

Abstract
The Lactamase Engineering Database (www.LacED.uni-stuttgart.de) was developed to facilitate the classification and analysis of TEM β-lactamases. The current version contains 474 TEM variants. Two hundred fifty-nine variants form a large scale-free network of highly connected point mutants. The network was divided into three subnetworks which were enriched by single phenotypes: one network with predominantly 2be and two networks with 2br phenotypes. Fifteen positions were found to be highly variable, contributing to the majority of the observed variants. Since it is expected that a considerable fraction of the theoretical sequence space is functional, the currently sequenced 474 variants represent only the tip of the iceberg of functional TEM β-lactamase variants which form a huge natural reservoir of highly interconnected variants. Almost 50% of the variants are part of a quartet. Thus, two single mutations that result in functional enzymes can be combined into a functional protein. Most of these quartets consist of the same phenotype, or the mutations are additive with respect to the phenotype. By predicting quartets from triplets, 3,916 unknown variants were constructed. Eighty-seven variants complement multiple quartets and therefore have a high probability of being functional. The construction of a TEM β-lactamase network and subsequent analyses by clustering and quartet prediction are valuable tools to gain new insights into the viable sequence space of TEM β-lactamases and to predict their phenotype. The highly connected sequence space of TEM β-lactamases is ideally suited to network analysis and demonstrates the strengths of network analysis over tree reconstruction methods.
INTRODUCTION
TEM β-lactamases cause resistance of their host organisms against β-lactam based antibiotics, such as penicillin, by catalyzing the hydrolysis of the β-lactam ring. Since the discovery of the first TEM β-lactamase, TEM-1, in 1963, over 200 variants have been found (1). An annotated library of these sequences, further referenced as the TEM mutation table, is maintained by the Lahey clinic (2). β-Lactamases are a major concern in modern health care due to their efficient inactivation of many β-lactams and their high variability in biochemical properties. The selective pressure by the widespread use of antibiotics has been assumed to lead to the development of new variants and to allow existing variants to surface (3, 4). Even though the currently known sequences often vary by only a few amino acids, the minor changes allow for a variety of different substrate spectra and resistances and can be broadly classified into four phenotypes. According to the classification of β-lactamases done by Bush and Jacoby, the TEM family contains proteins that confer broad- or extended-spectrum activity (phenotypes 2b and 2be, respectively), inhibitor resistance (2br), or a combination of extended-spectrum activity and inhibitor resistance (2ber) (5, 23). Sequences at the TEM mutation table are classified mostly according to these categories. This makes the family of TEM β-lactamases one of the largest families with high microdiversity and with thorough documentation of the effects these variations have on their biochemical properties. Since most variants differ by only a single amino acid, which can alter the substrate spectrum or allow for inhibitor resistance, the unambiguous annotation of TEM variants and their mutations is of the utmost importance. This fact has been recognized by the scientific community and has led to the creation of a numbering and naming scheme (2, 6).
Since not all TEM β-lactamases are featured in the TEM mutation table, the Lactamase Engineering Database (LacED; www.LacED.uni-stuttgart.de) for TEM β-lactamases has been established (7, 8) to combine reliable information of the TEM mutation table with the large amount of sequences that can be found in the NCBI protein database. Since the NCBI is publicly accessible for submission, some protein entries show inconsistencies, such as the use of a name that has already been assigned to another protein, the use of different names for the same protein, or the disregard of mutations in the sequence that would warrant a new name (7, 9). The LacED was updated by collecting currently available sequences of TEM β-lactamases, which were analyzed by a network approach. By excluding wrongly annotated sequences, nontranslated regions, and fragments, a highly connected network of TEM β-lactamases was created and systematically analyzed.
Networks were chosen as a representation method for the sequence relationships instead of phylogenetic trees. It has been discussed by Fitch that the representation through networks has advantages over phylogenetic trees for different reasons (10). For sequence families like TEM β-lactamases, where differences between sequences are very small and/or sequences are very similar, this approach is appropriate, since usually there are multiple equally probable evolutionary paths between variants which cannot be adequately displayed in a phylogenetic tree.
Another successful analysis of TEM β-lactamases with the help of networks was done by Guthrie et al. (11). In their network, nodes represented mutated positions in the protein sequence. The links between two mutations stood for the number of times they were present together in a sequence. They used this network, based on the combinations of two mutations, to extrapolate paths of three mutations and their corresponding sequences. They found that they could identify sequences with this method that conferred an increased resistance to cefotaxime. However, since each node represented a single mutation, the analysis lacked information about the specific background present in each variant and the various combinations of mutations. Therefore, we decided to use complete sequences in our network approach so mutations could always be analyzed in their specific background.
MATERIALS AND METHODS
Database update.
First, the TEM database of the LacED was updated. For this, a BLAST search (12) was performed against the nonredundant protein database of NCBI using the TEM-1 β-lactamase (AAP20891) as the seed sequence and an E value of 1 × 10−105. USEARCH 5.1 (13) was used to compare the results. Fragments were sorted to longer sequences if possible. Sequences that share a sequence identity of 1 but originate from different source organisms were grouped as proteins. Multiple-sequence alignments were generated with ClustalW 2.1 (14) and used to identify and remove other β-lactamases, such as Raoultella planticola and K. pneumonia β-lactamases, from the database. Sequences declared as fusion proteins also were deleted. This was done because the additional residues would outweigh any changes to the sequence of the TEM β-lactamase.
Only one seed sequence (TEM-1) was used in the first BLAST search. This could result in missing TEM β-lactamase sequences that were not close enough to TEM-1. Because of that, a second BLAST search was performed. This time all proteins of the previously updated database were used as seed sequences, and an E value of 1 × 10−100 was applied. Again fusion proteins were removed. This version of the database will be referred to further as LacED v2.1. A pairwise alignment of each sequence with TEM-1 and the sulfhydryl variable protein SHV-1 (AAD37412) was performed using the Needleman-Wunsch algorithm (EMBOSS:6.4 [32]). Sequences with a higher sequence identity to SHV-1 than to TEM-1 also were removed from the database. Mutation profiles were created for each protein using TEM-1 as a reference, as done for LacED v1.0.
