

Abstract
The objective of this study is to assess the relationship between inflammatory disease in granulomatosis with polyangiitis (GPA, Wegener’s) with the development of subclinical atherosclerosis.
46 adult patients with GPA were enrolled. Disease status was measured by Birmingham Vasculitis Assessment Scores as modified for GPA (BVAS-WG), Vasculitis Damage Index (VDI), disease duration and number of relapses. Classic atherosclerotic risk factors, platelet aggregation responses and circulating microparticles (MP) levels were recorded. All patients underwent carotid artery intima media thickness (IMT) measurement as outcome for subclinical atherosclerosis.
In univariate analyses, systolic and diastolic blood pressure, creatinine, and age were significantly associated with higher IMT [rho values: 0.37, 0.38, 0.35 and 0.054 respectively (p < 0.02 for all)]. In a multiple regression model, greater number of relapses, older age at onset of disease, and higher diastolic blood pressure were found to be associated with higher IMT (p values 0.003, <0.001 and 0.031 respectively). MP counts and platelet reactivity correlated well with disease activity in GPA. Furthermore, MP were found to activate vascular endothelial cells and platelets in vitro.
The cumulative burden of systemic inflammation in GPA correlated with development of subclinical atherosclerosis. The correlation with subclinical atherosclerosis could be due to glucocorticoid use and not the inflammatory process in GPA, giving the inherent bias that exits with the use of glucocorticoid with each relapse.
The findings of elevated levels of circulating leukocyte-derived MP and enhanced platelet reactivity during relapse suggest possible roles for MP and platelets in disease pathogenesis and support a growing literature that links inflammation, atherosclerosis, and platelet activation.
This hypothesis is further substantiated by our demonstration that MP isolated from plasma of GPA patients can activate platelets and vascular endothelial cells.
Key Indexing Terms: Atherosclerosis, Wegener, Granulomatosis with polyangiitis, Microparticles, endothelium, platelets
INTRODUCTION
Granulomatosis with polyangiitis (GPA) is characterized by necrotizing granulomatous inflammation and vasculitis of the small to medium sized blood vessels (1). Although the survival of patients with GPA has improved with immunosuppressive therapy, atherosclerosis has now emerged as a significant morbidity (2–4) that is independent of traditional cardiovascular risk factors. Coronary artery disease, stroke, and peripheral arterial occlusive disease occur more frequently in GPA compared to healthy controls when assessed by intima-media thickness (IMT) of the carotid artery (3, 5) and plaque burden in the carotid arteries, aorta, and/or femoral arteries (2). The link between GPA and atherosclerosis is not well characterized at a mechanistic level, but the association raises the possibility that persistently active vasculitis, or nonspecific inflammation, play a role in early atherosclerosis.
An increasing body of evidence has suggested that in the course of systemic inflammation cell-derived microparticles ( MP ), which are membrane bound vesicles that bud off normal cells including leukocytes, platelets, and vascular endothelial cells, during either activation or apoptosis, may be prognostic markers for thrombosis, atherosclerotic vascular disease, and systemic inflammation (6). Elevated levels of circulating MP have been shown to be associated with cardiovascular risk and are indicative of a poor clinical outcome (7). Circulating MP levels are increased in patients with cardiovascular risk factors (8, 9). Moreover, MP levels are demonstrated to be an independent marker of subclinical carotid atherosclerosis in asymptomatic subjects (8) and may be more valuable for mortality prognosis than usual biological markers of myocardial infarction (9). Circulating MP derived from endothelial cells also express markers of cellular injury, suggesting that levels of endothelial MP could reflect the degree of endothelial dysfunction (10). It has been demonstrated in vitro that statins impair monocyte and endothelial MP formation (11) perhaps contributing to their anti-atherosclerotic activity.
Several studies point to MP as effectors of vascular wall inflammation (12, 13). MP may contain pro-inflammatory mediators, such as IL1, and can interact directly with activated vascular endothelial cells, triggering monocyte arrest in inflamed and atherosclerotic endothelium, suggesting a novel mechanism for platelet-dependent monocyte recruitment in inflammation and atherosclerosis (14). In rheumatic conditions such as rheumatoid arthritis (15, 16), systemic lupus erythematosus (17), systemic vasculitides (18), and specifically in GPA (19), MP levels potentially served as important markers of disease activity. The strong evidence that MP are involved in the pathogenesis of atherosclerosis and that circulating MP levels are elevated in patients with GPA, along with the lack of association of traditional risk factors with accelerated atherosclerosis in GPA, led to our pilot investigation of the role MP in the accelerated atherosclerosis seen in this disease.
METHODS
Subject recruitment
Eligible consenting adult patients, age ≥ 18 years, who met the classification criteria for GPA (20) were enrolled from the Cleveland Clinic Center for Vasculitis Care and Research. Control populations included volunteers from patients’ acquaintances as well as other healthy controls (total number of controls is 28). Exclusion criteria included history of coronary heart disease, heart failure, stroke, peripheral vascular diseases, acute or chronic infection within the past 3 months, use of NSAID or aspirin within one week, a diagnosis of another chronic inflammatory or autoimmune disease, myeloproliferative diseases, active malignancy, venous thromboembolic disease, or known thrombophilia. Data pertinent to atherosclerotic risk factors and other co-morbidities were collected at enrollment, as were markers of disease chronicity (GPA disease duration, number of relapses over time of observation). Further, the extent of organ damage secondary to GPA was quantified by the Vasculitis Damage Index (VDI). Disease activity was determined by the Birmingham Vasculitis Assessment Scores as modified for GPA (BVAS-WG) (21). Using this instrument, disease remission is defined as a BVAS-WG score=0. In our Center, the BVAS-WG and VDI are collected and recorded on all patients with GPA, at every visit. Further, we collected the number of disease relapses for every patient from their initial diagnosis to the date of their enrollment. Data on current treatment and use of immunosuppressive agents in patients with GPA was also recorded.
