

Abstract
Francisella tularensis is the causative agent of tularemia and a category A potential agent of bioterrorism, but the pathogenic mechanisms of F. tularensis are largely unknown. Our previous transposon mutagenesis screen identified 95 lung infectivity-associated F. tularensis genes, including those encoding the Lon and ClpP proteases. The present study validates the importance of Lon and ClpP in intramacrophage growth and infection of the mammalian host by using unmarked deletion mutants of the F. tularensis live vaccine strain (LVS). Further experiments revealed that lon and clpP are also required for F. tularensis tolerance to stressful conditions. A quantitative proteomic comparison between heat-stressed LVS and the isogenic Lon-deficient mutant identified 29 putative Lon substrate proteins. The follow-up protein degradation experiments identified five substrates of the F. tularensis Lon protease (FTL578, FTL663, FTL1217, FTL1228, and FTL1957). FTL578 (ornithine cyclodeaminase), FTL663 (heat shock protein), and FTL1228 (iron-sulfur activator complex subunit SufD) have been previously described as virulence-associated factors in F. tularensis. Identification of these Lon substrates has thus provided important clues for further understanding of the F. tularensis stress response and pathogenesis. The high-throughput approach developed in this study can be used for systematic identification of the Lon substrates in other prokaryotic and eukaryotic organisms.
INTRODUCTION
Francisella tularensis is a highly infectious Gram-negative, facultative intracellular bacterium that causes tularemia in many species, including humans (1). F. tularensis is divided into four subspecies or biotypes: F. tularensis subsp. tularensis (type A), F. tularensis subsp. holarctica (type B), F. tularensis subsp. mediasiatica, and F. tularensis subsp. novicida. While the F. tularensis subspecies are all genetically similar and capable of causing lethal infection in mice, only the type A and B strains are associated with human disease (2). Because of its high virulence, extreme infectivity, and ability to disseminate by aerosols, F. tularensis is categorized as a category A bioterrorism agent (3). An attenuated live vaccine strain (LVS) derivative of a type B strain has been used extensively as a model organism to study tularemia pathogenesis due to the ability of LVS to cause a lethal infection in mice that resembles human tularemia (4, 5). However, the molecular and genetic bases of F. tularensis infectivity and virulence are not completely understood.
Several high-throughput in vivo mutagenesis screens have identified many putative virulence-associated genes of F. tularensis (6–8). Our signature-tagged mutagenesis (STM) study identified 95 lung infection-associated genes in F. tularensis LVS (8), particularly those encoding the Lon and ClpP proteases. Lon and ClpP are ATP-dependent proteases that are found widely in both prokaryotic and eukaryotic organisms (9, 10). Lon is a hexameric serine protease with a carboxyl-terminal catalytic domain and an amino-terminal ATPase domain composed of Walker A and Walker B motifs, required for hydrolysis and ATP binding, respectively. ClpP is a serine protease that is composed of ClpP (protease subunit) and ATPases (ClpX or ClpA in Gram-negative bacteria and ClpX and ClpC in Gram-positive bacteria). The catalytic sites of these proteases are located in the central chamber formed by the barrel-shaped organization of the subunits. Substrate recognition of Lon and ClpP is specified by the ATPase subunits through direct interaction with target proteins and/or indirect association with other adaptor proteins. Substrates need to be unfolded and translocated by the associated ATPases into the protease chambers in response to physiological conditions of the cells (9).
The main functions of Lon and ClpP are to degrade damaged or incompletely synthesized proteins and to regulate the abundance and activity levels of the selected cellular proteins in response to environmental and cellular conditions (9, 11). Lon regulates a wide range of bacterial functions, including encapsulation (11, 12), motility (13), competence (14), heat shock response (15), DNA replication and repair (16, 17), drug resistance (18), and production of virulence factors (19). Significant attenuation in fitness and/or virulence has been reported for the lon mutants of many pathogens, including Brucella abortus (20), Pseudomonas aeruginosa (21), Salmonella enterica serovar Typhimurium (22, 23), and Yersinia pestis (24). ClpP is involved in the proteolytic regulation of the heat shock response (25, 26), cell division (27–29), competence (26, 30), and biofilm formation (31). Disrupting ClpP also leads to impaired in vivo survival in many pathogens, such as Helicobacter pylori (32), Mycobacterium tuberculosis (33), and Staphylococcus aureus (34). Lastly, ClpP is a promising target for antimicrobials. ClpP-activating acyldepsipeptides have been shown to effectively kill both actively growing bacteria (35) and persisters (36).
