

Abstract
TAR DNA-binding protein of 43 kDa (TDP-43) has been identified as the major component of ubiquitin-positive neuronal and glial inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Aggregation of TDP-43 to amyloid-like fibrils and spreading of the aggregates are suggested to account for the pathogenesis and progression of these diseases. To investigate the molecular mechanisms of TDP-43 aggregation, we attempted to identify the amino acid sequence required for the aggregation. By expressing a series of deletion mutants lacking 20 amino acid residues in the C-terminal region in SH-SY5Y cells, we established that residues 274–313 in the glycine-rich region are essential for aggregation. In vitro aggregation experiments using synthetic peptides of 40 amino acids from this sequence and adjacent regions showed that peptides 274–313 and 314–353 formed amyloid-like fibrils. Transduction of these fibrils induced seed-dependent aggregation of TDP-43 in cells expressing wild-type TDP-43 or TDP-43 lacking nuclear localization signal. These cells showed different phosphorylated C-terminal fragments of TDP-43 and different trypsin-resistant bands. These results suggest that residues 274–353 are responsible for the conversion of TDP-43 to amyloid-like fibrils and that templated aggregation of TDP-43 by seeding with different peptides induces various types of TDP-43 pathologies, i.e. the peptides appear to act like prion strains.
Keywords: aggregation, amyloid, neurodegenerative disease, prion, TAR DNA-binding protein 43 (TDP-43) (TARDBP)
Introduction
Frontotemporal lobar degeneration (FTLD)2 is the second most common dementing disorder in patients under 65 years of age and is characterized by progressive atrophy of the frontal and temporal lobes in the brain. Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterized by progressive motor neuron degeneration. In 2006, TAR DNA-binding protein of 43 kDa (TDP-43) was identified as the major component of tau-negative, ubiquitin-positive inclusions in these diseases (1, 2). TDP-43 is a heterogeneous nuclear ribonucleoprotein of 414 amino acids, originally identified as a transcriptional repressor that binds to the transactivation-responsive region of the HIV-1 gene (3). It is expressed ubiquitously and is localized mainly in nuclei, functioning in exon splicing, gene transcription, regulation of mRNA stability and biosynthesis, and the formation of nuclear bodies. For example, it has been reported that TDP-43 binds to the junctional region between exons 9 and 10 of the gene coding cystic fibrosis transmembrane conductance regulator and causes skipping of exon 9 (4). Structurally, TDP-43 is characterized by two RNA-recognition motifs, a C-terminal tail that contains a glycine-rich region and a glutamine/asparagine (Gln/Asn)-rich region.
Discovery of missense mutations in the TARBDP gene in patients with familial and sporadic ALS and FTLD (5–12) demonstrated a direct link between genetic lesions and development of TDP-43 pathology. Importantly, most of the mutations are localized in the carboxyl-third of the molecule, which shows sequence similarity to prion proteins. Furthermore, biochemical and histological analyses demonstrated accumulation of full-length and C-terminal fragments (CTFs) in hyperphosphorylated and fibrillar forms in brain and spinal cord of patients (13). Furthermore, the CTF-banding patterns are different between the diseases and are closely related to the neuropathological phenotypes, which can be classified into at least four groups (types A–D) (13, 14). The CTF-banding patterns are considered to reflect the protease resistance of the assembled protein structures (15). It was recently demonstrated that the pathological TDP-43 has prion-like activity and can convert normal TDP-43 to an abnormal form in cultured cells (16). Interestingly, the CTF-banding patterns of converted host proteins resemble those of the pathological TDP-43 used as seeds, suggesting that the conversion is template-dependent.
These findings strongly suggest that aggregation of C-terminal regions of TDP-43 plays a central role in the pathogenesis and progression of ALS and FTLD with TDP-43 pathologies (FTLD-TDP). The aggregation of CTFs of TDP-43 has also been demonstrated in experimental cellular models; for example, our previous study showed that expression of CTF(162–414) of TDP-43 with a GFP tag in SH-SY5Y cells induced cytosolic mislocalization of TDP-43 in phosphorylated and ubiquitinated aggregates, recapitulating the TDP-43 pathology in brains (17).
In this study, we investigated aggregation of TDP-43 in cultured cells expressing a series of deletion constructs to identify the sequences responsible for the aggregation. Then, self-aggregation of synthetic peptides derived from the identified sequence and surrounding sequences was examined. Synthetic fibrillar aggregates of peptides from the identified sequence were found to work as seeds, converting normal TDP-43 into abnormal aggregates in cultured cells.
Experimental Procedures
Construction of Plasmids
To construct a series of deletion mutants lacking sequences of 20-amino acid residues in TDP-CTF with a GFP tag, we conducted site-directed mutagenesis of pEGFP-TDP(162–414) (17). PCR was carried out using a site-directed mutagenesis kit (Stratagene) with the following primers: Del 1 (lacking 214–233), forward, 5′-GAGTTCTTCTCTCAGTTTGCAGATGATCAG-3′, and reverse, 5′-CTGATCATCTGCAAACTGAGAGAAGAACTC-3′; Del 2 (lacking 234–253), forward, 5′-TTTGCCTTTGTTACAAGCGTTCATATATCC-3′, and reverse, 5′-GGATATATGAACGCTTGTAACAAAGGCAAA-3′; Del 3 (lacking 254–273), forward, 5′-ATCATTAAAGGAATCGGAAGATTTGGTGGT-3′, and reverse, 5′-ACCACCAAATCTTCCGATTCCTTTAATGAT-3′; Del 4 (lacking 274–293), forward, 5′-CAGTTAGAAAGAAGTGGGGGTGGAGCTGGT-3′, and reverse, 5′-ACCAGCTCCACCCCCACTTCTTTCTAACTG-3′; Del 5 (lacking 294–313), forward, 5′-TTTGGTAATAGCAGAGGTGCGTTCAGCATT-3′, reverse, 5′-AATGCTGAACGCACCTCTGCTATTACCAA-3′; Del 6 (lacking 314–333), forward, 5′-GGTGGGATGAACTTTTGGGGTATGATGGGC-3′, and reverse, 5′-GCCCATCATACCCCAAAAGTTCATCCCACC-3′; Del 7 (lacking 334–353), forward, 5′-GCACTACAGAGCAGTCAAAACCAAGGCAAC-3′, and reverse, 5′-GTTGCCTTGGTTTTGACTGCTCTGTAGTGC-3′; Del 8 (lacking 354–373), forward, 5′-CCATCGGGTAATAACTATAGTGGCTCTAAT-3′, and reverse, 5′-ATTAGAGCCACTATAGTTATTACCCGATGG-3′; Del 9 (lacking 374–393), forward, 5′-TCTGGAAATAACTCTGGCAGTGGTTTTAAT-3′, and reverse, 5′-ATTAAAACCACTGCCAGAGTTATTTCCAGA-3′. Deletion mutants lacking 40 amino acid residues were also constructed from GFP-tagged TDP-CTF(162–414), full-length TDP-43, and TDP-43(ΔNLS(187–192)). Twenty amino acid-long deletions of Del 2, 4, and 5 were induced in pEGFP-TDP(162–414), pcDNA3-TDP-43, and pcDNA3-TDP-43(ΔNLS(187–192)) (18) as described above, and PCR was conducted with the following primers: Del(234–273), Del 2(234–253) was used as a template, and the primers were forward, 5′-TTTGCCTTTGTTACAGGAAGATTTGGTGGT-3′, and reverse, 5′-ACCACCAAATCTTCCTGTAACAAAGGCAAA-3′; Del(274–313), Del 4(274–293) was used as a template, and the primers were forward, 5′-CAGTTAGAAAGAAGTGGTGCGTTCAGCATT-3′, and reverse, 5′-AATGCTGAACGCACCACTTCTTTCTAACTG-3′; Del(314–353), Del 6(314–333) was used as a template, and the primers were forward, 5′-GGTGGGATGAACTTTCAAAACCAAGGCAAC-3′, and reverse, 5′-GTTGCCTTGGTTTTGAAAGTTCATCCCACC-3′.
