

Abstract
Drug addiction is a complex condition of compulsive drug use that results in devastating physical and social consequences. Drosophila melanogaster has recently emerged as a valuable genetic model for investigating the mechanisms of addiction. Drug tolerance is a measurable endophenotype of addiction that can be easily generated and detected in animal models. The counter-adaptive theory for drug dependence postulates that the homeostatic adaptations that produce drug tolerance become counter-adaptive after drug clearance, resulting in symptoms of dependence. In flies, a single sedation with ethanol or with an organic solvent anesthetic (benzyl alcohol) induces functional tolerance, an adaptation of the nervous system that reduces the effect of these neural depressants. Here we review the role of the BK channel gene (slo) and genes that encode other synaptic proteins in the process of producing functional tolerance. These proteins are predicted to be part of an orchestrated response that involves specific interactions across a highly complex synaptic protein network. The response of the slo gene to drug exposure and the consequence of induced slo expression fit nicely the tenets of the counter-adaptive theory for drug tolerance and dependence. Induction of slo expression represents an adaptive process that generates tolerance because it enhances neuronal excitability, which counters the sedative effects of the drugs. After drug clearance, however, the increase in slo expression leads to an allostatic withdrawal state that is characterized by an increase in the susceptibility for seizure. Together, these results demonstrate a common origin for development of drug tolerance and withdrawal hyperexcitability in Drosophila.
I. Introduction
Drug addiction is a complex neurobiological condition that is characterized by compulsive and escalating drug use in spite of the social and physical harm that it causes. While addiction is a direct product of repeated drug use, its development and expression are strongly influenced by a number of genetic, psychosocial, and environmental factors. Historically, addiction has been defined in psychological terms with an emphasis placed on the motivational and behavioral aspects of the symptoms. More recently, it has begun to be defined in physiological terms as adaptations to the effects of a drug (Littleton and Little, 1994; Koob and Bloom, 1988). These adaptations persist following drug clearance and are thought to lead to withdrawal phenotypes. The diagnosis of addiction or alcoholism in humans includes negative changes in social behavior and status, which complicates its modeling in experimental systems. While the constellation of symptoms that represent the addicted state has not been captured in any model organism, the endophenotypes of addiction do lend themselves to study. An endophenotype of addiction is a drug response that has a clear genetic or physiological basis. It is believed that the molecular mechanisms that generate the endophenotype overlap with those that produce the addicted state. By describing the molecular basis of the endophenotype, we come to understand a portion of the molecular basis of the addicted state. A current challenge for neurobiological research is to identify and understand the neuroadaptive mechanisms that at the molecular, cellular, and systems levels generate the behavioral manifestations of addiction.
A. Tolerance, dependence, and withdrawal: A recipe for drug addiction
Two common manifestations of the physiological neuroadaptations to an addictive drug are the endophenotypes of tolerance and physical dependence. Tolerance refers to a diminishing effect of a drug as a consequence of prior drug exposure, while physical dependence is a product of the physical necessity for a drug. Withdrawal refers to the symptoms of dependence that appear during abstinence following drug use (Koob and Bloom, 1988).
The concept of physical drug dependence dates back to the early 1940's when C. K. Himmelsbach (1941) first described the morphine abstinence syndrome. Himmelsbach defined physical drug dependence as “a state in which certain physiological processes have become so conditioned to the effects of a drug that its presence is requisite to the maintenance of homeostasis.” In his studies with morphine users, Himmelsbach also noted that a state of dependency could only be diagnosed after removal of the drug in which patients exhibit characteristic withdrawal symptoms. These symptoms, he describes, “possibly reflect the degree to which mechanisms for the maintenance of homeostasis have been affected by the drug.” Under this definition, the manifestation of drug dependence is equated with the appearance of withdrawal symptoms.
This homeostatic view of dependence has since expanded into a comprehensive view of addiction and has become a theoretical framework for understanding the relationship between physical dependence and drug tolerance. In the homeostatic and redundancy theory of tolerance and dependence, W. R. Martin (1968) proposed that tolerance and dependence are part of the same phenomena. He postulated that the neuroadaptive mechanisms that counteract an effect of the drug to produce drug tolerance persist even after the drug has been cleared. Once uncovered by the extinction of the drug effects, the counter-adaptive changes translate into withdrawal symptoms—the physical manifestation of dependence (Figure 1).
Figure 1. Homeostatic counter-adaptive model of drug tolerance and dependence.

In the counter-adaptive model of drug tolerance and dependence, the carefully controlled balance between excitation and inhibition of neural activity in the brain (Initial state) is significantly altered by exposure to a psychoactive drug, creating a state of activity imbalance (Drug state). In an attempt to restore the balance of neural activity, homeostatic neuroadaptive mechanisms are activated (Tolerant state). After drug clearance, the homeostatic neuroadaptation is exposed, resulting in an opposing unbalanced state (Withdrawal state), leading back to the requirement of the drug to restore balance (Dependent state). Continuing use of the drug leads to further adaptation and an intensified requirement for the drug, resulting in a spiraling negative feed-forward cascade. Figure adapted from Littleton (1998).
The psychological drive for drug use is believed to have a direct connection to the physiological processes that underlie tolerance and dependence. The `opponent process theory' of acquired motivation provides a context for understanding the interrelationship between drug tolerance and dependence. This is an independent inception of the counter-adaptive theory couched in the syntax of psychology (Solomon and Corbit, 1974). This theory posits that homeostatic processes (the opponent processes) that counter the effects of a drug continue during abstinence to produce a state of dysregulation that can only be appeased by further drug use (Koob and Le Moal, 2006; Solomon, 1980; Littleton, 1998). Thus, the opponent process is responsible for both the tolerance to the pleasurable effects of a drug and the production of withdrawal symptoms that lead to a negative affective state and produce the feed-forward behavioral changes that promote addiction (Koob, 1996).
Animal models are crucial for understanding the neuroadaptive mechanisms responsible for the development of addiction. Unfortunately, the difficulty of measuring affective states in animals makes it impossible to generate complete models of the disease. However, tolerance and withdrawal are measurable endophenotypes that define both physiological and behavioral aspects of addiction and can be easily quantified in animal models.
B. Drug tolerance vs. drug resistance
In common parlance, tolerance and resistance are often considered to be synonymous. However, it is important to note that in a pharmacological context, drug tolerance differs significantly from drug resistance. Resistance is the term that describes the innate or baseline magnitude of drug responsivity. Therefore, resistance should be reported only for drug-naive animals. Drug tolerance is synonymous with inducible resistance triggered by prior drug exposure and implies a change from the pre-existing state. Sensitization on the other hand, is the opposite of tolerance. Sensitization describes a drug-induced reduction in resistance to an effect of the drug as a consequence of prior drug exposure. Both tolerance and sensitization are plastic phenomena.
A popular strategy for investigating the genetics of drug abuse is to measure the innate resistance of an organism to the effects of a particular drug. The rationale for this approach originated from the observation that humans with elevated behavioral resistance to ethanol have a fourfold increased probability of future alcoholism (Schuckit, 1994). Resistance is a risk factor for alcoholism because while these individuals are resistant to alcohol intoxication, they are not necessarily resistant to the addictive effects of the drug, and these individuals drink more to achieve the desired degree of intoxication. The mechanism of resistance is believed to arise at least in part from differences in the responsivity of the direct targets of ethanol (Mayfield et al., 2008). Thus, the identification of allelic variants or mutations that enhance resistance should identify medically relevant targets of ethanol. The identification of mutations that decrease resistance is also potentially valuable, with the caveat that mutations that merely reduce vigor might also decrease drug resistance. Tolerance is a drug-induced change that precedes and overlaps the genesis of addiction to many drugs. The changes that produce tolerance are believed to be a subset of those directly responsible for the addicted state. Therefore, a genetic study of tolerance offers a tractable approach for identifying genes that underlie the changes that cause addiction.
In mammals, there are two mechanistically distinct types of tolerance: metabolic tolerance and functional tolerance. Metabolic tolerance, also called pharmacokinetic tolerance, results from increased clearance of a drug and/or reduced drug uptake. These changes reduce the drug concentration or the time of exposure that cells experience. Tolerance caused by a reduction in the cellular response to a given concentration of drug is called functional tolerance or pharmacodynamic tolerance (Julien, 2004). For the nervous system, functional tolerance has been conceptualized in terms of homeostatic adaptations that attempt to restore more-normal neuronal excitability or activity and in terms of changes that resemble those involved in learning and memory (Koob and Le Moal, 2006; Hyman et al., 2006; Young and Goudie, 1995). It is not clear that these events would necessarily involve different mechanisms.
Tolerance is also categorized based on how it was induced. Acute tolerance refers to a decrease in sensitivity to the effect of a drug that develops during the course of a single drug exposure. The other extreme is chronic tolerance, which refers to a decrease in sensitivity that results from repetitive or sustained exposure to a drug. Finally, rapid tolerance describes the tolerance that exists after a single dose of the drug has been metabolically cleared (Young and Goudie, 1995).
C. Modeling addiction in Drosophila
Evidence gathered from familial studies of drug addiction points toward a significant genetic component associated with the risk for substance abuse (Kendler et al., 2003; Goldman et al., 2005). Identification of the genes behind the risk factors linked to addictive behaviors should uncover the mechanisms underlying the physiological neuroadaptions that ultimately lead to addiction. Because the ability to study genetics in humans is limited, the use of a genetically tractable organism is crucial for dissecting the endophenotypes of addiction. With an extraordinarily malleable genome and with a high degree of gene homology with mammalian genomes, the fruit fly Drosophila melanogaster has become a powerful genetic model system for studying addiction. After more than a century of genetic progress, Drosophila sports the most sophisticated genetic toolbox of any metazoan (Bellen et al., 2010; Duffy, 2002).