The study population included men and women regardless of ethnicities and races, in proportion to the prevalence rates of those variables in GPA. The research study was carried out according to The Code of Ethics of the World Medical Association (Declaration of Helsinki. The study was approved by the Institutional Review Board (IRB) and informed consent was obtained. Normal healthy platelet donors were recruited through the NIH-funded Specialized Centers of Clinically Oriented Research in hemostasis and thrombosis (SCCOR) at Cleveland Clinic.
Intima-media thickness (IMT)
The common carotid (CCA) segments of the carotid arteries bilaterally were scanned with a duplex ultrasound to quantify IMT as a biomarker for sub-clinical atherosclerosis (See supplementary file for details). For statistical analysis the higher of the left and right mean IMT was used. IMT were performed in patients with GPA only.
MP measurement and characterization of cellular origin
Blood was obtained in sodium citrate and processed within 90 min at room temperature. MP counts were performed from fresh samples across the whole study. Samples were centrifuged for 10 min at 150xg and the supernatant, designated platelet-rich plasma (PRP, was then centrifuged at 13000xg for 2 min to pellet the platelets. The resulting supernatant, designated platelet-poor plasma (PPP) contained MP. A 100μl aliquot of PPP was used to assess MP by flow cytometry and the remainder was centrifuged at 17000xg for 45 min to pellet MP. The MP pellet was re-suspended in modified Tyrode’s buffer containing 0.35% BSA and stored at −70°C. MP were analyzed by flow cytometry [Becton-Dickinson LSRI (with upgraded light scatter detectors present on the machine) BD Biosciences] assessing forward and side scatter, and fluorescence using FITC-conjugated annexin V (BD Biosciences) and antibodies specific for endothelial cells ( PE-conjugated CD105 from eBioscience and PE-conjugated CD144 from BD Biosciences ), platelets (FITC-conjugated CD41 from BD Biosciences), leukocytes (PE-conjugated CD18 from BD Biosciences), neutrophils (PE-conjugated CD16b from BD Biosciences) and monocytes (FITC-conjugated CD14 from BD Biosciences). Each marker was counted separately (see supplementary data for gating strategy).
Platelet activation studies
Platelets in PRP were counted in a Z2 particle counter (Coulter) and platelet number was adjusted to 2×108/ml. Platelet aggregation was assessed turbidometrically with a dual channel instrument using graded doses of ADP from 1–20μM and collagen (0.5–10 μg/ml) under constant stirring conditions. The change in light transmission after addition of agonist was recorded and expressed as a percentage of deflection compared to PPP.
In some studies platelet activation was assessed by flow cytometry using PE-conjugated PAC-1 IgM, a monoclonal antibody specific for the activated form of the α2bβ3 integrin. In these studies platelets were stained with calcein, an intracellular green fluorophore and then pre-incubated with GPA patient-derived MP with a ratio of 10:1 MP/platelet, as described previously by our group (31), prior to addition of 1μM ADP.
MP - endothelial cell interaction
Human dermal microvascular endothelial cells (huDMVEC, Clonetics CAMBREX) were cultured in EGm-2MV media from Lonza. The media was changed to low serum EGm-2 for 24h prior to the experiments. MP from GPA patients were then added at various ratios of MP to huDMVEC and incubated for timed periods. Cells were then detached using trypsin/EDTA, washed in PBS, and re-suspended in PBS containing 5% fetal bovine serum. To detect endothelial cell activation, cells were incubated with anti-ICAM-1 IgG or isotope-matched control IgG and bound antibody quantified by flow cytometry.
Statistical analysis
Categorical variables were summarized using frequency, distributions and contigency tables, and continuous variables were summarized using mean and standard deviation. Univariate correlations among variables were assessed using 2-sample t-test, Wilcoxon-rank-sum test, Chi-square test, or Spearman correlation. A multivariable linear regression model with backward selection procedure was used to identify variables associated with IMT. All variables were initially included in the multivariable regression model and the variables with p >0.05 were dropped during the model selection process until all remaining variables had p ≤ 0.05. All first and second order interactions among the variables retained in the final model were checked. No significant first or second order interaction was found to be present among the retained variables. Bootstrap bagging methods (22) were used to verify the final model. Specifically, 1000 datasets with the same sample size as the original dataset were generated from the original dataset by random sampling with replacement. Variable selections were performed for each of the 1000 bootstrap samples. Selection percentage of each variable was calculated as the frequency of the variable was selected. Unless otherwise defined, the significance level was set at 0.05 for all analyses. All analyses were performed using SAS 9.2 software.
