

Abstract
Influenza A (H7N9) virus induced high mortality since 2013. It is important to elucidate the potential genetic variations that contribute to virus infection susceptibilities. In order to identify genetic mutations that might increase host susceptibility to infection, we performed exon sequencing and validated the SNPS by Sanger sequencing on 18 H7N9 patients. Blood samples were collected from 18 confirmed H7N9 patients. The genomic DNA was captured with the Agilent SureSelect Human All Exon kit, sequenced on the Illumina Hiseq 2000, and the resulting data processed and annotated with Genome analysis Tool. SNPs were verified by independent Sanger sequencing. The DAVID database and the DAPPLE database were used to do bioinformatics analysis. Through exon sequencing and Sanger sequencing, we identified 21 genes that were highly associated with H7N9 influenza infection. Protein-protein interaction analysis showed that direct interactions among genetic products were significantly higher than expected (p = 0.004), and DAVID analysis confirmed the defense-related functions of these genes. Gene mutation profiles of survived and non-survived patients were similar, suggesting some of genes identified in this study may be associated with H7N9 influenza susceptibility. Host specific genetic determinants of disease severity identified by this approach may provide new targets for the treatment of H7N9 influenza.
During the 2013 H7N9 influenza outbreak in southeast China, there were 139 serologically confirmed cases and 48 deaths1,2. The H7N9 viruses were generated by the subsequent reassortment of H7 viruses with enzootic H9N2 viruses3,4. H7N9 infection was likely mediated by exposure to poultries because about 55.9% of H7N9 influenza patients had a clearly defined poultry exposure5. In addition, closing the poultry trading markets in China was coincided with control of the outbreak. Although many people may have contacted with these poultries, only a minority became sick6,7, suggesting that there may be genetic determinants of both host susceptibility and severity of infection8. The acute onset and rapid progress to severe pneumonia and acute respiratory distress syndrome of H7N9 infection with high mortality highlight the importance of identifying genetic polymorphisms that might predict host response to H7N9 infection. Knowledge of these polymorphisms might help predict both susceptibility to infection and the severity of host response during potential influenza outbreaks in future.
Material and Methods
Experimental Design
This study has been approved by Ethics Committee of Fudan University and all experiments were performed in accordance with relevant guidelines and regulations of Ethics Committee of Fudan University. Due to budget limitation, we collected blood samples from 18 H7N9 infected patients during pandemic outbreak period and patients follow up after hospital discharge. We performed exon sequencing on 8 patients of the 12 survivors after H7N9 and verified all the gene mutation in all the 18 blood samples including the non-survivors (Table 1). Control used in house data from BGI-Shenzhen9.
Table 1. The clinical information of the 18 patients.
| Number | Survived | Exon sequencing | Poultry exposure | Admitted date | Onset date | Virus postive date |
|---|---|---|---|---|---|---|
| 1 | N | N | N | 2013.4.7 | 2013.3.17 | 2013.4.7 |
| 2 | Y | Y | N | 2013.4.13 | 2013.3.13 | 2013.4.13 |
| 3 | Y | N | Y | 2013.4.11 | 2013.4.4 | 2013.4.10 |
| 4 | Y | N | N | 2013.4.13 | 2013.4.4 | 2013.4.13 |
| 5 | Y | Y | N | 2013.4.7 | 2013.3.30 | 2013.4.6 |
| 6 | Y | N | N | 2013.4.16 | 2013.4.11 | 2013.4.16 |
| 7 | Y | N | N | 2013.4.9 | 2013.4.4 | 2013.4.8 |
| 8 | Y | Y | N | 2013.4.10 | 2013.4.1 | 2013.4.9 |
| 9 | N | N | Y | 2013.4.6 | 2013.3.31 | 2013.4.6 |
| 10 | N | N | N | 2013.4.17 | 2013.4.10 | 2013.4.16 |
| 11 | Y | Y | N | 2013.4.13 | 2013.4.4 | 2013.4.13 |
| 12 | N | N | Y | 2013.4.18 | 2013.4.4 | 2013.4.10 |
| 13 | N | N | N | 2013.4.17 | 2013.4.11 | 2013.4.16 |
| 14 | Y | Y | N | 2012.4.9 | 2013.4.1 | 2013.4.9 |
| 15 | N | N | N | 2013.4.4 | 2013.4.2 | NA |
| 16 | Y | Y | unknown | 2013.4.21 | 2013.4.13 | 2013.4.20 |
| 17 | Y | Y | N | 2013.4.20 | 2013.4.13 | 2013.4.19 |
| 18 | Y | Y | Y | 2013.4.6 | 2013.4.1 | 2013.4.6 |
Sample Collection
After informed consents were obtained from all subjects, blood samples were collected from 18 H7N9 infected patients, while exon sequencing was performed on 8 patients. Patients were considered to have H7N9 pneumonia if the following criteria were fulfilled: (1) nasopharyngeal swab positive for H7N9 and; (2) Chest X-ray or CT showing pulmonary infiltrates; (3) clinical symptom of fever and cough. This study was approved by ethical committee of Shanghai Public Health Clinical Center.
Blood collection and DNA extraction
After consent, 10 ml venous blood was drawn from each patient in an EDTA-containing tube. Samples were immediately centrifuged at 500 g for 10 min and plasma was removed and stored for future measurement. Genomic DNA was extracted from the remaining cell pellet using the SQ Blood DNA Kit II (omegabiotek D0714-250). Briefly, cells were lysed and then cell nuclei and mitochondria were separated by centrifugation. The isolated nuclei were resuspended in XL Buffer (supplied by omegabiotek) which contains chaotropic salt and proteinase to remove contamination. Lastly, genomic DNA was purified by isopropanol precipitation.
