

Abstract
Maternal obesity predisposes offspring to metabolic and reproductive dysfunction. We have shown previously that female rat offspring born to mothers fed a high-fat (HF) diet throughout pregnancy and lactation enter puberty early and display aberrant reproductive cyclicity. The mechanisms driving this reproductive phenotype are currently unknown thus we investigated whether changes in ovarian function were involved. Wistar rats were mated and randomized to: dams fed a control diet (CON) or dams fed a HF diet from conception until the end of lactation (HF). Ovaries were collected from fetuses at Embryonic Day (E) 20, and neonatal ovaries at Day 4 (P4), prepubertal ovaries at P27 and adult ovaries at P120. In a subset of offspring, the effects of a HF diet fed postweaning were evaluated. The present study shows that fetuses of mothers fed a HF diet had significantly fewer oocytes at E20, and in neonates, have reduced AMH signaling that may facilitate an increased number of assembled primordial follicles. Both prepubertally and in adulthood, ovaries show increased follicular atresia. As adults, offspring have reduced FSH responsiveness, low expression levels of estrogen receptor alpha (Eralpha), the oocyte-secreted factor, Gdf9, oocyte-specific RNA binding protein, Dazl, and high expression levels of the granulosa-cell derived factor, AMH, in antral follicles. Together, these data suggest that ovarian compromise in offspring born to HF-fed mothers may arise from changes already observable in the fetus and neonate and in the long term, associated with increased follicular atresia through adulthood.
Keywords: atresia, maternal nutrition, ovary, primordial follicle, reproduction
INTRODUCTION
Excessive gestational weight gain (GWG) is a major clinical challenge; it is a risk factor for C-section, maternal postpartum weight retention, urinary tract infections, gestational diabetes, pre-eclampsia and hypertensive disorders during pregnancy [1, 2]. Moreover, it is associated with adverse fetal, neonatal and childhood outcomes [3, 4] including increased risk of later-life obesity [1, 5–7] and its associated co-morbidities [8]. Studies now show that these effects are transmitted into the second and third generations of offspring, independent of lifestyle and genetic factors, resulting in the multigenerational perpetuation of health deficits [9–11]. Equally maternal obesity, that is maternal BMI > 30 pre-pregnancy, has similar negative outcomes in the mother and fetus [2]. However, it is important to make the distinction between excessive GWG and maternal obesity. Despite similarities in phenotypic outcome, the mechanistic pathways that mediate these outcomes are inherently different [12]. Nevertheless, it is apparent that an “obesogenic” environment during pregnancy imposes a substantive burden on healthcare systems in both the short and long term.
Despite this alarming public health scenario, our understanding of the link between maternal excessive GWG and offspring disease risk is not clear. Even less is known about the transmittance of early life adversity to second and third generations of offspring, although increased risk of obesity and related disorders in first generation offspring are thought to be mediated by perturbations in fetal developing organ systems [2]. Given that the fetal ovary and its primordial germ cells are vulnerable to gestational environmental insults [13–15], it seems likely that maternal excessive GWG may impact on primordial germ cell development with the potential for negative reproductive consequences. Because germ cells within the developing fetal ovary of first generation offspring will eventually give rise to the second generation (grand-offspring), the link between in utero adversity as a result of excessive GWG, and multigenerational disease risk could lie in the developing ovaries and the oocytes within them.
Central to healthy ovarian development and function later in life is the establishment of the primordial follicle pool. Oocytes within primordial follicles originate from primordial germ cells (PGCs) that have migrated from the hindgut to the gonadal ridge during embryonic life [16, 17]. Upon arrival at the gonadal anlagen, PGCs undergo mitosis with a proportion of these germ cells transitioning into meiosis I, becoming arrested in the diplotene stage and are thereafter termed primary oocytes [18]. From Postnatal Day (P) 1 to P4 in rodents [19, 20] (approximately 15 to 20 wk gestation in humans) [21, 22], a subset of primary oocytes become completely surrounded by a single layer of flattened granulosa cells and are thereafter termed primordial follicles. The number of primordial follicles established early in life largely dictates the reproductive potential and lifespan of mammals as this is the pool from which oocytes that may eventually ovulate are drawn from. Once this pool is depleted, reproductive success declines or ceases.
Primordial follicles established early in life are recruited to join the growing follicle pool beginning at approximately P3/P4 in rodents [20], corresponding to 22 wk gestation in humans [23]. Once recruited, follicles grow in a characteristic manner, acquiring successive layers of granulosa and theca cells, a fluid-filled cavity known as an antrum, and ultimately reaching a stage where ovulation is possible. Most growing follicles at this age however, undergo endocrine-controlled apoptotic degeneration, or atresia [24], and are eliminated from the growing follicle pool at the antral follicle stage [25, 26]. Whether or not a follicle survives to undergo ovulation largely depends on the secretion of gonadotropins, which act as pro-survival factors from the primary follicle stage onwards [27]. Hypothalamic gonadotropin releasing hormone (GnRH) stimulates the release of luteinizing hormone (LH) or follicle stimulating hormone (FSH) [28-30] to induce follicular 17-β-estradiol (E2) production that not only aids in follicle development, but also negatively feeds back to the hypothalamus and anterior pituitary gonadotropes to regulate production of GnRH and LH/FSH, respectively [29, 31]. This coordination of gonadotropin and E2 secretion is critical for healthy follicle growth and if impaired, could manifest in reproductive deficits.
We have previously shown in a rat model of diet-induced excessive GWG, that offspring born to mothers fed a HF diet during pregnancy and lactation enter puberty early and display irregular reproductive cycles as adults characterized by persistent estrus [32]. Whether or not this impaired reproductive phenotype is associated with changes in follicular dynamics however, is not known. Given that crucial aspects of ovarian development occur perinatally and these milestones are vulnerable to environmental insults, it is likely that fetuses and offspring may also show signs of follicular defects as a result of maternal excess GWG (in the form of HF diet intake). Thus, we hypothesize that excessive GWG induced by a HF diet will have deleterious effects on fetal ovarian development and offspring ovarian function. Therefore, the present study sought to determine the impact of maternal excessive GWG on ovarian follicular growth and function in offspring at discrete time points in development: during fetal life at Embryonic Day 20 (E20), during neonatal life at P4, prepubertally at P27 and in adulthood (P120). These correspond to critical time points in rat ovarian development when full migration of PGCs to the developing gonad has occurred (E20), primordial follicle assembly has ceased (P4), follicle growth occurs without ovulation (P27), and reproductive maturity is established (P120).
MATERIALS AND METHODS
Animal Model
All animal experiments were approved by the McMaster University Animal Research Ethics Board (Animal Utilization Protocol 12-10-38), in accordance with the guidelines of the Canadian Council of Animal Care and the Animal Ethics Committee at the University of Auckland (Approval R402). We used our established model of HF diet-induced maternal excessive GWG to generate mothers and offspring [32, 33], and in all cases sample sizes refer to the litter (mother) as a biological replicate. Therefore, each sample represents one offspring from a different litter (mother). All tissue collection was performed at approximately 0900 h. In brief, female Wistar rats (aged 110 days) were time mated, and after confirmation of mating (sperm in vaginal smear), rats were housed individually in standard rat cages with free access to water. All rats were kept with a constant temperature maintained at 25°C and a 12L:12D cycle. Pregnant dams were randomly assigned to either a control (CON) diet (14% kcal fat, 3.0 kcal/gm; Diet 8604, Harlan Teklad) or a high-fat (HF) diet (45% kcal of fat, 4.73 kcal/g; product D12451, Research Diets) ad libitum throughout pregnancy and lactation. At E20, a subset of dams from each group (CON n = 8, HF n = 5) were fasted overnight, anesthetized with 2% isoflurane (product CP0406V2; Pharmaceutical Partners of Canada), and killed by decapitation. Retroperitoneal fat mass was weighed as a measure of body fat mass as previously described [32, 33]. Fetuses were sexed and weighed. A second subset of dams was set to deliver, and at birth, pups were weighed and sexed. At P4, litter size was adjusted to eight pups per litter (4 males and 4 females) to ensure standardized nutrition until weaning. Remaining pups were killed by decapitation. At P4, both ovaries were removed and fixed in 10% (v/v) buffered formalin for later processing and embedding (CON n = 8, HF n = 8) (McMaster University Health Science Centre Pathology Research Services).
In order to investigate prepubertal ovarian phenotypes, a subset of female offspring from CON and HF groups were fed a control diet from weaning and fasted overnight at P27, anesthetized with sodium pentobarbitone (60 mg·kg−1), and killed by decapitation. Trunk blood samples were taken for later biochemical analyses. Ovaries were dissected, weighed and one ovary was fixed in Bouin solution and paraffin embedded for histological analyses while the other ovary was snap frozen in liquid nitrogen for molecular analyses (CON n = 7, HF n = 6).