RESULTS
Number of disease relapses, diastolic blood pressure, and age at disease onset independently correlated with IMT in patients with GPA
A total of 46 patients were included in this study, with 38 patients in remission and 8 patients with active disease as defined by the BVAS-WG. The characteristics and demographics of the cohort are summarized in Table 1. Disease duration of patients was 87± 48 months. IMT was measured on patients with GPA only. The mean IMT of GPA patients was 0.72 (sd: 0.14 and range 0.50–1.09). IMT results were matched to age, sex and race historical control based on the ARIC and Bogalusa studies (23, 24). The ARIC and Bogalusa data were used to determine IMT percentile for ages>40 years, and ages 40 years and younger, respectively. IMT percentile of 75 % and above was considered abnormal. Sixty three percent (29/46) of the patients with GPA had an abnormal IMT.
Table I.
Demographic and disease status of the cohort
| Total N= 46 |
Relapse N= 8 BVAS-WG > 1 |
Remission N = 38 BVAS-WG =0 |
P value | |
|---|---|---|---|---|
| Mean Age (Year) | 55 ± 13 | 61 ±13 | 54 ±13 | 0.20 |
| Female (%) | 22 (49) | 5 (63) | 17 (45) | 0.45 |
| Mean creatinine (mg/dl) | 0.98 ± 0.27 | 1.19 ± 0.41 | 0.94 ± 0.22 | 0.17 |
| Mean VDI | 2.03 ± 1.98 | 3.00 ± 2.83 | 1.97 ± 1.97 | 0.70 |
| Mean Disease Duration (Month) | 87 ± 48 | 70 ± 65 | 90 ± 44 | 0.45 |
Abbreviations: BVAS-WG, Birmingham Vasculitis Assessment Scores as modified for GPA; GPA, granulomatosis with polyangiitis; VDI, vasculitis damage index
Univariate analyses revealed that GPA patients with higher IMT were more likely to have higher systolic and diastolic blood pressures, higher serum creatinine, and be older (Table 2). Presence of anti-neutrophil cytoplasmic antibodies (ANCA) did not correlate with IMT (p=0.77), however the number of patients without ANCA was small (8/46) and this could have affected the results. There was no association between IMT and disease damage as measured by VDI (Odds ratio 0.88, p = 0.48). There was no correlation between IMT and MP counts (rho −0.18, p 0.23) or platelet aggregation (rho 0.25, p 0.11).
Table II.
Correlation of IMT with atherosclerotic risk factors and disease variables
| Outcome | Variable | N | rho | 95%CI | P value |
|---|---|---|---|---|---|
| IMT | BMI | 44 | 0.19 | (−0.12,0.49) | 0.23 |
| IMT | Systolic blood pressure | 45 | 0.37 | (0.09,0.66) | 0.012 |
| IMT | Diastolic blood pressure | 45 | 0.38 | (0.09,0.66) | 0.011 |
| IMT | Creatinine | 45 | 0.35 | (0.06,0.64) | 0.019 |
| IMT | HDL | 45 | −0.21 | (−0.51,0.10) | 0.18 |
| IMT | LDL | 45 | −0.19 | (−0.50,0.11) | 0.2 |
| IMT | FBG | 45 | 0.12 | (−0.18,0.43) | 0.42 |
| IMT | Age at onset of disease | 45 | 0.54 | (0.28,0.80) | <0.001 |
| IMT | Disease Duration | 45 | 0.23 | (−0.07,0.53) | 0.13 |
| IMT | Azathioprine Duration | 45 | −0.24 | (−0.53,0.06) | 0.12 |
| IMT | Methotrexate Duration | 45 | 0.11 | (−0.20,0.41) | 0.48 |
| IMT | Cyclophosphamide Duration | 45 | −0.13 | (−0.43,0.18) | 0.41 |
| IMT | Cumulative Dose of cyclophosphamide | 45 | −0.15 | (−0.45,0.16) | 0.34 |
Abbreviations: BMI, bosy mass index; CI, confidence interval; FBG, Fasting blood glucose; HDL, high density lipoprotein; IMT, Intima-media thickness; LDL, low density lipoprotein; N, number; ρ, Spearman’s rank correlation coefficient. Bold indicates statistical significant values.
We also used multivariable regression model to examine disease related factors that correlate with IMT. All variables in table 2 were considered in the models and the number of relapses was added into the model, after adjusting for other significant factors. Variables with a p > 0.05 were dropped during the model selection process until all remaining variables had a p < 0.05. Independent correlates with IMT included larger number of disease relapses, older age at disease onset, and higher diastolic blood pressure (Table 3). All first and second order interactions among the three variables were not statistically significant. The overall p value for testing all first and second order interactions simultaneously is 0.43, which indicates no significant first or second order interaction among the three variables. Bootstrap bagging confirmed that larger number of disease relapses and older age at disease onset were reliable predictors of IMT, with both selected more than 50% of times out of 1000 bootstrap samples. However, the diastolic blood pressure didn’t appear as stable as the other two variables (selection percentage=39%).
Table III.