Exome capture, library preparation and sequencing
The isolated genomic DNA from 8 patients was fragmented into DNA strands with lengths of 150 to 200 bp by Covaris technology, and then adapters were ligated to both ends of the resulting fragments. The adapter-ligated templates were purified by the AgencourtAMPure SPRI beads and fragments with the insert size of about 200 bp were excised. Extracted DNA was amplified by ligation-mediated polymerase chain reaction (LM-PCR), purified, and hybridized to Agilent SureSelect Human All Exon (50 M) human exome array for enrichment. Hybridized fragments were bound to strepavidin beads whereas non-hybridized fragments were washed out after 24 h. Captured LM-PCR products were subjected to Agilent 2100 Bio-analyzer to estimate the magnitude of enrichment. Each captured library was then loaded on Hiseq2000 platform, and high-throughput sequencing for each captured library was performed. Raw image files were processed by Illumina base calling Software 1.7 for base calling with default parameters and the sequences of each individual were generated as 90 bp paired-end reads (Table 2).
Table 2. Exon sequencing data summary.
| Exome capture statistics | Sample |
|||||||
|---|---|---|---|---|---|---|---|---|
| P1 | P2 | P3 | P4 | P5 | P6 | P7 | P8 | |
| Initial bases on target | 51543125 | 51543125 | 51543125 | 51543125 | 51543125 | 51543125 | 51543125 | 51543125 |
| Total effective reads | 75899756 | 79734481 | 86586748 | 77653615 | 88308120 | 59564257 | 81497876 | 101930390 |
| Total effective yield (Mb) | 6765.23 | 7106.16 | 7730.69 | 6923.94 | 7869.24 | 5310.08 | 7276.14 | 9093.9 |
| Average read length (bp) | 89.13 | 89.12 | 89.28 | 89.16 | 89.11 | 89.15 | 89.28 | 89.22 |
| Number of reads uniquely mapped to target | 37836108 | 36730995 | 37985759 | 37747936 | 38384211 | 35029879 | 37527575 | 38836426 |
| Number of reads uniquely mapped to genome | 67787246 | 71481223 | 77694661 | 69327742 | 79613569 | 52915432 | 72767207 | 91803083 |
| Mismatch rate in all effective sequence (%) | 0.22 | 0.23 | 0.21 | 0.22 | 0.21 | 0.19 | 0.22 | 0.22 |
| Average sequencing depth on target (X) | 61.64 | 60.27 | 62.23 | 61.71 | 62.31 | 57.39 | 61.78 | 63.2 |
| Coverage of target region (%) | 98.6 | 99.2 | 99.1 | 99.1 | 98.9 | 98.6 | 99.3 | 99.2 |
| Fraction of target covered > = 20X (%) | 79.4 | 79.9 | 81.5 | 89.4 | 80.1 | 77.5 | 81.1 | 80.6 |
| Fraction of target covered > = 10X (%) | 88.8 | 89.6 | 90.4 | 89.7 | 89.4 | 87.9 | 90.2 | 89.9 |
| Fraction of target covered > = 4X (%) | 94.9 | 95.9 | 96.2 | 95.8 | 95.5 | 94.6 | 96.2 | 96 |
P, patient. The average sequencing depth on target were all more than 57X in eight samples, Coverage of target region were over 98%.
Read mapping and variation detection
After removing reads containing sequencing adapters and low-quality reads, high-quality reads were aligned to the NCBI human reference genome (hg19/GRCh37) using BWA (Burrows-Wheeler Aligner, v0.5.9-r16) with default parameters. Low-quality read was defined as more than half of a read was constituted with low quality bases (less than or equal to 5) or a read in which unknown bases were more than 10%. Picard (v1.54) (http://picard.sourceforge.net/) was used to mark duplicates. Subsequently, BAM files (sequence alignment/map format) were compressed to SAM files (the binary files of BAM files). SNPs (Single-nucleotide polymorphism) and InDels (Small insertions/deletions) were detected by module Unified Genotyper of GATK (Genome Analysis Toolkit v1.0.6076). And then ANNOVAR was used to do annotation and classification for SNPs and InDels respectively. Our data have been identified by dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi), 1000 human genomes database (www.1000genomes.org/) and BGI’s inhouse control database. We used BGI’s inhouse control, most controls coming from a Whole Exome Sequencing based study of genetic risk for psoriasis which has been published9, and the controls comprised 800 normal people across the whole country. These inhouse control cases were specially used for analysis of rare diseases. Considering only 33 patients had confirmed H7N9 infection in Shanghai, and large population exposed to risk factors, the H7N9 infection was a low possibility case, and could be considered as rare disease, so this control data could be used in this study. We collected 40 genes which were correlated with avian influenza from HuGE Navigator by keyword search with “influenza” and extracted 89 exonic SNVs (single nucleotide variations) (Supplementary Dataset 1) located in 27 genes (Table 3) of the 40 genes from SNPs result.
Table 3. 21 virus infection relevant genes identified from exon sequencing.