In spite of the perceived simplicity of the fly's anatomy and physiology, the fly maintains a surprising conservation of genetic architecture with its mammalian counterparts. Most important for the neurobiologist is that most mammalian genes associated with synaptic transmission and neural plasticity have homologs in Drosophila. These include the genes encoding ion channels, synaptic proteins, and the receptors for the major neurotransmitters used in mammals (Lloyd et al., 2000; Littleton and Ganetzky, 2000). At the behavioral level, Drosophila also show a remarkable degree of homology with higher organisms. Flies not only display basic behaviors—such as foraging, courtship, sleeping, and fighting—that are similar to those of mammals (Sokolowski, 1980; Spieth, 1974; Hendricks et al., 2000; Chen et al., 2002), but they can also remember, learn from experience, and modify their behavior in a goal-oriented manner (Quinn et al., 1974; Pick and Strauss, 2005). For more than two decades, the fruit fly has been the subject of a large number of studies modeling behavioral responses to a variety of potentially addictive psychoactive drugs. As a model organism, genetic studies with Drosophila have made substantial contributions toward the understanding of responses to psychostimulants, organic solvent anesthetics, and ethanol.
As in mammals, in flies psychostimulant drugs such as cocaine, nicotine, caffeine, and amphetamines suppress sleep and promote hyperactive behavior characterized by increased arousal and increased locomotion. Acute exposure to volatilized cocaine or nicotine produce clear stereotypic behaviors such as increased grooming, aberrant walking patterns, and bursts of hyperactivity that are characterized by fast uncontrolled movements. In mammals, the action of these drugs on dopaminergic signaling is central to their effects on arousal and reinforcement. In flies, these drugs enhance dopaminergic signaling, and this signaling has been shown to directly contribute to the drug-induced behavior (Rothenfluh and Heberlein, 2002; Wolf and Heberlein, 2003; Atkinson, 2009).
In flies, volatile organic solvent anesthetics, such as halothane and isoflurane, induce a state of sedation characterized by complete immobility and the loss of the ability to maintain postural control or respond to stimuli (Krishnan and Nash, 1990; Allada and Nash, 1993). Less-volatile organic solvents, such as ethanol and benzyl alcohol, have been shown to induce a biphasic response characterized by a brief initial increase in locomotor activity followed by sedation (Moore et al., 1998; Scholz et al., 2000; Parr et al., 2001; Cowmeadow et al., 2005; Ghezzi et al., 2004). Similarly, in humans, organic solvents elicit a wide range of behavioral and physiological responses that range from hyperactivity to profound sedation. Low or moderate exposure to volatile solvents can have excitatory effects, while higher levels of exposure produce sedation and anesthesia (Flanagan and Ives, 1994).
Volatile organic solvent anesthetics, inhalants, and ethanol are intriguing in that they are “dirty drugs” that alter the properties of many different molecules that participate in electrical signaling (Harris, 1999; Kopp Lugli et al., 2009). There is clear evidence that organic solvents have cellular and behavioral consequences related to those produced by more-conventional drugs of abuse. These consequences include modulating specific neurotransmitter receptor subtypes and triggering dopamine release in the nucleus accumbens to generate behavioral reinforcement (Lubman et al., 2008). In the rat hippocampus, volatile anesthetics have been shown to modulate both excitatory and inhibitory synaptic transmission culminating in a depression of synaptic transmission (Wakasugi et al., 1999; Harris et al., 1995). Gamma-aminobutyric acid (GABA) receptors are the best-established candidate for the drug target that mediates the sedative action of ethanol and volatile anesthetics in mammals (Harris, 1999; Beckstead et al., 2000). In flies, the inhibitory GABAB receptor has been implicated in the sedative effects of alcohol (Dzitoyeva et al., 2003), whereas the initial stimulating effects of ethanol have been linked to dopaminergic signaling (Bainton et al., 2000; Kong et al., 2010). The BK Ca2+-activated K+ channel has also been shown to be involved in organic solvent/ethanol responses in Caenorhabditis, Drosophila, and mammals (discussed below).
The conservation of drug-induced behaviors and targets from Drosophila to mammals is indeed encouraging. In light of these similarities, it is reasonable to expect that Drosophila genetics can be used to identify the genes that underlie previously undescribed drug responses and neuroadaptations that contribute to addiction. Ethanol tolerance was first described in Drosophila by Scholz and colleagues (2000). These authors show that adult flies acquire functional tolerance but not metabolic tolerance. Since then, several labs have used flies for investigating the mechanisms underlying this adaptation (reviewed in Atkinson, 2009 and Rodan and Rothenfluh, 2010). A focus of this review will be the role of the BK-type Ca2+-activated K+ channel and other synaptic proteins in the production of anesthetic and ethanol tolerance and dependence in Drosophila melanogaster.
II. Behavioral analysis of rapid drug tolerance in Drosophila
In flies, inhalation of benzyl alcohol vapor or ethanol vapor results in similar behavioral responses. At low, non-sedating doses, both drugs induce a gradual increase in locomotor activity accompanied by erratic movements. At higher doses that culminate in sedation, the hyperactive phase is observed only in the initial minutes of exposure and decays gradually, transitioning into a phase marked by low mobility levels, ataxia, and the loss of postural control. Eventually, flies will completely sedate, showing no signs of movement and remaining immobile on their backs. After the flies are moved to a drug-free vial, recovery from sedation is also gradual and can span between ~5 to 60 minutes, depending on the dose. While still on their backs, flies first begin to move their legs in a twitchy, spasmodic manner. These spasms last noticeably longer in ethanol treated flies. Subsequently, flies regain postural control and then pass through a period of standing immobility followed by ever more successful attempts to walk or climb (Cowmeadow et al., 2005; Ghezzi et al., 2004; Rothenfluh and Heberlein, 2002; Singh and Heberlein, 2000).
A. Benzyl alcohol tolerance
Our introduction to benzyl alcohol came while we were examining the slo gene for homeostatic responses to the insecticide imidacloprid. Imidacloprid is a partial nicotinic-acetylcholine receptor agonist (Matsuda et al., 1998). A sample of imidacloprid, dissolved in an undisclosed solvent, was provided to us by a drug company. We thought that the slo gene responded to this mixture in an important way but when we requested the name of the solvent we were denied by a rather protective company representative. A brief analysis by mass spectrometry and NMR revealed that the solvent was benzyl alcohol. To our surprise, it was this carrier solvent that produced most of the effects that originally intrigued us (Bohm, 2000). Benzyl alcohol proved to be a near-ideal organic solvent for sedating flies. We continued to study it to help describe the neuronal responses to organic solvent sedation.
Intentional exposure to benzyl alcohol in humans arises primarily from its use as an injectable local anesthetic (Fleisher and Ludwig, 2010) and because it is the first breakdown product of toluene (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 1999). Toluene is both an abused inhalant and a solvent whose commercial use is widespread (Dinwiddie, 1994). The metabolism of toluene probably represents the primary source of human exposure to benzyl alcohol.
Benzyl alcohol is an organic solvent anesthetic that is easy to administer and is extremely well tolerated by flies. Analysis of behavior showed that low doses of benzyl alcohol acted as a stimulant, while large doses produced sedation. Our standard animal for behavioral analysis is a female fly, aged 3–5 days old, with a Canton S wild-type genetic background. To sedate flies with benzyl alcohol, we coat vials with benzyl alcohol in acetone (usually 0.4% BA). Because of the difference in vapor pressures of the solvents, the acetone rapidly evaporates and leaves behind a fine coat of benzyl alcohol. Flies placed in the vial quickly succumb to the intoxicating effects of benzyl alcohol and are sedated within 10 to 15 minutes. Recently, we have used the vapor from small cups of benzyl alcohol positioned below the vials to sedate the flies. A vapor-permeable KimWipe® barrier is used to keep the flies from falling into the solvent. The emanating vapor sedates the flies with a similar time course and produces the same responses as the original method (Ghezzi et al., 2004; Ghezzi et al., 2010).
Because flies are negatively geotactic, they spend much of their time climbing the walls of their vial. During recovery from benzyl alcohol sedation, flies return to this pastime shortly after the return of the ability to stand. The return of wall climbing is a reliable indicator that flies have recovered from sedation with benzyl alcohol. This behavior lends itself to computer monitoring. We use a simple image subtraction program paired with consumer digital cameras to record the recovery from benzyl alcohol sedation (Ramazani et al., 2007).
In response to a single sedation, flies acquire functional tolerance that can be easily scored in an assay that measures the period of sedation. For tolerance experiments, flies are placed in the vials until sedated, moved to a benzyl alcohol–free vial to recover, and then incubated with food for 24 hours. Control animals are mock sedated in vials that were handled in the same manner but with the omission of the benzyl alcohol. The next day both groups are sedated simultaneously with benzyl alcohol vapor, switched to a fresh-air environment, and the duration of sedation is monitored. Flies recover more rapidly from their second benzyl alcohol sedation than from their first sedation; that is, they acquire tolerance to the drug. Examples of this behavior are shown in Figures 2 and 3.
Figure 2. Benzyl alcohol tolerance assay.