Multiple regression model of Independent risk factors of atherosclerosis in GPA
| Outcome | Parameter | Estimate (%95 CI) | P value |
|---|---|---|---|
| IMT | Relapse sum | 0.0119 (0.0001, 0.0236) | 0.003 |
| Age at onset of disease | 0.005 (0.003,0.008) | <0.001 | |
| Diastolic blood pressure | 0.0033 (0.0003,0.0064) | 0.031 |
Abbreviations: GPA, granulomatosis with polyangiitis; IMT: Intima-media thickness
MP levels were elevated in GPA relapse and correlated with platelet reactivity
Since we have demonstrated that the number of disease relapses was an independent risk factor for development of atherosclerosis in GPA, we analyzed possible mechanisms that could relate relapse to atherogenesis, focusing on circulating MP because of the increasingly recognized role that MP play in inflammation, thrombosis and atherosclerosis. We found that during the time of GPA relapse circulating levels of leukocyte-derived MP were significantly higher compared to those seen during disease remission (the endothelial-derived MP count did not differ between the relapse and the remission groups) (Figure 1-A). The median number of CD18 expressing MP/ml in the relapse group (n =8) and remission group (n=38) were 55,769 and 13,671 respectively (p = 0.002) and the median number of CD14 expressing MP/ml were 103,509 and 54,033 respectively (p < 0.05). The median number of CD16 expressing MP/ ml were 39,494 and 12,152 respectively (p = 0.006). These data suggest that MP could play a significant role during a disease relapse. Of note, the remission and relapse groups did not consist of the same patients; however there was no difference among the remission and relapse groups in regard of age, sex, disease duration, disease damage or creatinine level. The medians of the above variables were compared with 21 age- and gender- matched healthy population. The median number of CD14, CD 16 and CD18 expressing MP/ ml was 45,570, 12, 369 and 10,199 respectively. There was no difference among the remission group vs. the controls. CD 18 MP count was significantly higher in the relapse group vs. the control group (p= 0.009).
Figure 1.

Microparticle counts (A) and platelet aggregation (B) correlate with disease activity in granulomatosis with polyangiitis.
Platelet reactivity (Figure 1-B), assessed as aggregation response to graded doses of ADP, was significantly higher during disease relapse compared to during remission. Further analysis revealed that platelet aggregation correlated well with circulating levels of leukocyte-derived MP (r = 43% p = 0.006 for CD14 MP, r = 47% p = 0.002 for CD16 MP and r = 64% p < 0.001 for CD18 MP) consistent with a possible pathogenetic link between MP and platelet reactivity. Indeed, we found that MP isolated from plasma of GPA patients (in remission) enhanced reactivity of platelets from healthy donors. When platelets from healthy donors were pre-incubated with patient-derived MP and then exposed to low doses of ADP (1μM), surface expression of activated α2bβ3 integrin, assessed by flow cytometry using PAC-1 antibody, was significantly enhanced (Figure 2).
Figure 2.
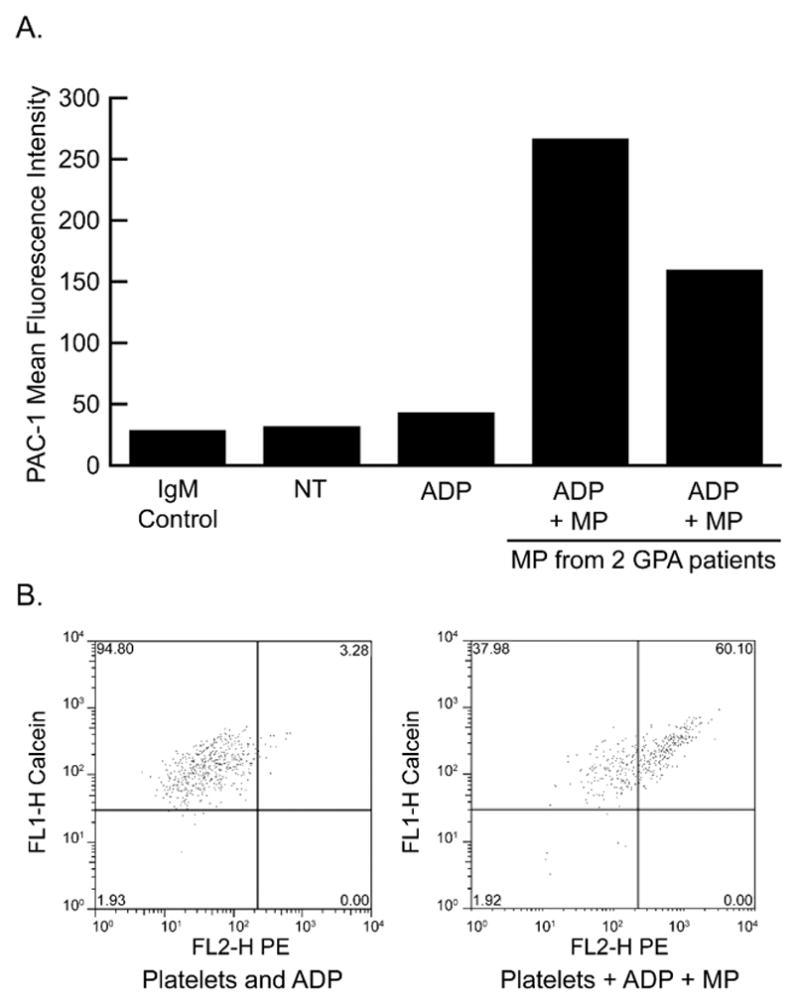
MPs from GPA patients increase platelet reactivity to ADP. (A) Calcein-labeled platelets were exposed to GPA patient-derived MPs at an MP-to-platelet ratio of 10:1 and then treated with ADP (1uM) prior to assessment of activation by PAC-1 immunostaining. Mean fluorescence intensity is shown using 2 patient samples. (B) Flow cytometry scattergrams from the 2 patient sample studies showing PAC-1 fluorescence on X axis and calcein fluorescence on Y-axis. ADP, adenosine diphosphate; FL2-H, fluorescence label; GPA, granulomatosis with polyangiitis; IgM, immunoglobulin M; MP, microparticle; NT, non reated; PAC-1, platelet activation clone.