Antibodies
A monoclonal antibody specific for GFP was purchased from MBL. An antibody specific for TDP-43 (monoclonal and polyclonal) was obtained from Proteintech. Monoclonal and polyclonal antibodies specific for abnormal phosphorylation of TDP-43 (pS409/410) and a polyclonal antibody specific for the TDP-43 C-terminal region (405–414) were prepared as described previously (13, 19), which are available from Cosmo Bio. As secondary antibodies, biotin-labeled secondary antibody was purchased from Vector Laboratories for use in the avidin-biotin complex method, and goat anti-mouse IgG (H+L)-HRP conjugate was obtained from Bio-Rad for use in the enhanced chemiluminescence (ECL) method.
Cell Culture and Transfection of Plasmids
SH-SY5Y cells were cultured in Dulbecco's modified Eagle's medium (DMEM)/F-12 medium (Sigma) supplemented with 10% fetal calf serum, minimum Eagle's medium nonessential amino acid solution (Gibco), and penicillin/streptomycin/glutamine (Gibco). Cells were maintained at 37 °C under a humidified atmosphere of 5% CO2 in a culture chamber. Cells were grown to 50% confluence in six-well culture dishes, and transfection of expression plasmids into cells was carried out using FuGENE 6 (Roche Applied Science) according to the manufacturer's instructions.
Immunoblot Analysis
Transfected SH-SY5Y cells were incubated in six-well plates for the indicated time and harvested in A68 buffer (10 mm Tris-HCl (pH 7.5), 10% sucrose, 0.8 m NaCl, 1 mm EGTA) containing 1% Sarkosyl (Sar). Cell lysate was centrifuged at 100,000 × g for 20 min at 25 °C, and the supernatant was recovered as the Sar-soluble fraction. The remaining pellets were lysed in SDS-sample buffer and heated at 100 °C for 5 min to prepare the Sar-insoluble fraction. The Sar-insoluble fraction was also prepared from brains of patients with FTLD or ALS as described (13). Trypsin digestion of the Sar-insoluble fraction was performed by incubation in 30 mm Tris-HCl buffer (pH 7.4) containing 0.01 mg/ml trypsin at 37 °C for 30 min. Protein concentration was estimated using the BCA protein assay kit (Pierce). Samples were loaded on SDS-12.5% polyacrylamide gels electrophoresed with the Tris-glycine buffer system. Proteins in gels were transferred onto a polyvinylidene difluoride membrane (Millipore) and blocked with 3% gelatin. The blots were incubated overnight with the indicated primary antibodies in 10% calf serum at an appropriate dilution (1:1000–5000) at room temperature. Membranes were washed and incubated for 1 h with a biotin-labeled secondary antibody (Vector Laboratories) or a horseradish peroxidase-labeled secondary antibody (Bio-Rad) at room temperature. Signals were detected using an avidin-biotin complex staining kit (Vector Laboratories) or ECL Prime Western blotting Detection System (GE Healthcare).
Fluorescence Microscopic Analysis
SH-SY5Y cells transfected with expression plasmids were grown on 35-mm glass dishes (Iwaki) for 2 days. To observe intracellular aggregates of GFP-tagged TDP-CTF (they show intense fluorescence of GFP), cells were examined with a fluorescence microscope (DIAPHOTO, Nikon) set at 488 nm. Quantitation of aggregate formation was performed as follows; nine images were acquired using a fluorescence microscope (BioZero, KEYENCE), using a sliding field of view. The nine images were combined to form a single image using BZ-2 Analyzer (KEYENCE) and luminance extraction was conducted to quantify the fluorescence of aggregates in the image. The lower limit threshold of luminance for extraction was adjusted to exclude the fluorescence of non-aggregated protein (which is weaker than that of aggregated protein), so that only luminance of aggregated TDP-CTF was detected and quantified.
In Vitro Aggregation of Synthetic Peptides
Synthetic 40-mer peptides with the following partial sequences of TDP-43 (234–273, FADDQIAQSLCGEDLIIKGISVHISNAEPKHNSNRQLERS; 274–313, GRFGGNPGGFGNQGGFGNSRGGGAGLGNNQGSNMGGGMNF; 314–353, GAFSINPAMMAAAQAALQSSWGMMGMLASQQNQSGPSGNN; and 353–392, QNQGNMQREPNQAFGSGNNSYSGSNSGAAIGWGSASNAGS) and control generated randomly (NHYVKMILKKALSRYPNRRNLPCVDLRYKRKSIILQRKYS) were dissolved in DMSO and diluted with Tris-HCl (pH 7.5) to the indicated concentrations. These peptide solutions were incubated on a shaker at 37 °C, and 10-μl aliquots were added to 300 μl of 20 mm MOPS buffer containing 5 μm thioflavin S at the indicated time. After 30 min of incubation at room temperature, 200-μl aliquots were loaded on a 96-well black plate, and fluorescence of thioflavin S (λex = 436 nm and λem = 535 nm) was measured using a luminescence plate reader (Chameleon, HIDEX).
Transmission Electron Microscopy
For electron microscopy, peptide solutions were incubated on a shaker for 8 days, placed on collodion-coated 300-mesh copper grids, and stained with 2% (v/v) phosphotungstate. Micrographs were recorded on a JEOL 1200EX electron microscope.
Induction of TDP-43 Aggregation in Cells by Peptide Fibrils
Aliquots of 100 μl of TDP-43 peptide solutions were incubated for 8 days on a shaker, diluted with 500 μl of 30 mm Tris-HCl (pH 7.5), and centrifuged at 100,000 × g for 20 min. After centrifugation, the supernatant was removed, and the remaining pellets (peptide fibrils) were dissolved in 30 mm Tris-HCl (pH 7.5). Aliquots of 5 μl of peptide fibril solution were added to 95 μl of 6 m guanidine HCl solution, and peptide fibrils were quantified by means of HPLC. For introduction into cells, 5 μg of peptide fibrils was mixed with 120 μl of Opti-MEM, and 62.5 μl of Multifectam (Promega) was added. After incubation for 30 min at room temperature, 62.5 μl of Opti-MEM was added. The mixture was further incubated for 5 min and added to cells immediately after transfection of expression plasmids of TDP-43. The treated cells were further incubated for 2 days in a CO2 incubator.
Immunocytochemistry
Transfection of expression plasmids and induction of peptide fibrils were conducted as described above, using SH-SY5Y cells grown on coverslips. After incubation for 2 days in a CO2 incubator, cells were fixed with 4% paraformaldehyde and stained with the indicated primary antibodies at 1:1000 dilution. After incubation for 1 h, cells were washed and treated with secondary antibodies (anti-rabbit IgG-conjugated Alexa-568 and anti-mouse IgG-conjugated Alexa-488, Invitrogen) and TO-PRO-3 (Invitrogen) to counterstain nuclear DNA. After 1 h, the cells were mounted and analyzed using an LSM5 Pascal confocal microscope (Carl Zeiss).
Cell Death Assay
Cell death assays were conducted using a CytoTox 96 non-radioactive cytotoxicity assay kit (Promega).
Statistical Analysis
All values in figures are means ± S.E. Biochemical data were statistically analyzed using the unpaired two-tailed Student's t test. A p value of ≤0.05 was considered statistically significant.