Shown are sequential pictures of two vials of flies taken at different time points of sedation and recovery from 0.4 % benzyl alcohol exposure. The vial on the left in every picture contains naïve flies, which have never been treated before; this is their first exposure (1st). The vial on the right contains flies that were previously sedated (24 hours earlier) with a similar dose of benzyl alcohol; this is their second exposure (2nd). The time point at which each picture was taken is indicated under each picture in minutes after start of the treatment. Time points from 1 to 9 minutes (white text over black) are in the presence of the solvent. The solvent has been removed at 10 minutes. Time points from 10 to 30 minutes (black text over white) are during the recovery in a solvent-free tube. Although both groups knock down simultaneously, flies recovering from the 2nd sedation recover negative geotaxis at earlier time points than flies recovering from their 1st sedation do.
Figure 3. Quantification of benzyl alcohol tolerance.
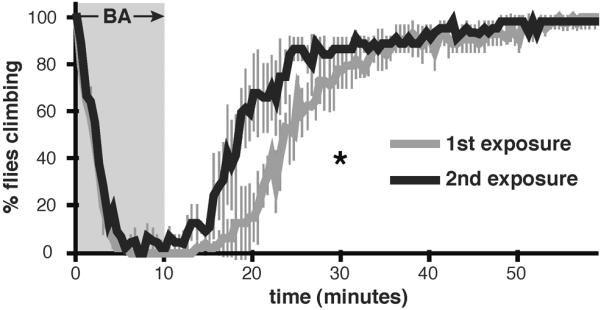
Shown are knock-down and recovery curves of wild-type flies after one (1st exposure) or two (2nd exposure) exposures to 0.4% benzyl alcohol. Values are plotted as percentage of flies climbing during sedation with benzyl alcohol (gray background) and during recovery from sedation (white background). Time between exposures is 24 hours. Significant difference is determined by the log-rank test (Error bars are SEM for each data point).
B. Ethanol tolerance and cross-tolerance
Flies also acquire tolerance to ethanol sedation. The ethanol tolerance assay is also a two-day protocol. On the first day, a population of three- to five-day-old females is divided into two groups. One group is sedated with a saturated ethanol vapor stream produced by ethanol bubblers, and the other group is mock sedated. After the flies recover from sedation, both groups are returned to food. Twenty-four hours after the first sedation, both groups are sedated, moved to a fresh-air environment for recovery, and the rate of recovery is recorded. A major difference between the ethanol and benzyl alcohol responses is that flies delay climbing after ethanol sedation even though they are obviously performing other types of normal behavior. The population returns to climbing rather asynchronously. Therefore, for the ethanol tolerance assays we visually monitor the return of postural control (standing) as a sign of recovery from sedation. Flies recover from a second ethanol sedation (delivered 24 hours after the first sedation) more rapidly than from their first ethanol sedation (Cowmeadow et al., 2005).
The molecular mechanism that produces ethanol tolerance appears to be completely or substantially the same as the mechanism that produces benzyl alcohol tolerance. This conclusion is based on the fact that both drugs induce mutual cross-tolerance and that the capacity to acquire tolerance to either drug is blocked by the same genetic mutations (Cowmeadow et al., 2005; Ghezzi et al., 2004; Al-Hasan et al., 2011; Krishnan et al., 2011).
C. Statistical analysis of tolerance
When describing these tolerance assays, we have not mentioned the magnitude of tolerance but only whether it occurs. When we began work on this topic, we rationalized that the magnitude of tolerance would show day-to-day variations caused by events that we could not control or could not imagine. For example, Drosophila show changes in behavior in response to barometric pressure (a factor that also affects solvent vaporization) (Ankney, 1984). Variability of this kind can be avoided if we treat tolerance as a binary phenomenon that either occurs or does not. Therefore, we compare a population's recovery from first sedation to its recovery from second sedation and ask only if the rates differ significantly from one another. Certainly, tolerance in a wild-type fly must be a quantitative trait that is produced by the concerted contributions of a number of gene products. Viewing tolerance as a binary phenomenon means that one can identify only mutations that have such a strong effect on the production of tolerance that the separation of the first and second sedation recovery curves is no longer statistically significant. While this simplification makes the assay very robust, it is limiting in that it does not detect the effect of mutations that make smaller incremental contributions to the production of tolerance. Fortunately, other investigators have screened for mutations that reduce the magnitude of tolerance, and therefore such genes will not be overlooked.
To determine whether the first-sedation and second-sedation groups recover at a significantly different rate, we use the log-rank test for equality of survival. Survival statistics are ideal for evaluating time to a specific event (Hosmer et al., 2002, 2008). Although this statistical test is most commonly applied to test for changes in the rate of deaths, disease, marriage, divorce, or failure of a mechanical part, it works well for determining whether the two groups recover from sedation at significantly different rates. Using this statistical test for tolerance, we observe that benzyl alcohol tolerance and ethanol tolerance each persist for about ten days (Cowmeadow et al., 2005; Ghezzi, unpublished). Thus, functional tolerance in flies is a long-term neuroadaptive response.
III. BK channels and the production of rapid tolerance
The mechanisms of action of ethanol and volatile anesthetics on the nervous system are well characterized. In both mammals and Drosophila, substantial evidence suggests that these drugs suppress neural function at least in part through interactions with voltage- and ligand-gated ion channels (Evans and Balster, 1991; Covarrubias and Rubin, 1993; Leibovitch et al., 1995; Harris, 1999; Nash, 2002). In general, organic solvent sedation is mediated by the inhibition of excitatory ion channels and the potentiation of inhibitory ion channels to produce a generalized depression of the nervous system. From a homeostatic perspective, it is expected that the adaptive mechanisms that lead to tolerance involve changes in neural activity that strive to restore normal neural excitability. In neurons, electrical excitability is an emergent property arising from the activity of and electrical interaction between an assortment of ion channel proteins. Therefore, likely regulators of a homeostatic adaptation are the ion channel proteins themselves.
A. Homeostatic modulation of BK channel gene expression mediates drug tolerance
The slo gene encodes BK-type Ca2+-activated K+ channels and is widely expressed throughout excitable tissues. BK channel activity plays a major role in shaping neuronal excitability, regulating synaptic activity, modulating smooth muscle tone, and controlling neuroendocrine secretion (Gribkoff et al., 2001). Our data suggested that slo expression was homeostatically regulated in response to drug exposure and that rapid functional tolerance to benzyl alcohol or ethanol sedation was dependent on slo expression in the nervous system. We observed that a single benzyl alcohol or ethanol sedation caused a slight increase in neuronal slo mRNA abundance concomitant with the appearance of tolerance. Conversely, low doses of benzyl alcohol that acted as a stimulant caused a reduction in neuronal slo mRNA abundance and produced behavioral sensitization. Animals homozygous for slo4 are healthy but do not acquire either benzyl alcohol tolerance or ethanol tolerance in our duration-of-sedation assays. The slo4 mutant allele had been shown to be a null mutation by behavioral, molecular, and electrophysiological analysis. In addition, we used a slo allele (ash218) that eliminates only slo expression from the nervous system to show that slo-dependent tolerance is a neural response. Finally, tolerance could be phenocopied by transgenic induction of slo expression (Ghezzi et al., 2004; Cowmeadow et al., 2005; Cowmeadow et al., 2006).
The observation that increased slo expression countered the sedating properties of two drugs led to the hypothesis that increased BK channel activity must act as a neural excitant. At the time that this hypothesis was proposed (Ghezzi et al., 2004), the dogmatic belief that K+ channel activity must always be inhibitory was still widely held, in spite of evidence accumulating to the contrary. How can increased expression of BK channels enhance neural excitability? In some cells, BK channel activity has been shown to shorten the refractory period, thereby increasing neural excitability in the form of an increased capacity for repetitive firing. The refractory period is the period of time that must elapse before the neuron can fire again. The refractory period is shortened because the high-conductance BK channels cause the rapid repolarization of the synapse, thereby preventing the activation of other classes of K+ channels that would otherwise occur. If these secondary K+ channels are activated they produce a long-lasting hyperpolarization that reduces the maximum firing rate (Lovell and McCobb, 2001; Pattillo et al., 2001; Van Goor et al., 2001; Brenner et al., 2005).
To determine if increased slo expression reduced the neural refractory rate, we examined the response of the giant fiber visual escape response pathway to benzyl alcohol sedation and to increased slo expression. In the giant fiber preparation, two stimulating tungsten electrodes are placed in the eyes, and a recording electrode is placed in an indirect flight muscle. A depolarizing stimulus is delivered to the eyes. From there the stimulus propagates through the brain and activates the giant fiber. The giant fiber axons extend from the brain to the thoracic ganglion where they are electrically coupled to an interneuron that activates a motor neuron that terminates on a thoracic flight muscle. At high stimulation potentials, the giant fiber is directly stimulated (in essence, the entire brain is depolarized by the stimulus), and the flight muscles report the firing pattern of the giant fiber pathway. This is called the short latency visual escape response pathway. A total of three neurons are involved in transmitting the signal to the flight muscles. Recordings from flight muscle report the firing pattern of this three-neuron circuit (Tanouye and Wyman, 1980).
In the fly giant fiber preparation, medical inhalation anesthetics had been shown to produce a dose-dependent increase in the neural refractory period that culminated in a failure of the neuron to respond to stimulation (Lin and Nash, 1996). Once the drug is removed, the firing capacity gradually recovers, in parallel with behavioral recovery. It was an exciting moment when we realized that the proposed reduction of the refractory period by increased BK channel activity could directly counter an effect of the anesthetic.