Our data suggest that the inflammatory burst that occurs during each GPA relapse may have a direct role in the pathogenesis of atherosclerosis. Given that MP levels were elevated during relapse and correlated with platelet reactivity we next determined whether MP could contribute to vascular endothelial cell activation. As shown in Figure 3-A, GPA patient (in relapse) -derived MP when incubated for 4h with huDMVEC induced surface expression of ICAM-1. MP-poor PPP, from the same sample, was used as a control and did not influence ICAM-1 expression. ICAM-1 induction by MP was blocked by cycloheximide (Figure 3-B) showing that new protein synthesis was required and that the ICAM-1 was not transferred to the EC by the MP.
Figure 3.
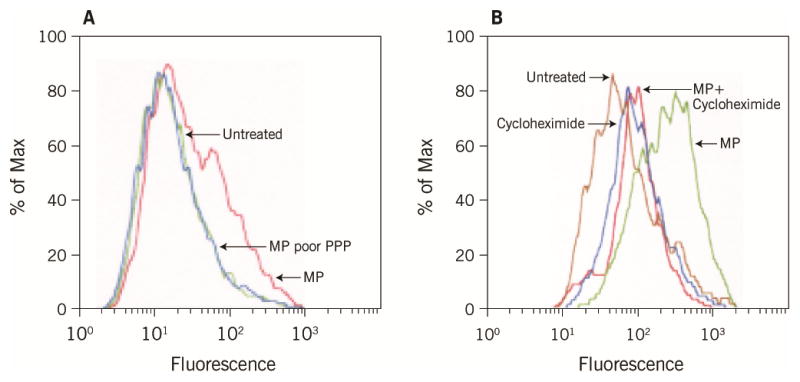
MPs induce ICAM expression on huDMVECs. (A) Flow cytometry histogram showing ICAM-1 immunostaining of huDMVEC after incubation with MP from GPA patient or MP-depleted plasma from the same patient. Untreated cells are shown as a control. (B) Cells treated as in panel A were pre-incubated with cycloheximide prior to incubation with MP. GPA, granulomatosis with polyangiitis; huDMVECs, human dermal microvascular endothelial cells; ICAM, intercellular adhesion molecule; MPs, microparticles; PPP, platelet-poor plasma.
Interestingly, MP isolated from control subjects or cultured THP-1 cells (see supplementary data) exerted a similar activating effect on huDMVEC. These data show that induction of ICAM-1 by MP does not depend on the cell of origin or method of MP generation, and suggest that it is the level of circulating MP that is most important in determining outcome.
DISCUSSION
A major novel finding from our study is that carotid IMT correlated well with the total number of disease relapses rather than the duration of disease. This suggests that in remitting relapsing inflammatory disorders such as GPA, it is the cumulative duration of active inflammatory insult that contributes most to the risk of development of atherosclerosis. Interestingly, the creatinine level was not associated with a higher number of IMT in the multivariate analysis, which indicate that variables other than creatinine levels contributed to the development of subclinical atherosclerosis in this cohort or it could be because our cohort had a mean creatinine level within the normal range,
Abundant experimental studies in humans and in animal and in vitro models have identified inflammation and oxidant stress as critical components of the atherosclerotic process. This is supported by various large scale prospective epidemiological studies showing that increased serum levels of inflammatory markers, such as C-reactive peptide and myeloperoxidase, are predictive of future cardiovascular disease (25). Our finding of elevated levels of leukocyte-derived MP in the plasma of GPA patients during disease flares is consistent with the published data (18, 26) and with the concept that relapses are associated with “bursts” of a systemic pro-inflammatory state. Furthermore, at a mechanistic level, we showed that MP isolated from patients with GPA have the capacity to activate vascular endothelial cells and platelets, cell types that are critical mediators of atherosclerosis. This is the first report to our knowledge showing that MP isolated from human patient plasma can activate vascular endothelial cells, although several studies have studied MP generated in vitro and demonstrated that they stimulate expression of the adhesion molecules in endothelial cells (27–29).