Results
Expression and Aggregation of Deletion Mutants Lacking 20 Amino Acids in SH-SY5Y Cells
To determine the sequences responsible for aggregation of CTFs of TDP-43, we constructed a series of deletion mutants lacking 20-amino acid residues in TDP-CTF with a GFP tag: Del 1(Δ214–233); Del 2(Δ234–253); Del 3(Δ254–273); Del 4(Δ274–293); Del 5(Δ294–313); Del 6(Δ314–333); Del 7(Δ334–353); Del 8(Δ354–373); and Del 9(Δ374–393) (Fig. 1, A and B). Aggregation of TDP-43 in cells transfected with a deletion or wild-type construct was evaluated by biochemical analysis of Sar-insoluble TDP-43 (Fig. 2). The Sar-soluble TDP-43 CTF levels were similar among the deletions as detected with anti-GFP (Fig. 2, left panel), but remarkable decreases of Sar-insoluble TDP-43 were detected with anti-GFP or pS409/410 in cells expressing Del 4, 5, or 8 (Fig. 2, middle and right panels). The results of band quantitation are shown in the lower panels. These data suggest that deletion of residues 274–293, 294–313, or 354–373 reduced the TDP-43 aggregation.
FIGURE 1.
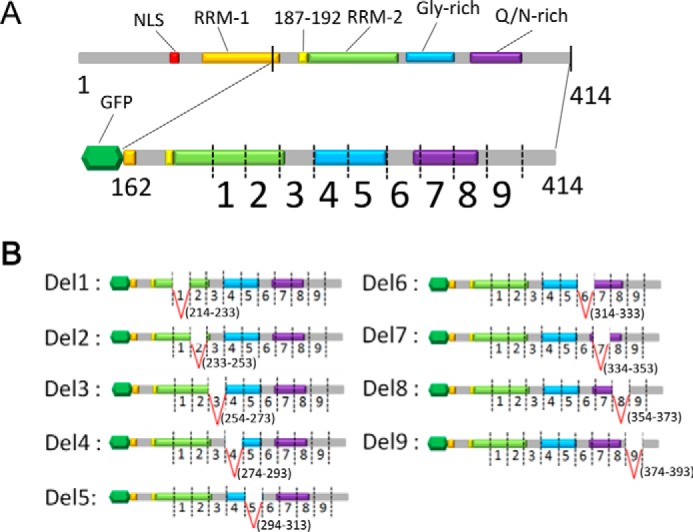
Construction of a series of partial deletion mutants of TDP-43 C-terminal fragment. A, schematic diagrams of full-length TDP-43 and GFP-tagged TDP-CTF(162–414). Deletion sites (1–9) were designed on the 214–393 region of TDP-CTF. B, schematic diagrams of deletion mutants of TDP-CTF (Del 1–9). Each mutant possesses one of nine deletions. Del 1 and 2 have deletions in RNA-recognition motif 2, Del 4 and 5 have deletions in the glycine-rich domain, and Del 7 and 8 have deletions in the Gln/Asn-rich domain. Deletion sites of Del 3 and 6 were designed to lie within gaps of domains, and Del 9 was closest to the C terminus.
FIGURE 2.

Immunoblot analysis of Sar-insoluble fraction of cells expressing TDP-CTF deletion mutants. After 48 h of incubation, SH-SY5Y cells transfected with TDP-CTF deletion mutants and WT (without deletion) were lysed in A68 buffer containing 1% Sar and sequentially fractionated into Sar-soluble (sup, supernatant) and Sar-insoluble (ppt, pellet) by centrifugation. These fractions were subjected to SDS-PAGE, and proteins in gels were transferred to PVDF membrane. Bands were detected with anti-GFP and anti-pS409/410 antibodies, and relative intensity (versus WT) is shown in the plot. Data are means ± S.E. (n = 3). *, p < 0.05 by Student's t test against the value of WT.
Aggregation of the deletion mutants of TDP-43 was also evaluated by means of fluorescence microscopy (Fig. 3). In cells expressing Del 1, 2, or 3, many small dot-like aggregates were observed, which were morphologically distinct from those in cells expressing wild-type CTF. In contrast, only small amounts of TDP-43 aggregates, or none, were observed in cells expressing Del 4–8, compared with those in cells expressing wild-type or Del 9. Intracellular aggregates of GFP-tagged TDP-43 CTF had much more intense fluorescence than diffusely expressed non-aggregated GFP-tagged TDP-43 CTF. Therefore, to quantify TDP-43 aggregates by luminance extraction, we adjusted the lower limit (threshold) of luminance value so that only the aggregates were detected. As shown in Fig. 3B, markedly reduced aggregate formation was observed in cells expressing Del 4–8, confirming the visual observations. These results indicate that sequences in the region between 274 and 373 are responsible for the aggregation of TDP-CTF, although there is some discrepancy between the results of these morphological observations and those of biochemical analyses shown in Fig. 2. It is possible that amorphous aggregates, which are different from the amyloid-like aggregates, but are nevertheless insoluble and phosphorylated, are formed in cells due to deletion of the sequence 314–353. Such aggregates might be diffusely localized in the cytoplasm of cells and would not be detected as aggregates by fluorescence microscopy.
FIGURE 3.
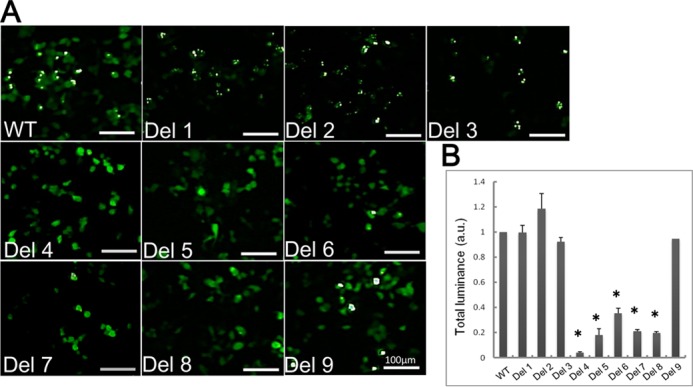
Fluorescence microscopic analysis of aggregate formation of TDP-CTF deletion mutants expressed in cells. A, fluorescence microscopic images of SH-SY5Y cells transfected with deletion mutants of TDP-CTF and WT. The fluorescence of GFP, which is fused to TDP-CTF, was detected. Aggregated TDP-CTF is distinguishable from diffusely expressed TDP-CTF, appearing as round or dot-like structures with much more intense GFP fluorescence. B, luminance extraction was conducted on acquired fluorescence microscopic images to quantify aggregate formation. To exclude fluorescence of non-aggregated protein, the lower limit of detectable GFP luminance level was adjusted so that only fluorescence of aggregated TDP-CTF was detected. Measured luminance was calculated as a relative value with respect to TDP-CTF WT. Data are means ± S.E. (n = 3). *, p < 0.05 by Student's t test against the value of WT.
Expression and Aggregation of Deletion Mutants Lacking 40 Amino Acids in SH-SY5Y Cells
To further analyze the effect of these sequences on TDP-43 aggregation, we constructed another series of deletion mutants lacking 40 amino acid residues in GFP-tagged TDP-CTF and an untagged full-length TDP-43 mutant (ΔNLS(187–192)), which lacks both the NLS (78–84) and a similar sequence (187–192), and forms aggregates in cells (18). Remarkable reduction of aggregation was observed both biochemically and morphologically in cells expressing the Del 4 and Del 5 mutants described above. The sequences deleted in these mutants correspond to the glycine-rich region of TDP-43. Therefore, 40-amino acid deletion constructs were made by deleting Del 4–5 and the adjacent Del 2–3, Del 6–7, and Del 8–9. They were expressed in cells, and aggregate formation was investigated by quantitation of Sar-insoluble TDP-43 (Fig. 4). Deletion of residues 234–273 (Del 2–3) in GFP-CTF caused a significant increase of Sar-insoluble phosphorylated TDP-43. In contrast, insoluble TDP-43 was dramatically reduced by deletion of residues 274–313 (Del 4–5), although no reduction was detected in response to deletion of residues 314–353 (Fig. 4A). Deletion of residues 354–393 (Del 8–9) also showed significant reduction of insoluble TDP-43 on the blot with pS409/410 but not with anti-GFP antibody. Similar results were observed in a cellular aggregation model using untagged full-length mutant (ΔNLS(187–192)) (Fig. 4B), suggesting that residues 274–313 and 354–393 are important for aggregation of not only CTF but also full-length TDP-43. Aggregate formation of these deletion mutants was also investigated by means of fluorescence microscopy. In cells expressing undeleted GFP-tagged CTF, Del 2–3(234–273), and Del 8–9(354–393), distinct aggregates were observed, whereas diffuse cytoplasmic localization of GFP was observed in cells expressing Del 4–5(274–313) or Del 6–7(314–353). The results of quantitation of aggregates (Fig. 4, C and D) suggested that residues 314–353 may also be important for aggregation of TDP-CTF.