To determine if slo induction enhances the neural capacity for repetitive firing, we compared the following-frequency profile of the giant fiber pathway before and after induction of slo gene expression. In one experiment, a single 15–20 minute benzyl alcohol sedation was used to induce slo expression. When tested 24 hours after sedation, the capacity of the giant fiber to follow high-frequency stimulation in the absence of the drug was significantly enhanced (reduced refractory period) compared to the capacity of the untreated control, indicative of an adaptive process that overlaps with the development of tolerance (Figure 4). As is the case with tolerance, the enhancement in following frequency is slo dependent, in that mutations that block only neural expression of slo prevent it. Most important, the increase in following frequency can be produced in the absence of drugs by activating an inducible slo transgene. These results show that an increase in slo neural expression is both necessary and sufficient for the increase in the capacity for repetitive firing (Ghezzi et al., 2010).
Figure 4. Model for how homeostatic regulation of BK channels contributes to both drug tolerance and withdrawal.

A) In a naïve, no drug state, neurons of the giant fiber pathway in Drosophila exhibit a basal capacity for repetitive firing when evoked by high frequency stimulation (Normal). B) The capacity for repetitive firing is significantly inhibited by exposure to sedative drugs (Sedation), leading to the overall depression of neural activity characteristic of sedation. C) Drug exposure induces expression of presynaptic BK channels as part of a homeostatic response to sedation. Increased BK channel activity enhances the capacity for repetitive firing, leading to a reduced effect of the anesthetic on neural firing during a subsequent exposure (Tolerance). D) After drug clearance, however, another effect of increased BK channel expression is unmasked, resulting in an enhanced neural excitability in the form of increased basal firing capacity and an increased susceptibility for seizures (Withdrawal). The electrophysiological traces shown here are schematic representations of hypothetical data, not real traces.
B. Modulation of the BK channel gene produces a withdrawal symptom
The stereotypical image of a person in alcohol withdrawal is that of a person who has shaking hands, cannot sleep, suffers bouts of delirium, and may have spontaneous seizures. At least three of these symptoms are probably indicative of an increased baseline of neural excitability. In humans, the symptoms of alcohol withdrawal were experimentally documented with prisoners who were allowed to drink large amounts alcohol (averaging 256 ml to 489 ml of 95% ethanol per day) for long periods of time (weeks to months) (Isbell et al., 1955). The connection between alcohol withdrawal and alcohol withdrawal seizures has been well reviewed in Porter et al. (1990) and Rogawski (2005). The counter-adaptive theory of tolerance/dependence nicely account for these symptoms in that the adaptation (presumed increased excitability) that counters an effect of a neural depressant is uncovered during abstinence and could lead to these symptoms.
As described above, increased BK channel activity enhances the capacity for repetitive neural firing—a hallmark of seizure. Recent studies have tightly linked enhanced BK channel activity with specific forms of epilepsy. A human mutation in the pore-forming subunit of the BK channel that increases channel open probability is the cause of generalized epilepsy and paroxysmal dyskinesia (Du et al., 2005). Increased activity of BK channels caused by a mutation in the β4 BK channel auxiliary subunit increases high-frequency firing and leads to temporal lobe seizures (Brenner et al., 2005).
Flies have been used as a model organism for studying epilepsy for many years (See the review in this volume by Parker et al.). We appropriated an electrophysiological method used to study epileptic-like seizures in flies to determine whether tolerance to benzyl alcohol sedation came at the cost of a reduction in the seizure threshold. Stimulation of the giant fiber pathway at high frequency (200 Hz) is electroconvulsive and induces a seizure with a characteristic pattern. The magnitude of the voltage required to elicit seizure activity represents an accurate measure of seizure susceptibility (Kuebler and Tanouye, 2000). We demonstrated that flies that have become tolerant to benzyl alcohol by prior exposure and have increased slo expression, displayed seizure activity at lower voltages than control flies did. Similarly, artificial induction of the slo from a transgene also reduced the threshold voltage for eliciting seizures (Ghezzi et al., 2010). Finally Kuebler et al (2001) have demonstrated that null mutations in slo had the opposite effect and increased the seizure threshold.
C. How does the slo gene sense drug sedation?
We wanted to understand how the slo gene sensed drug sedation. To do so requires an understanding of how the slo gene is regulated. The slo transcriptional control region is large and complex. The 7 kb transcriptional control region contains at least five tissue-specific transcription start sites (core promoters). This control region includes the first intron after the translation start site, which has been shown to contain important regulatory elements (Brenner and Atkinson, 1996). The two most 5' promoters appear to be responsible for all neural expression, the next two promoters have been shown to be active in epithelial cells of the digestive system, and the 3'-most promoter is responsible for muscle and tracheal cell expression (Figure 5A). Each transcription start site begins expression with a unique 5' UTR that will be spliced to a common exon (Bohm et al., 2000; Brenner et al., 1996; Brenner and Atkinson, 1997; Chang et al., 2000).
Figure 5. Drug-induced chromatin remodeling at the slo transcriptional control region.

A) Shown is the 7 kb transcriptional control region of the slo gene. This gene has at least five alternative tissue-specific promoters: two neuronal promoters (C0 and C1), two midgut promoters (C1b and C1c), and one muscle cell and tracheal cell specific promoter (C2). In addition, this region contains several DNA elements that are highly conserved across different Drosophila species (4b, 6b, 55b) and two CREB response elements (cre). B) Dynamic histone H4 acetylation changes across the slo transcriptional control region after benzyl alcohol sedation. No change in acetylation is detected 30 minutes after sedation. At 4 hours, an increase in acetylation is detected with a peak centered over the 55b element. This event is dependent on binding of phospho-CREB at the two CRE sites. After six hours, the acetylation peak relocates to the neural promoter area with a small peak around C0 and a broad peak around C1 and mRNA expression from these promoters become evident. After 24 hours, the histone acetylation peak becomes focused at the 6b element, and mRNA expression decays. By 48 hours, histone acetylation and mRNA expression return to non-sedated control level.
One of the first steps in gene activation is generally held to be the alteration of chromatin structure. Transcription factor–induced chemical modification of histones can influence the accessibility of the underlying DNA and alter the affinity of the transcription pre-initiation complex for the chromatin (Berger, 2007). Most activiting transcription factors recruit histone acetyl transferases to the transcriptional control region to acetylate a variety of histones. Histone acetylation is tightly correlated with making the underlying DNA more accessible and with transcription activation. To identify slo DNA enhancer elements that respond to drug sedation, we elected to map drug-induced histone acetylation footprints left behind by transcription factors. We began our survey by monitoring histone H4 acetylation. In general, histone H4 acetylation relaxes the association between nucleosomes and DNA, prevents the cross-linking of consecutive histones, and is correlated with increased gene expression (Lee et al., 1993; Allis et al., 2007).
An antibody that recognizes all forms of histone H4 lysine acetylation (K5, K8, K12, K16) was used in the chromatin immunoprecipitation (ChIP) assay to generate a profile of histone H4 acetylation across the slo transcriptional control region at different times following sedation. The H4 acetylation spikes were mapped across the transcriptional control region and correlated with the tissue-specific transcription start sites and various evolutionarily conserved elements (Figure 5B). Four hours after sedation, an acetylation spike appeared centered over conserved element 55b. This spike preceded detectable gene induction that was first detectable at 6 h post sedation. At 6 h post sedation, the region encompassing both neural promoters was hyperacetylated. This broad acetylation peak is probably a side effect of increased transcription from the two neural promoters. At 24 h after sedation, slo expression remained elevated above baseline but appeared to be in decline. At this time, only the histones localized over conserved element 6b (~60 n) remained hyperacetylated. Finally, at 48 h after sedation slo gene expression and the acetylation state of the transcription control region had returned to baseline (Figure 5B) (Wang et al., 2007).
Histone acetylation is the product of transcription factor activity. It was suspected that the CREB transcription factor contributed to this process because CREB is known to recruit a histone acetylase and because the location of the first acetylation spike, the 55b element, is flanked by two CRE consensus sites. Furthermore, the 55b element carried a motif that differed in one base from a consensus CRE site. Using antibodies specific for the CREB DNA binding domain and for phospho-CREB, we showed that benzyl alcohol sedation increased phosphoCREB occupancy at all three sites: the two CRE consensus sites and CRE-like element in 55b.. In Drosophila, there are two CREB genes that are idiosyncratically most often referred to as dCrebA and dCREB2 (aka CrebB-17A or CREB-B) (Usui et al., 1993; Smolik et al., 1992). Prior benzyl alcohol sedation activated expression from dCrebA and altered the splicing of dCREB2 transcripts to reduce expression of a repressor isoform. These changes were predicated to induce transcriptional activation by CREB. A CRE-luciferase reporter gene confirmed that benzyl alcohol sedation activated the CREB signaling pathway. Further implicating CREB was the demonstration that expression of an inducible dominant-negative dCREB2b transgene blocked histone acetylation across the slo promoter region, blocked slo induction, and blocked the development of tolerance. The dCREB2S182 hypomorphic mutation was also shown to prevent slo induction and to block the acquisition of tolerance (Wang et al., 2007; Wang et al., 2009). It has not yet been determined whether dCrebA mutations also disrupt the capacity to acquire tolerance.
D. Role of BK channels in mammalian responses to ethanol
The rat hypothalamic-neurohypophysial model system has also been used to study the role of BK channels in alcohol tolerance. The magnocellular neurons (MCN) of this system release arginine-vasopressin and oxytocin, triggered by the influx of Ca2+ through voltage-gated Ca2+ channels and effectively terminated by BK channel activity. Acute ethanol exposure decreases the open probability of L-type Ca2+ channels and enhances the open probability of the BK channels of the neurohypophysial terminals, which suppresses the firing of these cells and the resultant release of the hormones. Chronic ethanol exposure produces tolerance that is manifested as a reduction in the capacity of ethanol to inhibit hormone release. Tolerance is the product of reduction in sensitivity of these channels to ethanol (Knott et al., 2002). Ethanol-mediated regulation of BK channel activity has been extensively studied in this system. At pharmacologically meaningful concentrations, ethanol enhances the activity of BK channels in neurohypophysial terminals within minutes by enhancing the open probability of BK channels without affecting ion selectivity or unitary conductance (Pietrzykowski et al., 2004). Additionally, BK channel variants from human brain continue to show potentiation by ethanol when incorporated into planar lipid bilayers lacking other cellular components (Crowley et al., 2003). This demonstrates that ethanol potentiation of channel activity is an intrinsic response of at least some neuronally expressed BK channels.