| No | Genes | Full name | Chromosome loci |
|---|---|---|---|
| 1 | FCGR2A | Fc fragment of IgG | 1 |
| 2 | CPT2 | carnitine palmitoyltransferase 2 | 1 |
| 3 | TLR2 | toll-like receptor 2 | 4 |
| 4 | TLR3 | toll-like receptor 3 | 4 |
| 5 | HLA-DRB1 | MHC class II DLA DRB1 beta chain | 6 |
| 6 | HLA-DQA1 | MHC class II DLA DRA1 beta chain | 6 |
| 7 | HLA-DQB1 | major histocompatibility complex, class II, DQ beta 1 | 6 |
| 8 | NOS3 | nitric oxide synthase 3 | 7 |
| 9 | LEP | leptin | 7 |
| 10 | ABCB1 | ATP-binding cassette, sub-family B | 7 |
| 11 | TLR4 | toll-like receptor 4 | 9 |
| 12 | IFNAR1 | interferon (alpha, beta and omega) receptor 1 | 21 |
| 13 | IL10RB | interleukin 10 receptor | 21 |
| 14 | MBL2 | mannose-binding lectin (protein C) 2 | 10 |
| 15 | ZNF365 | zinc finger protein 365 | 10 |
| 16 | IFITM3 | interferon induced transmembrane protein 3 | 11 |
| 17 | KLRC2 | killer cell lectin-like receptor subfamily C, member 2 | 12 |
| 18 | CES1 | carboxylesterase 1 | 16 |
| 19 | RPAIN | RPA interacting protein | 17 |
| 20 | DNMT1 | DNA (cytosine-5-)-methyltransferase 1 | 19 |
| 21 | MX1 | MX dynamin-like GTPase 1 | 21 |
Gene mutation verification
The 89 mutations were verified in all the 18 patients by Sanger sequencing. 47 fragments of each patient were amplified from their genomic DNA by PrimeSTAR® HS (Premix) (Takara R040A) to verify the 89 mutated sites. The PCR products were subjected to 1% agarose gel electrophoresis and then purified from the gel by QIAquick Gel Extraction Kit (QIAGEN No. 28706). The purified PCR products were subjected to Sanger sequencing (ABI 3730).
Bioinformatics analysis
A protein-protein interactions (PPI) network of the resulting genes was constructed using the Disease Association Protein-Protein Link Evaluator (DAPPLE, http://www.broadinstitute.org/mpg/dapple/dapple.php)8 with 1000 permutations selected and 2 interacting binding degree as a cutoff. And Database for Annotation, Visualization and Integrated Discovery (DAVID, http://david.abcc.ncifcrf.gov/)10, a bioinformatics tool that can identify the biological processes, in which a group of genes are involved, were used for functional annotation.
Results
Mutational Analysis of Genes from 18 H7N9 Infected patients
We have admitted 18 H7N9 infected patients around 10 days after disease onset and a series of clinical manifestation, laboratory examinations and prognosis were carried out for the following 15 weeks. 6 of the 18 patients died and we found the increased plasma CRP (Creactive protein), PCT (Procalcitonin) and virus positive days were associated with mortality11. After exon sequencing of 8 survivors, 64 exonic SNPs, located in 21 genes, were found to be enriched in the H7N9 patients compared to controls from the NCBI human genome (hg19) (Supplementary Dataset 1 and Table 4). These mutations were found in genes encoding proteins responsible for multiple key host defense mechanisms, including cytokine production, airway epithelium barrier function and pathogen associated molecular pattern signaling pathway, suggesting biological plausibility (Table 2).
Table 4. The 64 validated exonic SNVs and their distribution between different groups.
| Gene Information |
Comparison between survived and deceased |
Comparison between the first 8 patients and the additional 10 pateints |
Comparison between case and control |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Chr | Pos | Ref | Alt | Mutation in 12 survivors | Mutation in 6 decedents | P-value | Mutation in first 8 patients | Mutation in the other 10 pateints | P-value | 1000 Genome | Inhouse control | dbSNP | P-value between case and inhouse control |
