

Abstract
The tumor suppressor PTEN dephosphorylates PIP3 to inhibit PI3K signaling in cells. Altering PTEN intracellular signaling can therefore significantly affect cell behavior. Two novel mechanisms of PTEN regulation including the secretion and entry of the translational variant PTEN-L, and enzymatic inhibition by the interacting protein P-REX2, have been shown to modulate PI3K signaling, cellular proliferation and survival, and glucose metabolism. Here, we review the methods used to identify and validate the existence of both PTEN-L and the P-REX2–PTEN complex, to determine their effects on PTEN phosphatase activity, and to examine their role in cellular physiology.
Keywords: PTEN, PTEN-L, P-REX2, Secretion
1. Introduction
The tumor suppressor phosphatase and tensin homolog deleted from chromosome 10 (PTEN) is a dual specificity phosphatase whose main substrate is phosphatidylinositol 3,4,5-trisphosphate (PIP3) [1–3]. PTEN dephosphorylates PIP3, the major secondary messenger in the phosphoinositide 3-kinase (PI3K) signaling pathway, to generate phosphatidylinositol 4,5-bisphosphate, therefore inhibiting downstream PI3K signaling, including membrane recruitment and activation of the serine/threonine kinase AKT [4–6]. Deregulation of PTEN has been implicated in diseases including cancer and diabetes. Germline mutations in PTEN are found in patients with Cowden syndrome [7], and PTEN deletion or inactivating mutations are found in a high percent of sporadic tumors [8,9]. Furthermore, patients with Cowden syndrome display increased insulin sensitivity [10].
Altering the PTEN signal in the cell, whether through the modification of the cellular dose or direct inhibition of its enzymatic activity, can greatly affect cellular behavior. This is clearly demonstrated by several recent publications produced from our lab in which we identify and characterize two novel and distinct mechanisms regulating intracellular PTEN signaling. We first identified a novel translational variant of PTEN, named initially PTEN-Long, and subsequently PTEN-L, which alters cellular PTEN dosage through its secretion and entry into cells (Fig. 1) [11,12]. We first suspected the existence of PTEN-L by observing a 75 kilodalton band on Western blots of numerous cell lines and tissues probed with PTEN antibody. Further analysis of the PTEN transcript revealed that an in-frame alternative translational initiation codon was present 519 base pairs upstream of the canonical initiation codon, resulting in the expression of protein containing an evolutionary conserved 173–amino acid domain at its N terminus followed by the classical 403 amino acids of PTEN. Also included in the PTEN-L upstream sequence is a secretion signal sequence and cleavage site, which allows for PTEN-L secretion outside of the cell. The N-terminal sequence also contains a poly-arginine stretch similar to the poly-basic residues of the HIV transactivator of transcription (TAT) protein, which allows PTEN-L to enter cells [11,13,14]. Treatment with exogenous PTEN-L negatively regulates PI3K signaling and triggers apoptosis and inhibits the growth of mouse xenograft models [11]. We also discovered a novel PTEN interacting protein, phosphatidylinositol-3,4,5-trisphosphate-dependent Rac exchange factor 2 (P-REX2) [15]. This Rac guanine nucleotide exchange factor [16,17] inhibits PTEN activity through its PH domain both in vitro and in vivo, and regulates insulin stimulated PI3K signaling and glucose metabolism (Fig. 1) [18]. In this article we thoroughly detail the common methods involved in the identification, verification, and functional analysis of these two distinct mechanisms of regulating intracellular PTEN signaling (Fig. 2).
Fig. 1.
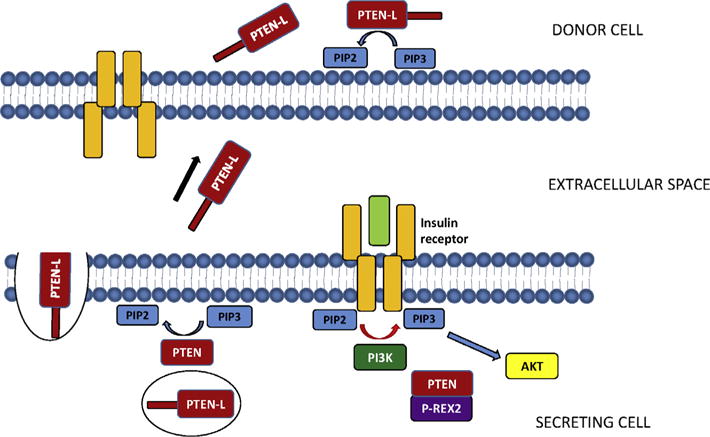
Regulation of intracellular PTEN signaling by PTEN-L and P-REX2. PTEN-L behaves similarly to canonical PTEN, dephosphorylating PIP3 to PIP2. In addition, PTEN-L is also packaged into secretory vesicles and released into the extracellular space. PTEN-L can then enter other cells, increasing the intracellular dose of PTEN in that cell and decreasing PIP3 levels (11). PTEN is also regulated by P-REX2 in response to insulin signaling. P-REX2 binds PTEN and inhibits its phosphatase activity toward PIP3, thus enhancing PI3K signaling in the cell (15, 18).
Fig. 2.
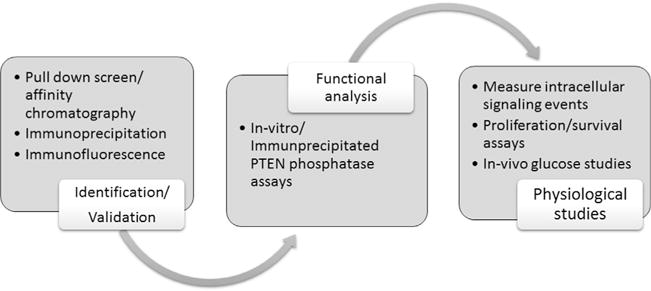
Schematic of methods used to identify PTEN-L and the PTEN-P-REX2 complex, characterize their enzymatic function, and uncover their physiological significance in cells.