In the hypothalamic-neurohypophysial model system there is strong evidence that BK channel activity is modulated by ethanol exposure in ways that could underlie chronic tolerance. Twenty-four hours of ethanol exposure results in internalization of some pre-existing BK channels in the nerve terminal and a declustering of those that remain (Pietrzykowski et al., 2004). The reorganization of channels is coupled with a change in gene expression that produces replacement channels that are refractory to ethanol. Within minutes after ethanol exposure, expression of the miRNA miR-9 is induced, causing the destruction of slo mRNA splice variants encoding ethanol-sensitive BK channels. Splice variants that encode more ethanol resistant BK channels persist to produce replacement channels. These changes produce a reduction in BK channel current density and ethanol sensitivity. The regulation of BK channel expression by miR-9 is not limited to this model system but has also been observed in medium spiny neurons (Pietrzykowski et al., 2008). These changes in BK channel activity, expression, and localization nicely correlate with cellular tolerance. While other gene products may make important contributions, it appears that the BK channels play a major role in chronic tolerance.
Acute tolerance has also been physiologically demonstrated for BK channels in the hypothalamic-neurohypophysial neurons, although a cellular correlate of the changed channel activity has not been reported. Twelve minutes after exposure to ethanol, the capacity of BK channels to be potentiated by ethanol is dramatically reduced (Pietrzykowski et al., 2004). This fits the definition of acute tolerance. This reduction may occur through the dephosphoryaltion of a single CaMKII site in the alpha subunit of the channel. This was demonstrated by the observation that the progressive phosphorylation of bovine BK channel subunits by CaMKII converts them from channels that can be potentiated by ethanol to channels that are inhibited by ethanol (Liu et al., 2006). Acute molecular tolerance to the effects of ethanol has also been shown to be influenced by accessory channel subunits that associate with the pore-forming subunit. In mice, channels that include the beta4 subunit do not show acute tolerance to ethanol exposure (Martin et al., 2008).
In this rat model system, only post-transcriptional events have been documented; changes in transcription have not been observed. It is possible that transcriptional regulation does not play a role in the mammalian system and that post-transcriptional regulation does not play a role in the Drosophila system. However, this dichotomy is more likely to be a product of the specific properties or experimental advantages of each model system. Important regulatory responses tend to be controlled at many levels—as exemplified in the MCN system in which ethanol regulation of the channel activity occurs by phosphorylation, miRNA degradation of specific splice variants, channel internalization, and declustering.
IV. The synaptic connection
The efficient control of synaptic vesicle availability is crucial for the regulation of synaptic excitability. In the synaptic vesicle cycle, newly formed or recycled vesicles are filled with neurotransmitters and stored in a reserve pool as clusters that reversibly attach to the actin cytoskeleton through Synapsins. Upon phosphorylation of Synapsins, the filled vesicles move from the reserve pool to a readily accessible pool where they are available for docking at the active zone. The vesicles in the readily releasable pool fuse with the presynaptic cell membrane in a Ca2+-dependent process to release neurotransmitter into the inter-synaptic space. Released neurotransmitters activate ligand-gated receptor ion channels at the postsynaptic cell to trigger electrical and second-messenger responses in the postsynaptic cell. After fusion, presynaptic vesicles must be recycled by endocytosis for reuse (Sudhof, 2004) (Figure 6).
Figure 6. Synaptic model of drug tolerance.

Shown is a schematic representation of a synapse current knowledge of putative interactions between synaptic proteins. Synaptic proteins that have been implicated in the development of tolerance to alcohol and anesthetics are displayed and labeled in black. These proteins include Synapsins (Syn), Dynamin (Dyn), Homer, Integrins, BK channels (BK), Syntaxin 1A (Syx), and the GABAB receptor (GABABR). Proteins and structures in gray are included to provide context. Thin dotted lines with arrows denote known protein-protein interactions. Figure adapted from Gorini et al. (2010).
BK channels are the largest conductance ion channels that a neuron encodes, and in the presynaptic terminal a small change in their activity can dictate the membrane potential of the terminal and the influx of Ca2+ through voltage-gated Ca2+ channels. This one-two punch of controlling membrane potential and Ca2+ influx enables BK channels to have a strong influence on synaptic release. In addition to BK channels, other synaptic proteins have also been implicated in the response to ethanol and organic solvent anesthetics (Figure 6). These include the presynaptic proteins Dynamin, Syntaxin 1A, and Synapsin (Al-Hasan et al., 2011; Krishnan et al., 2011; Godenschwege et al., 2004); the transmembrane cell adhesion integrin subunit βPS and αPS3 (Bhandari et al., 2009); the postsynaptic GABAB receptor (Dzitoyeva et al., 2003); and the postsynaptic scaffolding protein Homer (Urizar et al., 2007). BK channels have recently been shown to be in intimate physical contact with some of these proteins. Both mass spectrometry and co-immunoprecipitation have shown that BK channels are physically associated with both Syntaxin 1A and Dynamin 1 (Cibulsky et al., 2005; Gorini et al., 2010). Syntaxin 1A is involved in synaptic release, whereas Dynamin is a large GTPase that performs a key step in vesicle recycling.
Syntaxin 1A is a t-SNARE (soluble NSF attachment receptor) protein that mediates targeted vesicular fusion at the synapse. In C. elegans, a mutation in the Syntaxin 1A ortholog causes strong resistance to the volatile anesthetics isoflurane and halothane (van Swinderen et al., 1999), and in rats these anesthetic compounds have been shown to bind to Syntaxin 1A resulting in structural alterations of the protein (Nagele et al., 2005). In Drosophila, a mutation in the Syntaxin 1A gene disrupts the capacity to acquire ethanol tolerance (Krishnan et al., 2011).
In mammals, the Dynamins comprise a family of three different genes, two of which (Dynamin 1 and Dynamin 3) are expressed widely in the brain and localized to the synapse. While Dynamin 1 is well known for its function in rapid vesicle recycling and concentrates primarily in the presynaptic compartment (Takei et al., 1996; Gray et al., 2003), Dynamin 3 is best known for mediating clathrin-mediated uptake of surface receptors and concentrates in the brain within the postsynaptic density of dendrites (Gray et al., 2003).
In Drosophila, the multiple forms of Dynamin are all thought to be encoded by a single gene called shibire (Chen et al., 1991). The shits1 and shits2 are temperature-sensitive mutant alleles that cause temperature-dependent paralysis. At the restrictive temperature (~29°C), the mutant Shibire protein stops functioning, leading to a rapid and reversible cessation of synaptic vesicle recycling and synaptic signaling (Kosaka and Ikeda, 1983; van der Bliek and Meyerowitz, 1991). At the permissive temperature (~22°C), the mutant animals have been described as being essentially normal.
In a recent study, it has been shown that animals carrying the shits1 or the shits2 mutant alleles were unable to acquire benzyl alcohol tolerance at the permissive temperature. That is—with regard to benzyl alcohol tolerance—these mutant alleles did not behave as conditional mutations (Al-Hasan et al., 2011). With regard to ethanol tolerance, the story was more involved. The shits1 mutation again blocked the acquisition of tolerance in a non-conditional way. The shits2 mutation, however, blocked tolerance only at the restrictive temperature and only if the temperature-induced blockade in neural signaling overlapped the ethanol exposure. If the shibire blockade was evoked 5 or 23 hours after ethanol exposure tolerance, then the functional tolerance assayed at 24 h after the exposure appeared normal. This indicates that at the restrictive temperature, the shits2 mutation interferes with the acquisition but not the maintenance of functional tolerance. The difference between benzyl alcohol and ethanol responses probably indicates that, with regard to the induction of tolerance, that ethanol is a more effective drug.
One might expect that any event producing a neural blockade would also block the acquisition of tolerance. However, this does not appear to be the case. A temperature-sensitive mutation affecting the neuronal voltage-gated sodium channel was also tested. This mutation, parats1, blocks the production of neuronal action potentials and therefore produces a neural blockade by a different method. Even though it effectively blocked neuronal activity, the parats1 mutation did not block tolerance (Krishnan et al., 2011). This may mean that the shibire-encoded Dynamin protein is a trigger for the production of tolerance. However, at this time, one cannot rule out that when Dynamin changes conformation at the restrictive temperature, it disturbs another closely associated protein(s) and that this change blocks the acquisition of tolerance. Because it has been shown that Dynamin touches the BK channel, it is possible that this hypothetical protein is the BK channel.
The close physical interaction between two entities, BK channels and Dynamin, that have been genetically shown to be important for the acquisition of tolerance is particularly interesting since in mammalian systems Pietrzykowski and colleagues (Pietrzykowski et al., 2004) have shown that ethanol tolerance initiates with the declustering and internalization of BK channels. It is thus possible that the process underlying the repositioning of BK channels from the synaptic membrane is facilitated by an interaction of the channel with the endocytic Dynamin and that the same process occurs in Drosophila.