MP may have pro-inflammatory activities related to diverse mechanisms. MP can fuse with or be internalized by cells, including phagocytes, and in so doing may render cells susceptible to new stimuli by mediating intercellular transfer of receptors from their cell of origin to the plasma membranes of target cells, or they can “deliver” pro-inflammatory cytokines, chemokines, eicosanoids and microRNA to target cells. MP express cell surface molecules from their cells of origin can then act as ligands for pro-inflammatory receptors on target cells (28, 29). A recent study, for example, showed that MP isolated from synovial fluid of rheumatoid arthritis patients were capable of stimulating production of cytokines and chemokines, including IL-6 and IL-8 by fibroblast-like synoviocytes (30). Our laboratory showed that MP bind to platelets via a specific receptor, CD36, and thereby initiate an intracellular signaling cascade that enhances platelet reactivity, linking MP to thrombosis as well as inflammation (31). MP also represent important thrombotic protagonists because of the ability of their abundant surface phosphatidylserine (PS) to participate in the assembly and activation of coagulation enzyme complexes and because some MP subsets express tissue factor (TF) procoagulant. Recent data by Eleftheriou et al provided an important role of cell-derived microparticles in thrombosis complicating systemic vasculitis of the young.(32) In the current study we showed that platelet reactivity, assessed by aggregation response to graded doses of ADP, correlated well with GPA disease activity. This finding is consistent with numerous studies linking platelets to the pathogenesis of chronic inflammatory diseases. In rheumatoid arthritis, for example, platelets were found to amplify inflammation via collagen-dependent MP production (30) and in an autoimmune arthritis model (33) platelets were found to contribute actively to synovitis via production of proinflammatory prostacyclin. Recently, levels of circulating proteins secreted by activated platelets were found to correlate with GPA disease activity (34).
Moreover, platelets were found to contribute to the inflammatory response through production of MP that promote endothelial cell activation (35). Moreover, the role of MP in the pathogenesis of systemic vasculitides has been recently demonstrated (36). In this study, a role of neutrophil MP in the pathogenesis of ANCA-associated vasculitis was shown; polyclonal ANCAs isolated from patients and chimeric proteinase3-ANCA were able to induce the release of MP from primed neutrophils. These MP bound endothelial cells via a CD18-mediated mechanism and induced an increase in endothelial intercellular adhesion molecule-1 expression, production of endothelial reactive oxygen species, and release of endothelial IL-6 and IL-8. Interestingly, removal of the neutrophil MP abolished MP-mediated endothelial activation. In addition, these MP promoted thrombin generation (36).
Our study also confirmed previously published data from the European Vasculitis Study Group (EUVAS) trials and the Wegener’s Granulomatosis Etanercept Trial (WGET) cohort (37) showing that diastolic blood pressure and older age were independent risk factors in GPA patients for development of atherosclerosis. Our cohort was relatively small and therefore the study has potential limitations. Unlike the EUVAS and WGET studies, we did not see a correlation of disease activity with ANCA, but this could be related to the low number of subjects in our cohort who were ANCA negative.
The use of glucocorticoids at relapse and the inability to accurately assess their dosage in our cohort are potential confounding variables that could be contributing to subclinical atherosclerosis. The correlation with subclinical atherosclerosis could be due to glucocorticoid use and not the inflammatory process in GPA, giving the inherent bias that exits with the use of glucocorticoid with each relapse. We acknowledge this limitation of the study and with the cross sectional design of the study, adjusting for this bias would be very challenging. However, studies in other inflammatory diseases have not shown significant correlations between use of immunosuppressive medications and atherosclerosis (38). In our cohort we did not detect any correlation between use of immunosuppressive medications other than glucocorticoids and subclinical atherosclerosis (Table-1).
We acknowledge that this study has several limitations; first, the burden of inflammation, in this cohort, was measured by the sum of relapses which may not reflect an accurate measurement of the degree of inflammation. Multiple serial IMT measurements on the same patients and correlation with the severity and the number of relapses will be the ideal scenario; however, this requires a more challenging design and data from current study will be still considered essential to obtain as a preliminary step before designing a larger study.
Second, the data presented on the interaction of MP and endothelial cells are very preliminary and only included few patients; moreover it will be interesting to know whether MP derived from patients with inactive disease has the same effect on the endothelial cells as MP derived with active disease. Although we have demonstrated that MP derived from healthy controls does activate endothelial cells, but further experiments are needed. Evaluation with larger numbers of samples and experiments that include MP derived from patients with different disease activity and inflammatory diseases will be the goal of a future study.
In conclusion, we have demonstrated that the cumulative number of relapses in patients with GPA correlated with development of subclinical atherosclerosis. The findings of elevated levels of circulating leukocyte-derived MP and enhanced platelet reactivity during relapse suggest possible roles for MP and platelets in disease pathogenesis and support a growing literature linking inflammation, atherosclerosis, and platelet activation. This hypothesis is further substantiated by our demonstration that MP isolated from plasma of GPA patients can activate platelets and vascular endothelial cells. This data is considered preliminary giving the limitations of our study and additional studies with larger number of patients are needed to validate our findings in GPA and other inflammatory conditions.
Supplementary Material
Abbreviations
- GPA
Granulomatosis with polyangiitis
- IMT
intima-media thickness
- MP
microparticles
- CCA
common carotid
- VDI
Vasculitis Damage Index
- BVAS-WG
Birmingham Vasculitis Assessment Scores as modified for GPA
Footnotes
Potential conflicts of interest Acknowledgements: All authors have read the journal’s authorship agreement and policy on disclosure of potential conflicts of interest and disclosed no financial or personal relationship with organizations that could potentially be perceived as influencing the described research interest.
Authorship agreement: The article has been reviewed by and approved by all named authors
Disclosure of funding source: this study was supported by a grant from the American Heart Association [0885014D to Rula Hajj-Ali]; and by the National Institute of Health SCCOR program [P50 HL81011to Roy Silverstein].