All TEM sequences of the TEM mutation table were reconstructed using TEM-1 as the template sequence. The sequences then were compared to the updated LacED. Thirty-two sequences of the TEM mutation table were not found in the NCBI database and also were parsed into the database, resulting in the final version of the database (LacED v2.2). Additionally, information about the phenotype of the sequence was extracted from the TEM mutation table.
All sequences were compared to the reconstructed sequences of the TEM mutation table. If there was a match, the existing TEM number was assigned to a variant. The remaining sequences were compared with the old version of the LacED and were assigned the respective TEM-like (TEML) numbers. Variants found in neither database were assigned a new TEML number, starting with TEML-164.
Network analysis. (i) Building the adjacency matrix.
An adjacency matrix was built by aligning the variants against each other using the Needleman-Wunsch algorithm and assigning a score of 1 to each matrix element if the two variants differ by exactly one amino acid; otherwise a score of 0 was assigned. From the adjacency matrix, a network was built by connecting all variants that differ by one amino acid. A multisequence alignment showed that variants often were missing the first 23 residues, which make up the signal peptide, and the last 6 residues. For the network analysis, only sequence information in the core region (positions 24 to 280; numbering is according to that of TEM-1) was analyzed. Forty-nine variants differ only outside the core regions and were omitted from the analysis. Therefore, all variants that were identical after removal were assigned to a representative variant (see Table S1 in the supplemental material). Seventy variants are fragments and therefore were omitted as well.
(ii) Cluster search based on betweenness.
For the cluster search, the algorithm from Girvan et al. (15) was adapted. Their algorithm was used to find edges with a high betweenness centrality and remove them. This step was based on the assumption that edges with a high betweenness lie between higher connected clusters rather than in them. For this work, the algorithm was adapted to remove nodes rather than edges. Therefore, the betweenness of each node was calculated as described above, and then the node with the highest betweenness centrality was removed. The betweenness centrality for each node in the resulting network was calculated again. The node with the highest betweenness was removed again. This was repeated until the highest betweenness centrality in the network was zero. The betweenness centrality was calculated using the function betweenness_centrality implemented in the MatlabBGL package and then normalized by dividing the betweenness by the respective compartment size.
(iii) Quartet identification.
A quartet is a structure in the network consisting of four variants. Each variant has a sequence identity of 1 to two of the other variants and a sequence identity of 2 to the fourth variant. To find quartets in the network, variants were checked for their adjacent neighbors. Each of those neighbors then was compared to the others for neighbors they had in common. If there was such a node, that node was checked to determine whether it was the same node as one of the three already selected nodes. If this was not the case, the four variants were accepted as a quartet.
(iv) Quartet prediction.
To construct hypothetical variants, sequences with a difference of 2 mutations were combined into two new variants. For this, the two existing variants were aligned. The residues at the differing positions of the first variant then were extracted. These residues then were exchanged, one at a time, in the second variant. To avoid producing already existing variants, the hypothetical variants were checked against both the already existing variants and the already created variants. The hypothetical variants then were given a name consisting of the prefix TEM-X and a number.
RESULTS
Update of the Lactamase Engineering Database.
The Lactamase Engineering Database (LacED) (7) was updated using sequences from the NCBI protein database and the TEM mutation table (2) (www.lahey.org/Studies/temtable.asp). Sequence fragments of different lengths were assigned to a single protein entry, and the longest sequence was used as a representative variant for further analysis of β-lactamase variants. The updated database contains 474 variants and is publicly accessible at www.LacED.uni-stuttgart.de. One hundred ninety-nine variants originate from the TEM mutation table; therefore, TEM numbers were assigned. For 149 of these TEM β-lactamases, a matching entry in the NCBI protein database was found. Two hundred seventy-five variants originate from the NCBI protein database but were not found in the mutation table. Therefore, a TEM-like (TEML) number was assigned.
Data inconsistencies and reconciliation.
For all TEM variants in the LacED, mutation profiles were created based on the comparison of the respective sequence against the reference sequence of TEM-1 (AAP20891). Of the 50 variants in the TEM mutation table which did not match with a sequence entry in the NCBI protein database, 18 have a GenBank accession code, but the respective sequence entry in the NCBI protein database differed from the mutation profile in the TEM mutation table. For these 18 variants, the sequence from GenBank was parsed into the LacED and the respective TEM number from the TEM mutation table was assigned (Table 1). For 10 of these 18 variants, the NCBI protein database entry is not of full length (TEM-42, TEM-75, TEM-89, TEM-117, TEM-118, TEM-187, TEM-191, TEM-192, and TEM-199). For 5 variants, the NCBI protein database entry has additional mutations compared to sequences in the TEM mutation table (TEM-60, TEM-98, TEM-108, TEM-123, and TEM-124). For 2 variants, the NCBI protein database entry is missing a mutation and also shows an additional mutation at a different position (TEM-148 and TEM-193). For one variant, the NCBI protein database entry has a mutation at the same position but to a different amino acid (TEM-209).
TABLE 1.