V. Closing remarks
Drosophila has emerged as a valuable biological model for understanding the molecular mechanisms underlying drug abuse endophenotypes. Genes that have been identified are involved in second-messenger signaling, stress responses, olfaction, metabolism, transcriptional regulation, cytoskeletal organization, and memory formation and include proteins such as transcription factors, proteases, ion channels, synaptic proteins, neurotransmitter receptors, and biosynthetic enzymes (reviewed in Atkinson, 2009 and Rodan and Rothenfluh, 2010).
Tolerance and withdrawal are two key ingredients in the recipe for addiction. Together they provide a motivation for increased drug consumption. In Drosophila, aspects of both endophenotypes originate with the same gene. Whether the genes that underlie these endophenotypes directly contribute to pathological and compulsive drug use is not known. However, the recent development of Drosophila voluntary alcohol drinking assays may enable Drosophila geneticists to help resolve this question (Ja et al., 2007; Devineni and Heberlein, 2009).
In the field of developmental biology, Drosophila has provided many of the molecular keys for understanding mammalian development. Drug abuse research using Drosophila is a relatively young field that has already made important contributions toward understanding drug responses. In a sense, the experimental limitations of any given model system force one to have a distinct experimental perspective that makes possible the discovery of different facets of drug addiction. We believe that drug abuse research using Drosophila has well-breached the tipping point of discovery, making it a major contributor to our understanding of the addictive process.
Acknowledgements
We thank Jascha Pohl, Rudi Bohm, Brooks Robinson, Ben Troutwine, Rosie Robles, Harish Krishnan, and Jane Kirschman for critical reading of the manuscript. This work was supported by the National Institutes of Health grants RO1 DA022219 and R01 AA018037 to NSA.
References
- Al-Hasan YM, Krishnan HR, Ghezzi A, Prado FJ, 3rd., Robles RB, Atkinson NS. Tolerance to Anesthesia Depends on Synaptic Proteins. Behav. Genet. 2011 doi: 10.1007/s10519-011-9451-8. DOI 10.1007/s10519-011-9451-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allada R, Nash HA. Drosophila melanogaster as a model for study of general anesthesia: the quantitative response to clinical anesthetics and alkanes. Anesth. Analg. 1993;77:19–26. doi: 10.1213/00000539-199307000-00005. [DOI] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T, Reinberg D, Caparros M-L. Epigenetics. Cold Spring Harbor Laboratory Press; Woodbury, NY: 2007. [Google Scholar]
- Ankney PF. A note on barometric pressure and behavior in Drosophila pseudoobscura. Behav. Genet. 1984;14:315–317. doi: 10.1007/BF01065549. [DOI] [PubMed] [Google Scholar]
- Atkinson NS. Tolerance in Drosophila. J. Neurogenet. 2009;23:293–302. doi: 10.1080/01677060802572937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainton RJ, Tsai LT, Singh CM, Moore MS, Neckameyer WS, Heberlein U. Dopamine modulates acute responses to cocaine, nicotine and ethanol in Drosophila. Curr. Biol. 2000;10:187–194. doi: 10.1016/s0960-9822(00)00336-5. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Weiner JL, Eger EI, 2nd., Gong DH, Mihic SJ. Glycine and gamma-aminobutyric acid(A) receptor function is enhanced by inhaled drugs of abuse. Mol. Pharmacol. 2000;57:1199–1205. [PubMed] [Google Scholar]
- Bellen HJ, Tong C, Tsuda H. 100 years of Drosophila research and its impact on vertebrate neuroscience: a history lesson for the future. Nat. Rev. Neurosci. 2010;11:514–522. doi: 10.1038/nrn2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- Bhandari P, Kendler KS, Bettinger JC, Davies AG, Grotewiel M. An assay for evoked locomotor behavior in Drosophila reveals a role for integrins in ethanol sensitivity and rapid ethanol tolerance. Alcohol. Clin. Exp. Res. 2009;33:1794–1805. doi: 10.1111/j.1530-0277.2009.01018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohm RA. Ph.D. thesis. The University of Texas at Austin; Austin, TX: 2000. Transcriptional Control of slowpoke, a Calcium Activated Potassium Channel Gene. [Google Scholar]
- Bohm RA, Wang B, Brenner R, Atkinson NS. Transcriptional control of Ca(2+)-activated K(+) channel expression: identification of a second, evolutionarily conserved, neuronal promoter. J. Exp. Biol. 2000;203:693–704. doi: 10.1242/jeb.203.4.693. [DOI] [PubMed] [Google Scholar]
- Brenner R, Atkinson NS. Calcium-activated potassium channel gene expression in the midgut of Drosophila. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1997;118:411–420. doi: 10.1016/s0305-0491(97)00167-3. [DOI] [PubMed] [Google Scholar]
- Brenner R, Chen QH, Vilaythong A, Toney GM, Noebels JL, Aldrich RW. BK channel beta4 subunit reduces dentate gyrus excitability and protects against temporal lobe seizures. Nat. Neurosci. 2005;8:1752–1759. doi: 10.1038/nn1573. [DOI] [PubMed] [Google Scholar]
- Brenner R, Thomas TO, Becker MN, Atkinson NS. Tissue-specific expression of a Ca(2+)-activated K+ channel is controlled by multiple upstream regulatory elements. J. Neurosci. 1996;16:1827–1835. doi: 10.1523/JNEUROSCI.16-05-01827.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Atkinson N. Developmental and Eye-specific transcriptional control elements in an intronic region of a Ca2+-activated K+ channel gene. Dev. Biol. 1996;177:536–543. doi: 10.1006/dbio.1996.0183. [DOI] [PubMed] [Google Scholar]
- Chang WM, Bohm RA, Strauss JC, Kwan T, Thomas T, Cowmeadow RB, Atkinson NS. Muscle-specific transcriptional regulation of the slowpoke Ca(2+)-activated K(+) channel gene. J. Biol. Chem. 2000;275:3991–3998. doi: 10.1074/jbc.275.6.3991. [DOI] [PubMed] [Google Scholar]
- Chen MS, Obar RA, Schroeder CC, Austin TW, Poodry CA, Wadsworth SC, Vallee RB. Multiple forms of dynamin are encoded by shibire, a Drosophila gene involved in endocytosis. Nature. 1991;351:583–586. doi: 10.1038/351583a0. [DOI] [PubMed] [Google Scholar]
- Chen S, Lee AY, Bowens NM, Huber R, Kravitz EA. Fighting fruit flies: a model system for the study of aggression. Proc. Natl. Acad. Sci. USA. 2002;99:5664–5668. doi: 10.1073/pnas.082102599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulsky SM, Fei H, Levitan IB. Syntaxin-1A binds to and modulates the Slo calcium-activated potassium channel via an interaction that excludes syntaxin binding to calcium channels. J. Neurophysiol. 2005;93:1393–1405. doi: 10.1152/jn.00789.2004. [DOI] [PubMed] [Google Scholar]
- Covarrubias M, Rubin E. Ethanol selectively blocks a noninactivating K+ current expressed in Xenopus oocytes. Proc. Natl. Acad. Sci. USA. 1993;90:6957–6960. doi: 10.1073/pnas.90.15.6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowmeadow RB, Krishnan HR, Atkinson NS. The slowpoke gene is necessary for rapid ethanol tolerance in Drosophila. Alcohol. Clin. Exp. Res. 2005;29:1777–1786. doi: 10.1097/01.alc.0000183232.56788.62. [DOI] [PubMed] [Google Scholar]
- Cowmeadow RB, Krishnan HR, Ghezzi A, Al'Hasan YM, Wang YZ, Atkinson NS. Ethanol tolerance caused by slowpoke induction in Drosophila. Alcohol. Clin. Exp. Res. 2006;30:745–753. doi: 10.1111/j.1530-0277.2006.00087.x. [DOI] [PubMed] [Google Scholar]
- Crowley JJ, Treistman SN, Dopico AM. Cholesterol antagonizes ethanol potentiation of human brain BKCa channels reconstituted into phospholipid bilayers. Mol. Pharmacol. 2003;64:365–372. doi: 10.1124/mol.64.2.365. [DOI] [PubMed] [Google Scholar]
- Devineni AV, Heberlein U. Preferential ethanol consumption in Drosophila models features of addiction. Curr. Biol. 2009;19:2126–2132. doi: 10.1016/j.cub.2009.10.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinwiddie SH. Abuse of inhalants: a review. Addiction. 1994;89:925–939. doi: 10.1111/j.1360-0443.1994.tb03348.x. [DOI] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, Kotagal P, Luders HO, Shi J, Cui J, Richerson GB, Wang QK. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 2005;37:733–738. doi: 10.1038/ng1585. [DOI] [PubMed] [Google Scholar]
- Duffy JB. GAL4 system in Drosophila: a fly geneticist's Swiss army knife. Genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- Dzitoyeva S, Dimitrijevic N, Manev H. gamma-Aminobutyric acid B receptor 1 mediates behavior-impairing actions of alcohol in Drosophila: Adult RNA interference and pharmacological evidence. Proc. Natl. Acad. Sci. USA. 2003;100:5485–5490. doi: 10.1073/pnas.0830111100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans EB, Balster RL. CNS depressant effects of volatile organic solvents. Neurosci. Biobehav. Rev. 1991;15:233–241. doi: 10.1016/s0149-7634(05)80003-x. [DOI] [PubMed] [Google Scholar]