2. Identification of PTEN-P-REX2 complex and PTEN-L
2.1. Pull down screen using affinity chromatography
Pull down experiments using affinity chromatography and mass spectrometry is a powerful tool used to uncover novel protein interactions. We initially observed the PTEN-P-REX2 complex using this method to screen for PTEN regulators [15]. Pull down assays use affinity purified protein to capture binding partners. We used GST tagged recombinant PTEN produced from bacteria bound to glutathione Sepharose beads to screen for interacting proteins (Fig. 3). To produce recombinant GST-PTEN and GST, BL21(DE3) pLysE bacteria (Invitrogen) are transformed with plasmids encoding GST and GST-PTEN. This chemically competent E. coli strain contains a T7 RNA polymerase gene controlled by the lacUV5 promoter. Protein expression is induced after a 4 L bacterial culture reaches exponential growth by adding 0.1 mM isopropyl-1-thio-β-D-galactopyranoside (IPTG, Sigma), which induces high level protein expression from a T7 promoter present in our expression vector. It is important to optimize the proper induction conditions, as proteins can degrade at different temperatures and time points. We found that incubation at 16 degrees allowed for robust and stable expression of GST-PTEN. Protein is extracted from bacteria by sonication at a cycle of 5 s on, 5 s off for 40 min in 40 mL lysis buffer containing 400 mM NaCl, 50 mM Tris pH 7.2, 1% Triton X-100, 1 mM EDTA. The lysate is centrifuged at 20,000g for 1 h, filtered through a 0.45 micron filter, and then incubated with glutathione-Sepharose beads (GE Life Technologies) for a final concentration of 10 mg/1 mL glutathione Sepharose. The beads are then washed six times with BC500 buffer (25 mM Tris·HCl buffer, pH 7.4, 500 mM KCl, 0.2% Triton X-100, 1 mM EDTA, and 10% (vol/vol) glycerol), followed by two washes with BC200 buffer (25 mM Tris HCl buffer, pH 7.4, 200 mM KCl, 0.2% Triton X-100, 1 mM EDTA, and 10% (vol/vol) glycerol).
Fig. 3.
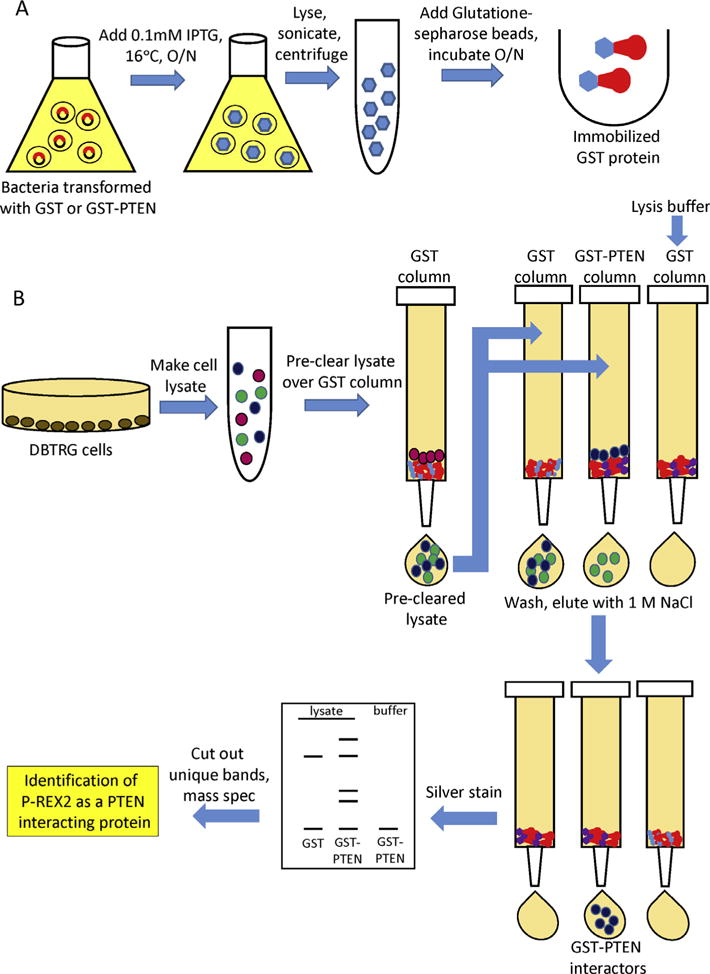
GST-PTEN pull down screen (15). (A) Generating immobilized GST and GST-tagged PTEN. BL21(DE3) pLysE bacteria transformed with plasmids encoding GST and GST-PTEN are grown to exponential growth and protein expression is induced with 0.1 mM IPTG. After incubation at 16 degrees overnight, cells are sonicated for 40 min in 40 mL lysis buffer and centrifuged at 20,000g for 1 h, and filtered. Glutathione–Sepharose beads are then added for a final concentration of 10 mg per 1 mL glutathione Sepharose. The beads are then washed six times with BC500 buffer followed by two washes with BC200 buffer. (B) GST-PTEN pull-down screen. DBTRG cell lysates are generated as described in Section 2.1. Lysates are pre-cleared by passing them over a column containing GST beads. Pre-cleared lysates are then incubated with either GST-PTEN or GST beads overnight at 4 °C, along with a mock column of GST-PTEN and lysis buffer. The beads are washed 10 times with BC200 buffer wash buffer, and proteins are eluted with hypotonic buffer containing 1 M NaCl. Elutions are resolved by SDS–PAGE and visualized by silver stain, and protein bands specific to the GST-PTEN column are excised and peptide sequenced by MALDI-TOF mass spectrometry.