Editorial support: None
Contributor Information
J Major, Email: majorj1@ccf.org.
CA Langford, Email: langfoc@ccf.org.
GS Hoffman, Email: hoffmag@ccf.og.
T Clark, Email: clarkt@ccf.org.
L Zhang, Email: Li.Zhang@ucsf.edu.
Z Sun, Email: sunz@ccf.org.
RL Silverstein, Email: rsilverstein@mcw.edu.
References
- 1.Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Annals of internal medicine. 1992 Mar 15;116(6):488–98. doi: 10.7326/0003-4819-116-6-488. [DOI] [PubMed] [Google Scholar]
- 2.Chironi G, Pagnoux C, Simon A, et al. Increased prevalence of subclinical atherosclerosis in patients with small-vessel vasculitis. Heart (British Cardiac Society) 2007 Jan;93(1):96–9. doi: 10.1136/hrt.2006.088443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Leeuw K, Sanders JS, Stegeman C, et al. Accelerated atherosclerosis in patients with Wegener’s granulomatosis. Annals of the rheumatic diseases. 2005 May;64(5):753–9. doi: 10.1136/ard.2004.029033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swets BP, Brouwer DA, Tervaert JW. Patients with systemic vasculitis have increased levels of autoantibodies against oxidized LDL. Clinical and experimental immunology. 2001 Apr;124(1):163–7. doi: 10.1046/j.1365-2249.2001.01488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.ZAENKER M. Accelerated atherosclerosis in Wegener’s granulomatosis: a sonographic case-control study on intima media thickness. Arthritis Rheum. 2002:S185. [Google Scholar]
- 6.Boulanger CM, Amabile N, Tedgui A. Circulating microparticles: a potential prognostic marker for atherosclerotic vascular disease. Hypertension. 2006 Aug;48(2):180–6. doi: 10.1161/01.HYP.0000231507.00962.b5. [DOI] [PubMed] [Google Scholar]
- 7.Morel O, Ohlmann P, Morel N, et al. Microparticles and cardiovascular disease. Archives des maladies du coeur et des vaisseaux. 2005 Mar;98(3):226–35. [PubMed] [Google Scholar]
- 8.Chironi G, Simon A, Hugel B, et al. Circulating leukocyte-derived microparticles predict subclinical atherosclerosis burden in asymptomatic subjects. Arteriosclerosis, thrombosis, and vascular biology. 2006 Dec;26(12):2775–80. doi: 10.1161/01.ATV.0000249639.36915.04. [DOI] [PubMed] [Google Scholar]
- 9.Mallat Z, Benamer H, Hugel B, et al. Elevated levels of shed membrane microparticles with procoagulant potential in the peripheral circulating blood of patients with acute coronary syndromes. irculation. 2000 Feb 29;101(8):841–3. doi: 10.1161/01.cir.101.8.841. [DOI] [PubMed] [Google Scholar]
- 10.Horstman LL, Jy W, Jimenez JJ, et al. Endothelial microparticles as markers of endothelial dysfunction. Front Biosci. 2004 May 1;9:1118–35. doi: 10.2741/1270. [DOI] [PubMed] [Google Scholar]
- 11.Tramontano AF, O’Leary J, Black AD, et al. Statin decreases endothelial microparticle release from human coronary artery endothelial cells: implication for the Rho-kinase pathway. Biochemical and biophysical research communications. 2004 Jul 16;320(1):34–8. doi: 10.1016/j.bbrc.2004.05.127. [DOI] [PubMed] [Google Scholar]
- 12.Morel O, Hugel B, Jesel L, et al. Procoagulant membranous microparticles and atherothrombotic complications in diabetics. Archives des maladies du coeur et des vaisseaux. 2004 Oct;97(10):1006–12. [PubMed] [Google Scholar]
- 13.VanWijk MJ, VanBavel E, Sturk A, et al. Microparticles in cardiovascular diseases. Cardiovascular research. 2003 Aug 1;59(2):277–87. doi: 10.1016/s0008-6363(03)00367-5. [DOI] [PubMed] [Google Scholar]
- 14.Mause SF, von Hundelshausen P, Zernecke A, et al. Platelet microparticles: a transcellular delivery system for RANTES promoting monocyte recruitment on endothelium. Arteriosclerosis, thrombosis, and vascular biology. 2005 Jul;25(7):1512–8. doi: 10.1161/01.ATV.0000170133.43608.37. [DOI] [PubMed] [Google Scholar]
- 15.Berckmans RJ, Nieuwland R, Kraan MC, et al. Synovial microparticles from arthritic patients modulate chemokine and cytokine release by synoviocytes. Arthritis research & therapy. 2005;7(3):R536–44. doi: 10.1186/ar1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knijff-Dutmer EA, Koerts J, Nieuwland R, et al. Elevated levels of platelet microparticles are associated with disease activity in rheumatoid arthritis. Arthritis and rheumatism. 2002 Jun;46(6):1498–503. doi: 10.1002/art.10312. [DOI] [PubMed] [Google Scholar]
- 17.Pereira J, Alfaro G, Goycoolea M, et al. Circulating platelet-derived microparticles in systemic lupus erythematosus. Association with increased thrombin generation and procoagulant state. Thrombosis and haemostasis. 2006 Jan;95(1):94–9. [PubMed] [Google Scholar]
- 18.Brogan PA, Shah V, Brachet C, et al. Endothelial and platelet microparticles in vasculitis of the young. Arthritis and rheumatism. 2004 Mar;50(3):927–36. doi: 10.1002/art.20199. [DOI] [PubMed] [Google Scholar]