FIGURE 4.

Effect of 40-amino acid deletions on aggregate formation of TDP-CTF and TDP-43(ΔNLS(187–192)). Deletion mutants of GFP-tagged TDP-CTF(162–414) and TDP-43(ΔNLS(187–192)) were constructed with 40-amino acid deletions at 234–273, 274–313, 314–353 and, 354–393 and expressed in SH-SY5Y cells. TDP-43 deletion mutants collected in Sar-soluble (sup) and Sar-insoluble (ppt) fractions were detected with anti-GFP and anti-pS409/410 antibodies for GFP-tagged TDP-CTF series (A), and with anti-TDP-43 and anti-pS409/410 for TDP-43 (ΔNLS(187–192)) series (B), respectively. Cells expressing GFP-tagged TDP-CTF deletion mutants were also observed by fluorescence microscopy (C), and aggregate formation was quantified by luminance extraction of GFP fluorescence of aggregated TDP-CTF (D). Data are means ± S.E. (n = 3). *, p < 0.05 by Student's t test against the value of WT.
In Vitro Aggregation of Synthetic Peptides of TDP-43
Next, we investigated whether or not these 40 amino acid peptides can themselves form amyloid-like fibrillar aggregates. Synthetic peptides corresponding to sequences 234–273, 274–313, 314–353, and 353–392 of TDP-43 and a control peptide (Fig. 5A) were dissolved in 30 mm Tris-HCl buffer at various concentrations (0.05, 0.1, and 0.2 mm), and incubated at 37 ° C with shaking. Amyloid-like aggregate formation was analyzed in terms of thioflavin S (ThS) fluorescence. The peptide 234–273 showed an increase of ThS fluorescence at day 4 at a concentration of 0.2 mm, but no increase was observed even by day 8 at lower concentrations. In contrast, the peptides 274–313 and 314–353 showed markedly increased ThS fluorescence at day 1 at most concentrations studied (Fig. 5B). No ThS increase was observed with the peptide 353–392 or the control peptide. The amyloid-like aggregates formed by the above peptides at a concentration of 0.2 mm were negatively stained with phosphotungstate on a grid and observed by electron microscopy. As shown in Fig. 5C, short fibrillar aggregates were observed in the peptide 234–273 solution. In contrast, relatively long, straight, or twisted fibrils (10–15 nm diameter) were observed in the 274–313 and 314–353 peptide solutions. These results indicate that the TDP-43 peptides 274–313 and 314–353 have a high propensity to form amyloid-like fibrils, whereas 234–273 and 354–393 peptides have a low propensity.
FIGURE 5.

In vitro aggregation of synthetic TDP-43 peptides (40 amino acid residues). A, schematic diagrams of deletion sites on TDP-43 and sequences of synthetic peptides. B, TDP-43 peptide solutions of 0.05, 0.1, and 0.2 mm were prepared and incubated in 37 °C on a shaker. At the indicated time point, aliquots of peptide solution were added to 5 μm ThS solution, and ThS fluorescence was measured. Data are means ± S.E. (n = 3). C, peptide solutions after 8-day incubation at 37 °C on a shaker were loaded on a grid and negatively stained with phosphotungstate. Samples were observed under a transmission electron microscope. Magnification of acquired micrographs is ×40,000, and scale bar is 100.0 nm.
Synthetic Peptide Fibrils Can Serve as a Seed to Convert Intracellular TDP-43 into Abnormal Aggregates
Prion-like propagation of intracellular abnormal proteins has been demonstrated in cellular and animal models (20–22). It has also been shown that the Sar-insoluble TDP-43 fibrils prepared from the brains of patients with ALS/FTLD have prion-like properties and can seed-dependently convert normal TDP-43 to abnormal forms (16). Therefore, we investigated whether fibrils made of these synthetic peptides have the ability to convert normal TDP-43 into abnormal aggregates. SH-SY5Y cells were transiently transfected with wild-type TDP-43 and then transduced with these peptide monomers or the fibrils (234–273, 274–313, and 314–353), and then Sar-soluble and -insoluble TDP-43 was analyzed by immunoblotting (Fig. 6, A and B). In cells transduced with the monomeric peptides, no insoluble phosphorylated TDP-43 was detected. In contrast, pS409/410-positive insoluble TDP-43 of 45 kDa was detected in cells transduced with fibrils of peptide 274–313 or peptide 314–353, suggesting that these synthetic peptide fibrils converted wild-type full-length TDP-43 to abnormal aggregates. We also investigated the effect of phosphorylation on seed-dependent TDP-43 aggregation. Mutant TDP-43 (S403A/S404A/S409A/S410A), in which the four major phosphorylation sites Ser-403, Ser-404, Ser-409 and Ser-410 were substituted by alanine, was expressed in cells, which were treated with peptide fibrils. The amount of insoluble TDP-43 was compared with that in cells expressing wild-type TDP-43 by using anti-TDP-43 and anti-TDP-43(405–414), because pS409/410 could not be used in this case. Immunoblot analysis showed no differences in aggregation between mutant TDP and WT-TDP (Fig. 6C), strongly suggesting that phosphorylation of TDP-43 is not necessary for aggregation of TDP-43, but it most likely occurs as a secondary event in seed-dependent TDP-43 aggregation.
FIGURE 6.
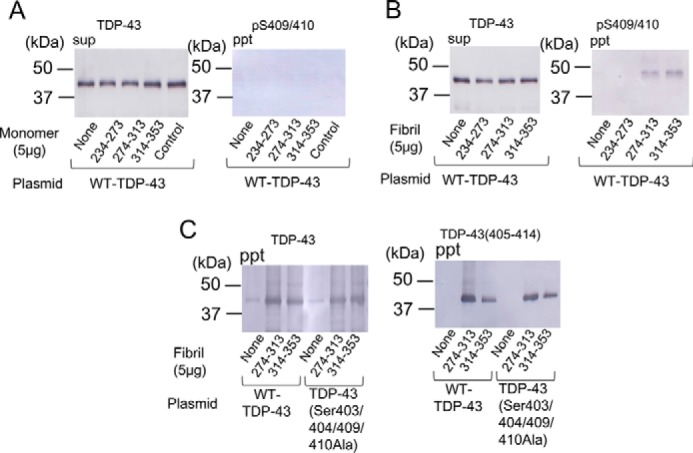
Synthetic peptide fibrils function as seeds in SH-SY5Y cells expressing full-length TDP-43. TDP-43 peptide monomers (5 μg) without incubation (A) and fibrils prepared as described above (B) were introduced into cells immediately after transfection of full-length TDP-43. After incubation for 2 days, Sar-soluble (sup) and Sar-insoluble (ppt) fractions of cell lysate were prepared, and immunoblot analysis using anti-TDP-43 and anti-pS409/410 antibodies was conducted. Black arrowhead indicates abnormally phosphorylated TDP-43 of 45 kDa. C, cells transfected with WT-TDP-43 and TDP-43 (S403A/S404A/S409A/S410A) were treated with peptide fibrils of 274–313 and 314–353 and then immunoblotted using anti-TDP-43 and anti-TDP-43(405–414).