Next, the source of potential interacting proteins is prepared. We decided to use the PTEN-deficient cell line DBTRG-05MG as the protein source due to the lack of competitive binding with endogenous PTEN. To prepare cytoplasmic extracts, cells from 36 15 cm dishes were collected in PBS and spun down at 1000g for 5 min. We treated the cells with 40 mL hypotonic buffer containing 10 mM HEPES pH 7.4, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and protease inhibitor cocktail (Sigma) for 15 min. to swell the cells, followed by lysis with NP-40 at a final concentration of 0.5%. Debris was pelleted by centrifugation at 3000 rpm for 5 min at 4 °C. Lysates were then equilibrated to 200 mM NaCl, incubated on ice for 1 h and then centrifuged at 25,000g for 1 h at 4 °C.
When performing pull down experiments, it is important to exclude non-specific interactors, and therefore purified GST alone is used as a control. To eliminate non-specific binding to GST itself, cell lysates were first passed over a column containing GST beads. This lysate was then divided into two aliquots, and incubated with either GST-PTEN or GST beads overnight at 4 °C. Lysis buffer is also incubated with GST-PTEN and serves as an additional control for bacterial proteins bound to the protein column. To further eliminate background, careful consideration must be made to the wash conditions, as the salt concentration should to be high enough to eliminate non-specific interactors, but not excessively high that desired weaker binding proteins are washed away. We washed the beads 10 times with BC200 buffer wash buffer, and eluted proteins in buffer containing 1 M NaCl. Elutions are resolved by SDS–PAGE and visualized by silver stain, and protein bands specific to the GST-PTEN column are excised and peptide sequenced by MALDI-TOF mass spectrometry.
2.2. Immunoprecipitation
2.2.1. Endogenous protein
Immunoprecipitation allows for the isolation of a protein from a cellular lysate using an antibody that specifically recognizes that protein. We verified the existence of endogenous PTEN-L in cells using this technique [11]. The immunoprecipitation of endogenous proteins is an important tool because it does not depend upon exogenous overexpression of tagged-proteins that may behave differently from endogenous proteins. However, successful immunoprecipitation of endogenous protein often proves difficult due to a lack of specific antibodies and low levels of protein expression in cells. To verify that PTEN-L is a translational variant of PTEN expressed in cells, we immunoprecipitated PTEN-L from HEK293 cells, which express both variants. The cells are lysed in buffer containing 150 mM NaCl, 25 mM Tris pH 7.5, 0.1% Triton X-100 with protease inhibitor cocktail, vortexed and centrifuged at 16,000g for 1 h. Following cell lysis, it is convenient to pre-clear the lysate 1 h at 4 °C by incubation with protein A/G agarose beads (Santa Cruz) and IgG specific to the antibody used for immunoprecipitation. This greatly reduces non-specific binding of proteins to agarose beads and antibody immunoglobulins. Pre-cleared lysate is then incubated with an antibody recognizing the protein of interest. To discriminate PTEN from PTEN-L, we generated an antibody produced in rabbit that recognizes the N-terminal PTEN-L sequence PRHQQLLPSLSSFFFSHRLPD not found in the normal PTEN protein. We performed reciprocal immunoprecipitations of PTEN and PTEN-L by incubating lysate with either 5 μL of PTEN 138G6 antibody (Cell Signaling) or PTEN-L antibody along with 40 μL protein A/G-agarose beads overnight at 4 °C. To decrease background, the beads are washed extensively with lysis buffer before eluting protein in 40 μL 2× Laemmli buffer. PTEN-L is then visualized by Western blot. One problem that often arises when detecting immunoprecipitated proteins is the reactivity of secondary antibodies with immunoglobulins from the antibody used in the IP. Therefore, we often take advantage of secondary antibodies that detect immunoglobulin light chain or heavy chain specifically, therefore removing background bands. Also, many primary antibodies exist that are directly conjugated to HRP, such as PTEN A2B1 from Santa Cruz Biotechnology, that eliminates the need for a secondary antibody altogether.
We also verified the existence of the PTEN-P-REX2 complex by co-immunoprecipitating the endogenous protein complex from both cells and tissue [15,18]. We used mouse embryonic fibroblasts (MEFs) and mouse liver tissue as a protein source, as they are sensitive to P-REX2 inhibition of PTEN. To generate mouse liver lysates for the immunoprecipitation, fresh livers are homogenized for 30 s in lysis buffer containing 150 mM NaCl, 50 mM Hepes, 1 mM EDTA, 1% Nonidet P-40, 0.025% deoxycholate, 1 mM NaF, 1 mM NaVO4, and protease inhibitor mixture using a Tissuemiser, and then centrifuged at 16,000g for 1 h at 4 °C. MEFs were washed with PBS and lysed with the same lysis buffer, and centrifuged at 16,000g for 1 h at 4 °C. We immunoprecipitated PTEN with PTEN N-19 antibody (4 μg/500 μg total protein; Santa Cruz Biotechnology) and 40 μL protein A/G beads overnight at 4 °C. We used this N-terminal PTEN antibody because a C-terminal antibody may have blocked the interaction between P-REX2 and the C-terminal PDZ-binding domain of PTEN. We also generated our own P-REX2 antibody produced in rabbit, which recognizes the sequence VQLDSRKHNSHDKE, to probe for endogenous P-REX2 in human and mouse. The beads were washed 5 times with lysis buffer, proteins were eluted with 40 μL 2 Laemmli buffer and the protein complex was analyzed by Western blot.
2.2.2. Exogenous protein immunoprecipitation
In addition to endogenous protein, we also immunoprecipitated exogenously expressed protein to examine potential protein function. We used this method to show that PTEN-L can be immunoprecipitated from conditioned media and therefore is secreted from cells via a mechanism that is dependent on its signal sequence [11]. To overexpress PTEN-L and analyze the properties of its N-terminal sequence, we generated several mammalian expression vectors encoding the PTEN-L open reading frame, as well as a mutant in which six alanines from the predicted signal sequence are deleted. Before immunoprecipitation can be performed, the DNA vectors are transfected into HEK293 cells. We use Lipofectamine 2000 (Life Technologies) for our transfections, as it results in reproducible and high transfection efficiency and is well tolerated by the cells. Conditioned media is collected after 18 h, which is long enough to ensure adequate protein overexpression while minimizing the release of endogenous proteins into the media due to cell death. The media is passed through a 0.22 micron filter, and PTEN-L is immunoprecipitated from the collected media using PTEN 138G6 antibody as described in the previous section.