In order to determine adult ovarian phenotypes at P120, females in the diestrus stage (determined by vaginal smearing) were fasted overnight, anesthetized with sodium pentobarbitone (60 mg/kg−1), and killed by decapitation. Trunk blood samples were collected and processed for later biochemical analyses. Ovaries were dissected, weighed and one ovary was fixed in Bouin solution and paraffin embedded for histological analyses while the other ovary was snap frozen in liquid nitrogen for molecular analyses (CON n = 9, HF n = 6).
Finally, in a subgroup of offspring, we investigated the effect of a postweaning HF diet. At weaning (P21) a subset of female offspring were randomly assigned to receive either a control (CON) diet or a high-fat (hf) diet (as detailed above) ad libitum, with free access to water for the remainder of the study. This created four groups of adult offspring: CON-con (n = 9), CON-hf (n = 5), HF-con (n = 6), and HF-hf (n = 5) with the first abbreviation in the diet code indicating maternal diet and the second indicating post-weaning diet. These data are presented in supplemental data. (Supplemental Fig. S1; all Supplemental Data are available online at www.biolreprod.org).
Morphometric Analyses of Oocyte and Follicle Populations
Embryonic Day (E) 20, P4, P27, and P120 paraffin-embedded ovaries were serially sectioned; E20, P4, and P27 ovaries were sectioned at 4 μm and P120 ovaries were sectioned at 8 μm. Every fifth (E20, P4, and P120) or 10th (P27) ovarian section was processed for hematoxylin and eosin staining as before [15, 34], dried overnight, and mounted prior to morphometric analyses.
All follicle counts were performed by a blinded experimenter. Only oocytes or follicles containing an oocyte with a visible nucleus were counted. All follicles were classified according to Hirshfield and Midgley [35], as before [15, 34], as follows: primordial follicle, an oocyte surrounded by one layer of flattened granulosa cells; transitioning follicle, an oocyte surrounded by more than 2 cuboidal granulosa cells and at least one flattened granulosa cell; primary follicles, an oocyte surrounded by one to fewer than two complete layers of cuboidal granulosa cells; secondary follicle, an oocyte surrounded by greater than one layer of cuboidal granulosa cells, with no visible antrum; antral follicle, an oocyte surrounded by multiple layers of cuboidal granulosa cells and containing one or more antral spaces; type II atretic follicle [36] an oocyte (with or without a nucleus) undergoing degeneration or cytoplasmic blebbing and retraction of the granulosa cell layer with a fluid filled cavity present. The area (μm2) of each ovarian section was measured using a Nikon DS-L3 camera control unit. Total volume of each section was calculated (area of the section X thickness of the section) and oocyte/follicle counts for each animal were normalized to the total volume (μm3) of ovarian tissue counted per animal as before [15]. In addition, the area (μm2) of each secondary/antral follicle was measured using a Nikon DS-L3 camera control unit.
AMH and AMH Receptor Type II Immunolocalization
Sections that were not used for follicle counts were used for immunohistochemistry. Briefly, ovarian sections were rehydrated and then immersed in 1% H2O2 (product H325-500; Fisher Chemical) in PBS to inhibit endogenous peroxidase activity. Antigen retrieval was performed by incubating sections in citrate buffer (10 mM Na3C6H5O7; 0.05% Tween-20, pH 6.0) at 90°C for 12 min, followed by 20 min at room temperature. Nonspecific binding was blocked for 30 min with 5% bovine serum albumin (BSA), fraction V (product BP1600-100; Fisher BioReagents) in PBS at room temperature (RT) and incubated overnight with AMH (1:50 dilution; product; AF1446; R&D Systems) or AMHRII (1:50 dilution; product AF1618; R&D Systems) made up in 1% BSA overnight at 4°C in a humidity chamber. The next day, ovarian sections were incubated with a biotinylated secondary antibody (1:1000 dilution; product 605-706-125; Rockland) in 1% BSA in PBS for 2 h at RT, then incubated with avidin/horseradish peroxidase (HRP) complex (Vectastain Elite ABC kit; product PK-6101; Vector Laboratories) for 1 h at RT. Sections were then incubated with 3,3′-diaminobenzidine (DAB; 1 mg/ml H2O, 0.1% H2O2, DAB Easy tablets; product AC32800-5000; Acros Organics) for 5 min. Following DAB incubation, slides were counterstained with hematoxylin stain 2-gill method (product CS-401-1D; Fisher Chemical). All experiments had one ovarian section (negative control) that underwent all steps except the primary antibody incubation (1% BSA in PBS overnight at 4°C). Ovarian images were obtained using an Eclipse Ni microscope, equipped with a 20× objective. Density of oxidized DAB (brown color) was determined by pixel classification using Nikon NIS-Elements AR version 4.20.01 software, and semiquantitative data are expressed as a proportion of immunopositive (brown) area per total ovarian area analyzed.
Determination of Follicular Apoptosis; Immunolocalization of Activated Caspase-3
Detection of apoptotic cells in follicles waccording toformed as previously described [37] in fetal, neonatal, prepubertal, and adult ovarian sections (n = 3 per group). Endogenous peroxidase activity inhibition and antigen retrieval waccording toformed as above. Nonspecific binding was blocked with 5% BSA for 10 min at RT and incubated overnight at 4°C with anti-rabbit cleaved caspase-3 antibody from Cell Signaling (catalog number 9661) at 1:100 dilution overnight at 4°C. The following day, tissue sections were incubated at RT for 2 h with anti-rabbit biotinylated secondary antibody (1:200 dilution; Sigma-Aldrich) and then incubated with ExtrAvidin (1:50 dilution; Sigma-Aldrich) for 1 hour. Activated caspase-3 was visualized with 3,3′-diaminobenzidine (DAB tablets; product D4293; Sigma Aldrich), and all sections were counterstained with Gills 2 hematoxylin (product GHS216; Sigma Aldrich). For determination of apoptotic cells, activated caspase-3 immunopositive cells were measured in six fields of view per section at ×250 magnification (n = 3 per group). Image analysis was performed using an Olympus BX-61 microscope and integrated MetaMorph software. Data are caspase-3 immunopositive area as the proportion of total ovarian area analyzed.
Circulating Plasma Levels of FSH, LH, and E2
Circulating plasma FSH, LH, and E2 levels were measured by commercially available rat ELISA (FSH using product CEA830Ra; Uscn Life Science Inc.; LH using product CEA441Ra; and E2 using product ES180S-100 Mouse/Rat; Calbiotech) according to the manufacturers' specifications in P27 and P120 offspring. Intra-assay coefficients of variability for FSH, LH, and E2 were 5.0, 7.3, and 7.7%, respectively.
Molecular Analyses
RNA extraction and reverse transcription.
Total RNA was extracted from ∼5 mg of frozen, ground ovarian tissue, using the RNeasy mini kit (product 74104; Qiagen) with 350 μl of RLT buffer (1:100 dilution [v:v]; β-mercaptoethanol; product M3148; Sigma-Aldrich) using the manufacturer's instructions. RNA quantity and purity were analyzed using a NanoDrop 2000 spectrophotometer; Thermo Scientific). RNA integrity was assessed using the ratio of absorbance at 260-to-280 nm (A260:A280 nm) and A260:A230 nm for each sample (>2.0 and >1.5, respectively). All RNA samples were stored at −80°C until cDNA generation.
Two micrograms of total RNA were used for first-strand cDNA synthesis, using Superscript VILO cDNA synthesis kit (Life Technologies) according to the manufacturer's instructions and using a standard thermocycler (product C1000 Touch; Bio-Rad). cDNA was stored at −20°C until quantitative PCR (qPCR) assays were performed.
qPCR Assays
A quantitative PCR assay was performed as previously described [15] using the Lightcycler 480 system (Roche Diagnostics) and LightCycler 480 SYBR Green I Master (catalog number 04707516001; Roche Diagnostics). Primers for all genes were designed using Primer BLAST software, available at NCBI (website: blast.ncbi.nlm.nih.gov). Primers were manufactured by Life Technologies or Integrated DNA Technologies, and sequences are listed in Supplemental Table S1. Optimal primer conditions were adjusted to the following cycling conditions: length: 20 bp (range: 17–23 bp); melting temperature (Tm): 60°C (range: 58–62°C); and amplicon length: 50–200 bp. Dissociation analyses were performed to ensure specificity and samples producing a single peak in dissociation curves were used.