Previous studies have identified many proteins that are specifically degraded by ClpP in Escherichia coli (37), Caulobacter crescentus (28), and S. aureus (38, 39) by a substrate-trapping method using inactive forms of ClpP or ClpC. Raju et al. identified 132 and 107 putative ClpP substrates in M. tuberculosis and Mycobacterium smegmatis, respectively, by quantitative proteomic profiling of the wild-type and ClpP knockdown strains (40). The two ClpP homologs (ClpP1 and ClpP2) in M. tuberculosis and M. smegmatis form the ClpP proteolytic core and are essential for mycobacterial survival in vitro and during infection of mice (33, 40, 41). However, systematic identification of the Lon substrates has not been reported. Bissonnette et al. previously mentioned the identification of approximately 200 putative Lon substrates in E. coli by a substrate-trapping method as reported in a personal communication (15), but no details of this study have been published.
The Francisella Lon and ClpP proteases are encoded by the protease locus, which consists of four genes (tig, clpP, clpX, and lon) (Fig. 1A). The available full genome sequences reveal virtually identical sequences in the locus among the strains from the four subspecies of F. tularensis. Although transposon insertions in each of tig, clpP, clpX, and lon led to significant attenuation of F. tularensis LVS in the murine model of respiratory tularemia in our previous study (8), the results could not define the precise contributions of individual genes to the F. tularensis physiology and pathogenesis because of potential polar effects of the transposon insertions on the downstream genes. In this work, we demonstrated the importance of Lon and ClpP in F. tularensis stress tolerance and infection of mammalian hosts. Surprisingly, the clpX deletion mutant of LVS had no obvious phenotype. Lastly, we identified five authentic substrates of F. tularensis Lon by a combination of phenotypic enrichment of the potential Lon substrates, tandem mass spectrometry (MS/MS)-based isobaric multiplexed quantitative proteomics, and protein degradation.
FIG 1.

Transcription of the F. tularensis protease locus. (A) Schematic illustration of the gene arrangement in the protease locus. The gene orientations and promoters identified in panel C are marked by straight and bent arrows, respectively. (B) Cotranscription of the protease locus genes determined by RT-PCR. FTL890-tig, tig-clpP, clpP-clpX, clpX-lon, and lon-hupB junctions were amplified using cDNA, RNA, or DNA as a template. A 1,200-bp internal fragment of lon was amplified as a positive control. The sizes of molecular markers are indicated at the sides of the panel in kilobases. (C) Detection of promoter activity of the 5′ noncoding sequence of each gene in the protease locus with the β-galactosidase reporter. The 5′ noncoding sequence (∼500 bp) immediately upstream of each gene in the protease locus was placed in front of a promoterless β-galactosidase gene in the pMP633 shuttle plasmid. The LVS derivatives containing individual reporter constructs were grown to an OD600 of 0.4 in MHB in the presence of hygromycin (200 μg/ml) at 37°C before being lysed to measure β-galactosidase activity. The constructs for the E. coli pTAC and F. tularensis clpB promoters were used as negative and positive controls, respectively. Bars represent the average β-galactosidase activity ± SEM (n = 3) from one of three independent experiments. (D) Transcription of the protease locus genes in wild-type (WT) LVS and the isogenic tig (tig::Tn) and clpP (clpP::Tn) transposon insertion mutants. The mRNA levels of the protease locus genes, as well as hupB, were quantified by qRT-PCR and normalized on the basis of the DNA helicase gene (FTL1656). The fold changes of the mRNA level in the mutants were converted to percentages of the wild-type value. Bars represent the average ± SEM (n = 3) from one of three independent experiments. (E) Heat-inducible transcription of the protease locus genes in LVS under physiological (37°C) or heat stress (44°C) conditions. The transcripts of the target genes with the total RNA extracts from the LVS culture grown at 37°C or 44°C were quantified and presented as in panel D. (F) Heat-inducible expression transcription of lon from the tig promoter but not from its own promoter. The lon transcripts in LVS (WT) and isogenic tig and clpP transposon insertion mutants were measured with the total RNA extracts from the cultures grown at 37°C or 44°C as for panel D.