We also used immunoprecipitation of exogenously expressed proteins to characterize the P-REX2-PTEN interaction in detail. We generated many different deletion constructs for both PTEN and P-REX2 that also contained either FLAG or V5 epitope tags. By co-expressing different domains of P-REX2 and PTEN, detailed interactions can be easily mapped by utilizing the epitope tags for immunoprecipitation. We co-expressed different combinations of PTEN and P-REX2 domains in 10 cm plates of HEK293 cells by transfection with 60 μL of Lipofectamine 2000. Cells were lysed 36 h after transfection in buffer containing 150 mM NaCl, 25 mM Tris pH 7.4, 0.1% Triton X-100, 1 mM EDTA, and protease inhibitor cocktail, and spun down at 16,000g for 30 min at 4 °C. To eliminate non-specific interactions with mouse IgG, lysates were pre-cleared with mouse IgG plus Protein A/G beads for 1 h at 4 °C. Protein complexes were then immunoprecipitated by incubation with 3 μL V5 antibody (Invitrogen) or Flag antibody (Sigma) plus 40 μL Protein A/G beads overnight at 4 °C. Beads were washed five times with lysis buffer and proteins were eluted with 40 μL 2 Laemmli buffer.
2.3. Fluorescence imaging
Cellular fluorescence imaging is a popular method used to visualize the localization of endogenous or exogenous proteins inside of a live or fixed cell. This technique was useful for validating PTEN-L entry into cells [11], and for observing co-localization of PTEN and P-REX2 in cells [15]. Proteins can be tagged with a fluorescent protein domain, allowing for direct visualization in live cells. Proteins either in living or fixed cells can be fluorescently labeled directly using fluorophore conjugated primary antibodies or indirectly using fluorophore conjugated secondary antibodies.
2.3.1. Purification of RFP-tagged PTEN-L for cell uptake studies
To show PTEN-L entry into cells, we took the direct approach and generated PTEN-L protein with a C-terminal red fluorescent protein (RPF)-tag. The expressed protein also contains a C-terminal V5/His tag, which allows for its purification from bacteria by using an AKTA-FPLC (fast protein liquid chromatography), a chromatography system used for the rapid and effective purification of proteins. A two-step purification process is performed using two different chromatography columns (Fig. 4). PTEN-L expression was induced in 400 mL cultures of BL21(DE3) pLysE bacterial cells with 0.1 mM IPTG for 4.5 h at 21 °C. Bacterial cells were then lysed with 10 mL lysis buffer, sonicated, and centrifuged as described in Section 2.1 Bacterial lysates were then passed over a HisTrap nickel column (GE Life Sciences) with running buffer containing 500 mM NaCl 25 mM Tris pH 7.6, 20 mM Imidazole. The HisTrap column contains cross-linked agarose beads with an immobilized chelating group to generate charged Ni2+ ions, which form complexes with the histidines present on the His-tagged protein. Proteins were eluted with high concentrations of imidazole buffer (500 mM NaCl 25 mM Tris pH 7.5, 500 mM imidazole), which competes for binding to nickel. To further purify PTEN-L, the eluted protein was then passed over a HiTrap heparin column (GE Life Sciences, running buffer contains 25 mM Tris pH 7.5 25 mM NaCl), and eluted with a high salt buffer containing 25 mM Tris pH 7.5 and 1 M NaCl. The resulting eluates were concentrated using Amicon Ultra 50KDa exclusion columns and quantified by coomassie stain.
Fig. 4.
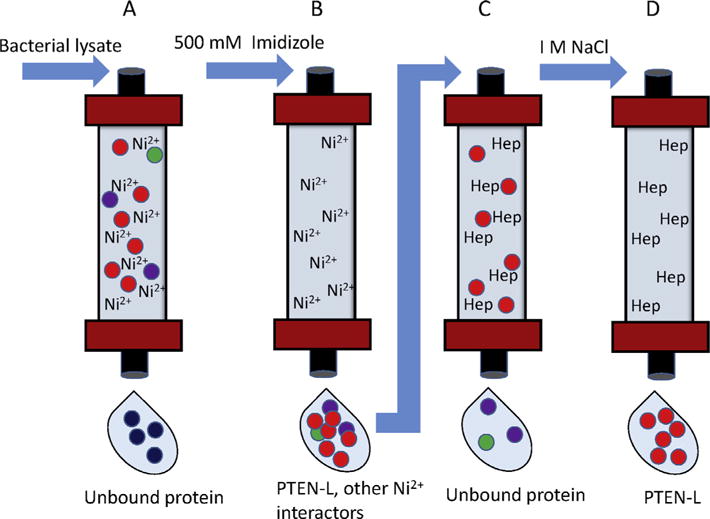
Purification of RFP-tagged PTEN-L (11). (A) Bacterial lysates expressing PTEN-L containing both an RFP and V5/His tag are passed over a HisTrap nickel column. Ni2+ ions form complexes with the histidines present on the His-tagged protein, and unbound protein passes through the column. (B) Proteins bound to the column are eluted with buffer containing 500 mM imidazole, which competes for binding to nickel. (C) The eluted protein containing PTEN-L is passed over a HiTrap heparin column. Heparin binding proteins, including PTEN-L, bind to the column, while unbound proteins are washed away. (D) PTEN-L is eluted with buffer containing 1 M NaCl.