To confirm the aggregate formation morphologically, we performed immunocytochemical analyses of cells expressing WT-TDP-43 or ΔNLS-TDP-43 and treated with these fibrils (Fig. 7A). No phosphorylated and aggregated TDP-43 was seen in cells expressing WT-TDP-43 or ΔNLS-TDP-43 alone. In contrast, dot-like structures positive for pS409/410 were found in WT-TDP-43-transfected cells treated with synthetic peptide fibrils of 274–313 or 314–353. The inclusions were also positive for anti-TDP-43 antibody, which recognizes the middle region of TDP-43. Most of the inclusions appeared to be localized in the cytoplasm, not in the nuclei, and this was confirmed by image analysis (Fig. 7B; representative data for cells transfected with WT-TDP-43 + fibril 274–313 are shown). Interestingly, the nuclear staining of endogenous TDP-43 was lost in the cells with cytoplasmic TDP-43 aggregates, recapitulating an important neuropathological feature of TDP-43 proteinopathy. Together with the immunoblot analyses, these results indicate that full-length WT-TDP-43 expressed in cells became aggregated and phosphorylated after introduction of the synthetic peptide fibrils of 274–313 and 314–353 but not with peptide fibrils of 234–273. More cytoplasmic inclusions of TDP-43, positive for both anti-pS409/410 and anti-TDP-43, were detected in cells expressing ΔNLS-TDP-43 and treated with these peptide fibrils. There were no apparent differences in morphology of the inclusions formed in cells treated with peptide fibrils 274–313 and peptide fibrils 314–353.
FIGURE 7.
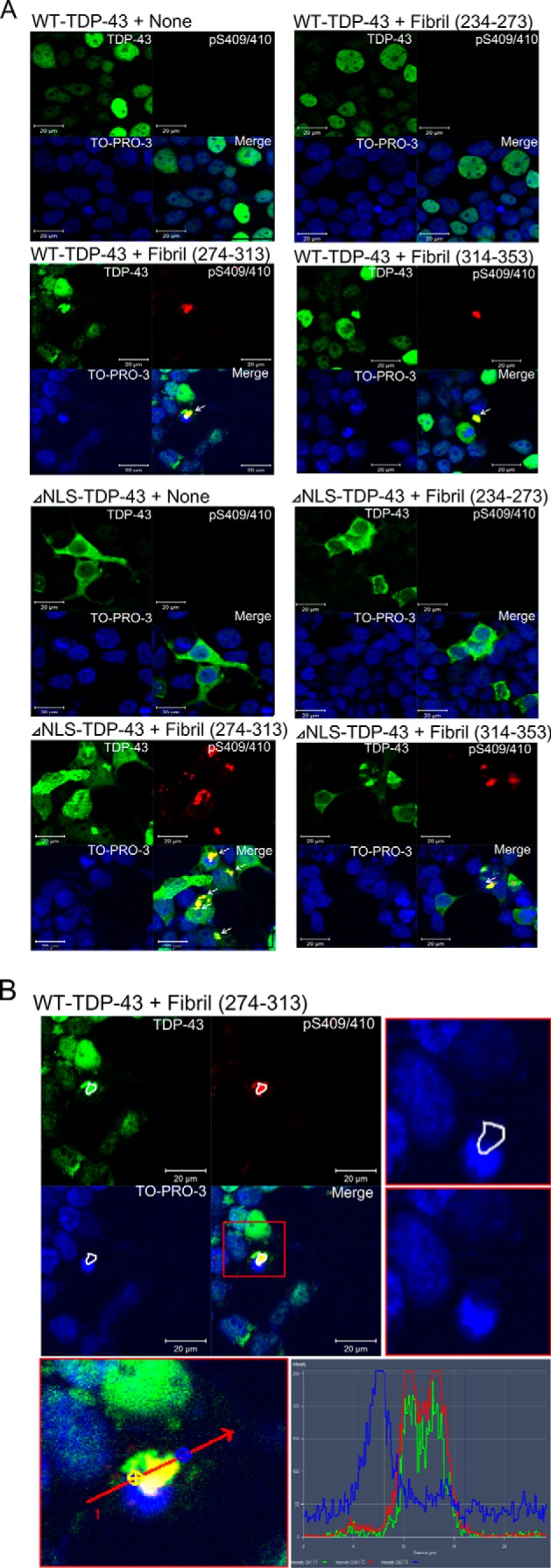
Immunocytochemical analysis of intracellular aggregation of TDP-43 induced by peptide fibrils. Cells cultured on coverslips were transfected with expression plasmids of WT-TDP-43 and ΔNLS-TDP-43 and treated with TDP-43 peptide fibrils (234–273, 274–313, and 314–353). After 2 days of incubation, cells were fixed and stained with antibodies and nuclear staining dye (green, anti-TDP-43; red, anti-pS409/410; blue, TO-PRO-3). A, white arrows indicate abnormally phosphorylated cytoplasmic TDP-43 aggregates. Scale bar, 20 μm. B, cytoplasmic localization of abnormally phosphorylated TDP-43 aggregates in WT-TDP-43 + fibril (274–313) cells was confirmed by image analysis. The locations of aggregates on single and merged images are surrounded by white lines (upper left panel). A magnified TO-PRO-3 image of the vicinity of the aggregates is shown in the upper right panel (with and without the white lines to indicate the location of aggregates). The lower panel and graph show the intensity distribution profiles of TDP-43 (green line), phosphorylated TDP-43 (red line), and TO-PRO-3 (blue line) along the indicated line.
Distinct Conformational Changes Are Induced by Different Peptide Fibrils
We further investigated these inclusions biochemically to determine whether there is any difference between the aggregates induced by the 274–313 fibrils and those induced by the 314–353 fibrils. Immunoblot analyses of the Sar-insoluble fractions of cells transfected with WT-TDP-43 or ΔNLS-TDP-43 showed that full-length TDP-43 with an apparent molecular mass of 45 kDa is accumulated in response to treatment with fibrils of peptide 274–313 or peptide 314–353. In addition to the intensely labeled 45-kDa band, some weak bands were also detected in the upper and lower molecular weight regions with both anti-TDP-43 and pS409/410 antibodies (Fig. 8A). Interestingly, the banding patterns at lower molecular weight were different between the treatments with the 274–313 fibrils and the 314–353 fibrils. This was the case both in cells expressing WT-TDP-43 and in cells expressing ΔNLS-TDP-43. This finding may indicate that the different peptide fibrils converted WT-TDP-43 into conformationally distinct aggregates. The finding that these fibrils converted wild-type full-length TDP-43 to abnormal aggregates is important, and to confirm this observation, we treated the Sar-insoluble fractions of these cells with trypsin and then analyzed the trypsin-resistant fragments by immunoblotting with pS409/410. It should be noted that trypsin-resistant banding patterns are different between different disease phenotypes and the patterns are useful for biochemical classification of TDP-43 proteinopathy (13, 15). Upon treatment of the Sar-insoluble fractions with trypsin, the full-length TDP-43 band disappeared, and bands of 16–24 kDa appeared in the cases of cells treated with the 273–313 fibrils and the 314–353 fibrils (Fig. 8B). The banding pattern of the 273–313 fibril-treated cells (several major bands at 18, 19, and ∼23 kDa) was different from that of the 314–353 fibril-treated cells (one major band at 18 kDa and one minor band at 16 kDa) (Fig. 8B). To link these results to the molecular pathogenesis of TDP-43 proteinopathy, we compared the banding patterns of TDP-43 aggregates formed in cultured cells by peptide-fibril introduction to those in the brains of patients. TDP-43 proteinopathy is subclassified into at least three types based on the predominant TDP-43-positive structures: type A is mainly seen in FTLD-TDP with PGRN mutations; type B corresponds to ALS and FTLD with motor neuron disease and type C is a representative feature of sporadic FTLD-TDP with impairment of semantic memory (14). Each type is characterized biochemically by specific patterns of insoluble TDP-43 CTFs detected with anti-pS409/410 (13, 15). In this study, Sar-insoluble fractions were prepared from brains of typical cases with FTD (type A) and ALS (type B) (Fig. 8C). As shown in Fig. 8C, the trypsin-resistant bands of insoluble TDP-43 from FTD and ALS were very similar to those in cells treated with 274–313 and 314–353 fibrils, respectively, in terms of both patterns and molecular sizes. These results strongly suggest that extracellular introduction of different TDP-43 peptide fibrils induced formation of different types of intracellular TDP-43 aggregates via seeded conversion of full-length WT-TDP-43 and ΔNLS-TDP-43. We also conducted lactate dehydrogenase assay to examine cytotoxicity in cells containing intracellular aggregates following peptide-fibril introduction. No significant increase of lactate dehydrogenase was detected in this experiment (Fig. 8D), suggesting that toxicity of the aggregates is below the detectable limit in this assay.