| CPT2 | chr1 | 53676401 | T | G | 5 | 2 | 1 | 4 | 3 | 1 | 0.06 | 0.2104 | rs2229291 | 0.352872195 |
| CPT2 | chr1 | 53676448 | G | A | 12 | 4 | 0.4 | 8 | 8 | 0.7 | 0.5 | 0.669 | rs1799821 | 0.107976012 |
| CPT2 | chr1 | 53679028 | G | A | 1 | 0 | 0.7 | 1 | 0 | 0.7 | 0.0008 | 0.019710021 | ||
| CPT2 | chr1 | 53679229 | A | G | 2 | 3 | 1 | 4 | 1 | 1 | 0.13 | 0.1368 | rs1799822 | 1 |
| FCGR2A | chr1 | 161479745 | A | G | 6 | 3 | 0.7 | 3 | 6 | 0.4 | 0.43 | 0.4777 | rs1801274 | 0.023221855 |
| IFITM3 | chr11 | 320649 | G | A | 0 | 2 | 0.2 | 0 | 2 | 0.2 | 0.2488 | rs11553885 | 0.140015157 | |
| IFITM3 | chr11 | 320772 | A | G | 12 | 5 | 0.1 | 8 | 9 | 0.2 | 0.21 | 0.3101 | rs12252 | 0.110209014 |
| RPAIN | chr17 | 5326145 | C | G | 12 | 5 | 0.4 | 8 | 9 | 0.4 | 0.43 | 0.6017 | rs12761 | 0.036500478 |
| TLR3 | chr4 | 187004074 | C | T | 9 | 2 | 0.1 | 6 | 5 | 0.4 | 0.25 | 0.3324 | rs3775291 | 0.028998842 |
| TLR3 | chr4 | 187004217 | C | T | 5 | 4 | 1 | 3 | 9 | 0.1 | 0.28 | 0.3702 | rs3775290 | 0.192232214 |
| TLR2 | chr4 | 154624656 | T | C | 5 | 5 | 0.7 | 5 | 5 | 0.7 | 0.43 | 0.4474 | rs3804099 | 0.622004107 |
| TLR2 | chr4 | 154625259 | A | G | 1 | 0 | 0.7 | 1 | 0 | 0.7 | 0.0023 | 0.0239 | rs144038898 | 0.32478413 |
| TLR2 | chr4 | 154625409 | T | C | 4 | 5 | 1 | 4 | 5 | 1 | 0.12 | 0.4378 | rs3804100 | 0.801147802 |
| HLA-DQA1 | chr6 | 32605284 | G | A | 1 | 0 | 0.2 | 1 | 0 | 0.2 | 0.06 | 0.0766 | rs12722039 | 0.350824196 |
| HLA-DQA1 | chr6 | 32605309 | A | G | 1 | 0 | 0.2 | 1 | 0 | 0.2 | 0.06 | 0.0622 | rs12722042 | 0.263992352 |
| HLA-DQA1 | chr6 | 32609094 | C | T | 2 | 5 | 0.4 | 2 | 5 | 0.4 | 0.35 | 0.2225 | rs1129737 | 0.54610042 |
| HLA-DQA1 | chr6 | 32609147 | A | T | 0 | 2 | 1 | 0 | 2 | 1 | 0.19 | 0.2679 | rs12722051 | 0.262995374 |
| HLA-DQA1 | chr6 | 32609173 | C | G | 2 | 5 | 0.2 | 2 | 5 | 0.2 | 0.4 | 0.2656 | rs10093 | 0.264047871 |
| HLA-DQA1 | chr6 | 32609195 | G | A | 1 | 5 | 1 | 1 | 5 | 1 | 0.09 | 0.1675 | rs36219699 | 1 |
| HLA-DQA1 | chr6 | 32609813 | T | C | 5 | 8 | 0.4 | 5 | 8 | 0.4 | 0.66 | 0.2919 | rs707951 | 0.174782886 |
| HLA-DQA1 | chr6 | 32610436 | T | C | 6 | 8 | 0.4 | 6 | 8 | 0.4 | 0.61 | 0.2679 | rs1048372 | 0.262995374 |
| HLA-DQA1 | chr6 | 32610535 | A | C | 6 | 8 | 0.4 | 6 | 8 | 0.4 | 0.57 | 0.2703 | rs1130116 | 0.262188523 |
| HLA-DQB1 | chr6 | 32629129 | T | C | 1 | 2 | 0.4 | 1 | 2 | 0.4 | 0.19 | 0.2416 | rs1130432 | 0.384909932 |
| HLA-DQB1 | chr6 | 32629155 | C | A | 1 | 2 | 0.4 | 1 | 2 | 0.4 | 0.23 | 0.2153 | rs17412886 | 0.545857944 |
| HLA-DQB1 | chr6 | 32629847 | A | G | 7 | 9 | 0.7 | 7 | 9 | 0.7 | 0.8 | 0.3541 | rs1049133 | 0.444388806 |
| HLA-DQB1 | chr6 | 32629859 | A | G | 6 | 9 | 0.7 | 6 | 9 | 0.7 | 0.65 | 0.3421 | rs1049130 | 0.59863218 |
| HLA-DQB1 | chr6 | 32629868 | A | G | 2 | 5 | 1 | 2 | 5 | 0.1 | 0.18 | 0.134 | rs1049088 | 1 |
| HLA-DQB1 | chr6 | 32629889 | G | A | 5 | 7 | 0.4 | 5 | 7 | 0.4 | 0.44 | 0.2392 | rs1049087 | 0.385697708 |
| HLA-DQB1 | chr6 | 32629904 | A | G | 5 | 6 | 0.4 | 5 | 6 | 0.4 | 0.6 | 0.2656 | rs1049086 | 0.264047871 |
| HLA-DQB1 | chr6 | 32629936 | C | T | 1 | 2 | 0.4 | 1 | 2 | 0.4 | 0.21 | 0.2057 | rs1049107 | 0.548651412 |
| HLA-DQB1 | chr6 | 32629963 | C | T | 1 | 2 | 0.4 | 1 | 2 | 0.4 | 0.2 | 0.177 | rs1049100 | 0.752136224 |
| HLA-DQB1 | chr6 | 32634313 | C | G | 1 | 2 | 0.4 | 1 | 2 | 0.4 | 0.21 | 0.0885 | rs1049059 | 0.646223667 |
| HLA-DQB1 | chr6 | 32634369 | C | A | 1 | 2 | 0.4 | 1 | 2 | 0.4 | 0.21 | 0.0981 | rs1049056 | 0.664995324 |
| HLA-DRB1 | chr6 | 32549596 | T | C | 8 | 4 | 0.4 | 8 | 4 | 0.4 | 0.311 | rs111823233 | 0.171375278 | |
| NOS3 | chr7 | 150695726 | T | C | 8 | 9 | 0.7 | 8 | 9 | 0.7 | 0.78 | 0.6699 | rs1549758 | 0.182947056 |