2.3.2. Immunofluorescence
Purified RFP-tagged PTEN-L is added directly to U87-MG cells that are plated on gelatin coated glass coverslips. The cells incubate for 30 min, are washed with PBS, and mounted on slides with Prolong Gold Antifade with DAPI. A Nikon Eclipse 80i with a CoolSnap HQ2 CCD camera is used to image the cells. When imaging cells, it is important to limit the intensity and time period of light exposure, as photobleaching may reduce the fluorescent signal.
To show P-REX2 and PTEN co-localization in cells, we used indirect immunofluorescence. Unlike direct immunofluorescence, which uses primary antibodies conjugated to a fluorophore, indirect immunofluorescence required two antibodies; a primary antibody that binds the target protein, and a fluorescent secondary antibody. Because more than one secondary antibody can bind to the primary antibody, indirect immunofluorescence usually results in a higher degree of sensitivity. There is also greater flexibility when choosing an appropriate secondary antibody. However, one must be aware of the possibility of cross-reactivity of the secondary antibody and the need to use primary antibodies raised from different species when labeling more than one protein. High levels of background fluorescence may also be present in samples with high levels of endogenous immunoglobulins; therefore it is always important to include a negative control to avoid potential false positives. To show co-localization of P-REX2 and PTEN in cells, double immunofluorescence labeling was performed, where antibodies from different specifies are used to probe PTEN and P-REX2 so that different fluorophores can be used. Like PTEN-L, we used U87-MG cells due to their lack of endogenous PTEN. U87-MG cells are plated on gelatin coated glass coverslips and transfected with PTEN and V5-tagged P-REX2. Twenty-four hours after transfection, cells are washed twice with PBS, then fixed in 2% paraformaldehyde for 20 min. Cells were washed four times with PBS, and then permeabilized with 0.1% Triton X-100 in PBS plus 10% goat serum for 30 min. We incubated cells with anti-V5 antibody produced in mouse at a 1:500 dilution and PTEN 138G6 antibody produced in rabbit at a 1:200 dilution in 0.1% Triton X-100 in PBS overnight. The cells were washed three times with PBS, and then incubated with mouse and rabbit secondary antibody at a 1:500 dilution in 0.1% Triton X-100 in PBS for 1 h at room temperature. Cells were again washed with PBS three times, and coverslips were mounted on the slides with Prolong Gold Antifade with DAPI and visualized with a Nikon Eclipse 80i with a CoolSnap HQ2 CCD camera.
3. Analysis of PTEN-L and P-REX2 effect on lipid phosphatase activity
3.1. PTEN lipid phosphatase activity assays
3.1.1. In vitro phosphatase assay with bacterially produced protein
The next crucial step was to elucidate the function of PTEN-L and the PTEN-P-REX2 complex. In the case of PTEN-L, we sought to show that it is a functional lipid phosphatase with enzymatic activity toward PIP3 [11]. To do this, we utilized an in vitro assay in which quantified levels of free phosphate is used as a readout for PTEN lipid phosphatase activity. PTEN lipid phosphatase assays are performed using water soluble di-C8-D-myo-Phosphatidylinositol 3,4,5 trisphosphate (PIP3) as the lipid source and Malachite Green (Biomol) as the colorimetric readout. Malachite Green forms a colored complex with liberated phosphate that can be quantified by reading the absorbance at 620 nm [19]. PTEN-L was purified from bacteria as described in Section 2.3 and assayed along with canonical PTEN, which served as a positive control. Due to the high sensitivity of this assay, any reagents contaminated with free phosphate will result in false positive readouts. Therefore, all reagents should be tested for free phosphate before the start of the assay by incubating them with Malachite Green. PIP3 was diluted to various concentrations ranging from 20 μM to 100 μM in phosphatase buffer (25 mM Tris and 100 mM NaCl pH 7.6) and added to purified PTEN or PTEN-L and phosphatase buffer for a final volume of 50 μL. It is important that the concentration of PTEN-L used in the assay is within a linear range of detection. Therefore, the activity of various dilutions of PTEN protein were assayed before the experiment to determine a linear concentration. The reaction incubated for 30 min at 37 °C, and was stopped by the addition of 50 μL Malachite green. The resulting absorption was read at 620 nm after incubation at room temperature for 15 min in a Micro-QUANT microplate spectrophotometer. A standard curve was also generated using phosphate standards in order to quantify the phosphate released by each sample.
3.1.2. In vitro phosphatase assay with protein purified from mammalian cells
The Malachite Green assay was also used to measure the noncompetitive inhibition of PTEN lipid phosphatase activity by P-REX2 [15]. There are limiting factors to consider when performing in vitro phosphatase assays, most notably being the source of purified protein. Here, we used PTEN and P-REX2 purified from HEK293 cells due to their purity and the presence of post-translational modifications. Protein purified from bacteria is not post-translationally modified, and therefore may not be regulated in the same manner as protein purified from mammalian cells. For instance, P-REX2 does not inhibit bacterially-produced PTEN unless it contains phospho-mimicking mutations in its regulatory C-terminal tail domain [18].
To purify PTEN and P-REX2 for in vitro phosphatase assays, plasmids encoding C-terminally V5-tagged PTEN or P-REX2 were transfected into 10 15 cm plates of HEK293 cells using 60 μL Lipofectamine 2000 per plate. The cells were lysed in buffer containing 150 mM NaCl, 25 mM Tris pH 7.4, 1% Triton X-100, 1 mM EDTA, and protease cocktail inhibitor 36 h after transfection. Lysates were vortexed vigorously and ultracentrifuged at 100,000g for 2 h. The lysate was then pre-cleared with 100 μL mouse IgG conjugated to agarose beads for 1 h at 4 °C, and V5-tagged protein was immu-noprecipitated with 100 μL V5-agarose (Sigma) for 4 h at 4 °C. The beads were washed once with lysis buffer and four times with phosphatase buffer. Protein was eluted with 100 μL of V5 peptide (150 μg/mL) in phosphatase buffer, which competes off the V5-tagged proteins from the beads, and quantified by coomassie staining.
