K. Long-range chromatin contacts in embryonic stem cells reveal a role for pluripotency factors and polycomb proteins in genome organization. Cell Stem Cell. 2013;13:602–616. doi: 10.1016/j.stem.2013.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, et al. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015;518:331–336. doi: 10.1038/nature14222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Richmond TA, Arnaout RA, Selzer RR, Lee WL, Honan TA, Rubio ED, Krumm A, Lamb J, Nusbaum C, et al. Chromosome Conformation Capture Carbon Copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eminli S, Utikal J, Arnold K, Jaenisch R, Hochedlinger K. Reprogramming of neural progenitor cells into induced pluripotent stem cells in the absence of exogenous Sox2 expression. Stem Cells. 2008;26:2467–2474. doi: 10.1634/stemcells.2008-0317. [DOI] [PubMed] [Google Scholar]
- Ficz G, Hore TA, Santos F, Lee HJ, Dean W, Arand J, Krueger F, Oxley D, Paul YL, Walter J, et al. FGF signaling inhibition in ESCs drives rapid genome-wide demethylation to the epigenetic ground state of pluripotency. Cell Stem Cell. 2013;13:351–359. doi: 10.1016/j.stem.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habibi E, Brinkman AB, Arand J, Kroeze LI, Kerstens HH, Matarese F, Lepikhov K, Gut M, Brun-Heath I, Hubner NC, et al. Whole-genome bisulfite sequencing of two distinct interconvertible DNA methylomes of mouse embryonic stem cells. Cell Stem Cell. 2013;13:360–369. doi: 10.1016/j.stem.2013.06.002. [DOI] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, Jaenisch R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503:290–294. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagey MH, Newman JJ, Bilodeau S, Zhan Y, Orlando DA, van Berkum NL, Ebmeier CC, Goossens J, Rahl PB, Levine SS, et al. Mediator and cohesin connect gene expression and chromatin architecture. Nature. 2010;467:430–435. doi: 10.1038/nature09380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, Kim J, Aryee MJ, Ji H, Ehrlich LI, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell. 2007;128:1231–1245. doi: 10.1016/j.cell.2006.12.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijger PH, Di Stefano B, de Wit E, Limone F, van Oevelen C, de Laat W, Graf T. Cell-of- Origin-Specific 3D Genome Structure Acquired during Somatic Cell Reprogramming. Cell Stem Cell. 2016 doi: 10.1016/j.stem.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics. 2010;Chapter 11(Unit 11):17. doi: 10.1002/0471250953.bi1107s32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lujan E, Zunder ER, Ng YH, Goronzy IN, Nolan GP, Wernig M. Early reprogramming regulators identified by prospective isolation and mass cytometry. Nature. 2015;521:352–356. doi: 10.1038/nature14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks H, Kalkan T, Menafra R, Denissov S, Jones K, Hofemeister H, Nichols J, Kranz A, Stewart AF, Smith A, et al. The transcriptional and epigenomic foundations of ground state pluripotency. Cell. 2012;149:590–604. doi: 10.1016/j.cell.2012.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peric-Hupkes D, Meuleman W, Pagie L, Bruggeman SW, Solovei I, Brugman W, Graf S, Flicek P, Kerkhoven RM, van Lohuizen M, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38:603–613. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, et al. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013;153:1281–1295. doi: 10.1016/j.cell.2013.04.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polo JM, Liu S, Figueroa ME, Kulalert W, Eminli S, Tan KY, Apostolou E, Stadtfeld M, Li Y, Shioda T, et al. Cell type of origin influences the molecular and functional properties of mouse induced pluripotent stem cells. Nat Biotechnol. 2010;28:848–855. doi: 10.1038/nbt.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope BD, Ryba T, Dileep V, Yue F, Wu W, Denas O, Vera DL, Wang Y, Hansen RS, Canfield TK, et al. Topologically associating domains are stable units of replication-timing regulation. Nature. 2014;515:402–405. doi: 10.1038/nature13986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. doi: 10.1038/nature12587. [DOI] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Akutsu H, Fukuda A, Follett P, Natesan S, Kono T, Shioda T, Hochedlinger K. Aberrant silencing of imprinted genes on chromosome 12qF1 in mouse induced pluripotent stem cells. Nature. 2010;465:175–181. doi: 10.1038/nature09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell Stem Cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tanabe K, Nakamura M, Narita M, Takahashi K, Yamanaka S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proc Natl Acad Sci U S A. 2013;110:12172–12179. doi: 10.1073/pnas.1310291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berkum NL, Dekker J. Determining spatial chromatin organization of large genomic regions using 5C technology. Methods Mol Biol. 2009;567:189–213. doi: 10.1007/978-1-60327-414-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Z, Gao F, Kim S, Yang H, Lyu J, An W, Wang K, Lu W. Klf4 organizes long-range chromosomal interactions with the oct4 locus in reprogramming and pluripotency. Cell Stem Cell. 2013;13:36–47. doi: 10.1016/j.stem.2013.05.010. [DOI] [PubMed] [Google Scholar]
- Ying QL, Wray J, Nichols J, Batlle-Morera L, Doble B, Woodgett J, Cohen P, Smith A. The ground state of embryonic stem cell self-renewal. Nature. 2008;453:519–523. doi: 10.1038/nature06968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Jiao W, Sun L, Fan J, Chen M, Wang H, Xu X, Shen A, Li T, Niu B, et al. Intrachromosomal looping is required for activation of endogenous pluripotency genes during reprogramming. Cell Stem Cell. 2013a;13:30–35. doi: 10.1016/j.stem.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wong CH, Birnbaum RY, Li G, Favaro R, Ngan CY, Lim J, Tai E, Poh HM, Wong E, et al. Chromatin connectivity maps reveal dynamic promoter-enhancer long-range associations. Nature. 2013b;504:306–310. doi: 10.1038/nature12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.