| NOS3 | chr7 | 150696111 | T | G | 8 | 9 | 0.4 | 8 | 9 | 0.4 | 0.8 | 0.8732 | rs1799983 | 1 |
| NOS3 | chr7 | 150704250 | C | G | 4 | 1 | 1 | 4 | 1 | 1 | 0.42 | 0.5096 | rs2566514 | 0.04531093 |
| ABCB1 | chr7 | 87138645 | A | G | 7 | 8 | 0.7 | 7 | 8 | 0.7 | 0.6 | 0.6507 | rs1045642 | 0.110998732 |
| ABCB1 | chr7 | 87160618 | A | T or C | 7 | 5 | 0.7 | 7 | 5 | 0.7 | 0.1244 | rs2032582 | 0.709435825 | |
| ABCB1 | chr7 | 87179601 | A | G | 3 | 4 | 0.7 | 3 | 4 | 0.7 | 0.58 | 0.4617 | rs1128503 | 0.040826503 |
| LEP | chr7 | 127892124 | A | G | 1 | 0 | 0.7 | 1 | 0 | 0.7 | 0.0005 | 0.0024 | rs148407750 | 0.039055387 |
| TLR4 | chr9 | 120470894 | C | G | 1 | 0 | 0.7 | 1 | 0 | 0.7 | – | 0.00990099 | ||
| MBL2 | chr10 | 54528266 | G | C | 8 | 9 | 1 | 8 | 9 | 1 | 0.77 | 0.6388 | rs930507 | 0.194023134 |
| MBL2 | chr10 | 54531235 | C | T | 3 | 3 | 1 | 3 | 3 | 1 | 0.12 | 0.2057 | rs1800450 | 1 |
| ZNF365 | chr10 | 64159333 | G | T | 7 | 7 | 0.7 | 7 | 7 | 0.7 | 0.49 | 0.7967 | rs3758490 | 0.11468297 |
| ZNF365 | chr10 | 64415184 | A | G | 8 | 9 | 1 | 8 | 9 | 1 | 0.85 | 0.9115 | rs7076156 | 1 |
| ZNF365 | chr10 | 64416220 | C | T | 0 | 1 | 0.7 | 0 | 1 | 0.7 | 0.02 | 0.0359 | rs76895268 | 0.444340031 |
| KLRC2 | chr12 | 10587111 | A | G | 2 | 9 | 1 | 2 | 9 | 1 | 0.76 | 0.4234 | rs1141715 | 0.003533467 |
| KLRC2 | chr12 | 10588530 | C | G | 6 | 5 | 1 | 6 | 5 | 1 | 0.26 | 0.4785 | rs34195537 | 0.004729536 |
| CES1 | chr16 | 55844509 | T | C | 2 | 0 | 0.7 | 2 | 0 | 0.7 | – | 0.00990099 | ||
| CES1 | chr16 | 55853545 | C | A | 0 | 4 | 0.7 | 0 | 4 | 0.7 | 0.04 | 0.0407 | rs115629050 | 0.488479286 |
| CES1 | chr16 | 55855361 | G | T | 1 | 0 | 0.7 | 1 | 0 | 0.7 | 0.04 | 0.0311 | rs2307227 | 0.396649049 |
| DNMT1 | chr19 | 10251572 | G | C | 1 | 0 | 0.7 | 1 | 0 | 0.7 | 0.0037 | 0.0072 | rs144675407 | 0.112914886 |
| DNMT1 | chr19 | 10265312 | T | C | 8 | 9 | 0.4 | 8 | 9 | 0.4 | 0.99 | 0.7297 | rs721186 | 0.00938611 |
| DNMT1 | chr19 | 10265333 | A | G | 1 | 0 | 0.7 | 1 | 0 | 0.7 | – | 0.00990099 | ||
| DNMT1 | chr19 | 10265372 | C | T | 1 | 0 | 0.7 | 1 | 0 | 0.7 | 0.0005 | – | rs140376680 | 0.00990099 |
| DNMT1 | chr19 | 10267077 | T | C | 8 | 7 | 0.4 | 8 | 7 | 0.4 | 0.54 | 0.6388 | rs2228611 | 0.194023134 |
| DNMT1 | chr19 | 10291181 | T | C | 0 | 2 | 0.2 | 0 | 2 | 0.2 | 0.06 | 0.3756 | rs16999593 | 0.437380251 |
| IFNAR1 | chr21 | 34715699 | G | C | 4 | 7 | 0.1 | 4 | 7 | 0.1 | 0.21 | 0.5239 | rs2257167 | 0.129919992 |
| MX1 | chr21 | 42812891 | C | T | 1 | 2 | 1 | 1 | 2 | 1 | 0.36 | 0.177 | rs467960 | 0.332738289 |
| MX1 | chr21 | 42817930 | G | A | 1 | 1 | 1 | 1 | 1 | 1 | 0.43 | 0.4474 | rs469390 | 0.001564732 |
| MX1 | chr21 | 42821113 | T | C | 4 | 2 | 0.4 | 4 | 2 | 0.4 | 0.35 | 0.3278 | rs2070229 | 0.422944246 |
| MX1 | chr21 | 42824661 | A | G | 4 | 4 | 0.7 | 4 | 4 | 0.7 | 0.27 | 0.5215 | rs1050008 | 0.042066821 |
| IL10RB | chr21 | 34640788 | A | G | 8 | 1 | 0.1 | 8 | 1 | 0.1 | 0.35 | 0.7392 | rs2834167 | 1 |
Bioinformatics analysis
The resulting genes with exonic SNPs were uploaded to the online tool DAPPLE for PPI network analysis. The results indicate that the PPI network was statistically significant. There were 5 disease proteins participating in the direct network with 3 direct interactions in total expected direct interactions = 0.347, p = 0.004 (Fig. 1, Table 5). Moreover, there were 13 genes participating in the indirect network under the same condition (Fig. 2).
Figure 1. Direct connections among gene products from exome sequencing result.
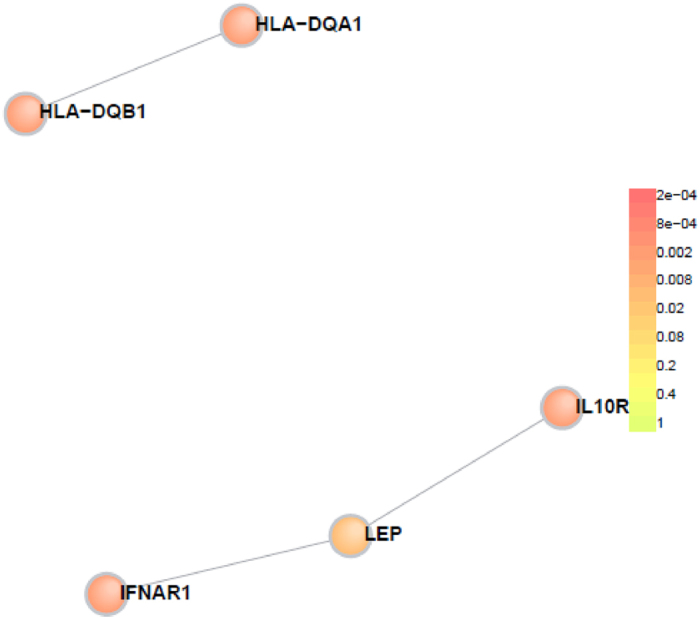
Colours indicate significance of participation in the PPI network.