To measure P-REX2 inhibition of PTEN phosphatase activity, 40 nM of purified PTEN was incubated with 10 nM or 20 nM purified P-REX2, PIP3 at concentrations ranging from 20 μM to 100 μM, and phosphatase buffer to a final reaction volume of 50 μL. The reaction proceeded for 30 min at 37 °C, and was stopped by the addition of 50 μL Malachite green. The resulting absorption was read at 620 nm after incubation at room temperature for 15 min in a Micro-QUANT microplate spectrophotometer.
3.1.3. Immunoprecipitated PTEN
Measuring the enzymatic activity of purified PTEN in vitro gives you little information as to its intracellular regulation. We therefore performed phosphatase assays on endogenous PTEN protein complexes immunoprecipitated from cells or mouse tissues to determine how it is being modulated in a physiological setting (Fig. 5). Specifically, we used his method to show that P-REX2 inhibits PTEN activity in cells in response to insulin stimulation [18]. We immunoprecipitated PTEN from starved and insulin stimulated P-rex2+/+ and P-rex2−/− MEFs and mouse liver lysates with PTEN 138G6 antibody as described in Section 2.2. The pre-clear step is critical when performing phosphatase assays, as nonspecific binding of phosphatases other than PTEN may also dephosphorylate PIP3. It is also important to include a negative control immunoprecipitation to eliminate the possibility that contaminating proteins are dephosphorylating PIP3 in addition to PTEN. Following immunoprecipitation, the protein-bound beads are washed extensively with phosphatase buffer to remove nonspecific interactions as well as free phosphate, and are then incubated with 20 μM soluble di-C8-D-myo-PIP3 and brought up to a final volume of 50 μL with phosphate free buffer. This reaction proceeds for 30 min at 37 °C. The beads are removed from the reaction mixture by low speed centrifugation for 5 min at room temperature, and the supernatant is added to 50 μL Malachite Green reagent, and absorbance read at 620 nm after 15 min.
Fig. 5.
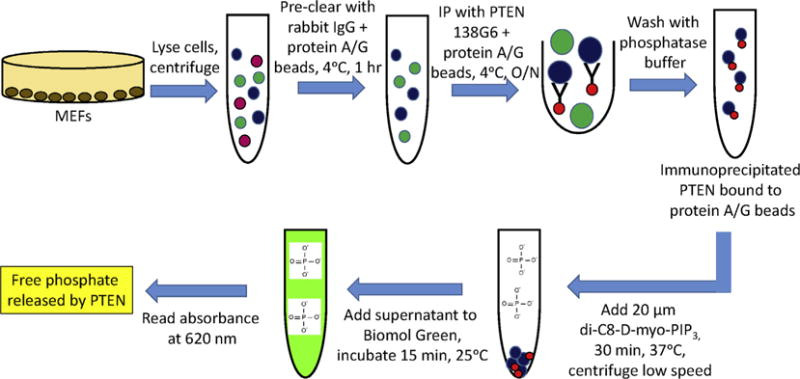
Immunoprecipitation PTEN phosphatase assays (18) MEFs are washed in PBS, lysed, and centrifuged at 16,000g for 30 min at 4 °C. Lysates are pre-cleared with 2 μg rabbit IgG plus 40 μL protein A/G beads for 1 h at 4 °C. PTEN is immunoprecipitated with 138G6 antibody plus 40 μL protein A/G beads overnight at 4 °C. Beads are washed with phosphatase buffer and incubated with 20 μM soluble di-C8-D-myo-PIP3 for 30 min at 37 °C. The beads are removed from the reaction mixture by low speed centrifugation, and the supernatant is added to 100 μL Biomol Green reagent for 15 min. The absorbance level at 620 nm is the measure of free phosphate released by PTEN.
4. Effect on cellular physiology
The previous methods revealed that PTEN-L is a secreted lipid phosphatase that can re-enter cells, while P-REX2 binds to and inhibits the lipid phosphatase activity of PTEN in vitro and in vivo. We utilized the next set of methods to show that modulating intracellular PTEN activity via PTEN-L or P-REX2 effects cell signaling, survival, and proliferation [11].
4.1. Measuring PI3K signaling in cells and in mice
4.1.1. Immunoblotting
We have reported that addition of purified PTEN-L to the media of cells in culture leads to decreased PI3K signaling, while P-REX2 inhibition of PTEN leads to increased PI3K signaling specifically in response to insulin and IGF-1. One of the most effective ways to analyze PI3K signaling transduction in cells is by immunoblotting cell lysates for activated and phosphorylated kinases downstream of PI3K, including AKT, GSK3β, FOXO1/2/3, and PRAS40. Probing a variety of downstream targets is important to understand the signal. To examine the effect of exogenous treatment of PTEN-L in cells on PI3K signaling, we chose to use the cell lines U87-MG and MDA-MB-468, which do not express PTEN and therefore likely to be responsive to changes intracellular PTEN. Cells were incubated with serum free media for 30 min. to eliminate contaminating PTEN-L present in serum. Cells were then exposed to serum free media containing various concentrations of purified PTEN-L and harvested at different time points in 2X Laemmli buffer. Western blots were probed for antibodies recognizing phospho-AKT, and the AKT target proteins PRAS40 and FOXO (Cell Signaling) as a measure of downstream PI3K signaling.