Inconsistencies between 18 mutation profiles of the TEM mutation table and the LacED
| TEM variant | Mutation profile in TEM mutation table | GenBank accession no. | Mutation profile in LacED (derived from GenBank entry)a |
|---|---|---|---|
| TEM-42 | Q39K, A42V, G238S, E240K, T265M | X98047 | N-3, Q39K, A42V, G238S, E240K, T265M, C-2 |
| TEM-59 | Q39K, S130G | AF062386 | Q39K, S130G, C-9 |
| TEM-60 | Q39K, L51P, E104K, R164S | AF047171 | Q39K, L51P, E104K, R164S, A187R, S223C, F230L |
| TEM-75 | L21F, R164H, T265M | AY130284 | N-12, L21F, R164H, T265M, C-13 |
| TEM-89 | Q39K, E104K, S130G, G238S | AY039040 | Q39K, E104K, S130G, G238S, C-8 |
| TEM-98 | Empty | AF397068 | S4D, I5P, H289L |
| TEM-108 | V80E, G196S, N276S | AF506748 | S4D, I5P, V80E, G196S, N276S |
| TEM-117 | L21F | AY130282 | N-12, L21F, C-19 |
| TEM-118 | R164H, T265M | AY130285 | N-12, R164H, T265 M, C-13 |
| TEM-123 | Q6K, E104K, G238S | AY327539 | Q6K, E104K, G238S, A248-, R275A |
| TEM-124 | Q6K, E104K, M182T | AY327540 | Q6K, E104K, M182T, A248-, R275A |
| TEM-148 | T189K | AM087454 | T188K |
| TEM-187 | L21F, R164H, A184V, T265M | HM246246 | L21F, R164H, A184V, T265M, C-1 |
| TEM-191 | E240K | JF949916 | N-10, E240K, C-24 |
| TEM-192 | M68I | JF949915 | N-10, M68I, C-25 |
| TEM-193 | N136H, L138F, R164C, E166G, E168K, I173T, N175H, M186V, T188N, L220I | JN935135 | N136H, L138F, R164C, E166G, E168K, N170T, N175H, M186V, T188N, L220I |
| TEM-199 | Q39K, E104K, M155I, G238S | JX050178 | N-3, Q39K, E104K, M155I, G238S |
| TEM-209 | G41A | KF240808 | G41D |
A248-, deletion at position 248. The following abbreviations for differences were used: N-3, missing 3 amino acids at the N terminus; C-2, missing 2 amino acids at the C terminus; Q39K, mutation Q-K at position 39.
Thirty-two variants had no GenBank accession code. Therefore, sequences were generated from the mutation profiles and entered into the LacED.
Construction of the TEM β-lactamase network.
In order to construct the TEM β-lactamase network, only proteins with a complete sequence in the core region from position 24 to 280 were analyzed. For 354 β-lactamase sequences with different core regions, a similarity matrix was constructed by counting the number of different residues for each pair of sequences (see data set S1 in the supplemental material).
Since the majority of TEM β-lactamase variants differ by fewer than 5 positions, they were analyzed by a network rather than by a phylogenetic tree (Fig. 1). From the similarity matrix, an adjacency matrix was constructed by linking all variant pairs that differ by exactly one residue. From this a network was constructed, where each node represents a variant and edges connect two variants that differ by one residue.
FIG 1.
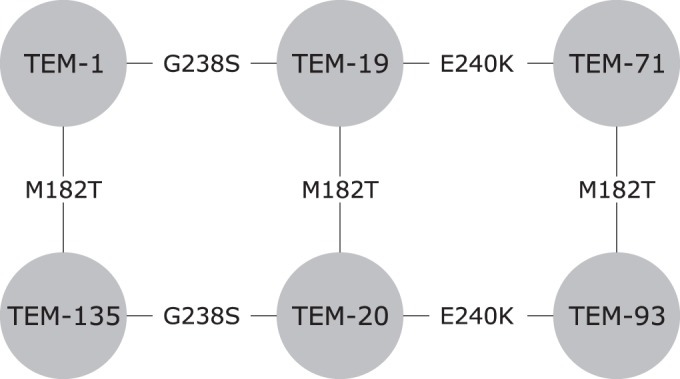
Multiple possible paths that lead from TEM-1 to TEM-93 with the mutation profile M182T, G238S, and E240K using existing variants.
Two hundred fifty-nine variants are clustered into a single connected network (Fig. 2). Additionally, 4 clusters with two nodes each are formed, containing the sequences TEML-302 and TEML-329, TEML-206 and TEML-242, TEML-157 and TEML-159, and TEML-171 and TEML-307. These 4 clusters, and 87 variants that were not connected to any other variant, were omitted from the network analysis (see Table S2 in the supplemental material).
FIG 2.
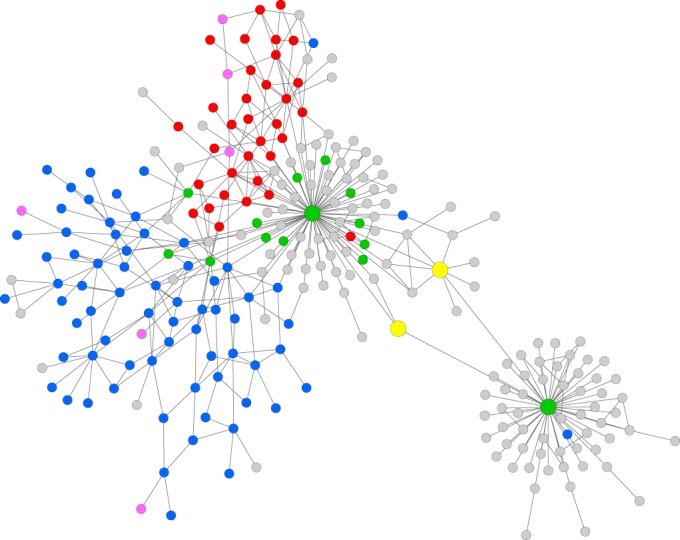
Network based on the adjacency matrix. Nodes are colored based on their phenotype (2b, green; 2be, blue; 2br, red; 2ber, pink; unknown, gray). The big green node in the left cluster represents TEM-1, and the big green node in the right cluster represents TEM-116. The two yellow nodes represent TEM-171 and TEM-181 (phenotype unknown), which form a bridge between the two clusters.
Characteristics of the TEM β-lactamase network.
The network consists of two visually separate clusters: cluster 1, with TEM-1 as a central variant, and cluster 2, with TEM-116 as a central variant (Fig. 2). The 200 variants in cluster 1 are highly interconnected, resulting in 328 edges. Cluster 2 consists of 59 variants and has a lower connectivity. Because most of the variants are exclusively connected to TEM-116, cluster 2 includes only 75 edges. The two central variants TEM-1 and TEM-116 differ by two mutations, V84I and A184V. Each of the two variants, TEM-171 and TEM-181, forms a bridge between TEM-1 and TEM-116. Thus, there are two alternative paths between TEM-1 and TEM-116 contributing the two mutations, V84I and A184V, respectively.