Table 5. The PPI network statistics.
| PARAMETER | OBSERVED | EXPECTED | P_VALUE |
|---|---|---|---|
| Direct Edges Count | 3 | 0.347 | 0.003996004 |
| Seed Direct Degrees Mean | 1.2 | 0.3024 | 0.014985015 |
| Seed Indirect Degrees Mean | 17.25 | 3.732515889 | 0.000999001 |
| CI Degrees Mean | 2.096491 | 2.067364797 | 0.265734266 |
Figure 2. Indirect connections among gene products from exome sequencing result.
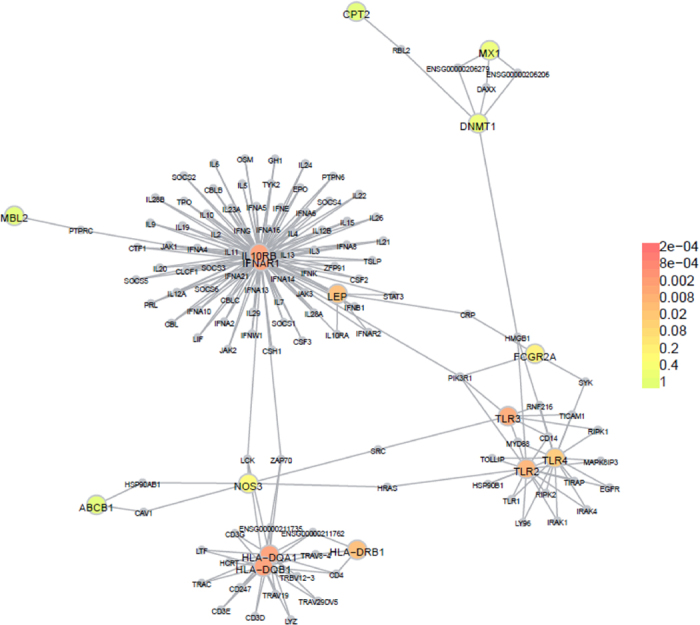
We further confirmed the functions of these candidate genes using the online tool DAVID. The genes were significantly enriched for defense-related processes such as response to stimulus (p = 1.81 × 10−8), immune response (p = 8.85 × 10−7), immune system process (p = 1.16 × 10−6), response to biotic stimulus (p = 5.48 × 10−6) and modulation by symbiont of host immune response (p = 1.53 × 10−5) (Table 6).
Table 6. Top 10 go term analysis results.
| Term | Count | percentage | P-Value | Genes |
|---|---|---|---|---|
| GO:0050896:response to stimulus | 17 | 80.95238095 | 1.81E-08 | HLA-DQB1, MBL2, KLRC2, CES1, HLA-DRB1, IFITM3, TLR2, TLR3, ABCB1, TLR4, HLA-DQA1, IFNAR1, LEP, RPAIN, IL10RB, NOS3, MX1 |
| GO:0006955:immune response | 9 | 42.85714286 | 8.85E-07 | HLA-DQB1, MBL2, HLA-DRB1, IL10RB, IFITM3, TLR2, TLR3, TLR4, HLA-DQA1 |
| GO:0002376:immune system process | 10 | 47.61904762 | 1.16E-06 | HLA-DQB1, MBL2, HLA-DRB1, IL10RB, IFITM3, TLR2, TLR3, TLR4, HLA-DQA1, IFNAR1 |
| GO:0009607:response to biotic stimulus | 7 | 33.33333333 | 5.48E-06 | IFITM3, TLR2, TLR3, NOS3, TLR4, MX1, IFNAR1 |
| GO:0052553:modulation by symbiont of host immune response | 3 | 14.28571429 | 1.53E-05 | TLR2, TLR3, TLR4 |
| GO:0052556:positive regulation by symbiont of host immune response | 3 | 14.28571429 | 1.53E-05 | TLR2, TLR3, TLR4 |
| GO:0052166:positive regulation by symbiont of host innate immunity | 3 | 14.28571429 | 1.53E-05 | TLR2, TLR3, TLR4 |
| GO:0052306:modulation by organism of innate immunity in other organism during symbiotic interaction | 3 | 14.28571429 | 1.53E-05 | TLR2, TLR3, TLR4 |
| GO:0052305:positive regulation by organism of innate immunity in other organism during symbiotic interaction | 3 | 14.28571429 | 1.53E-05 | TLR2, TLR3, TLR4 |
| GO:0052555:positive regulation by organism of immune response of other organism during symbiotic interaction | 3 | 14.28571429 | 1.53E-05 | TLR2, TLR3, TLR4 |
Gene mutation distribution between different groups
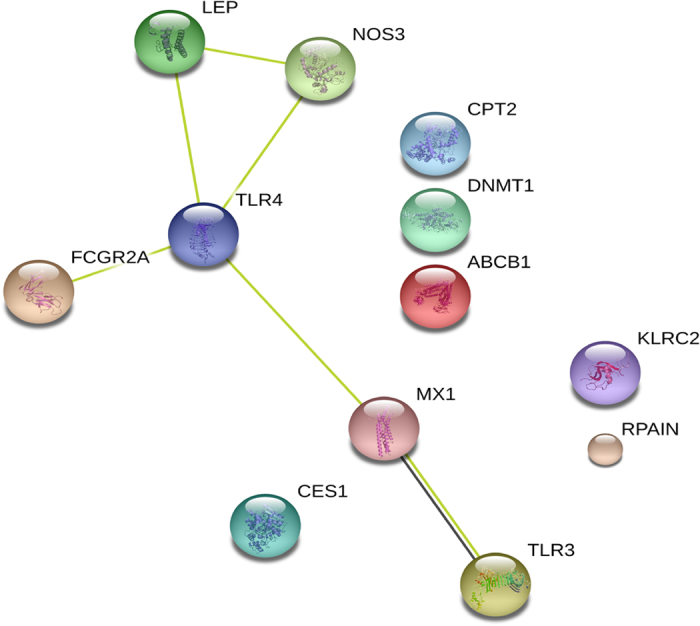
Whole exome sequencing was performed on 8 H7N9 patients and 89 exonic SNPs were identified. These SNPs were subjected to Sanger sequencing in all the 18 patients and 64 exonic SNVs were verified. We compared the mutation rate of the case and the inhouse control using the Fisher Exact Test and found significant statistical difference (Supplementary Dataset 1 and Table 4). There were 17 SNVs significantly different between the case and the inhouse control and we have validated 16 of them by Sanger sequencing (Table 7). The 16 validated SNVs were located in 12 genes, and the protein-protein interaction among them (Fig. 3) was consistent with the protein-protein interaction among the 21 genes done before (Fig. 2). It is more likely that both the genes identified from this study that showed statistical difference of mutation frequency and the genes with same mutation rate between patients and controls have participated in the pathogenesis of H7N9 virus infection. We also did Mann-Whitney U test between the first 8 patients and the additional 10 patients and none of P-value was significant (Table 4), which could prove the inhouse control data do not introduce any false signals. Moreover, We compared the mutation rate of death group and survival group and analysis the mutation rate by Mann-Whitney U test and no significant difference was found between the survival and non-survival group (Table 4), suggesting some of genes identified in this study may be associated with H7N9 influenza susceptibility.