- Flanagan RJ, Ives RJ. Volatile substance abuse. Bull. Narc. 1994;46:49–78. [PubMed] [Google Scholar]
- Fleisher GR, Ludwig S. Textbook of pediatric emergency medicine. Wolters Kluwer/Lippincott Williams & Wilkins Health; Philadelphia, PA: 2010. [Google Scholar]
- Ghezzi A, Al-Hasan YM, Larios LE, Bohm RA, Atkinson NS. slo K(+) channel gene regulation mediates rapid drug tolerance. Proc. Natl. Acad. Sci. USA. 2004;101:17276–17281. doi: 10.1073/pnas.0405584101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi A, Pohl JB, Wang Y, Atkinson NS. BK channels play a counter-adaptive role in drug tolerance and dependence. Proc. Natl. Acad. Sci. USA. 2010;107:16360–16365. doi: 10.1073/pnas.1005439107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godenschwege TA, Reisch D, Diegelmann S, Eberle K, Funk N, Heisenberg M, Hoppe V, Hoppe J, Klagges BR, Martin JR, Nikitina EA, Putz G, Reifegerste R, Reisch N, Rister J, Schaupp M, Scholz H, Schwarzel M, Werner U, Zars TD, Buchner S, Buchner E. Flies lacking all synapsins are unexpectedly healthy but are impaired in complex behaviour. Eur. J. Neurosci. 2004;20:611–622. doi: 10.1111/j.1460-9568.2004.03527.x. [DOI] [PubMed] [Google Scholar]
- Goldman D, Oroszi G, Ducci F. The genetics of addictions: uncovering the genes. Nat. Rev. Genet. 2005;6:521–532. doi: 10.1038/nrg1635. [DOI] [PubMed] [Google Scholar]
- Gorini G, Ponomareva O, Shores KS, Person MD, Harris RA, Mayfield RD. Dynamin-1 co-associates with native mouse brain BKCa channels: proteomics analysis of synaptic protein complexes. FEBS Lett. 2010;584:845–851. doi: 10.1016/j.febslet.2009.12.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray NW, Fourgeaud L, Huang B, Chen J, Cao H, Oswald BJ, Hemar A, McNiven MA. Dynamin 3 is a component of the postsynapse, where it interacts with mGluR5 and Homer. Curr. Biol. 2003;13:510–515. doi: 10.1016/s0960-9822(03)00136-2. [DOI] [PubMed] [Google Scholar]
- Gribkoff VK, Starrett JE, Jr., Dworetzky SI. Maxi-K potassium channels: form, function, and modulation of a class of endogenous regulators of intracellular calcium. Neuroscientist. 2001;7:166–177. doi: 10.1177/107385840100700211. [DOI] [PubMed] [Google Scholar]
- Harris RA. Ethanol actions on multiple ion channels: which are important? Alcohol. Clin. Exp. Res. 1999;23:1563–1570. [PubMed] [Google Scholar]
- Harris RA, Mihic SJ, Dildy-Mayfield JE, Machu TK. Actions of anesthetics on ligand-gated ion channels: role of receptor subunit composition. FASEB J. 1995;9:1454–1462. doi: 10.1096/fasebj.9.14.7589987. [DOI] [PubMed] [Google Scholar]
- Hendricks JC, Finn SM, Panckeri KA, Chavkin J, Williams JA, Sehgal A, Pack AI. Rest in Drosophila is a sleep-like state. Neuron. 2000;25:129–138. doi: 10.1016/s0896-6273(00)80877-6. [DOI] [PubMed] [Google Scholar]
- Himmelsbach CK. The morphine abstinence syndrome, its nature and treatment. Ann. Intern. Med. 1941;15:829–843. [Google Scholar]
- Hosmer DW, Lemeshow S, Sunny K. Applied Survival Analysis: Regression Modeling of Time to Event Data (Solutions Manual) Wiley-Interscience; New York, NY: 2002. [Google Scholar]
- Hosmer D, Lemeshow S, May S. Applied Survival Analysis: Regression Modeling of Time To Event Data. Wiley-Interscience; New York, NY: 2008. [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu. Rev. Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans . Re-evaluation of some organic chemicals, hydrazine and hydrogen peroxide. World Health Organization, International Agency for Research on Cancer; Lyon, France: 1999. [Google Scholar]
- Isbell H, Fraser HF, Wikler A, Belleville RE, Eisenman AJ. An experimental study of the etiology of rum fits and delirium tremens. Q. J. Stud. Alcohol. 1955;16:1–33. [PubMed] [Google Scholar]
- Ja WW, Carvalho GB, Mak EM, de la Rosa NN, Fang AY, Liong JC, Brummel T, Benzer S. Prandiology of Drosophila and the CAFE assay. Proc. Natl. Acad. Sci. USA. 2007;104:8253–8256. doi: 10.1073/pnas.0702726104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien RM. A Primer of Drug Action: A concise, nontechnical guide to the actions, uses, and side effects of psychoactive drugs. Worth Publishers Inc; New York, NY: 2004. [Google Scholar]
- Kendler KS, Jacobson KC, Prescott CA, Neale MC. Specificity of genetic and environmental risk factors for use and abuse/dependence of cannabis, cocaine, hallucinogens, sedatives, stimulants, and opiates in male twins. Am. J. Psychiatry. 2003;160:687–695. doi: 10.1176/appi.ajp.160.4.687. [DOI] [PubMed] [Google Scholar]
- Knott TK, Dopico AM, Dayanithi G, Lemos J, Treistman SN. Integrated channel plasticity contributes to alcohol tolerance in neurohypophysial terminals. Mol. Pharmacol. 2002;62:135–142. doi: 10.1124/mol.62.1.135. [DOI] [PubMed] [Google Scholar]
- Kong EC, Woo K, Li H, Lebestky T, Mayer N, Sniffen MR, Heberlein U, Bainton RJ, Hirsh J, Wolf FW. A pair of dopamine neurons target the D1-like dopamine receptor DopR in the central complex to promote ethanol-stimulated locomotion in Drosophila. PLoS ONE. 2010;5:e9954. doi: 10.1371/journal.pone.0009954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. Neurobiology of addiction. Elsevier/Academic Press; Boston, MA: 2006. [Google Scholar]
- Koob GF. Drug addiction: the yin and yang of hedonic homeostasis. Neuron. 1996;16:893–896. doi: 10.1016/s0896-6273(00)80109-9. [DOI] [PubMed] [Google Scholar]
- Koob GF, Bloom FE. Cellular and molecular mechanisms of drug dependence. Science. 1988;242:715–723. doi: 10.1126/science.2903550. [DOI] [PubMed] [Google Scholar]
- Kopp Lugli A, Yost CS, Kindler CH. Anaesthetic mechanisms: update on the challenge of unravelling the mystery of anaesthesia. Eur. J. Anaesthesiol. 2009;26:807–820. doi: 10.1097/EJA.0b013e32832d6b0f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosaka T, Ikeda K. Possible temperature-dependent blockage of synaptic vesicle recycling induced by a single gene mutation in Drosophila. J. Neurobiol. 1983;14:207–225. doi: 10.1002/neu.480140305. [DOI] [PubMed] [Google Scholar]
- Krishnan HR, Al-Hasan YM, Pohl JB, Ghezzi A, Atkinson NS. A Role for Dynamin in Triggering Ethanol Tolerance. Alcohol. Clin. Exp. Res. 2011 doi: 10.1111/j.1530-0277.2011.01587.x. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan KS, Nash HA. A genetic study of the anesthetic response: mutants of Drosophila melanogaster altered in sensitivity to halothane. Proc. Natl. Acad. Sci. USA. 1990;87:8632–8636. doi: 10.1073/pnas.87.21.8632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuebler D, Tanouye MA. Modifications of seizure susceptibility in Drosophila. J. Neurophysiol. 2000;83:998–1009. doi: 10.1152/jn.2000.83.2.998. [DOI] [PubMed] [Google Scholar]
- Kuebler D, Zhang H, Ren X, Tanouye MA. Genetic suppression of seizure susceptibility in Drosophila. J. Neurophysiol. 2001;86:1211–1225. doi: 10.1152/jn.2001.86.3.1211. [DOI] [PubMed] [Google Scholar]
- Lee DY, Hayes JJ, Pruss D, Wolffe AP. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Leibovitch BA, Campbell DB, Krishnan KS, Nash HA. Mutations that affect ion channels change the sensitivity of Drosophila melanogaster to volatile anesthetics. J. Neurogenet. 1995;10:1–13. doi: 10.3109/01677069509083455. [DOI] [PubMed] [Google Scholar]
- Lin M, Nash HA. Influence of general anesthetics on a specific neural pathway in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA. 1996;93:10446–10451. doi: 10.1073/pnas.93.19.10446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton J. Neurochemical mechanisms underlying alcohol withdrawal. Alcohol Health Res. World. 1998;22:13–24. [PMC free article] [PubMed] [Google Scholar]
- Littleton J, Little H. Current concepts of ethanol dependence. Addiction. 1994;89:1397–1412. doi: 10.1111/j.1360-0443.1994.tb03736.x. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Ganetzky B. Ion channels and synaptic organization: analysis of the Drosophila genome. Neuron. 2000;26:35–43. doi: 10.1016/s0896-6273(00)81135-6. [DOI] [PubMed] [Google Scholar]
- Liu J, Asuncion-Chin M, Liu P, Dopico AM. CaM kinase II phosphorylation of slo Thr107 regulates activity and ethanol responses of BK channels. Nat. Neurosci. 2006;9:41–49. doi: 10.1038/nn1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd TE, Verstreken P, Ostrin EJ, Phillippi A, Lichtarge O, Bellen HJ. A genome-wide search for synaptic vesicle cycle proteins in Drosophila. Neuron. 2000;26:45–50. doi: 10.1016/s0896-6273(00)81136-8. [DOI] [PubMed] [Google Scholar]