All qPCR assays were carried out with an initial denaturation at 95°C for 5 min, followed by amplification of the gene product through 45 successive cycles of 95°C for 10 sec, 60°C for 10 sec, and 72°C for 10 sec. Each plate for a gene of interest contained a standard curve (either 5 or 10-fold serial dilution) generated from a pooled sample of ovarian cDNA, while amplification and dissociation curves were generated for all standards and samples (LightCycler 480 system; Roche Diagnostics). Each sample was run in triplicate. All mRNA data relative to the geometric mean of seven different reference genes (Ywhaz, Ywhag, β-actin, B2M, SDHA, HPRT, and cyclophilin), the levels of which did not differ among groups.
Statistical Analyses
In all cases, biological replicates are from different litters (mothers). All maternal, E20, P4, and P27, and P120 data were analyzed by unpaired Student t-test (CON versus HF). When evaluating the effect of maternal and postweaning diet (see Supplemental data) on P120 ovaries, data were analyzed using a two-way factorial ANOVA. Where appropriate, Tukey post-hoc analysis was performed according to two-way factorial ANOVA (Supplemental Figs. S2 and S3). In all cases, data that were not normally distributed were log transformed in order to achieve normality. All data are mean ± SEM. Sex ratio was analyzed by binary logistic regression with maternal diet as the predictor variable. The relationship between maternal fat mass and fetal oocyte number was assessed through correlation analysis using the Pearson product-moment correlation coefficient. A P value of < 0.05 for all data sets, was considered statistically significant. All analyses were performed using SigmaPlot version 12.0 (Systat Software Inc.), Prism version 5.01 (GraphPad Software Inc.), and PASW Statistics version 18 (SPSS Inc.) software.
RESULTS
Maternal Phenotype
Consistent with our previously published reports [32, 33], dams fed a HF diet during gestation gained more weight from E1 to E20 compared to CON dams (CON: 101.8 ± 8.2 g; HF: 137.5 ± 12.6; P < 0.05). Maternal weight gain at E20 relative to maternal weight at E1 (expressed as a percentage of weight at E1) was significantly greater in dams fed a HF diet during gestation (CON: 33.9 ± 2.5%; HF: 43.9 ± 3.2; P < 0.05). At E20, HF-fed dams were hyperleptinemic (CON: 4.6 ± 0.3 ng/ml; HF: 7.4 ± 0.3; P < 0.0001) and had a greater retroperitoneal fat mass (CON: 3.6 ± 0.4 g; HF: 9.2 ± 0.9; P < 0.0001) than CON dams. Circulating fasting insulin (CON: 0.59 ± 0.12 ng/ml; 0.50 ± 0.03) and glucose (CON: 3.7 ± 0.2 mmol/L; 3.9 ± 0.27) concentrations were not significantly different between HF- and CON-fed dams at E20.
Offspring Phenotype
Maternal diet did not influence the fetal sex ratio at E20 (B = 0.159; P = 0.591; odds ratio = 1.173). Bodyweight and circulating leptin and insulin concentrations did not differ between groups at E20 or P4 (Table 1).
TABLE 1.
Phenotypic characteristics of offspring born to HF-fed dams.*
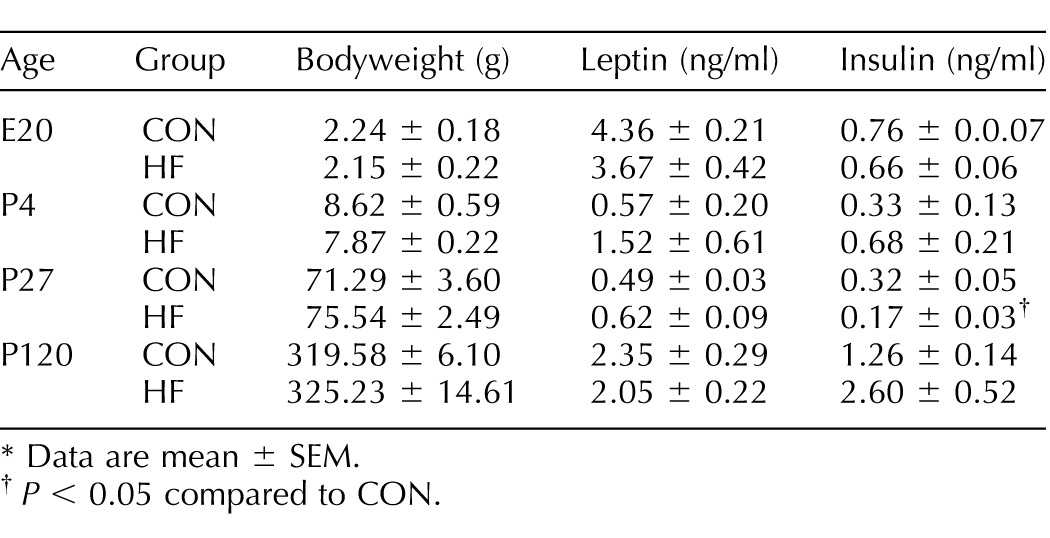
Data are mean ± SEM.
P < 0.05 compared to CON.
Maternal HF Diet Reduced Total Numbers of Oocytes in Fetuses at E20
Fetuses of HF-fed mothers demonstrated a significant (P < 0.01) decrease of 11% in total oocyte number at E20 (Fig. 1, a–c). Moreover, we observed a significant negative correlation between maternal fat mass (retroperitoneal fat depot) and fetal oocyte number (Pearson correlation; r = −0.57; P < 0.05) (Fig. 1d) and maternal plasma leptin concentrations and fetal oocyte number (Pearson correlation; r = −0.63; P < 0.05) (Fig. 1e). Furthermore, there was a significant increase in the proportion of activated caspase-3 immunopositive cells, indicative of increased apoptosis in the fetal ovary as a result of a maternal HF diet (Fig. 1f).
FIG. 1.

Maternal HF diet intake resulted in decreased number of oocytes in fetal ovaries at E20. Representative photomicrographs show fetal offspring ovaries from dams fed CON (a) and HF (b). Bars = 100 μm. c) Maternal HF diet during pregnancy resulted in an 11% decrease in the number of oocytes at E20 in fetal offspring ovaries (n = 8, CON; n = 5, HF). Maternal retroperitoneal fat mass (d) and maternal plasma leptin concentrations (e) negatively correlated (r = −0.57; P < 0.05; r = −0.63; P < 0.05, respectively) with the number of oocytes in fetal offspring ovaries. Solid line is the best fit, and dotted lines are 95% confidence intervals. f) Maternal HF diet during pregnancy resulted in significant increase in activated caspase-3 immunostaining, expressed as a proportion of positive immunostaining per total area analyzed (n = 5 CON; n = 5 HF). Data are mean ± SEM fold changes relative to CON offspring for oocyte counts. Student unpaired t-test, **P < 0.01; ***P < 0.001.
Maternal HF Diet Increased Numbers of Primordial and Transitioning Follicles in Neonates Through Suppression of AMH
Postnatal Day 4 is the approximate time when the primordial follicle pool is established in rats [38–40]. At P4, offspring ovaries demonstrated oocytes that had not yet assembled into follicles and remained in oocyte nests, and primordial and transitioning follicles (Fig. 2, a and b). Neonatal offspring born to HF-fed mothers demonstrated an increased number of both primordial (Fig. 2c) and transitioning follicles (Fig. 2d). This increase in primordial follicle assembly and recruitment was likely mediated by an almost complete downregulation of immunopositive AMH (Fig. 2, e–h) and its receptor AMHRII (Fig. 2, i–l). In addition to greater primordial and transitioning follicle numbers, there were also higher levels of activated caspase-3–positive immunostaining of follicular cells in offspring born to HF-fed mothers (Fig. 2m), suggesting that more follicles were undergoing atresia.
FIG. 2.

Maternal HF diet intake resulted in increased primordial follicle assembly and reduced AMH in offspring ovaries at P4. Representative photomicrographs of hematoxylin and eosin stained P4 offspring ovaries from dams fed CON (a) and HF (b). Maternal HF diet during pregnancy and lactation resulted in increases of 67% and 69%, respectively, in the number of primordial (c) and transitioning (d) follicles (n = 8, CON; n = 8, HF). Representative photomicrographs of AMH (e, f, g) and AMHRII (i, j, k) immunostaining of P4 offspring ovaries from CON (e, i, respectively) and HF (f, j, respectively) dams with negative controls (g, k, respectively). Maternal HF diet during pregnancy and lactation resulted in a significant decrease in positive AMH and AMHRII immunostaining, expressed as a proportion of immunostaining per total area analyzed (h, l, respectively) (n = 5, CON; n = 4, HF). Maternal HF diet during pregnancy and lactation resulted in a significant increase in activated caspase-3 immunostaining, expressed as a proportion of positive immunostaining per total area analyzed (m) (n = 3, CON; n = 3, HF). Data are mean ± SEM fold changes relative to CON offspring for follicle counts. Student unpaired t-test, *P < 0.05; **P < 0.01. Bars = 100 μm (a–c, e–g, and i–k).