Table 7. The 17 SNVs significantly different between case and control.
| Gene | Chr | Pos | Ref | Obs | caseRef | caseAlt | controlRef | controlAlt | FisherExactTestPvalue | Sanger sequencing validated |
|---|---|---|---|---|---|---|---|---|---|---|
| PPARG | chr3 | 12475632 | G | A | 15 | 1 | 1600 | 0 | 0.00990099 | No |
| CPT2 | chr1 | 53679028 | G | A | 15 | 1 | 1599 | 1 | 0.019710021 | Yes |
| FCGR2A | chr1 | 161479745 | A | G | 13 | 3 | 836 | 764 | 0.023221855 | Yes |
| RPAIN | chr17 | 5326145 | C | G | 2 | 14 | 638 | 962 | 0.036500478 | Yes |
| TLR3 | chr4 | 187004074 | C | T | 6 | 10 | 1069 | 531 | 0.028998842 | Yes |
| NOS3 | chr7 | 150704250 | C | G | 12 | 4 | 785 | 815 | 0.04531093 | Yes |
| ABCB1 | chr7 | 87179601 | A | G | 13 | 3 | 862 | 738 | 0.040826503 | Yes |
| LEP | chr7 | 127892124 | A | G | 15 | 1 | 1597 | 3 | 0.039055387 | Yes |
| TLR4 | chr9 | 120470894 | C | G | 15 | 1 | 1600 | 0 | 0.00990099 | Yes |
| KLRC2 | chr12 | 10587111 | A | G | 3 | 13 | 923 | 677 | 0.003533467 | Yes |
| chr12 | 10588530 | C | G | 14 | 2 | 835 | 765 | 0.004729536 | Yes | |
| CES1 | chr16 | 55844509 | T | C | 15 | 1 | 1600 | 0 | 0.00990099 | Yes |
| DNMT1 | chr19 | 10265312 | T | C | 0 | 16 | 433 | 1167 | 0.00938611 | Yes |
| chr19 | 10265333 | A | G | 15 | 1 | 1600 | 0 | 0.00990099 | Yes | |
| chr19 | 10265372 | C | T | 15 | 1 | 1600 | 0 | 0.00990099 | Yes | |
| MX1 | chr21 | 42817930 | G | A | 15 | 1 | 885 | 715 | 0.001564732 | Yes |
| chr21 | 42824661 | A | G | 12 | 4 | 766 | 834 | 0.042066821 | Yes |
Figure 3. The protein-protein interaction among the 12 genes significantly different between case and control.
Discussion
The 2013 Chinese H7N9 influenza outbreak lead to an estimated 48 deaths with 33% mortality and significant morbidity in patients who survived the virus. An important observation during the recent H7N9 outbreak in China was the wide variation in host response to infections, with some patients developing only mild upper respiratory tract infections, while other patients developed severe ARDS and died. Although several determinants of the host response to infection have been identified, many important genetic factors that dampen or exacerbate the host response to H7N9 infection likely remain undiscovered. Previous studies suggested that genetic mutations in the protein machinery that comprise key host defense mechanisms could impact outcomes of influenza infection12. The differential susceptibilities to influenza A(H7N9) were affected by functional variants of LGALS1 causing the expression variations13. The H7N9 influenza outbreak in China provides an unique opportunity to study mutations in this machinery, because many poultry workers were exposed to the virus, yet comparatively few became infected. This may suggest that genetic mutations in host defense mechanisms could be responsible for the selectivity of H7N9 infection. Others have identified genes that are protective during influenza infection, including MX1, NCR1, CCR5, IFITM3 and IL1014. Mutations in these genes may lead to increased host susceptibility to infection or to a heightened, and potentially deleterious, host response to infection. We hypothesized that the exome sequencing of these patients may reveal genetic mutations that increased susceptibility to viral infection, and that in the future, these mutations could provide information regarding risk of infection, especially poultry workers or family members of infected patients.
Using a variety of computational genetic techniques, we identified 21 genes that showed a high rate of mutation in patients infected with H7N9 when compared to the general population. Among these genes, some have been identified in prior studies of H7N9 susceptibility genes14. For example, Wang et al. reported that IFITM3 dysfunction is associated with increased cytokine production during H7N9 infection and is correlated with mortality14. IFITM3 (chr11, 320772, A > G) was reported to be enriched in patients hospitalized due to H1N1/09 infection15. Polymorphisms of CPT2, a carnitine palmitoyltransferase 2 protein, were found in patients suffering from influenza-associated encephalopathy; results of overexpression of CPT2 variants in vitro suggested that the variants were heat-labile and failed to perform optimally during fever16,17. Four disease outcome-associated SNPs were identified on chromosomes 17 (RPAIN and C1QBP), chromosome 1 (FCGR2A), and chromosome 3 (unknown gene). C1QBP and GCGR2A play roles in the formation of immune complexes and complement activation, suggesting that the severe disease outcome of H1N1 infection may result from an enhanced host immune response12,16.