The degree centrality of each node was determined by counting the number of its edges (i.e., the variants that differ by a single mutation). The characteristics of the network were analyzed by determining the number of nodes with a given degree centrality, and a monotonically decreasing power law function with the exponent −1.167 was found (Fig. 3). Thus, the TEM variants form a scale-free network rather than a random network, which would have resulted in a Poisson distribution. In a scale-free network, a few nodes (hubs) are connected to many other nodes, and many nodes are connected to a few other nodes. The exponent of the power law function characterizes the distribution. For an exponent of −1, there are 10-fold fewer nodes with a 10-fold higher degree centrality.
FIG 3.
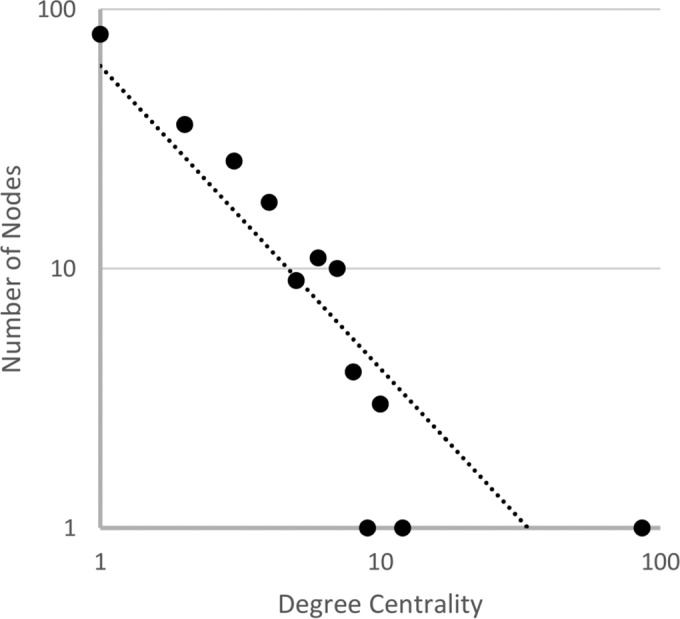
Number of nodes as a function of their degree centrality. The distribution of the degree centralities was approximated with a power function y = 60.401x−1.167, in which x is the degree centrality and y the number of nodes that have this degree centrality.
Clustering and phenotypes.
The highly connected network was clustered by iteratively removing nodes with the highest betweenness (see Table S3 in the supplemental material). Upon the removal of the highest ranking node (TEM-1), cluster 2, with TEM-116 as the central node, splits off (Fig. 4A). In the next round, TEM-116 was identified as the node with the highest betweenness. The removal of this node results in a disintegration of cluster 2 into 22 isolated sequences, 6 pairs of nodes, and 1 cluster of 10 nodes.
FIG 4.
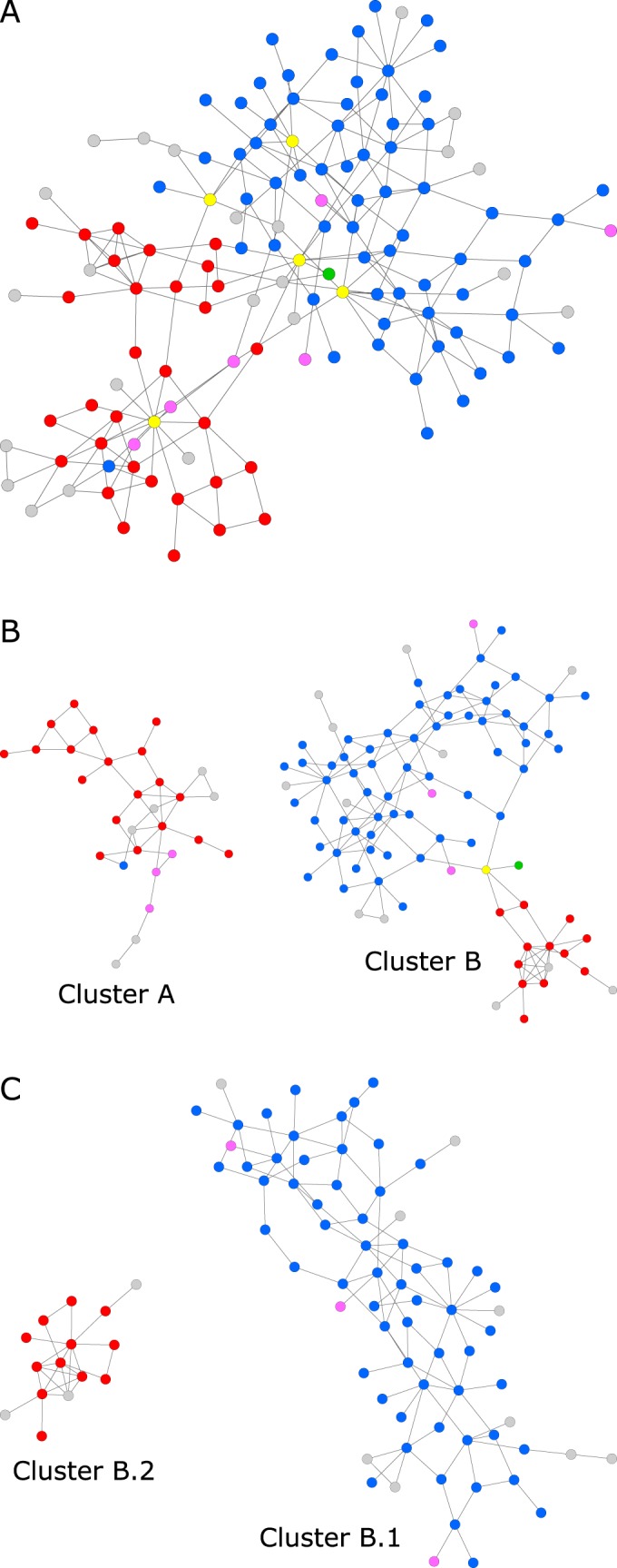
Recalculated networks after the subsequent removal of variants with the highest betweenness. Variants are colored according to their phenotype (2b, green; 2be, blue; 2br, red; 2ber, pink; unknown, gray). Variants with the highest betweenness are colored yellow. (A) Cluster 1 remaining after the removal of TEM-1. Cluster 2 with TEM-116 as the central variant is not shown. Variants marked with the highest betweenness are TEM-12, TEM-2, TEM-33, TEM-135, and TEM-63. (B) Cluster 1 split after the removal of variants into clusters A and B. The variant marked with the highest betweenness is TEM-110. (C) Cluster B split after the removal of variants into clusters B.1 and B.2.