Maternal HF Diet Resulted in Small Secondary Follicles and Increased Follicular Atresia in Prepubertal Offspring
Follicle numbers were similar between groups at P27 (Fig. 3a). Because we observed higher levels of follicles at P4, we hypothesized that these HF-exposed follicles were likely lost to accelerated atresia or recruitment during the prepubertal period resulting in primordial follicle numbers reaching control levels at P27. In support of this hypothesis, we observed a significant increase in the number of type II atretic follicles in HF prepubertal offspring compared to CON offspring (Fig. 3a). Activated caspase-3–positive immunostaining of follicular cells was increased (Fig. 3c) in HF offspring compared to those in CON offspring at P27, in agreement with our observed increase in atretic follicle number. HF offspring secondary follicles were smaller than those in CON offspring (Fig. 3b). HF offspring demonstrated significantly lower plasma concentrations of E2 (Fig. 3d), despite comparable circulating concentrations of plasma FSH (Fig. 3, d and e) and LH (Fig. 3f).
FIG. 3.

Maternal HF diet intake resulted in compromised ovarian follicle growth in prepubertal offspring at P27. P27 offspring born to HF-fed dams had similar numbers of primordial, transitioning, primary, secondary, and antral follicles (a); however, these offspring had more type II atretic follicles (a) and smaller secondary follicles (b) than offspring born to CON-fed dams (n = 7, CON; n = 6, HF). Maternal HF diet during pregnancy and lactation resulted in a significant increase in caspase-3 immunostaining, expressed as a proportion of positive immunostaining per total area analyzed (c) (n = 3, CON; n = 3, HF). Prepubertal offspring of HF-fed dams also had reduced circulating plasma E2 (d) concentrations, despite concentrations of plasma FSH (e) and LH (f) similar to those of offspring born to CON-fed dams (n = 7, CON; n = 6, HF). Data are mean ± SEM fold changes relative to CON offspring for follicle counts. Student unpaired t-test, *P < 0.05; **P < 0.01.
Maternal HF Diet Resulted in Impaired Ovarian Follicle Growth in Adult Offspring
Primordial and transitioning follicle number tended to be higher in HF offspring compared to CON but this difference did not reach statistical significance (P = 0.09) (Fig. 4a). Primary, secondary, and antral follicle numbers were similar between groups (Fig. 4a); however, HF offspring had higher numbers of type II atretic follicles compared to CON (Fig. 4a). Consistent with increased atresia, activated caspase-3–positive immunostaining was increased (Fig. 4b) in HF offspring compared to CON. HF offspring showed a significant increase in the FSH:E2 ratio (Fig. 4c); whereas plasma concentrations of FSH (Fig. 4d) were significantly increased despite comparable concentrations of E2 (Fig. 4e). Plasma LH was lower in HF compared to CON offspring (Fig. 4f).
FIG. 4.

Maternal HF diet intake increased follicular atresia due to apoptosis in adult offspring at P120. P120 offspring born to HF-fed dams had similar numbers of primordial, transitioning, primary, secondary, and antral follicles (a); however, these offspring had more type II atretic follicles (a) than offspring born to CON-fed dams (n = 9, CON; n = 6, HF). Maternal HF diet during pregnancy and lactation resulted in a significant increase in caspase-3 immunostaining, expressed as a proportion of positive immunostaining per total area analyzed in adult offspring (b) (n = 3, CON; n = 3, HF). HF offspring also demonstrated an increase in circulating FSH:E2 ratio (c), which was driven by a significant increase in circulating FSH (d), without a decrease in circulating E2 (e) in these offspring. Offspring of HF-fed dams had significantly lower circulating concentrations of LH (f) (n = 9, CON; n = 6, HF for plasma measurements). Data are mean ± SEM fold changes relative to CON offspring for follicle counts. Student unpaired t-test, *P < 0.05; **P < 0.01.
A greater proportion of antral follicles in HF offspring demonstrated positive AMH immunostaining than CON offspring (Fig. 5, a and b). HF offspring demonstrated significantly decreased ovarian mRNA expression levels of Gdf9, Dazl, and Erα compared to CON offspring (Fig. 5c). However, ovarian mRNA expression levels of Bmp15 and Erβ (Fig. 5c) and steroidogenic enzymes (StAR, Cyp11a1, Cyp19a1, Cyp17a1, 3βHSD, 17βHSD1) and gonadotropin receptors (Fshr, Lhr) were comparable between groups (data not shown).
FIG. 5.

Maternal HF diet intake resulted in increased antral follicle AMH immunostaining and decreased mRNA levels of key regulators of follicle growth in adult offspring at P120. Representative photomicrographs of AMH immunostaining in antral follicles in CON and HF offspring ovaries with negative control (a). Bars = 100 μm. P120 offspring born to HF-fed dams had increased positive AMH immunostaining in antral follicles (b). Adult HF offspring also demonstrated decreased mRNA expression of Gdf9, Dazl, and Erα (c) (n = 9, CON; n = 6, HF). No change in Bmp15 or Erβ mRNA expression was observed in adult HF offspring (c). Data are mean ± SEM fold changes relative to CON offspring for follicle counts. Student unpaired t-test, *P < 0.05; **P < 0.01.
Postweaning Diet Effects on Offspring Ovarian Outcomes
In a subset of animals, the impact of a postweaning HF diet (Supplemental Fig. S2) on adult ovarian outcome was investigated (at P120). A PW HF diet increased primordial follicle number and the number of atretic follicles found in CON adult offspring ovaries (Supplemental Fig. S2a), an effect that was similar to that found in HF animals fed a control postweaning diet. PW HF diet did little to further impact on follicle counts in HF offspring (albeit atretic follicle numbers were reduced) (Supplemental Fig. S2a). In both CON and HF adult offspring, a PW HF diet did not alter activated caspase-3–positive immunostaining (Supplemental Fig. S2b), circulating FSH (Supplemental Fig. S2c), E2 (Supplemental Fig. S2d), or circulating FSH:E2 ratio (Supplemental Fig. S2e); however, in CON offspring, a postweaning HF diet significantly decreased circulating LH (Supplemental Fig. S2f), to a level similar to that of HF animals. Additionally, in CON adults, ovarian antral follicle AMH was increased, to the same degree as a maternal HF diet exposure (Supplemental Fig. S3a). The combination of a maternal and postweaning HF diet did not further impact on ovarian antral follicle AMH (Supplemental Fig. S3, a and b), although we observed a significant interaction between maternal and postweaning diet, where ovarian immunopositive staining of AMH responses to a postweaning diet depended upon maternal dietary history. Finally, a postweaning HF diet did not impact ovarian mRNA expression of Gdf9, Bmp15, Dazl, Erα, or Erβ (Supplemental Fig. S3c).
DISCUSSION
In the present study, we show that exposure to a maternal HF diet during pregnancy and lactation results in follicular impairments that are present both before and after puberty and that these impairments likely stem from compromised oocyte/follicular development and function that occur during fetal and/or neonatal life. Although the exact mechanisms are unclear, it appears that increased apoptosis, and a loss of the AMH brake on follicular recruitment early in neonatal life, could underpin our observed changes in follicular numbers in prepubertal and adult ovaries.
Fetuses of HF mothers demonstrated a decrease in total oocyte number. It is not clear how fetal oocyte loss is induced. Our data suggest that it is likely that the oocyte pool in HF fetuses is subject to higher rates of apoptosis compared to CON offspring. Consistent with our findings, it has previously been shown in sheep that at mid-gestation, fetuses of over-nourished ewes have increased oocyte loss [41]. The mechanisms mediating this maternal-fetal oocyte relationship have yet to be determined; however, we speculate that increased maternal-fetal inflammation [42] may contribute to accelerated fetal oocyte loss as maternal obesity has been shown to induce inflammation in other developing fetal organs [43–45]. Alternatively, we show that both maternal retroperitoneal fat mass and maternal plasma leptin concentrations negatively correlate with total oocyte number thus fetal oocyte survival/loss may be mediated by the state of the mother. It has been proposed that placental and central leptin resistance in obese pregnancies may alter fuel partitioning in utero [46, 47] compromising fetal growth, potentially impairing healthy ovarian development. Furthermore, maternal obesity has been associated with fetal leptin resistance in some animal models [48]. Whether leptin receptors (ObRb) exist on fetal ovarian germ cells is unknown, although ObRb has been localized to primordial germ cells in the testis [49]. ObRb has been found in adult ovarian granulosa, and theca cells [50, 51] and leptin is known to influence follicular steroidogenesis [52] and oocyte maturation [49, 53]. Whether it is possible that primordial germ cells can be leptin resistant is even less clear. This could be addressed in future studies should ObRb localize in female PGCs. Given that oocytes and follicles are continuously lost to atresia, and that most oocytes undergo apoptosis during fetal life [54, 55], we hypothesize that there has been a premature acceleration of the normal rate of oocyte number decline in HF fetuses. This notion is consistent with our hypothesis that HF offspring experience accelerated primordial follicle recruitment (see below). However, whether this loss is mediated by leptin directly, or leptin-induced placental dysfunction or maternal-fetal inflammation remains to be determined.