- Lovell PV, McCobb DP. Pituitary control of BK potassium channel function and intrinsic firing properties of adrenal chromaffin cells. J. Neurosci. 2001;21:3429–3442. doi: 10.1523/JNEUROSCI.21-10-03429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubman DI, Yucel M, Lawrence AJ. Inhalant abuse among adolescents: neurobiological considerations. Br. J. Pharmacol. 2008;154:316–326. doi: 10.1038/bjp.2008.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GE, Hendrickson LM, Penta KL, Friesen RM, Pietrzykowski AZ, Tapper AR, Treistman SN. Identification of a BK channel auxiliary protein controlling molecular and behavioral tolerance to alcohol. Proc. Natl. Acad. Sci. USA. 2008;105:17543–17548. doi: 10.1073/pnas.0801068105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin WR. XVI. A homeostatic and redundancy theory of tolerance to and dependence on narcotic analgesics. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1968;46:206–225. [PubMed] [Google Scholar]
- Matsuda K, Buckingham SD, Freeman JC, Squire MD, Baylis HA, Sattelle DB. Effects of the alpha subunit on imidacloprid sensitivity of recombinant nicotinic acetylcholine receptors. Br. J. Pharmacol. 1998;123:518–524. doi: 10.1038/sj.bjp.0701618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield RD, Harris RA, Schuckit MA. Genetic factors influencing alcohol dependence. Br. J. Pharmacol. 2008;154:275–287. doi: 10.1038/bjp.2008.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U. Ethanol intoxication in Drosophila: Genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell. 1998;93:997–1007. doi: 10.1016/s0092-8674(00)81205-2. [DOI] [PubMed] [Google Scholar]
- Nagele P, Mendel JB, Placzek WJ, Scott BA, D'Avignon DA, Crowder CM. Volatile anesthetics bind rat synaptic snare proteins. Anesthesiology. 2005;103:768–778. doi: 10.1097/00000542-200510000-00015. [DOI] [PubMed] [Google Scholar]
- Nash HA. In vivo genetics of anaesthetic action. Br. J. Anaesth. 2002;89:143–155. doi: 10.1093/bja/aef159. [DOI] [PubMed] [Google Scholar]
- Parr J, Large A, Wang X, Fowler SC, Ratzlaff KL, Ruden DM. The inebri-actometer: a device for measuring the locomotor activity of Drosophila exposed to ethanol vapor. J. Neurosci. Methods. 2001;107:93–99. doi: 10.1016/s0165-0270(01)00357-0. [DOI] [PubMed] [Google Scholar]
- Pattillo JM, Yazejian B, DiGregorio DA, Vergara JL, Grinnell AD, Meriney SD. Contribution of presynaptic calcium-activated potassium currents to transmitter release regulation in cultured Xenopus nerve-muscle synapses. Neuroscience. 2001;102:229–240. doi: 10.1016/s0306-4522(00)00453-x. [DOI] [PubMed] [Google Scholar]
- Pick S, Strauss R. Goal-driven behavioral adaptations in gap-climbing Drosophila. Curr. Biol. 2005;15:1473–1478. doi: 10.1016/j.cub.2005.07.022. [DOI] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Friesen RM, Martin GE, Puig SI, Nowak CL, Wynne PM, Siegelmann HT, Treistman SN. Posttranscriptional regulation of BK channel splice variant stability by miR-9 underlies neuroadaptation to alcohol. Neuron. 2008;59:274–287. doi: 10.1016/j.neuron.2008.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrzykowski AZ, Martin GE, Puig SI, Knott TK, Lemos JR, Treistman SN. Alcohol tolerance in large-conductance, calcium-activated potassium channels of CNS terminals is intrinsic and includes two components: decreased ethanol potentiation and decreased channel density. J. Neurosci. 2004;24:8322–8332. doi: 10.1523/JNEUROSCI.1536-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter RJ, Mattson RH, Porter RJ, Mattson RH, Cramer JA. Alcohol and seizures: Basic mechanisms and clinical concepts. F.A. Davis; Philadelphia, PA: 1990. [Google Scholar]
- Quinn WG, Harris WA, Benzer S. Conditioned behavior in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA. 1974;71:708–712. doi: 10.1073/pnas.71.3.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramazani RB, Krishnan HR, Bergeson SE, Atkinson NS. Computer automated movement detection for the analysis of behavior. J. Neurosci. Methods. 2007;162:171–179. doi: 10.1016/j.jneumeth.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodan AR, Rothenfluh A. The genetics of behavioral alcohol responses in Drosophila. Int. Rev. Neurobiol. 2010;91:25–51. doi: 10.1016/S0074-7742(10)91002-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA. Update on the neurobiology of alcohol withdrawal seizures. Epilepsy Curr. 2005;5:225–230. doi: 10.1111/j.1535-7511.2005.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfluh A, Heberlein U. Drugs, flies, and videotape: the effects of ethanol and cocaine on Drosophila locomotion. Curr. Opin. Neurobiol. 2002;12:639–645. doi: 10.1016/s0959-4388(02)00380-x. [DOI] [PubMed] [Google Scholar]
- Scholz H, Ramond J, Singh CM, Heberlein U. Functional ethanol tolerance in Drosophila. Neuron. 2000;28:261–271. doi: 10.1016/s0896-6273(00)00101-x. [DOI] [PubMed] [Google Scholar]
- Schuckit MA. Low level of response to alcohol as a predictor of future alcoholism. Am. J. Psychiatry. 1994;151:184–189. doi: 10.1176/ajp.151.2.184. [DOI] [PubMed] [Google Scholar]
- Singh CM, Heberlein U. Genetic control of acute ethanol-induced behaviors in Drosophila. Alcohol. Clin. Exp. Res. 2000;24:1127–1136. [PubMed] [Google Scholar]
- Smolik SM, Rose RE, Goodman RH. A cyclic AMP-responsive element-binding transcriptional activator in Drosophila melanogaster, dCREB-A, is a member of the leucine zipper family. Mol. Cell. Biol. 1992;12:4123–4131. doi: 10.1128/mcb.12.9.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolowski MB. Foraging strategies of Drosophila melanogaster: a chromosomal analysis. Behav. Genet. 1980;10:291–302. doi: 10.1007/BF01067774. [DOI] [PubMed] [Google Scholar]
- Solomon RL. The opponent-process theory of acquired motivation: the costs of pleasure and the benefits of pain. Am. Psychol. 1980;35:691–712. doi: 10.1037//0003-066x.35.8.691. [DOI] [PubMed] [Google Scholar]
- Solomon RL, Corbit JD. An opponent-process theory of motivation. I. Temporal dynamics of affect. Psychol. Rev. 1974;81:119–145. doi: 10.1037/h0036128. [DOI] [PubMed] [Google Scholar]
- Spieth HT. Courtship behavior in Drosophila. Annu. Rev. Entomol. 1974;19:385–405. doi: 10.1146/annurev.en.19.010174.002125. [DOI] [PubMed] [Google Scholar]
- Sudhof TC. The synaptic vesicle cycle. Annu. Rev. Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, De Camilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. J. Cell Biol. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanouye MA, Wyman RJ. Motor outputs of giant nerve fiber in Drosophila. J. Neurophysiol. 1980;44:405–421. doi: 10.1152/jn.1980.44.2.405. [DOI] [PubMed] [Google Scholar]
- Urizar NL, Yang Z, Edenberg HJ, Davis RL. Drosophila homer is required in a small set of neurons including the ellipsoid body for normal ethanol sensitivity and tolerance. J. Neurosci. 2007;27:4541–4551. doi: 10.1523/JNEUROSCI.0305-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Smolik SM, Goodman RH. Isolation of Drosophila CREB-B: a novel CRE-binding protein. DNA Cell Biol. 1993;12:589–595. doi: 10.1089/dna.1993.12.589. [DOI] [PubMed] [Google Scholar]
- van der Bliek AM, Meyerowitz EM. Dynamin-like protein encoded by the Drosophila shibire gene associated with vesicular traffic. Nature. 1991;351:411–414. doi: 10.1038/351411a0. [DOI] [PubMed] [Google Scholar]
- Van Goor F, Li YX, Stojilkovic SS. Paradoxical role of large-conductance calcium-activated K+ (BK) channels in controlling action potential-driven Ca2+ entry in anterior pituitary cells. J. Neurosci. 2001;21:5902–5915. doi: 10.1523/JNEUROSCI.21-16-05902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Swinderen B, Saifee O, Shebester L, Roberson R, Nonet ML, Crowder CM. A neomorphic syntaxin mutation blocks volatile-anesthetic action in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 1999;96:2479–2484. doi: 10.1073/pnas.96.5.2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakasugi M, Hirota K, Roth SH, Ito Y. The effects of general anesthetics on excitatory and inhibitory synaptic transmission in area CA1 of the rat hippocampus in vitro. Anesth. Analg. 1999;88:676–680. doi: 10.1097/00000539-199903000-00039. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ghezzi A, Yin JC, Atkinson NS. CREB regulation of BK channel gene expression underlies rapid drug tolerance. Genes Brain Behav. 2009;8:369–376. doi: 10.1111/j.1601-183X.2009.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Krishnan HR, Ghezzi A, Yin JC, Atkinson NS. Drug-induced epigenetic changes produce drug tolerance. PLoS Biol. 2007;5:2342–2353. doi: 10.1371/journal.pbio.0050265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf FW, Heberlein U. Invertebrate models of drug abuse. J. Neurobiol. 2003;54:161–178. doi: 10.1002/neu.10166. [DOI] [PubMed] [Google Scholar]
- Young AM, Goudie AJ. Adaptive processes regulating tolerance to behavioral effects of drugs. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: Fourth Generation of Progress. Raven Press; New York, NY: 1995. pp. 733–742. [Google Scholar]