FIGURE 8.

Different peptide fibrils generated different band patterns of TDP-43 C-terminal fragments and protease-resistant fragments. Peptide fibrils of 274–313 and 314–353 were introduced into cells expressing WT-TDP-43 and ΔNLS-TDP-43, and aggregated TDP-43 contained in the Sar-insoluble fraction was detected by immunoblot analysis using anti-pS409/410 antibodies. A, 22–28-kDa band patterns were compared between 274–313 and 314–353 peptide fibril-induced cells. Schematic diagrams of band patterns are indicated below the blot. B, Sar-insoluble fractions were treated with 10 μg/ml trypsin for 30 min, and then the reaction was stopped by heating at 100 °C for 5 min. Immunoblot analysis using anti-pS409/410 detected remaining trypsin-resistant bands, and their pattern is indicated as a schematic diagram below the blot. C, comparison of TDP-43 in brains of patients with that in cells treated with peptide-fibril seeds. Sar-insoluble fractions prepared from brains of patients (one case of FTD and one case of ALS) and from cells treated with peptide fibrils of 274–313 and 314–353 were digested with trypsin (10 μg/ml at 37 °C for 30 min). Untreated and trypsin-treated samples were immunoblotted with pS409/410. D, extents of cell death in transfected and fibril-introduced cells were quantified by lactate dehydrogenase assay. Cell death assay was conducted 2 days after treatment with peptide fibrils. Data are means ± S.E. (n = 3).
Molecular Mechanism of Seed-dependent Aggregation of TDP-43
To investigate the molecular mechanism of the intracellular aggregation of TDP-43 induced by transduction of the peptide fibrils, we expressed deletion mutants of TDP-43 lacking these 40-amino acid sequences, namely TDP-43(del234–273), TDP-43(del274–313), and TDP-43(del314–353) in cells, and then treated the cells with the peptide fibrils. These deletion mutants were detected at ∼40 kDa, with apparently lower molecular mass than WT-TDP-43, in the Sar-soluble fraction (sup). Expression of WT-TDP-43 or deletion mutant TDP-43 (del274–313) or TDP-43 (del314–353) alone did not afford Sar-insoluble phosphorylated TDP-43 (Fig. 9A), but a phosphorylated insoluble 40-kDa band was detected in cells expressing TDP-43 (del234–273). These results indicate that deletion of residues 234–273 from full-length TDP-43 promoted aggregation, but deletion of the other 40-amino acid sequences did not affect aggregation.
FIGURE 9.

Seeding ability of peptide fibrils for TDP-43 deletion mutants. A, series of deletion mutants of WT-TDP-43 lacking a 40-amino acid sequence of the C-terminal region were constructed and expressed in SH-SY5Y cells. After 48 h of incubation, cells were fractionated into Sar-soluble (sup) and Sar-insoluble (ppt). Immunoblotting of sup was conducted using anti-TDP-43 and immunoblotting of ppt was conducted using anti-pS409/410 antibodies. B, TDP-43 peptide fibrils of 274–313 (B) and 314–353 (C) were introduced into cells expressing these WT-TDP-43 deletion mutants. The sup and ppt fractions were prepared and immunoblotted. Black arrowhead indicates endogenous full-length TDP-43, and white arrowhead indicates deletion mutant of TDP-43. D, based on a report of moderate similarity between C-terminal regions of TDP-43 and PrP (23), regions corresponding to TDP-43 (234–273, 274–313 and 314–353) are indicated on the alignment of human TDP-43 C-terminal region and human prion protein (PrP). *, identical amino acid residues.
Cells expressing these deletion mutants were treated with peptide fibrils and cultured for another 2–3 days. The Sar-insoluble fractions prepared from these cells were analyzed by immunoblotting with anti-TDP-43 and pS409/410. As shown in Fig. 9B, the Sar-insoluble phosphorylated TDP-43 induced by fibrils (274–313) was dramatically decreased in cells expressing TDP-43 lacking residues 274–313, compared with those in cells expressing WT-TDP-43 or the other deletion mutants. Similarly, the Sar-insoluble phosphorylated TDP-43 induced by the fibrils (314–353) was markedly decreased in cells expressing TDP-43 lacking residues 314–353, compared with those in cells expressing WT-TDP-43 or the other deletion mutants (Fig. 9C). Expression levels of WT and deletion mutants were almost the same as observed in immunoblot analysis of the soluble fractions (Fig. 9, B and C). The greater accumulation of insoluble TDP-43 in cells expressing TDP-43 (del 234–237) is probably due to its high propensity to form aggregates (Fig. 9A). Thus, these results demonstrate that seed-dependent conversion of intracellular TDP-43 into abnormal aggregates requires association of fibril seeds with the same sequence in the host protein.
Discussion
In this study, we have identified the region between residues 274 and 373 of TDP-43 as an essential sequence for aggregation of GFP-tagged TDP-CTF and untagged full-length TDP-43 lacking NLS, as well as the similar (ΔNLS(187–192)), by using two series of deletion mutants lacking 20 or 40 amino acids. In addition, synthetic peptides corresponding to residues 274–313 and 314–353 in this region have a high propensity to form amyloid-like fibrils.
The TDP-43 C-terminal domain (233–414) has a moderate level of sequence similarity to prion protein (23), and the two highly amyloidogenic 40-amino acid sequences (274–313 and 314–353) identified in this study cover ∼44% of this region. The former sequence 274–313 contains 17 Gly residues in the 40 amino acids, corresponding to the glycine-rich region of TDP-43, and 18 amino acids (17 Gly and 1 Asn) are identical and 7 amino acids are similar to those in the prion protein. The latter sequence 314–353 contains 9 Gln/Asn and 5 Met residues, covering half of the Gln/Asn-rich region of TDP-43. Within this sequence, 10 amino acids (4 Ala, 2 Gly, 2 Met, 1 Asn, 1 Leu, and 1 Ser) are identical and 4 amino acids are similar to those of the prion protein (Fig. 9D). Reported pathogenic mutations of ALS/FTLD are intensely focused in these regions, and it has been suggested that the A315T mutation increases the tendency of the protein to form a β-sheet structure. Because these two synthetic 40-amino acid peptides have a high propensity to form amyloid-like fibrils at a low concentration, these sequences are likely to be important for aggregation of full-length TDP-43. In fact, deletion of these sequences resulted in a dramatic decrease in the formation of dot-like intracellular inclusions in cells expressing GFP-tagged TDP-CTF or untagged full-length TDP-43 (ΔNLS(187–192)), although the Sar-insoluble phosphorylated TDP-43 was not reduced by deleting the sequence of 314–353.
It remains unknown why such inconsistent results were obtained between the biochemical analysis and the observations by fluorescence microscopy. One possible explanation is that the Del 6–7 mutant may form amorphous unstructured, Sarkosyl-insoluble aggregates rather than punctate amyloid-like aggregates. The sequence 314–353 may stabilize the TDP-43 molecule, and its deletion leads to formation of amorphous aggregates of oligomers, which are insoluble and phosphorylated, and are diffusely localized in cytoplasm; such structures would not be detected as aggregates by fluorescence microscopy. It remains to be clarified whether such diffusely accumulated TDP-43 has any toxic effect on cells and whether it has any prion-like ability to serve as a seed for TDP-43 aggregation.