Despite a significant decrease in oocyte number during late fetal life, neonates born to HF mothers demonstrated comparable numbers of oocytes at P4. More of these oocytes, however, were assembled into primordial follicles and more primordial follicles were transitioning to join the growing follicle pool. Accompanying this change was an almost complete down-regulation of AMH and its receptor, AMHRII. It is well documented that AMH treatment inhibits both primordial follicle assembly [40] and transition of primordial follicles to primary follicles [56–58]. Therefore, it appears that a lack of AMH signaling may have contributed to the increase in the number of primordial follicles assembled and the increase in transitioning follicle numbers in neonates born to HF-fed dams. We suggest that this is indicative of a premature induction of the normal recruitment pattern of primordial follicles during neonatal life.
Our data suggest that increased primordial follicle assembly and recruitment in HF ovaries may coincide with an increase in follicular atresia. Indeed, primordial follicle numbers in HF offspring experience a sharper decline during the neonatal-to-prepubertal period, reaching control levels by P27. This drop can be explained by the fact that at P4, neonates of HF mothers experienced increased primordial follicle recruitment and increased primordial follicle death, as evidenced by the increase in transitioning follicles and the increased immunostaining for activated caspase-3, respectively. This explanation is further corroborated by the observation that HF offspring demonstrate an increase in atretic follicles at P27 without concomitant changes in the numbers of other follicle subtypes. Thus it is possible that in HF offspring, the increase in primordial follicle recruitment during the prepubertal period is associated with a concomitant increase in atresia near the end-stages of follicle development.
This pattern of early life follicle dynamics is consistent with findings in the study by Flaws et al. [60] and Bristol-Gould et al. [59], who proposed that the prepubertal ovary has a “census” or “quorum sensing” mechanism to detect numbers of primordial follicles. If this number exceeds a certain threshold, excess primordial follicles are eliminated via atresia in order to meet a quota necessary for adult fertility [59]. Evidence from activin-treated neonatal mice and transgenic mice overexpressing the anti-apoptotic factor Bcl-2 support this hypothesis. Activin treatment during primordial follicle assembly in mice initially increased the number of primordial follicles, however, at P19, these mice showed comparable primordial follicle numbers compared to vehicle-treated controls [59]. Similarly, ovary specific overexpression of Bcl-2 in mice resulted in an increased number of primordial follicles at birth followed by a drop to wild type levels at P30 [60]. Bristol-Gould et al. [59] proposed that this mechanism functions to eliminate poor quality oocytes. In their study, P24 mice treated with activin during neonatal life had fewer ovulated oocytes that were able to advance to metaphase II; oocytes were smaller in diameter and had more spindle abnormalities than vehicle-treated control oocytes [59]. These observations suggest that excessive primordial follicle formation during the neonatal period results in growing follicles with poor quality oocytes. Based on our data, it is possible that exposure to a perinatal HF diet results in more follicles with reduced oocyte quality prior to puberty; in prepubertal HF offspring, we observed an increased number of atretic follicles and smaller secondary follicles. Corresponding to work in humans showing that likelihood of fertilization, and quality of embryos produced is negatively affected by smaller follicle size [61, 62].
Prepubertal HF offspring showed decreased concentrations of plasma E2 despite comparable levels of both circulating FSH and LH; which may be indicative of a reduced ovarian response to gonadotropins. Responsiveness to gonadotropins is a major factor in determining follicle survival [26, 27, 63] and decreased responsiveness results in follicles that are likely to undergo atresia. Moreover, P27 HF offspring demonstrated reduced circulating insulin concentrations. Because insulin has been shown to be an important follicle growth factor [64, 65], it is possible that a decrease in insulin signaling negatively affected follicular growth in HF animals. This is consistent with our previous observations [32] that offspring of HF-fed dams at P27 were hypoinsulinemic compared to offspring of CON-fed dams, although leptin levels remained unchanged (Table 1). Regardless, the fact that we see increased atresia in the face of similar follicle numbers could indicate that primordial follicles that are prematurely recruited into the growing pool are subsequently lost, resulting in no net loss (or gain) of antral follicles.
Similar to P27 offspring, adult offspring also demonstrated an increased number of atretic follicles. E2 signaling through ERα in theca cells has been shown to be necessary for antral follicle growth [66] and diminished local responsiveness to E2 through ERα in HF offspring may have reduced antral follicle survival. We found that mRNA levels of Erα were lower in adult HF offspring, which may have contributed to follicular demise. Impaired responsiveness to gonadotropins may also contribute to increased follicular atresia, given that circulating plasma FSH concentrations were significantly higher in HF offspring and there was no concomitant increase in circulating E2 concentrations compared to CON offspring.
In contrast to the decrease observed in neonatal ovaries, AMH-positive immunostaining in adult antral follicles was higher in HF than in CON offspring. High levels of AMH have been shown to reduce granulosa cell responsiveness to FSH by decreasing cAMP production, aromatase and FSHR expression [67–69]. Reduced antral follicle FSH responsiveness as a result of increased AMH may explain why high circulating concentrations of FSH were not associated with high levels of E2 in HF offspring. Paradoxically, it has also been shown that FSH can stimulate AMH through cAMP in cultured human granulosa cells [70]. Therefore, if FSH responsiveness in HF offspring is decreased by AMH through a decrease in cAMP mediated signaling, AMH's own expression should also be decreased. This apparent paradox may be due to species-specific differences [71], or it may be attributable to the fact that FSHR signaling is more complex than the simple activation of the cAMP pathway. Downstream AMH signaling could be altered by other inputs [72], including GDF9, an oocyte-secreted growth factor necessary for fertility [73]. Addition of GDF9 to cultured preantral follicles has been found to enhance FSH-induced follicle growth [74]. Conversely, injection of Gdf9 antisense oligonucleotides into the oocytes of preantral follicles to block translation inhibits FSH-induced follicular growth [74] demonstrating that levels of Gdf9 influence FSH responsiveness. HF offspring had lower mRNA levels of Gdf9 despite comparable numbers of primary, secondary, and antral follicles between groups. Therefore, it is possible that decreased Gdf9 may contribute to decreased FSH responsiveness in antral follicles of HF offspring.
mRNA levels of the oocyte-specific RNA binding protein DAZL were decreased in adult HF offspring. In vitro maturation of porcine antral follicles without FSH has been shown to decrease Dazl mRNA, suggesting that FSH stimulates Dazl transcription [75]. A decrease in Dazl mRNA in HF offspring, thus, is consistent with an inability of FSH to induce follicle growth and differentiation. Although, it has also been shown that Dazl heterozygous female mice have increased FSH sensitivity [76], suggesting that a lack of DAZL supports FSH-induced follicle growth. This implicates DAZL as a regulator of FSH sensitivity; where decreased Dazl in HF offspring is potentially a response that aims to increase FSH sensitivity and rescue growing follicles from atresia. DAZL has also been implicated in the apoptotic pathway, particularly in primordial germ cell development [77], and may act in this way to affect follicle growth.
In human studies, AMH has been used effectively as a marker of ovarian aging [78–80]. However, controversy exists as to whether AMH levels are reliable, given that it does not fulfill the requirements for an ideal screening test, and neither does ovarian aging fulfill the ideal requirements for a disease for which a screening test can be developed [81]. Nonetheless, AMH is still considered a good clinical indicator of ovarian aging [82]. It would thus follow that increased AMH immunostaining in adult HF offspring would be a positive outcome, in terms of ovarian age. Speculation aside, as the function of AMH and knowledge about how it is regulated has still not been completely elucidated, more studies are needed to understand the role of perinatal maternal HF diet on offspring ovarian function and how ovarian AMH signaling and regulation are involved in these effects.
Maternal High-Fat Diet Combined With Postweaning High-Fat Nutrition
We have demonstrated that maternal HF intake during pregnancy increased fetal oocyte loss and increased number of primordial follicles assembled and recruited during neonatal life. We speculate that these “excess” primordial follicles are of lower quality and are therefore lost to atresia during the prepubertal period and that this has negative consequences on follicle growth later in life. Although we investigated whether a postweaning HF diet interacted with the perinatal diet, we did not observe many interactions in our outcomes variables. In fact in many cases, a postweaning HF diet had the same effect as a perinatal HF diet; a postweaning HF diet increased primordial follicle number and the number of atretic follicles found in control adults to the same degree as a perinatal HF diet. Further, in control adults, ovarian antral follicle AMH was increased, to the same degree as a maternal HF diet exposure. The combination of a maternal and postweaning HF diet did not further impact on ovarian antral follicle AMH. These outcomes suggest that despite being fed a control postweaning diet, maternal HF exposure sets up the offspring for ovarian compromise that is comparable to that induced by an “unhealthy” postnatal lifestyle.