- 19.Erdbruegger U, Grossheim M, Hertel B, et al. Diagnostic role of endothelial microparticles in vasculitis. Rheumatology (Oxford, England) 2008 Dec;47(12):1820–5. doi: 10.1093/rheumatology/ken373. [DOI] [PubMed] [Google Scholar]
- 20.Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990 Aug;33(8):1101–7. doi: 10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 21.Stone JH, Hoffman GS, Merkel PA, et al. A disease-specific activity index for Wegener’s granulomatosis: modification of the Birmingham Vasculitis Activity Score. International Network for the Study of the Systemic Vasculitides (INSSYS) Arthritis and rheumatism. 2001 Apr;44(4):912–20. doi: 10.1002/1529-0131(200104)44:4<912::AID-ANR148>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 22.Breiman L. Bagging predictors. Machine Learning. 1996;24:123–40. [Google Scholar]
- 23.Howard G, Sharrett AR, Heiss G, et al. Carotid artery intimal-medial thickness distribution in general populations as evaluated by B-mode ultrasound. ARIC Investigators. Stroke; a journal of cerebral circulation. 1993 Sep;24(9):1297–304. doi: 10.1161/01.str.24.9.1297. [DOI] [PubMed] [Google Scholar]
- 24.Stein JH, Douglas PS, Srinivasan SR, et al. Distribution and cross-sectional age-related increases of carotid artery intima-media thickness in young adults: the Bogalusa Heart Study. Stroke; a journal of cerebral circulation. 2004 Dec;35(12):2782–7. doi: 10.1161/01.STR.0000147719.27237.14. [DOI] [PubMed] [Google Scholar]
- 25.Albert MA, Glynn RJ, Ridker PM. Plasma concentration of C-reactive protein and the calculated Framingham Coronary Heart Disease Risk Score. Circulation. 2003 Jul 15;108(2):161–5. doi: 10.1161/01.CIR.0000080289.72166.CF. [DOI] [PubMed] [Google Scholar]
- 26.Erdbrugger U, Grossheim M, Hertel B, et al. Endothelial microparticles are elevated in ANCA associated vasculits (AAV) Clinical and Experimental Rheumatology. 2007;25(suppl 44 1):S-86. [Google Scholar]
- 27.Curtis AM, Wilkinson PF, Gui M, et al. p38 mitogen-activated protein kinase targets the production of proinflammatory endothelial microparticles. J Thromb Haemost. 2009 Apr;7(4):701–9. doi: 10.1111/j.1538-7836.2009.03304.x. [DOI] [PubMed] [Google Scholar]
- 28.Forlow SB, McEver RP, Nollert MU. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood. 2000 Feb 15;95(4):1317–23. [PubMed] [Google Scholar]
- 29.Mack M, Kleinschmidt A, Bruhl H, et al. Transfer of the chemokine receptor CCR5 between cells by membrane-derived microparticles: a mechanism for cellular human immunodeficiency virus 1 infection. Nature medicine. 2000 Jul;6(7):769–75. doi: 10.1038/77498. [DOI] [PubMed] [Google Scholar]
- 30.Boilard E, Nigrovic PA, Larabee K, et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010 Jan 29;327(5965):580–3. doi: 10.1126/science.1181928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghosh A, Li W, Febbraio M, et al. Platelet CD36 mediates interactions with endothelial cell-derived microparticles and contributes to thrombosis in mice. The Journal of clinical investigation. 2008;118(5):1934–43. doi: 10.1172/JCI34904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Eleftheriou D, Hong Y, Klein NJ, et al. Thromboembolic disease in systemic vasculitis is associated with enhanced microparticle-mediated thrombin generation. Journal of thrombosis and haemostasis : JTH. 2011;9(9):1864–7. doi: 10.1111/j.1538-7836.2011.04434.x. [DOI] [PubMed] [Google Scholar]
- 33.Boilard E, Larabee K, Shnayder R, et al. Platelets participate in synovitis via Cox-1-dependent synthesis of prostacyclin independently of microparticle generation. J Immunol. 2011 Apr 1;186(7):4361–6. doi: 10.4049/jimmunol.1002857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomasson G, Lavalley M, Tanriverdi K, et al. Relationship between markers of platelet activation and inflammation with disease activity in Wegener’s granulomatosis. J Rheumatol. 2011 Jun;38(6):1048–54. doi: 10.3899/jrheum.100735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1beta-rich microparticles. J Immunol. 2011 May 1;186(9):5489–96. doi: 10.4049/jimmunol.1001623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hong Y, Eleftheriou D, Hussain AAK, et al. Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. Journal of the American Society of Nephrology : JASN. 2012;23(1):49–62. doi: 10.1681/ASN.2011030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011 Apr;63(4):588–96. doi: 10.1002/acr.20433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Roman MJ, Shanker BA, Davis A, et al. Prevalence and correlates of accelerated atherosclerosis in systemic lupus erythematosus. The New England journal of medicine. 2003 Dec 18;349(25):2399–406. doi: 10.1056/NEJMoa035471. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.