We also looked in vivo and found that treatment of mice with purified PTEN-L results in decreased PI3K signaling in mouse tissue and xenografted tumors. When xenografting cells into mice, many factors must be considered, including the cell type, cell number, and host animal. We use athymic nude mice in our tumor studies. Their lack of an effective immune system allows for the injection of human cells without rejection. We resuspended 1 × 106 PTEN null MDA-MB-468 human breast cancer cells in matrigel, which is a basement membrane protein mixture that supports the establishment and growth of tumor cells in mice, and were injected into the lower mammary fat pad of 8–10 week old mice. Following detection of a palpable tumor, mice were treated with PTEN-L or a mock protein prep by intraperitoneal injection. We chose this method as the preferred delivery method due to its reproducibility, ease, and effectiveness when compared to other delivery techniques including intravenous, subcutaneous, intramuscular, and intratumoral delivery. We first confirmed that purified V5-tagged PTEN-L injected intraperitoneally could be detected in various tissues in vivo. To do this, mice were treated twice with 10 mg/kg PTEN-L 24 h apart, and then euthanized via induction with CO2. Brain, lung, and liver tissues were collected and snap frozen. To make lysates for immunoblotting, tissues were homogenized in 25 mM Tris pH 7.5, 500 mM NaCl, centrifuged at 16,000 g for 30 min, and then mixed with Laemmli buffer. We probed with V5 antibody to detect exogenous PTEN-L. To measure changes in PI3K signaling, we probed with antibodies for various downstream PI3K targets including phosphorylated Akt, S6, and Foxo (Cell Signaling).
To show the physiological effect of P-REX2 on PI3K signaling in cells, we again used P-rex2+/+ and P-rex2−/− MEFs [18]. The MEFs were starved for 3 h and then incubated with a variety of growth factors including 10 μg/mL bovine insulin, 20 ng/mL IGF1, 20 ng/mL PDGF, or 20 ng/mL EGF, and lysates were collected over a time course of one hour. Collecting lysates at various time increments and treating with multiple growth factors provides a more complete analysis of the signaling response. Differences in pathway activation may only be observed with certain growth factors at early or late time points, and this may be overlooked if a thorough time course is not performed. After observing the effect that P-rex2 had on insulin and IGF-1 stimulated PI3K signaling, we then looked at insulin signaling in mouse tissue. Here, we chose to examine the role of P-rex2 on PI3K signaling in liver and fat, as these tissues are highly responsive to insulin stimulation. We therefore compared PI3K pathway activation in fasted and insulin stimulated P-rex2+/+ and P-rex2−/− mice. To do this, eight-week male mice were fasted overnight. For collection of starved tissue, fasted mice were sacrificed by cervical dislocation and liver, fat, and hind leg skeletal muscle were collected. For insulin stimulation, fasted mice were injected with 0.75 mU/g bovine insulin intraperitoneally and liver and fat were collected at various time points following stimulation. Tissues were homogenized for 30 s at high speed using a Tissuemiser in Triton-containing lysis buffer, and levels of phosphorylated insulin receptor, Akt, and Gsk3β were determined by Western blot.
4.1.2. Immunohistochemistry
Immunohistochemistry is also a useful tool used to visualize changes in protein expression in the cells of biological tissue by using antibodies conjugated to color-producing or fluorescent enzymes. Like immunofluorescence, it can be either direct by using a labeled primary antibody, or indirect through the use of labeled secondary antibodies. In our case, we used indirect IHC to show that treating mice with PTEN-L leads to a decrease in AKT phosphorylation in MDA-MB-468 human breast cancer xenografts [11]. Proper tissue preparation and fixation is critical for preserving tissue structure and cellular morphology, as well as maintaining the antigenicity of target proteins. Our tissue samples were fixed in 10% formalin for 24 h, and then placed in 70% ethanol for 24 h, followed by another incubation in fresh ethanol. The samples were then embedded in paraffin and sliced. It important to ensure that the epitope is accessible to the antibody. Since our samples are fixed in formalin, methylene bridges may form that cross-link proteins and hide the epitope site required for antibody binding. To break these methylene bridges, we used the Trilogy pretreatment solution from Cell Marque. Trilogy solution was warmed in a steamer for 25 min. The slides were then brought into the hot Trilogy solution for 25 min, and then cooled for 20 min. When choosing an appropriate primary antibody for immunohistochemistry, it is important to consider the species of the tissue. It is critical to use a primary antibody produced in another species due to the potential for cross-reactivity of the secondary antibody with endogenous immunoglobulins in the tissue itself, resulting in background staining. We used the Vectastain ABC (Avidin–Biotinylated enzyme Complexes) elite kit to stain our tissues. This kit utilizes biotin conjugated secondary antibodies, which bind strongly to avidin–biotinylated enzyme complexes. Proteins conjugated to antibodies were then detected using 3,3′-diaminobenzidine, which is oxidized in the presence of peroxidase, forming a brown stain.
4.2. Effect on cellular proliferation and survival
Changes in cellular proliferation and survival are a hallmark of deregulated PI3K signaling. Indeed, PTEN-L treatment results in the induction of apoptosis and decreased cellular viability [11], while overexpression of the PTEN inhibitor P-REX2 leads to increased proliferation and altered cell morphology in a 3-D culture [15]. To examine the effects of exogenous PTEN-L treatment on cell viability, we chose to look at U87-MG and MDA-MB-468 cells, both of which are PTEN null and therefore likely to be more sensitive to changes in intracellular PTEN dose. To measure cell viability, cells are treated with purified either PTEN-L or PTEN-LDR6, which is missing six arginines in the N-terminal sequence responsible for PTEN-L cellular entry. After 24 h, the cells are trypsinized and dead cells are stained with the diazo dye trypan blue. This is known as a dye exclusion method of staining, as trypan blue does not cross the selective membrane of living cells. We also used multiple xenografted tumor models to show that treatment with PTEN-L causes tumor regression in mice. We used a variety of human breast and colon tumor cell lines, most of which are PTEN null, and injected them into the fat pad of nude mice as described in Section 4.1. We monitor tumor progression by imaging with Xenogen Spectrum small animal imaging system 15 min after injection of Luciferin Substrate into the mice, or by caliper measurements. When tumors reached a size of ~0.2 cm3, 4 mg/kg of PTEN-L or a control preparation is injected intraperitoneally once a day, and tumors are monitored daily.