In conclusion, we demonstrate that maternal HF diet-induced excessive gestational weight gain resulted in increased fetal oocyte loss and an increased number of primordial follicles assembled and recruited during neonatal life. We speculate that these “excess” primordial follicles are of lower quality and are therefore lost to atresia during the prepubertal period. This appears to have negative consequences on follicle growth later in life as more follicles undergo atresia both before and after puberty. In adulthood, we speculate that follicular impairments are partially driven by decreased FSH responsiveness due to a decline in oocyte quality, demonstrated by the downregulation of the oocyte-secreted factor, Gdf9, and the oocyte-specific RNA binding protein, Dazl in HF offspring. This study suggests that the intrauterine environment has lasting consequences on female rat offspring follicle growth and may partially explain the estimated 15-30% of cases of unexplained infertility [83]. However, it is unclear what impact this impaired follicle growth has on oocyte quality, fertilization rates, and embryo development of the next generation. Given that multigenerational increases in disease risk exist [84], understanding how the fetal germline is impacted by the maternal condition is imperative as this may represent a critical mechanism modulating intergenerational disease risk transmission. Future studies are therefore needed to delineate the complex mechanisms at play so that they can be targeted with interventions to prevent the deleterious personal and societal costs of non-genetic, multigenerational inheritance of disease risk.
Supplementary Material
Footnotes
This study was funded in part by the Health Research Council of New Zealand and Gravida, the Canada Research Chair in Perinatal Programming to D.M.S., the Canadian Institutes for Health Research to J.P., and the Royal Society of New Zealand Gravida to M.H.V.
REFERENCES
- Poston L. Gestational weight gain: influences on the long-term health of the child. Curr Opin Clin Nutr Metab Care. 2012;15:252–257. doi: 10.1097/MCO.0b013e3283527cf2. [DOI] [PubMed] [Google Scholar]
- Poston L. Maternal obesity, gestational weight gain and diet as determinants of offspring long term health. Best Pract Res Clin Endocrinol Metab. 2012;26:627–639. doi: 10.1016/j.beem.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Crane JM, White J, Murphy P, Burrage L, Hutchens D. The effect of gestational weight gain by body mass index on maternal and neonatal outcomes. J Obstet Gynaecol Can. 2009;31:28–35. doi: 10.1016/s1701-2163(16)34050-6. [DOI] [PubMed] [Google Scholar]
- Gaillard R, Durmus B, Hofman A, Mackenbach JP, Steegers EA, Jaddoe VW. Risk factors and outcomes of maternal obesity and excessive weight gain during pregnancy. Obesity (Silver Spring) 2013;21:1046–1055. doi: 10.1002/oby.20088. [DOI] [PubMed] [Google Scholar]
- Lau EY, Liu J, Archer E, McDonald SM, Liu J. Maternal weight gain in pregnancy and risk of obesity among offspring: a systematic review. J Obes. 2014;2014:524939. doi: 10.1155/2014/524939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oken E, Taveras EM, Kleinman KP, Rich-Edwards JW, Gillman MW. Gestational weight gain and child adiposity at age 3 years. Am J Obstet Gynecol. 2007;196:322, e321–328. doi: 10.1016/j.ajog.2006.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starling AP, Brinton JT, Glueck DH, Shapiro AL, Harrod CS, Lynch AM, Siega-Riz AM, Dabelea D. Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start study. Am J Clin Nutr. 2015;101:302–309. doi: 10.3945/ajcn.114.094946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts VH, Frias AE, Grove KL. Impact of maternal obesity on fetal programming of cardiovascular disease. Physiology (Bethesda) 2015;30:224–231. doi: 10.1152/physiol.00021.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai M, Jellyman JK, Ross MG. Epigenomics, gestational programming and risk of metabolic syndrome. Int J Obes. 2015;39:633–641. doi: 10.1038/ijo.2015.13. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Bulnes A, Astiz S, Ovilo C, Lopez-Bote CJ, Sanchez-Sanchez R, Perez-Solana ML, Torres-Rovira L, Ayuso M, Gonzalez J. Early-postnatal changes in adiposity and lipids profile by transgenerational developmental programming in swine with obesity/leptin resistance. J Endocrinol. 2014;223:M17–M29. doi: 10.1530/JOE-14-0217. [DOI] [PubMed] [Google Scholar]
- Shasa DR, Odhiambo JF, Long NM, Tuersunjiang N, Nathanielsz PW, Ford SP. Multigenerational impact of maternal overnutrition/obesity in the sheep on the neonatal leptin surge in granddaughters. Int J Obes (Lond) 2015;39:695–701. doi: 10.1038/ijo.2014.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie GJ, Sloboda DM, Reynolds CM, Vickers MH. Timing of maternal exposure to a high fat diet and development of obesity and hyperinsulinemia in male rat offspring: same metabolic phenotype, different developmental pathways? J Nutr Metab. 2013;2013:517384. doi: 10.1155/2013/517384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banu SK, Stanley JA, Sivakumar KK, Arosh JA, Barhoumi R, Burghardt RC. Identifying a novel role for X-prolyl aminopeptidase (Xpnpep) 2 in CrVI-induced adverse effects on germ cell nest breakdown and follicle development in rats. Biol Reprod. 2015 doi: 10.1095/biolreprod.114.125708. 92:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe GJ, Lynch TJ, Albrecht ED. Regulation of baboon fetal ovarian development by placental estrogen: onset of puberty is delayed in offspring deprived of estrogen in utero. Biol Reprod. 2013;89:132. doi: 10.1095/biolreprod.112.107318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernal AB, Vickers MH, Hampton MB, Poynton RA, Sloboda DM. Maternal undernutrition significantly impacts ovarian follicle number and increases ovarian oxidative stress in adult rat offspring. PLoS One. 2010;5:e15558. doi: 10.1371/journal.pone.0015558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamsen LS, Brochner CB, Byskov AG, Mollgard K. The migration and loss of human primordial germ stem cells from the hind gut epithelium towards the gonadal ridge. Int J Dev Biol. 2012;56:771–778. doi: 10.1387/ijdb.120202lm. [DOI] [PubMed] [Google Scholar]
- Sánchez F, Smitz J. Molecular control of oogenesis. Biochim Biophys Acta. 2012;1822:1896–1912. doi: 10.1016/j.bbadis.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Kerr JB, Myers M, Anderson RA. The dynamics of the primordial follicle reserve. Reproduction. 2013;146:R205–R215. doi: 10.1530/REP-13-0181. [DOI] [PubMed] [Google Scholar]
- Pepling ME, Spradling AC. Mouse ovarian germ cell cysts undergo programmed breakdown to form primordial follicles. Dev Biol. 2001;234:339–351. doi: 10.1006/dbio.2001.0269. [DOI] [PubMed] [Google Scholar]
- Rajah R, Glaser EM, Hirshfield AN. The changing architecture of the neonatal rat ovary during histogenesis. Dev Dyn. 1992;194:177–192. doi: 10.1002/aja.1001940303. [DOI] [PubMed] [Google Scholar]
- Fulton N, Martins da Silva SJ, Bayne RAL, Anderson RA. Germ cell proliferation and apoptosis in the developing human ovary. J Clin Endocrinology Metab. 2005;90:4664–4670. doi: 10.1210/jc.2005-0219. [DOI] [PubMed] [Google Scholar]
- Zambrano E, Guzmán C, Rodríguez-González GL, Durand-Carbajal M, Nathanielsz PW. Fetal programming of sexual development and reproductive function. Mol Cell Endocrinol. 2014;382:538–549. doi: 10.1016/j.mce.2013.09.008. [DOI] [PubMed] [Google Scholar]
- Maheshwari A, Fowler PA. Primordial follicular assembly in humans – revisited. Zygote. 2008;16:285–296. doi: 10.1017/S0967199408004802. [DOI] [PubMed] [Google Scholar]