The significance of the abnormal phosphorylation in these intracellular abnormal proteins in relation to neurodegenerative diseases has been debated. Our present results clearly show that phosphorylation on Ser-403, Ser-404, Ser-409, and Ser-410 has no effect on seed-dependent aggregation of TDP-43, suggesting that phosphorylation may be a secondary event occurring after seed-dependent amyloid-like fibril formation. This may also be the case in other intracellular abnormal amyloid-like protein diseases.
Some research to identify aggregation-related sequences of TDP-43 has been reported. For example, Hu and co-workers (24) assayed the aggregation ability of a series of N- or C-terminally truncated TDP-43 (C-terminally GFP-tagged form) in an Escherichia coli overexpression system and proposed that 318–343 is essential for aggregation of TDP-43. A synthetic peptide corresponding to this sequence underwent structural transformation from α-helix to β-sheet during aggregation (24). Wang et al. (25) proposed that the truncated RRM2208–265) has a more important role than the glycine-rich region in TDP-43 aggregation, based on findings in Neuro2A cells overexpressing C-terminally GFP-tagged truncated RRM-2 or glycine-rich region. Saini et al. (26) identified 246–255 and 311–320 as core aggregation sequences of TDP-43 by evaluating the aggregation ability of a series of peptides derived from the TDP-43 C-terminal sequence (220–414). Furukawa et al. (27)reported that the C-terminal half of TDP-43 (i.e. TDP-432-C) can recapitulate the aggregation propensities of the full-length protein. Huang and co-workers (28, 29) showed that peptides in the sequence (287–322) have a propensity for aggregation, and this tendency was altered by substitution of glycines with prolines. The sequences reported in previous studies and identified in this study are summarized in Fig. 10. Our sequence 274–353 overlaps well with those reported by Huang and co-workers (28, 29), although other sequences were not identified in this study. The discrepancy may be due to the difference in the length of peptides used in in vitro aggregate formation assay (we prepared 40-mers in contrast with their short 8–12-mer peptides). Furthermore, we clearly demonstrated the seeding ability of our peptide fibrils, which had not been addressed previously.
FIGURE 10.
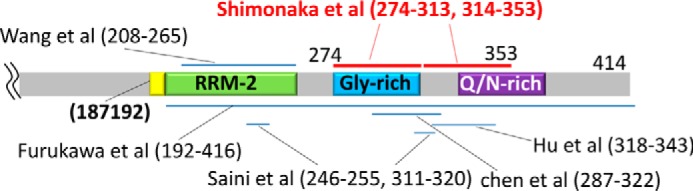
Aggregation-associated sequences of TDP-43 identified in this study and previous reports. Sequences are indicated by bars in a schematic diagram of the TDP-43 C-terminal region.
Emerging evidence suggests prion-like propagation or spreading of intracellular pathological proteins, including tau, α-synuclein, and TDP-43, in diseased brains. In fact, the inclusions in brains of patients are positive for thioflavin S fluorescence, and most of these pathological proteins are accumulated in filamentous or fibrous forms (30). In addition, the paired helical filaments prepared from Alzheimer disease brains and α-synuclein fibrils made of recombinant protein were demonstrated to have a cross-β structure (31, 32), which is the same as that of abnormal prion protein. Furthermore, the seed-dependent prion-like conversion of normal proteins into an abnormal form has been demonstrated experimentally in cellular and animal models (16, 20–22).
TDP-43 pathologies can be seen in various diseases and are thought to be the cause of neurodegeneration in these diseases, including ALS, FTLD, and related disorders, which are referred to as TDP-43 proteinopathy. Clinicopathologically, the TDP-43 proteinopathy can be classified to at least four subtypes (types A–D), and there is a close relation between the pathological subtypes of TDP-43 proteinopathy (14) and the immunoblot patterns of C-terminal fragments of phosphorylated TDP-43 (13, 15). Furthermore, protease-resistant banding patterns of pathologically insoluble TDP-43 are also useful for biochemical classification of the subtypes (15). However, the molecular mechanisms have been unclear. In this study, we demonstrated that fibrils made of synthetic peptides corresponding to the critical regions of TDP-43 have the ability to convert intracellular wild-type TDP-43 into abnormal aggregates, when they are introduced into cells. The TDP-43 aggregates induced by different peptide fibrils showed different CTF-banding patterns and different trypsin-resistant bands, strongly suggesting that different seeds induce assembly of TDP-43 into distinct amyloid-like fibrils. We also demonstrated that the interaction of the seeds with the same peptide sequence in the host protein is required for the seeded aggregation. The distinct seeds formed by assembly of different parts of the C-terminal regions of TDP-43 may convert normal TDP-43 into corresponding abnormal conformations and thus determine the clinicopathological subtypes of TDP-43 proteinopathy. Cell-to-cell spreading of a single species of pathological protein in brain may account for the relatively homogeneous pathology in individual brains, despite the fact that various types of conformational change or structural assembly can occur.
The sequences identified in this study and the seeded aggregation model that we have developed should be useful for establishing animal models of TDP-43 proteinopathy. The results obtained in this study may also contribute to our understanding of the pathogenesis and progression of TDP-43 proteinopathy.
Author Contributions
M. H. and S. S. designed the research and wrote the manuscript. S. S. performed most of the biochemical and immunofluorescence experiments. T. N. and G. S. provided key reagents and conducted the experiments of cellular models. S. H., T. N., S. S., and M. H. analyzed the data.
This work was supported by Ministry of Education, Culture, Sports, Science and Technology KAKENHI Grants 26117005 (to M. H.) and 26111730 (to T. N.), Japan Society for the Promotion of Science KAKENHI Grant 23228004 (to M. H.), Health and Labor Sciences Research Grant 12946221 from Ministry of Health, Labor, and Welfare of Japan (to M. H.), grant-in-aid for research on rare and intractable diseases, the Research Committee on Establishment of Novel Treatments for Amyotrophic Lateral Sclerosis, and the Brain Mapping by Integrated Neurotechnologies for Disease Studies (Brain/MINDS) from Japan Agency for Medical Research and Development, AMED (to M. H.). The authors declare that they have no conflicts of interest with the contents of this article.
- FTLD
- frontotemporal lobar degeneration
- TDP
- TAR-DNA-binding protein
- CTF
- C-terminal fragment
- ThS
- thioflavin S
- ALS
- amyotrophic lateral sclerosis
- NLS
- nuclear localization signal
- Sar
- Sarkosyl
- sup
- supernatant
- ppt
- pellet.