Inhibition of PTEN activity by P-REX2 also affects cell proliferation. We reported that overexpression of P-REX2 in the mammary epithelial cell line MCF10A leads to increased proliferation in reduced media conditions [15]. To measure cell proliferation, 4000 cells were plated per well in a 48-well plate. Cells were allowed to proliferate and then fixed and stained at desired time points with 0.05% crystal violet in 10% formalin. Crystal violet is a triphenylmethane dye commonly used for the Gram-staining of bacteria. Unlike dye exclusion viability assays, crystal violet is a reporter of cell growth through the quantification of dye taken up by all cells [20]. Following staining, each well was thoroughly washed with PBS to remove any residual dye. For relative quantification of cell density, the crystal violet was resolubilized in 10% acetic acid and the absorbance was read at 595.
4.3. Glucose metabolism
PTEN negatively regulates insulin stimulated PI3K signaling and therefore is a regulator of whole body glucose metabolism [21–23]. Therefore, we were not surprised to discover that modulating PTEN intracellular activity through PTEN-L or P-REX2 leads to changes in blood glucose levels and glucose uptake in mice. Specifically, intraperitoneal injection of PTEN-L results in an increase in blood glucose, whereas loss of P-REX2 causes a decrease in glucose uptake. To measure blood glucose levels, mice back crossed into C57/Bl were treated by intraperitoneal injection with 10 mg/kg of purified PTEN-L. We collected blood from the tail and measured blood sugar levels over a four hour time course using a OneTouch Ultra glucometer. To measure whole body glucose uptake, a glucose challenge test is utilized. Here, six week old male mice were starved overnight and injected intraperitoneally with 2 mg dextrose/g body weight. We collected blood from the tail and recorded blood glucose levels at 15 min intervals for 2 h.
5. Concluding remarks
This article reviews the methods used to identify PTEN-L and P-REX2 as novel modulators of intracellular PTEN signaling, as well as understanding how they regulate PI3K signaling, cell growth, and survival in various cellular contexts. However, it is important to appreciate that these methods are also widely used to study many different facets of PTEN including the effects of post-translational modifications and cancer mutations on PTEN function, PTEN protein phosphatase activity, and PIP3 independent functions of PTEN. Furthermore, new techniques and tools are constantly being introduced, leading to a rapidly increasing body of PTEN knowledge and a more complete understanding of its role in human disease.
References
- 1.Maehama T, Dixon JE. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 2.Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, et al. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 3.Leslie NR, Downes CP. J Biochem. 2004;382:1–11. doi: 10.1042/BJ20040825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stambolic V, Suzuk A, de la Pompa JL, Brothers GM, Mirtsos C, Sasaki T, et al. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 5.Sun H, Lesche R, Li DM, Liliental J, Zhang H, Gao J, et al. Proc Natl Acad Sci USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li J, Simpson L, Takahashi M, Miliaresis C, Myers MP, Tonks N, et al. Cancer Res. 1998;58:5667–5672. [PubMed] [Google Scholar]
- 7.Zhou XP, Marsh DJ, Hampel H, Mulliken JB, Gimm O, Eng C. Hum Mol Genet. 2000;9:765–768. doi: 10.1093/hmg/9.5.765. [DOI] [PubMed] [Google Scholar]
- 8.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, et al. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 9.Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, et al. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 10.Mitchell F. Nat Rev Endocrinol. 2012;8:698. doi: 10.1038/nrendo.2012.186. [DOI] [PubMed] [Google Scholar]
- 11.Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, et al. Science. 2013;341:399–402. doi: 10.1126/science.1234907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pulido R, Baker SJ, Barata JT, Carracedo A, Cid VJ, Chin-Sang ID, et al. Sci Signal. 2014;7:pe15. doi: 10.1126/scisignal.2005560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Subramanian T, Govindarajan R, Chinnadurai G. EMBO J. 1991;10:2311–2318. doi: 10.1002/j.1460-2075.1991.tb07768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 15.Fine B, Hodakoski C, Koujak S, Su T, Saal LH, Maurer M, et al. Science. 2009;4:1261–1265. doi: 10.1126/science.1173569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rosenfeldt H, Vázquez-Prado J, Gutkind JS. FEBS Lett. 2004;572:167–171. doi: 10.1016/j.febslet.2004.06.097. [DOI] [PubMed] [Google Scholar]
- 17.Donald S, Hill K, Lecureuil C, Barnouin R, Krugmann S, Coadwell W John, et al. FEBS Lett. 2004;572:172–176. doi: 10.1016/j.febslet.2004.06.096. [DOI] [PubMed] [Google Scholar]
- 18.Hodakoski C, Hopkins BD, Barrows D, Mense SM, Keniry M, Anderson KE, et al. Proc Natl Acad Sci USA. 2014;111:155–160. doi: 10.1073/pnas.1213773111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martin B, Pallen CJ, Wang JH, Graves DJ. J Biol Chem. 1985;260:14932–14937. [PubMed] [Google Scholar]; Mol Cell. 2013;50:43–55. doi: 10.1016/j.molcel.2013.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonnekoh B, Wevers A, Jugert F, Merk H, Mahrle G. Arch Dermatol Res. 1989;281:487–490. doi: 10.1007/BF00510085. [DOI] [PubMed] [Google Scholar]
- 21.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim Y, et al. Proc Natl Acad Sci USA. 2004;101:2082–2087. doi: 10.1073/pnas.0308617100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurlawalla-Martinez C, Stiles B, Wang Y, Devaskar SU, Kahn BB, Wu H. Mol Cell Biol. 2005;25:2498–2510. doi: 10.1128/MCB.25.6.2498-2510.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wijesekara N, Konrad D, Eweida M, Jefferies C, Liadis N, Giacca A, et al. Mol Cell Biol. 2005;25:1135–1145. doi: 10.1128/MCB.25.3.1135-1145.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]