- Kaipia A, Hsueh AJ. Regulation of ovarian follicle atresia. Annu Rev Physiol. 1997;59:349–363. doi: 10.1146/annurev.physiol.59.1.349. [DOI] [PubMed] [Google Scholar]
- Hirshfield AN. Size-frequency analysis of atresia in cycling rats. Biol Reprod. 1988;38:1181–1188. doi: 10.1095/biolreprod38.5.1181. [DOI] [PubMed] [Google Scholar]
- Hsueh AJ, Billig H, Tsafriri A. Ovarian follicle atresia: a hormonally controlled apoptotic process. Endocr Rev. 1994;15:707–724. doi: 10.1210/edrv-15-6-707. [DOI] [PubMed] [Google Scholar]
- Hsueh AJ, Kawamura K, Cheng Y, Fauser BC. Intraovarian control of early folliculogenesis. Endocr Rev. 2015;36:1–24. doi: 10.1210/er.2014-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayes FC, Britt JH, Esbenshade KL. Role of gonadotropin-releasing hormone pulse frequency in differential regulation of gonadotropins in the gilt. Biol Reprod. 1997;56:1012–1019. doi: 10.1095/biolreprod56.4.1012. [DOI] [PubMed] [Google Scholar]
- Popat VB, Prodanov T, Calis KA, Nelson LM. The menstrual cycle: a biological marker of general health in adolescents. Ann N Y Acad Sci. 2008;1135:43–51. doi: 10.1196/annals.1429.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsutsumi R, Webster NJ. GnRH pulsatility, the pituitary response and reproductive dysfunction. Endocrine Journal. 2009;56:729–737. doi: 10.1507/endocrj.k09e-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DM, Gore AC. Transgenerational neuroendocrine disruption of reproduction. Nat Rev Endocrinol. 2011;7:197–207. doi: 10.1038/nrendo.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor KL, Vickers MH, Beltrand J, Meaney MJ, Sloboda DM. Nature, nurture or nutrition? Impact of maternal nutrition on maternal care, offspring development and reproductive function. J Physiol. 2012;590:2167–2180. doi: 10.1113/jphysiol.2011.223305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie G, Sloboda D, Kamal T, Vickers M. Maternal nutritional history predicts obesity in adult offspring independent of postnatal diet. J Physiol. 2009;587:905–915. doi: 10.1113/jphysiol.2008.163477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan KA, Bernal AB, Vickers MH, Gohir W, Petrik JJ, Sloboda DM. Early life exposure to undernutrition induces ER stress, apoptosis, and reduced vascularization in ovaries of adult rat offspring. Biol Reprod. 2015;92:110. doi: 10.1095/biolreprod.114.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirshfield AN, Midgley AR., Jr Morphometric analysis of follicular development in the rat. Biol Reprod. 1978;19:597–605. doi: 10.1095/biolreprod19.3.597. [DOI] [PubMed] [Google Scholar]
- Osman P. Rate and course of atresia during follicular development in the adult cyclic rat. J Reprod Fertil. 1985;73:261–270. doi: 10.1530/jrf.0.0730261. [DOI] [PubMed] [Google Scholar]
- Greenaway J, Connor K, Pedersen HG, Coomber BL, LaMarre J, Petrik J. Vascular endothelial growth factor and its receptor, Flk-1/KDR, are cytoprotective in the extravascular compartment of the ovarian follicle. Endocrinology. 2004;145:2896–2905. doi: 10.1210/en.2003-1620. [DOI] [PubMed] [Google Scholar]
- Kezele P, Skinner MK. Regulation of ovarian primordial follicle assembly and development by estrogen and progesterone: endocrine model of follicle assembly. Endocrinology. 2003;144:3329–3337. doi: 10.1210/en.2002-0131. [DOI] [PubMed] [Google Scholar]
- Kezele P, Nilsson EE, Skinner MK. Keratinocyte growth factor acts as a mesenchymal factor that promotes ovarian primordial to primary follicle transition. Biol Reprod. 2005;73:967–973. doi: 10.1095/biolreprod.105.043117. [DOI] [PubMed] [Google Scholar]
- Nilsson EE, Schindler R, Savenkova MI, Skinner MK. Inhibitory actions of anti-Müllerian hormone (AMH) on ovarian primordial follicle assembly. PLoS One. 2011;6:e20087. doi: 10.1371/journal.pone.0020087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Da Silva P, Aitken RP, Rhind SM, Racey PA, Wallace JM. Effect of maternal overnutrition during pregnancy on pituitary gonadotrophin gene expression and gonadal morphology in female and male foetal sheep at day 103 of gestation. Placenta. 2003;24:248–257. doi: 10.1053/plac.2002.0897. [DOI] [PubMed] [Google Scholar]
- Aye IL, Lager S, Ramirez VI, Gaccioli F, Dudley DJ, Jansson T, Powell TL. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. 2014;90:129. doi: 10.1095/biolreprod.113.116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Yan X, Tong JF, Zhao J, Zhu MJ. Maternal obesity, inflammation, and fetal skeletal muscle development. Biol Reprod. 2010;82:4–12. doi: 10.1095/biolreprod.109.077099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murabayashi N, Sugiyama T, Zhang L, Kamimoto Y, Umekawa T, Ma N, Sagawa N. Maternal high-fat diets cause insulin resistance through inflammatory changes in fetal adipose tissue. Eur J Obstet Gynecol Reprod Biol. 2013;169:39–44. doi: 10.1016/j.ejogrb.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Yan X, Huang Y, Wang H, Du M, Hess BW, Ford SP, Nathanielsz PW, Zhu M-J. Maternal obesity induces sustained inflammation in both fetal and offspring large intestine of sheep. Inflam Bowel Dis. 2011;17:1513–1522. doi: 10.1002/ibd.21539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier DR, Ferraro ZM, Gruslin A. Role of leptin in pregnancy: consequences of maternal obesity. Placenta. 2013;34:205–211. doi: 10.1016/j.placenta.2012.11.035. [DOI] [PubMed] [Google Scholar]
- Farley DM, Choi J, Dudley DJ, Li C, Jenkins SL, Myatt L, Nathanielsz PW. Placental amino acid transport and placental leptin resistance in pregnancies complicated by maternal obesity. Placenta. 2010;31:718–724. doi: 10.1016/j.placenta.2010.06.006. [DOI] [PubMed] [Google Scholar]
- Mazzucco MB, Fornes D, Capobianco E, Higa R, Jawerbaum A, White V. Maternal saturated-fat-rich diet promotes leptin resistance in fetal liver lipid catabolism and programs lipid homeostasis impairments in the liver of rat offspring. J Nutr Biochem. 2016;27:61–69. doi: 10.1016/j.jnutbio.2015.08.019. [DOI] [PubMed] [Google Scholar]
- El-Hefnawy T, Ioffe S, Dym M. Expression of the leptin receptor during germ cell development in the mouse testis. Endocrinology. 2000;141:2624–2630. doi: 10.1210/endo.141.7.7542. [DOI] [PubMed] [Google Scholar]
- Karlsson C, Lindell K, Svensson E, Bergh C, Lind P, Billig H, Carlsson LM, Carlsson B. Expression of functional leptin receptors in the human ovary. J Clin Endocrinol Metab. 1997;82:4144–4148. doi: 10.1210/jcem.82.12.4446. [DOI] [PubMed] [Google Scholar]
- Archanco M, Muruzabal FJ, Llopiz D, Garayoa M, Gomez-Ambrosi J, Fruhbeck G, Burrell MA. Leptin expression in the rat ovary depends on estrous cycle. J Histochem Cytochem. 2003;51:1269–1277. doi: 10.1177/002215540305101003. [DOI] [PubMed] [Google Scholar]
- Agarwal SK, Vogel K, Weitsman SR, Magoffin DA. Leptin antagonizes the insulin-like growth factor-I augmentation of steroidogenesis in granulosa and theca cells of the human ovary. J Clin Endocrinol Metab. 1999;84:1072–1076. doi: 10.1210/jcem.84.3.5543. [DOI] [PubMed] [Google Scholar]
- Ryan NK, Woodhouse CM, Van der Hoek KH, Gilchrist RB, Armstrong DT, Norman RJ. Expression of leptin and its receptor in the murine ovary: possible role in the regulation of oocyte maturation. Biol Reprod. 2002;66:1548–1554. doi: 10.1095/biolreprod66.5.1548. [DOI] [PubMed] [Google Scholar]