References
- 1. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., and Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 2. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 3. Ou S. H., Wu F., Harrich D., García-Martínez L. F., and Gaynor R. B. (1995) Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 69, 3584–3596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Buratti E., Dörk T., Zuccato E., Pagani F., Romano M., and Baralle F. E. (2001) Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 20, 1774–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Daoud H., Valdmanis P. N., Kabashi E., Dion P., Dupré N., Camu W., Meininger V., and Rouleau G. A. (2009) Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 46, 112–114 [DOI] [PubMed] [Google Scholar]
- 6. Gitcho M. A., Baloh R. H., Chakraverty S., Mayo K., Norton J. B., Levitch D., Hatanpaa K. J., White C. L. 3rd, Bigio E. H., Caselli R., Baker M., Al-Lozi M. T., Morris J. C., Pestronk A., Rademakers R., et al. (2008) TDP-43 A315T mutation in familial motor neuron disease. Ann. Neurol. 63, 535–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kabashi E., Valdmanis P. N., Dion P., Spiegelman D., McConkey B. J., Vande Velde C., Bouchard J. P., Lacomblez L., Pochigaeva K., Salachas F., Pradat P. F., Camu W., Meininger V., Dupre N., and Rouleau G. A. (2008) TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 40, 572–574 [DOI] [PubMed] [Google Scholar]
- 8. Kühnlein P., Sperfeld A. D., Vanmassenhove B., Van Deerlin V., Lee V. M., Trojanowski J. Q., Kretzschmar H. A., Ludolph A. C., and Neumann M. (2008) Two German kindreds with familial amyotrophic lateral sclerosis due to TARDBP mutations. Arch. Neurol. 65, 1185–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rutherford N. J., Zhang Y. J., Baker M., Gass J. M., Finch N. A., Xu Y. F., Stewart H., Kelley B. J., Kuntz K., Crook R. J., Sreedharan J., Vance C., Sorenson E., Lippa C., Bigio E. H., et al. (2008) Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 4, e1000193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sreedharan J., Blair I. P., Tripathi V. B., Hu X., Vance C., Rogelj B., Ackerley S., Durnall J. C., Williams K. L., Buratti E., Baralle F., de Belleroche J., Mitchell J. D., Leigh P. N., Al-Chalabi A., et al. (2008) TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 319, 1668–1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Van Deerlin V. M., Leverenz J. B., Bekris L. M., Bird T. D., Yuan W., Elman L. B., Clay D., Wood E. M., Chen-Plotkin A. S., Martinez-Lage M., Steinbart E., McCluskey L., Grossman M., Neumann M., Wu I. L., et al. (2008) TARDBP mutations in amyotrophic lateral sclerosis with TDP-43 neuropathology: a genetic and histopathological analysis. Lancet Neurol. 7, 409–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yokoseki A., Shiga A., Tan C. F., Tagawa A., Kaneko H., Koyama A., Eguchi H., Tsujino A., Ikeuchi T., Kakita A., Okamoto K., Nishizawa M., Takahashi H., and Onodera O. (2008) TDP-43 mutation in familial amyotrophic lateral sclerosis. Ann. Neurol. 63, 538–542 [DOI] [PubMed] [Google Scholar]
- 13. Hasegawa M., Arai T., Nonaka T., Kametani F., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F., Morita M., Nakano I., Oda T., Tsuchiya K., and Akiyama H. (2008) Phosphorylated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Ann. Neurol. 64, 60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mackenzie I. R., Neumann M., Baborie A., Sampathu D. M., Du Plessis D., Jaros E., Perry R. H., Trojanowski J. Q., Mann D. M., and Lee V. M. (2011) A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 122, 111–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsuji H., Arai T., Kametani F., Nonaka T., Yamashita M., Suzukake M., Hosokawa M., Yoshida M., Hatsuta H., Takao M., Saito Y., Murayama S., Akiyama H., Hasegawa M., Mann D. M., and Tamaoka A. (2012) Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 135, 3380–3391 [DOI] [PubMed] [Google Scholar]
- 16. Nonaka T., Masuda-Suzukake M., Arai T., Hasegawa Y., Akatsu H., Obi T., Yoshida M., Murayama S., Mann D. M., Akiyama H., and Hasegawa M. (2013) Prion-like properties of pathological TDP-43 aggregates from diseased brains. Cell Rep. 4, 124–134 [DOI] [PubMed] [Google Scholar]
- 17. Nonaka T., Kametani F., Arai T., Akiyama H., and Hasegawa M. (2009) Truncation and pathogenic mutations facilitate the formation of intracellular aggregates of TDP-43. Hum. Mol. Genet. 18, 3353–3364 [DOI] [PubMed] [Google Scholar]
- 18. Nonaka T., Arai T., Buratti E., Baralle F. E., Akiyama H., and Hasegawa M. (2009) Phosphorylated and ubiquitinated TDP-43 pathological inclusions in ALS and FTLD-U are recapitulated in SH-SY5Y cells. FEBS Lett. 583, 394–400 [DOI] [PubMed] [Google Scholar]
- 19. Inukai Y., Nonaka T., Arai T., Yoshida M., Hashizume Y., Beach T. G., Buratti E., Baralle F. E., Akiyama H., Hisanaga S., and Hasegawa M. (2008) Abnormal phosphorylation of Ser409/410 of TDP-43 in FTLD-U and ALS. FEBS Lett. 582, 2899–2904 [DOI] [PubMed] [Google Scholar]
- 20. Nonaka T., Watanabe S. T., Iwatsubo T., and Hasegawa M. (2010) Seeded aggregation and toxicity of α-synuclein and tau: cellular models of neurodegenerative diseases. J. Biol. Chem. 285, 34885–34898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Masuda-Suzukake M., Nonaka T., Hosokawa M., Oikawa T., Arai T., Akiyama H., Mann D. M., and Hasegawa M. (2013) Prion-like spreading of pathological α-synuclein in brain. Brain 136, 1128–1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goedert M. (2015) NEURODEGENERATION. Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 349, 1255555. [DOI] [PubMed] [Google Scholar]
- 23. Guo W., Chen Y., Zhou X., Kar A., Ray P., Chen X., Rao E. J., Yang M., Ye H., Zhu L., Liu J., Xu M., Yang Y., Wang C., Zhang D., et al. (2011) An ALS-associated mutation affecting TDP-43 enhances protein aggregation, fibril formation and neurotoxicity. Nat. Struct. Mol. Biol. 18, 822–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang L. L., Che M. X., Zhao J., Zhou C. J., Xie M. Y., Li H. Y., He J. H., and Hu H. Y. (2013) Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 288, 19614–19624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang Y. T., Kuo P. H., Chiang C. H., Liang J. R., Chen Y. R., Wang S., Shen J. C., and Yuan H. S. (2013) The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 288, 9049–9057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Saini A., and Chauhan V. S. (2011) Delineation of the core aggregation sequences of TDP-43 C-terminal fragment. Chembiochem 12, 2495–2501 [DOI] [PubMed] [Google Scholar]
- 27. Furukawa Y., Kaneko K., Watanabe S., Yamanaka K., and Nukina N. (2011) A seeding reaction recapitulates intracellular formation of Sar-insoluble transactivation response element (TAR) DNA-binding protein-43 inclusions. J. Biol. Chem. 286, 18664–18672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen A. K., Lin R. Y., Hsieh E. Z., Tu P. H., Chen R. P., Liao T. Y., Chen W., Wang C. H., and Huang J. J. (2010) Induction of amyloid fibrils by the C-terminal fragments of TDP-43 in amyotrophic lateral sclerosis. J. Am. Chem. Soc. 132, 1186–1187 [DOI] [PubMed] [Google Scholar]
- 29. Sun C. S., Wang C. Y., Chen B. P., He R. Y., Liu G. C., Wang C. H., Chen W., Chern Y., and Huang J. J. (2014) The influence of pathological mutations and proline substitutions in TDP-43 glycine-rich peptides on its amyloid properties and cellular toxicity. PLoS ONE 9, e103644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bigio E. H., Wu J. Y., Deng H. X., Bit-Ivan E. N., Mao Q., Ganti R., Peterson M., Siddique N., Geula C., Siddique T., and Mesulam M. (2013) Inclusions in frontotemporal lobar degeneration with TDP-43 proteinopathy (FTLD-TDP) and amyotrophic lateral sclerosis (ALS), but not FTLD with FUS proteinopathy (FTLD-FUS), have properties of amyloid. Acta Neuropathol. 125, 463–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Berriman J., Serpell L. C., Oberg K. A., Fink A. L., Goedert M., and Crowther R. A. (2003) tau filaments from human brain and from in vitro assembly of recombinant protein show cross-β structure. Proc. Natl. Acad. Sci. U.S.A. 100, 9034–9038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serpell L. C., Berriman J., Jakes R., Goedert M., and Crowther R. A. (2000) Fiber diffraction of synthetic α-synuclein filaments shows amyloid-like cross-beta conformation. Proc. Natl. Acad. Sci. U.S.A. 97, 4897–4902 [DOI] [PMC free article] [PubMed] [Google Scholar]