In the third round, 5 nodes (TEM-12, TEM-2, TEM-33, TEM-135, and TEM-63) were removed, which results in a separation of the remaining network into 2 distinct clusters, A and B, and 8 isolated variants (Fig. 4B). Cluster A consists of 29 variants. Twenty-two of these variants share a mutation of the methionine at position 69. Thirteen variants share a mutation of asparagine at position 276. The phenotype is known for 23 of these variants: 19 variants have a 2br phenotype, 3 variants have a 2ber phenotype, and 1 variant has a 2be phenotype.
With the removal of the next variant, TEM-110, cluster B could be further separated into 2 clusters and 1 isolated variant (Fig. 4C). Cluster B.1 consists of 72 variants. The two most common mutations in this cluster are at positions 104 and 164, with 37 and 36 occurrences, respectively. The phenotype is known for 63 variants: 60 variants are of 2be phenotype, and 3 variants are of 2ber phenotype.
Cluster B.2 contains 15 variants. The variants in this cluster share a mutation of the arginine at position 244. Other mutations, such as Q39K or T265M, occurred no more than 2 times in this cluster. The phenotype is known for 12 variants in this cluster: all variants are 2br phenotypes.
By subsequent removal of further nodes, cluster B.1 splits into subclusters that accumulate certain mutations. Since the biochemical characterization of the TEM β-lactamases is limited to only four phenotypes, it is not obvious whether these 2be subclusters consist of variants with systematically different biochemical properties.
Quartet identification.
A quartet has two characterizing mutations; therefore, it consists of four variants where each variant is connected to two other variants by one mutation and to the third variant by two mutations (Fig. 1). The network contains 89 quartets, which are formed by 93 variants, 64 of them being part of more than one quartet. TEM-1 is part of 30 quartets; variants TEM-12 (R164S), TEM-17 (E104K), TEM-33 (M69L), and TEM-35 (M69L, and N276D) are part of 10 to 20 quartets. For 82 of the variants involved in quartets, the phenotype is known. Forty-eight quartets contain variants of the same phenotype (Fig. 5). In 13 quartets, three variants have the same phenotype and one variant has the 2b phenotype. Eleven quartets contain two pairs of adjacent variants with identical phenotypes inside the pair but different phenotypes between the two pairs (phenotypes 2b and 2br, 2b and 2be, and 2br and 2ber). Sixteen quartets contain at least one variant of an unknown phenotype. Most interestingly, one quartet (TEM-1, TEM-33, TEM-12, and TEM-154) contains one variant of each phenotype. Starting from TEM-1 (phenotype 2b), mutation M69L confers inhibitor resistance to TEM-33 (phenotype 2br), while mutation R164S confers extended-spectrum β-lactamase activity to TEM-12 (phenotype 2be). TEM-154 contains both mutations and is both inhibitor resistant and has ESBL activity (2ber). This observation supports the additivity of mutations M69L and R164S.
FIG 5.
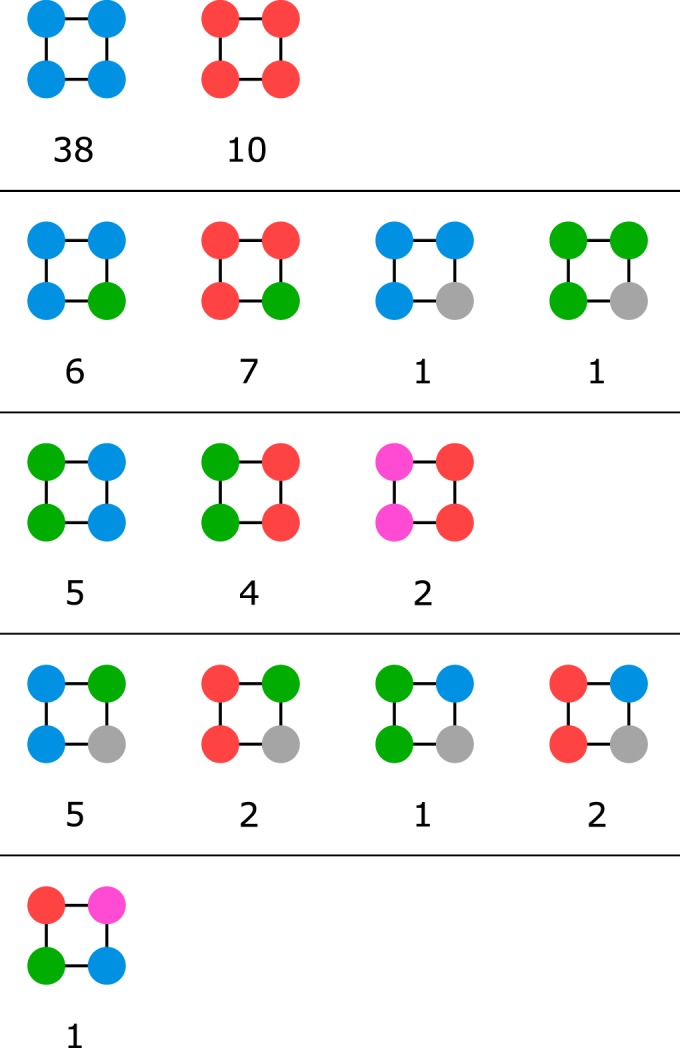
Distribution of phenotypes in the existing quartets (arranged into groups of 4 identical, 3 identical, 2-by-2 identical, 2 identical, and 4 different phenotypes). Quartets with more than 1 variant with an unknown phenotype are not listed. The coloring of the variants is according to the phenotype (2b, green; 2be, blue; 2br, red; 2ber, pink; unknown, gray).
Quartet prediction.