- Ghafari F, Gutierrez CG, Hartshorne GM. Apoptosis in mouse fetal and neonatal oocytes during meiotic prophase one. BMC Dev Biol. 2007;7:87. doi: 10.1186/1471-213X-7-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorne GM, Lyrakou S, Hamoda H, Oloto E, Ghafari F. Oogenesis and cell death in human prenatal ovaries: what are the criteria for oocyte selection? Mol Hum Reprod. 2009;15:805–819. doi: 10.1093/molehr/gap055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durlinger AL, Gruijters MJ, Kramer P, Karels B, Ingraham HA, Nachtigal MW, Uilenbroek JT, Grootegoed JA, Themmen AP. Anti-Müllerian hormone inhibits initiation of primordial follicle growth in the mouse ovary. Endocrinology. 2002;143:1076–1084. doi: 10.1210/endo.143.3.8691. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Visser JA, Themmen AP. Regulation of ovarian function: the role of anti-Müllerian hormone. Reproduction. 2002;124:601–609. doi: 10.1530/rep.0.1240601. [DOI] [PubMed] [Google Scholar]
- Nilsson E, Rogers N, Skinner MK. Actions of anti-Müllerian hormone on the ovarian transcriptome to inhibit primordial to primary follicle transition. Reproduction. 2007;134:209–221. doi: 10.1530/REP-07-0119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristol-Gould SK, Kreeger PK, Selkirk CG, Kilen SM, Cook RW, Kipp JL, Shea LD, Mayo KE, Woodruff TK. Postnatal regulation of germ cells by activin: the establishment of the initial follicle pool. Dev Biol. 2006;298:132–148. doi: 10.1016/j.ydbio.2006.06.025. [DOI] [PubMed] [Google Scholar]
- Flaws JA, Hirshfield AN, Hewitt JA, Babus JK, Furth PA. Effect of Bcl-2 on the primordial follicle endowment in the mouse ovary. Biol Reprod. 2001;64:1153–1159. doi: 10.1095/biolreprod64.4.1153. [DOI] [PubMed] [Google Scholar]
- Rosen MP, Shen S, Dobson AT, Rinaudo PF, McCulloch CE, Cedars MI. A quantitative assessment of follicle size on oocyte developmental competence. Fertil Steril. 2008;90:684–690. doi: 10.1016/j.fertnstert.2007.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ectors FJ, Vanderzwalmen P, Van Hoeck J, Nijs M, Verhaegen G, Delvigne A, Schoysman R, Leroy F. Relationship of human follicular diameter with oocyte fertilization and development after in-vitro fertilization or intracytoplasmic sperm injection. Hum Reprod. 1997;12:2002–2005. doi: 10.1093/humrep/12.9.2002. [DOI] [PubMed] [Google Scholar]
- Quirk SM, Cowan RG, Harman RM, Hu CL, Porter DA. Ovarian follicular growth and atresia: The relationship between cell proliferation and survival. J Anim Sci. 2004;82:E40–52. doi: 10.2527/2004.8213_supplE40x. [DOI] [PubMed] [Google Scholar]
- Willis D, Franks S. Insulin action in human granulosa cells from normal and polycystic ovaries is mediated by the insulin receptor and not the type-I insulin-like growth factor receptor. J Clin Endocrinol Metab. 1995;80:3788–3790. doi: 10.1210/jcem.80.12.8530637. [DOI] [PubMed] [Google Scholar]
- Chang RJ, Cook-Andersen H. Disordered follicle development. Mol Cell Endocrinol. 2013;373:51–60. doi: 10.1016/j.mce.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kang DW, Hudgins-Spivey S, Krust A, Lee EY, Koo Y, Cheon Y, Gye MC, Chambon P, Ko C. Theca-specific estrogen receptor-alpha knockout mice lose fertility prematurely. Endocrinology. 2009;150:3855–3862. doi: 10.1210/en.2008-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HM, Klausen C, Leung PC. AntiMüllerian hormone inhibits follicle-stimulating hormone-induced adenylyl cyclase activation, aromatase expression, and estradiol production in human granulosa-lutein cells. Fertil Steril. 2013;100:585–592. doi: 10.1016/j.fertnstert.2013.04.019. e581. [DOI] [PubMed] [Google Scholar]
- Durlinger AL, Kramer P, Karels B, de Jong FH, Uilenbroek JT, Grootegoed JA, Themmen AP. Control of primordial follicle recruitment by anti-Müllerian hormone in the mouse ovary. Endocrinology. 1999;140:5789–5796. doi: 10.1210/endo.140.12.7204. [DOI] [PubMed] [Google Scholar]
- Pellatt L, Rice S, Dilaver N, Heshri A, Galea R, Brincat M, Brown K, Simpson ER, Mason HD. Anti-Müllerian hormone reduces follicle sensitivity to follicle-stimulating hormone in human granulosa cells. Fertil Steril. 2011;96:1246–1251. doi: 10.1016/j.fertnstert.2011.08.015. e1241. [DOI] [PubMed] [Google Scholar]
- Taieb J, Grynberg M, Pierre A, Arouche N, Massart P, Belville C, Hesters L, Frydman R, Catteau-Jonard S, Fanchin R, Picard JY, Josso N, et al. FSH and its second messenger cAMP stimulate the transcription of human anti-Müllerian hormone in cultured granulosa cells. Mol Endocrinol. 2011;25:645–655. doi: 10.1210/me.2010-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffin CL, Vandevoort CA. Follicle growth, ovulation, and luteal formation in primates and rodents: a comparative perspective. Exp Biol Med (Maywood) 2013;238:539–548. doi: 10.1177/1535370213489437. [DOI] [PubMed] [Google Scholar]
- Landomiel F, Gallay N, Jégot G, Tranchant T, Durand G, Bourquard T, Crépieux P, Poupon A, Reiter E. Biased signalling in follicle stimulating hormone action. Mol Cell Endocrinol. 2014;382:452–459. doi: 10.1016/j.mce.2013.09.035. [DOI] [PubMed] [Google Scholar]
- Dong J, Albertini DF, Nishimori K, Kumar TR, Lu N, Matzuk MM. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature. 1996;383:531–535. doi: 10.1038/383531a0. [DOI] [PubMed] [Google Scholar]
- Orisaka M, Orisaka S, Jiang J-Y, Craig J, Wang Y, Kotsuji F, Tsang BK. Growth Differentiation factor 9 is antiapoptotic during follicular development from preantral to early antral stage. Mol Endocrinol. 2006;20:2456–2468. doi: 10.1210/me.2005-0357. [DOI] [PubMed] [Google Scholar]
- Hill SM, Frasch T, Xiang S, Yuan L, Duplessis T, Mao L. Molecular mechanisms of melatonin anticancer effects. Integr Cancer Ther. 2009;8:337–346. doi: 10.1177/1534735409353332. [DOI] [PubMed] [Google Scholar]
- McNeilly JR, Watson EA, White YAR, Murray AA, Spears N, McNeilly AS. Decreased oocyte DAZL expression in mice results in increased litter size by modulating follicle-stimulating hormone-induced follicular growth. Biol Reprod. 2011;85:584–593. doi: 10.1095/biolreprod.110.086264. [DOI] [PubMed] [Google Scholar]
- Chen HH, Welling M, Bloch DB, Munoz J, Mientjes E, Chen X, Tramp C, Wu J, Yabuuchi A, Chou YF, Buecker C, Krainer A, et al. DAZL limits pluripotency, differentiation, and apoptosis in developing primordial germ cells. Stem Cell Reports. 2014;3:892–904. doi: 10.1016/j.stemcr.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rooij IA, Broekmans FJ, te Velde ER, Fauser BC, Bancsi LF, de Jong FH, Themmen AP. Serum anti-Müllerian hormone levels: a novel measure of ovarian reserve. Hum Reprod. 2002;17:3065–3071. doi: 10.1093/humrep/17.12.3065. [DOI] [PubMed] [Google Scholar]
- Themmen AP. Anti-Müllerian hormone: its role in follicular growth initiation and survival and as an ovarian reserve marker. J Natl Cancer Inst Monogr. 2005:18–21. doi: 10.1093/jncimonographs/lgi026. [DOI] [PubMed] [Google Scholar]
- La Marca A, Volpe A. Anti-Müllerian hormone (AMH) in female reproduction: is measurement of circulating AMH a useful tool? Clin Endocrinol (Oxf) 2006;64:603–610. doi: 10.1111/j.1365-2265.2006.02533.x. [DOI] [PubMed] [Google Scholar]
- Loh JS, Maheshwari A. Anti-Müllerian hormone–is it a crystal ball for predicting ovarian ageing? Hum Reprod. 2011;26:2925–2932. doi: 10.1093/humrep/der271. [DOI] [PubMed] [Google Scholar]
- Nelson SM, Anderson RA, Broekmans FJ, Raine-Fenning N, Fleming R, La Marca A. Anti-Müllerian hormone: clairvoyance or crystal clear? Hum Reprod. 2012;27:631–636. doi: 10.1093/humrep/der446. [DOI] [PubMed] [Google Scholar]
- Quaas A, Dokras A. Diagnosis and treatment of unexplained infertility. Rev Obstet Gynecol. 2008;1:69–76. [PMC free article] [PubMed] [Google Scholar]
- Frias AE, Grove KL. Obesity: a transgenerational problem linked to nutrition during pregnancy. Semin Reprod Med. 2012;30:472–478. doi: 10.1055/s-0032-1328875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.