Among the 21 genes we identified, we use the online tool DAPPLE to performed a PPI analysis and found 5 proteins directly participates the PPI network. Those proteins include: LEP, IFNAR1, IL10RB, HLA-DQA1, HLA-DQB1. The PPI analysis suggested significant role of these proteins in influenza infection and may provide target for interventional therapy.
The primary limitation of this study is the relative small sample size. Only 18 patients were enrolled, and confirmation of these findings in subsequent studies will be needed. We are planning to collect more samples for next step sequencing.
Conclusion
Using comparative genetic analysis in 18 patients with confirmed H7N9 viral infection in China, we identified 21 genetic mutations that occurred at a higher rate in infected patients when compared to the general population. Many of the identified genes are involved in key host defense mechanisms, which gives strong biologic plausibility to the role of these genes in both host susceptibility to infection as well as host immune response related pathology. Further investigations into the function of these genes in host susceptibility may help identify individuals who are at high risk for infection. In addition, translational research into the function of the genes identified in this study may provide new potential therapeutic targets for influenza virus infection.
Additional Information
How to cite this article: Chen, C. et al. Multiple gene mutations identified in patients infected with influenza A (H7N9) virus. Sci. Rep. 6, 25614; doi: 10.1038/srep25614 (2016).
Supplementary Material
Acknowledgments
This research was supported by the National Natural Science Foundation of China (81170056, 81100046, 30930090), and by grant B115 from Shanghai Leading Academic Discipline Project. Dr. Yuanlin Song was supported by the State Key Basic Research Program (973) project (2015CB553404), Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and Key Medical grant from Shanghai Science and Technology Committee (11411951102, 12JC1402300), and supported by Doctoral Fund of Ministry of Education of China (20130071110044).
Footnotes
Author Contributions Y.S. and W.Z. designed the study. C.C. drafted the manuscripts and E.S. revised the MS and provided partial data analysis. C.C., M.W., X.T., L.Y., X.F., X.L. and Q.W. collected the blood and did exon sequencing as well as patient data acquisition. Z.Z. and Y.H. did the Sanger sequencing. X.X., T.L., S.L., C.B., C.D., Z.Z. and J.Q. did clinical service in these patients and helped in samples collection and process.
References
- Gao R. et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med 368, 1888–1897, doi: 10.1056/NEJMoa1304459 (2013). [DOI] [PubMed] [Google Scholar]
- Lam T. T. et al. The genesis and source of the H7N9 influenza viruses causing human infections in China. Nature 502, 241–244, doi: 10.1038/nature12515 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A. et al. Sequential reassortments underlie diverse influenza H7N9 genotypes in China. Cell host & microbe 14, 446–452, doi: 10.1016/j.chom.2013.09.001 (2013). [DOI] [PubMed] [Google Scholar]
- Qi X. et al. Probable person to person transmission of novel avian influenza A (H7N9) virus in Eastern China, 2013: epidemiological investigation. BMJ 347, f4752, doi: 10.1136/bmj.f4752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H. N. et al. Clinical findings in 111 cases of influenza A (H7N9) virus infection. N Engl J Med 368, 2277–2285, doi: 10.1056/NEJMoa1305584 (2013). [DOI] [PubMed] [Google Scholar]
- Watanabe T., Watanabe S., Maher E. A., Neumann G. & Kawaoka Y. Pandemic potential of avian influenza A (H7N9) viruses. Trends Microbiol 22, 623–631, doi: 10.1016/j.tim.2014.08.008 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T. et al. One family cluster of avian influenza A(H7N9) virus infection in Shandong, China. BMC Infect Dis 14, 98, doi: 10.1186/1471-2334-14-98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossin E. J. et al. Proteins encoded in genomic regions associated with immune-mediated disease physically interact and suggest underlying biology. PLoS Genet 7, e1001273, doi: 10.1371/journal.pgen.1001273 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. et al. A large-scale screen for coding variants predisposing to psoriasis. Nature genetics 46, 45–50, doi: 10.1038/ng.2827 (2014). [DOI] [PubMed] [Google Scholar]
- Sherman B. T. et al. DAVID Knowledgebase: a gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinformatics 8, 426, doi: 10.1186/1471-2105-8-426 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S. et al. Prognosis of 18 H7N9 avian influenza patients in Shanghai. PLoS One 9, e88728, doi: 10.1371/journal.pone.0088728 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga J. et al. Genetic variants associated with severe pneumonia in A/H1N1 influenza infection. Eur Respir J 39, 604–610, doi: 10.1183/09031936.00020611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. et al. Functional variants regulating LGALS1 (Galectin 1) expression affect human susceptibility to influenza A(H7N9). Sci Rep 5, 8517, doi: 10.1038/srep08517 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc Natl Acad Sci USA 111, 769–774, doi: 10.1073/pnas.1321748111 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everitt A. R. et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 484, 519–523, doi: 10.1038/nature10921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. Y. & Brass A. L. Host genetic determinants of influenza pathogenicity. Curr Opin Virol 3, 531–536, doi: 10.1016/j.coviro.2013.07.005 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak C. M. et al. Fatal viral infection-associated encephalopathy in two Chinese boys: a genetically determined risk factor of thermolabile carnitine palmitoyltransferase II variants. J Hum Genet 56, 617–621, doi: 10.1038/jhg.2011.63 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.