Because of the high connectivity of cluster 1, 93 of 200 variants are involved in a quartet. Therefore, we assume that quartets are ubiquitous. Thus, if two single mutations result in two active variants, then the combination of both mutations is expected to result in an active variant too. Therefore, variants that have not been found yet were predicted by constructing quartets from existing triplets. For each of the 4,134 triplets of the network, a variant was predicted which upgrades the triplet to a quartet (see Data set S2 in the supplemental material). In total, 3,916 variants were predicted to complete at least one triplet. Fifty-three variants complete 2 triplets each, 28 variants complete 3 triplets, 2 variants complete 4 triplets, and 4 variants complete 5 triplets each. We assume that the 4 variants that complete 5 triplets each have the highest probability of being active.
TEM-X1 has the mutations E104K, R164H, and G238S (Fig. 6A). Nine existing variants are involved in 5 quartets with TEM-X1. The 3 mutations present in TEM-X1 are present in 2 other variants in the cluster, TEM-107 and TEM-134, with the background mutations M182T and Q39K, respectively. The mutation combination E104K and R164H is present in 3 further variants, TEM-16, TEM-43, and TEM-6, with the background mutation M182T or Q39K or no background mutation, respectively. The mutation combination E104K and G238S is present in 3 further variants, TEM-52, TEM-3, and TEM-15, with the background mutation M182T or Q39K or no background mutation, respectively. The mutation E104K is present in 1 variant with no background mutations. TEM-X1 as a combination of three mutations (E104K, R164H, and G238S) very probably is active, because multiple quartets of pairs of these mutations exist as active variants. Interestingly, the combination of mutations R164H and G238S has not been observed yet, unless the stabilizing mutation M182T or the activity-increasing mutation Q39K was present.
FIG 6.

Predicted variants with the 5 completed quartets. Existing variants are colored according to their phenotype (2b, green; 2be, blue; 2br, red; 2ber, pink; unknown, gray). Predicted variants are colored black. (A) TEM-X1 with the mutations E104K, R164H, and G238S; (B) TEM-X2 with the mutations E104K and T265M; (C) TEM-X3 with the mutation E240K; (D) TEM-X4 with the mutations R164H and M182T.
TEM-X2 has the mutations E104K and T265M (Fig. 6B). Nine existing variants are involved in quartets with TEM-X2. The 2 mutations present in TEM-X2 are present in 2 variants in the cluster, TEM-9 and TEM-4, with the background mutations R164S and G238S, respectively. The mutation E104K is present in 3 variants, TEM-26, TEM-15, and TEM-17, with the background mutation R164S or G238S or no background mutation, respectively. The mutation T265M is present in 3 variants, TEM-102, TEM-25, and TEM-110, with the background mutation R164S or G238S or no background mutation, respectively. In addition, TEM-1 is part of the cluster.
TEM-X3 has the mutation E240K (Fig. 6C). Eleven existing variants are involved in quartets with TEM-X3. The mutation present in TEM-X3 also is present in 5 other variants in the cluster, including TEM-28, TEM-144, and TEM-10, with the background mutations R164H, R164C, and R164S, respectively, as well as TEM-189 and TEM-71, with the background mutations M69L and G238S, respectively.
TEM-X4 has the mutations R164H and M182T (Fig. 6D). Nine existing variants are involved in quartets with TEM-X2. The 2 mutations present in TEM-X4 also are present in 1 variant in the cluster, TEM-43, with the background mutation E104K. The mutation R164H is present in 2 variants, TEM-6 and TEM-29, with the background mutation E104K or no background mutation, respectively. The mutation M182T is present in 4 variants, TEM-106, TEM-63, TEM-165, and TEM-135, with the background mutation E104K, E104K/R164S, or R164S or no background mutation, respectively. The mutation R164S is present in TEM-12 without background mutation. In addition, TEM-1 is part of the cluster.
DISCUSSION
The sequence space of TEM β-lactamases is formed by a largely unexplored network.
By analyzing the TEM mutation table, maintained by the Lahey clinic (www.lahey.org/Studies/temtable.asp), and the NCBI protein database (16), sequence information of 474 unique TEM β-lactamase variants was collected. This number is expected to increase rapidly in the near future due to the decreasing costs of DNA sequencing (17). Two hundred thirty-seven of these variants form a highly connected, scale-free network formed mainly by mutations at 15 different positions. Because TEM β-lactamase variants confer antibiotic resistance and exhibit a broad range of biochemical properties, it is of the utmost interest to learn more about the sequence space of viable sequences and their substrate specificities and inhibition profiles. However, it is not clear which fraction of the 2010 to ≈1013 theoretical variants are viable and active. A comprehensive analysis of the known sequence space bears the promise of predicting yet-unknown viable variants and their properties.
To analyze a large number of globally similar sequences, two fundamentally different methods, phylogenetic trees and networks, can be applied. The construction of binary trees by distance-based or maximum likelihood methods (18, 19) assumes an evolutionary model with additivity of evolutionary distances and the existence of extinct ancestor sequences. The interpretation of phylogenetic trees is highly intuitive, and they are an appropriate model for assigning sequences to families, subtypes, or clades. However, phylogenetic tree analysis will fail if all members have similar distances from each other (20). This becomes especially apparent for variants forming multiple connected quartets which cannot be represented in a binary tree assuming additivity of distances (see Fig. S1 in the supplemental material). A hierarchical structure is not necessarily a given, as the evolution of a sequence could have taken multiple alternative paths with very small steps between them.
While phylogenetic trees are based on the assumption of unique ancestors, J. Maynard Smith suggested a model of protein evolution in a network of functional proteins where all viable proteins form a continuous network (21). Evolution is described as a walk in sequence space with multiple equally probable evolutionary paths between variants, in contrast to phylogenetic tree constructions (10). In a network description of sequence space, gaps in the contemporary sequence space separating sequence clusters are a consequence of our limited knowledge of sequence space rather than of ancient bifurcations resulting in separate clades (21). Therefore, the currently known networks are expected to be embedded in a highly connected network of mostly unknown TEM β-lactamase variants. The number of unicellular organisms has been estimated to be 1030 cells (22), which provides an enormous capacity for genetic diversity. All TEM β-lactamase variants included in the analysis of cluster 1 are highly similar and mainly differ in only 15 positions. Although the theoretical sequence space has an impressive size of 1013 variants, all variants could be completely encoded and expressed by the existing cells in the biosphere. However, the size and structure of the viable sequence space is difficult to predict from the small number of known variants. Many stable, catalytically active, and expressible variants are expected to exist in the biosphere without being detected yet, or they might develop in the future.
Sequence space and phenotype.
The cluster analysis of the TEM β-lactamase network identified subclusters which are formed predominantly by variants of the same phenotype. This applies especially to the 2be and 2br phenotypes (5, 23). The tight link between genotype and phenotype is useful for predicting the phenotype of unknown variants and for designing variants with desired properties. The identification of these highly connected clusters also sheds light on the evolutionary paths toward new TEM β-lactamase variants. Clusters B.1 and B.2 are populated predominantly by 2be and 2br variants, respectively. However, cluster B.1 also contains a small number of variants with new properties, namely, the 2ber type, which have emerged by a single mutation from variants of phenotype 2be of cluster B.1 (TEM-68, TEM-109, and TEM-121). While they are formally classified as variants of the 2ber phenotype, their inhibitor resistance is less pronounced than that of the 2br variants in cluster B.2 (23). Therefore, a more detailed analysis of these variants would be highly interesting in regard to the influence of single mutations on the phenotype.
Prediction of viable variants.
In general, the combination of existing mutations into a new variant does not guarantee that the new variant will be active. While the additivity of single mutations has been shown for distant mutations at rigid molecular interfaces (22, 24), other observations showed the nonadditivity of mutations due to long-range interactions, spatial constraints, stability-enhancing mutations, or other epistatic effects (25, 26). For TEM β-lactamases it could be shown that almost 50% of the naturally occurring variants are part of quartets, some of them even of multiple quartets. This demonstrates that a highly connected subgroup of sequences exists in the TEM β-lactamase network that tolerate the addition of two single mutations into a double mutation variant. In addition, the analysis of all naturally occurring quartets of a known phenotype showed that in 61 cases the additivity of mutations in regard to the phenotype exists. Thus, the iterative construction of quartets from known triplets offers a promising strategy to predict the still-obscure part of the viable sequence space of TEM variants.
During recent years, many new TEM variants have been published and integrated into the LacED. During the last 24 months, 4 variants emerged, which we identified in January 2014 by quartet prediction: (i) TEML-275 (A184V, E240G, and W290L; AIA18198) was predicted to complement the triplet consisting of TEM-1, TEM-181 (A184V), and TEM-207 (E240G); (ii) TEML-301 (M69L, R164S, and M182T; AJW77574), complementing triplet TEM-12 (R164S), TEM-154 (M69L and R164S), and TEM-165 (R164S and M182T); (iv) TEM-215 (H153R; AJO16045), complementing triplet TEM-1, TEM-112 (H153R and G238S), and TEM-19 (G238S); and (iv) TEML-327 (W165G and N276D; AJC64566), complementing triplet TEM-190 (M69L, W165G, and N276D), TEM-35 (M69L and N276D), and TEM-84 (N276D). These recent results strongly support the concept of quartet prediction as a promising method for the prediction of viable TEM variants which have not been discovered yet.
Role of the background.
The quartet prediction is based on the observations that the combination of two mutations that result in a stable and active TEM β-lactamase results in a stable and active enzyme and that the phenotype classification is consistent. However, this does not imply that the effects of mutations are strictly additive. While the mutations R164S and G238S increased the catalytic efficiency of TEM-1 toward cefotaxime 5- and 130-fold, respectively, the double mutation resulted in only a 5-fold increase due to local interactions between the mutated sites (25). However, the double mutant still is of the 2be phenotype, like the single-mutant variants R164S and G238S, while TEM-1 has the 2b phenotype.
The characterizing mutations for the existing quartets often were either stabilizing (M182T and T265M [26–28]) or showed a slight increase in enzyme activity (Q39K and E104K [29, 30]). Mutations that confer stability are not only required to compensate for the loss of stability caused by mutations that increase the substrate spectrum or confer inhibitor resistance (27) but also directly result in an increase of MIC, since increased protein stability results in an increased fraction of properly folded β-lactamase (31). Mutations that show only a slight increase in functionality were suggested to provide a slight advantage at low β-lactam concentrations, allowing the acquisition of new mutations (29). All 4 predicted sequences that completed 5 quartets completed at least one quartet that contains stabilizing (M182T and T265M) or activity-increasing (Q39K and E104K) mutations as characterizing mutations. Therefore, these predicted variants most probably exist. This criterion also could be applied to assess the predictive quality of the other variants generated by quartet prediction.
Thus, quartet prediction and TEM β-lactamase network analysis are valuable tools to gain new insights into the viable sequence space of TEM β-lactamases and to predict phenotypes. The highly connected sequence space of TEM β-lactamases is ideally suited for network analysis and demonstrates the strengths of network analysis over tree reconstruction methods. The currently known 474 TEM variants represent only the tip of an iceberg of functional variants which form a huge natural reservoir of enzymes with clinical relevance.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge funding by the Ministry of Science, Research, and the Arts of Baden-Württemberg (Biosynthesis netWork BW2) and by the German Research Foundation DFG (SFB716).
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AAC.02930-15.
REFERENCES
- 1.Datta N, Kontomichalou P. 1965. Penicillinase synthesis controlled by infectious R factors in Enterobacteriaceae. Nature 208:239–241. doi: 10.1038/208239a0. [DOI] [PubMed] [Google Scholar]
- 2.Bush K, Jacoby G. 1997. Nomenclature of TEM beta-lactamases. J Antimicrob Chemother 39:1–3. doi: 10.1093/jac/39.1.1. [DOI] [PubMed] [Google Scholar]
- 3.Lerbech AM, Opintan JA, Bekoe SO, Ahiabu M-A, Tersbøl BP, Hansen M, Brightson KTC, Ametepeh S, Frimodt-Møller N, Styrishave B. 2014. Antibiotic exposure in a low-income country: screening urine samples for presence of antibiotics and antibiotic resistance in coagulase negative staphylococcal contaminants. PLoS One 9:e113055. doi: 10.1371/journal.pone.0113055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luyt C-E, Bréchot N, Trouillet J-L, Chastre J. 2014. Antibiotic stewardship in the intensive care unit. Crit Care 18:480. doi: 10.1186/s13054-014-0480-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bush K, Jacoby GA, Medeiros AA. 1995. A functional classification scheme for β-lactamases and its correlation with molecular structure. Antimicrob Agents Chemother 39:1211–1233. doi: 10.1128/AAC.39.6.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambler RP. 1980. The structure of beta-lactamases. Philos Trans R Soc Lond B Biol Sci 289:321–331. doi: 10.1098/rstb.1980.0049. [DOI] [PubMed] [Google Scholar]
- 7.Thai QK, Bös F, Pleiss J. 2009. The Lactamase Engineering Database: a critical survey of TEM sequences in public databases. BMC Genomics 10:390. doi: 10.1186/1471-2164-10-390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thai QK, Pleiss J. 2010. SHV Lactamase Engineering Database: a reconciliation tool for SHV β-lactamases in public databases. BMC Genomics 11:563. doi: 10.1186/1471-2164-11-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Widmann M, Pleiss J, Oelschlaeger P. 2012. Systematic analysis of metallo-β-lactamases using an automated database. Antimicrob Agents Chemother 56:3481–3491. doi: 10.1128/AAC.00255-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fitch WM. 1997. Networks and viral evolution. J Mol Evol 44:S65–S75. doi: 10.1007/PL00000059. [DOI] [PubMed] [Google Scholar]
- 11.Guthrie VB, Allen J, Camps M, Karchin R. 2011. Network models of TEM β-lactamase mutations coevolving under antibiotic selection show modular structure and anticipate evolutionary trajectories. PLoS Comput Biol 7:e1002184. doi: 10.1371/journal.pcbi.1002184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST (supplemental material). Bioinformatics 26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 14.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 15.Girvan M, Newman MEJ. 2002. Community structure in social and biological networks. Proc Natl Acad Sci U S A 99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL. 2008. GenBank. Nucleic Acids Res 36:D25–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayden EC. 2014. The $1,000 genome. Nature 507:295. [DOI] [PubMed] [Google Scholar]
- 18.Nakhleh L, Jin G, Zhao F, Mellor-Crummey J. 2005. Reconstructing phylogenetic networks using maximum parsimony. Proc IEEE Comput Syst Bioinform Conf 2005:93–102. [DOI] [PubMed] [Google Scholar]
- 19.Fitch WM, Margoliash E. 1967. Construction of phylogenetic trees. Science 155:279–284. doi: 10.1126/science.155.3760.279. [DOI] [PubMed] [Google Scholar]
- 20.Widmann M, Pleiss J. 2014. Protein variants form a system of networks: microdiversity of IMP metallo-beta-lactamases. PLoS One 9:e101813. doi: 10.1371/journal.pone.0101813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith JM. 1970. Natural selection and the concept of a protein space. Nature 225:563–564. doi: 10.1038/225563a0. [DOI] [PubMed] [Google Scholar]
- 22.Zimmer DB, Eubanks JO, Ramakrishnan D, Criscitiello MF. 2013. Evolution of the S100 family of calcium sensor proteins. Cell Calcium 53:170–179. doi: 10.1016/j.ceca.2012.11.006. [DOI] [PubMed] [Google Scholar]
- 23.Bush K, Jacoby GA. 2010. Updated functional classification of beta-lactamases. Antimicrob Agents Chemother 54:969–976. doi: 10.1128/AAC.01009-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitman WB, Coleman DC, Wiebe WJ. 1998. Prokaryotes: the unseen majority. Proc Natl Acad Sci U S A 95:6578–6583. doi: 10.1073/pnas.95.12.6578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dellus-Gur E, Elias M, Caselli E, Prati F, Salverda MLM, de Visser JAGM, Fraser JS, Tawfik DS. 2015. Negative epistasis and evolvability in TEM-1 β-lactamase—the thin line between an enzyme's conformational freedom and disorder. J Mol Biol 427:2396–2409. doi: 10.1016/j.jmb.2015.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang W, Palzkill T. 1997. A natural polymorphism in beta-lactamase is a global suppressor. Proc Natl Acad Sci U S A 94:8801–8806. doi: 10.1073/pnas.94.16.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang X, Minasov G, Shoichet BK. 2002. Evolution of an antibiotic resistance enzyme constrained by stability and activity trade-offs. J Mol Biol 320:85–95. doi: 10.1016/S0022-2836(02)00400-X. [DOI] [PubMed] [Google Scholar]
- 28.Salverda MLM, De Visser JAGM, Barlow M. 2010. Natural evolution of TEM-1 β-lactamase: experimental reconstruction and clinical relevance. FEMS Microbiol Rev 34:1015–1036. doi: 10.1111/j.1574-6976.2010.00222.x. [DOI] [PubMed] [Google Scholar]
- 29.Blazquez J, Morosini MI, Negri MC, Gonzalez-Leiza M, Baquero F. 1995. Single amino acid replacements at positions altered in naturally occurring extended-spectrum TEM beta-lactamases. Antimicrob Agents Chemother 39:145–149. doi: 10.1128/AAC.39.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petit A, Maveyraud L, Lenfant F, Samama JP, Labia R, Masson JM. 1995. Multiple substitutions at position 104 of beta-lactamase TEM-1: assessing the role of this residue in substrate specificity. Biochem J 305:33–40. doi: 10.1042/bj3050033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jacquier H, Birgy A, Nagard Le H, Mechulam Y, Schmitt E, Glodt J, Bercot B, Petit E, Poulain J, Barnaud G, Gros P-A, Tenaillon O. 2013. Capturing the mutational landscape of the beta-lactamase TEM-1. Proc Natl Acad Sci U S A 110:13067–13072. doi: 10.1073/pnas.1215206110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rice P, Longden I, Bleasby A. 2000. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16:276–277. doi: 10.1016/S0168-9525(00)02024-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.