MATERIALS AND METHODS
Bacterial cultivation, plasmids, chemicals, and primers.
The bacterial strains and plasmids used in this work are listed in Table 1. For all experiments except those for growth assessment and protein induction, F. tularensis LVS and its derivatives were cultured in Mueller-Hinton broth (MHB) as described previously (8). Inducible production of proteins in LVS derivatives was carried out in Chamberlain's defined medium (CDM) (42). Where necessary, kanamycin (10 μg/ml) or hygromycin (200 μg/ml) was added to the medium. For measurement of the growth kinetics of F. tularensis LVS and its derivatives, the strains were individually cultured overnight in 5 ml of CDM at 37°C with aeration. The bacterial cells were diluted to an optical density at 600 nm (OD600) of approximately 0.05 with 5 ml of fresh CDM or MHB and incubated at 37°C with aeration. Growth kinetics was monitored by measuring the OD600 at the indicated time points. E. coli strains were grown in Luria-Bertani (LB) medium in the presence or absence of ampicillin (100 μg/ml), kanamycin (50 μg/ml), or hygromycin (200 μg/ml). Unless stated otherwise, all bacterial media and chemicals were purchased from Sigma (Shanghai, China). All enzymes for DNA cloning were supplied by New England BioLabs (NEB) (Beijing, China). The primers used in this study are listed in Table S1 in the supplemental material.
TABLE 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Description | Antibiotic resistancea | Reference or source |
|---|---|---|---|
| F. tularensis subsp. holarctica strains | |||
| LVS | Live vaccine strain | 8 | |
| ST1101 | LVS derivative, ΔmglA | 75 | |
| ST1116 | LVS derivative, ΔclpX | This study | |
| ST1118 | LVS derivative, ΔclpP | This study | |
| ST1120 | LVS derivative, Δlon | This study | |
| ST1284 | LVS derivative, tig::Tn, transposon insertion between nucleotides 1104 and 1105 in tig | Kanr | 8 |
| ST1292 | LVS derivative, clpP::Tn, transposon insertion between nucleotides 255 and 256 in clpP | Kanr | 8 |
| ST1796 | ST1118 (pST1760); ΔclpP mutant complemented in trans with the wild-type LVS clpP driven by native tig promoter | Hygr | This study |
| ST1889 | ST1120 (pST1864); LVS Δlon mutant complemented in trans with wild-type LVS lon driven by native tig promoter | Hygr | This study |
| E. coli strains | |||
| DH5α | F− endA1 glnV44 thi-1 recA1 relA1 gyrA96 deoR nupG ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 hsdR17(rK− mK+) λ− | Biomed (Beijing, China) | |
| TOP10F′ | F′ [lacIq Tn10(Tetr)] mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 deoR nupG recA1 araD139 Δ(ara-leu)7697 galU galK rpsL(Strr) endA1 λ− | Invitrogen | |
| BL21 | B F− dcm ompT hsdS(rB− mB−) gal | Biomed | |
| Rosetta | B F− dcm ompT hsdS(rB− mB−) gal dcm λ (DE3 [lacI lacUV5-T7 gene 1 ind1 sam7 nin5]) | Biomed | |
| ER2566 | F− λ− fhuA2 [lon] ompT lacZ::T7 gene 1 gal sulA11 Δ(mcrC-mrr)114::IS10R(mcr-73::miniTn10-Tets)2 R(zgb-210::Tn10)(Tets) endA1 [dcm] | NEB | |
| Plasmids | |||
| pACYCDuet-1 | E. coli protein expression vector | Cmr | Novagen |
| pBS | pBluescript II (SK−) | Ampr | Stratagene |
| pBAD18 | E. coli protein expression vector | Ampr | 53 |
| pDL | Bacillus subtilis integration vector with bgaB of Bacillus stearothermophilus as a reporter gene | Cmr Ampr | Bacillus Genetic Stock Center |
| pEDL17 | Francisella-E. coli shuttle plasmid | Hygr | 57 |
| pET28a | E. coli protein expression vector | Kanr | Novagen |
| pGEX-2T | E. coli protein expression vector | Ampr | GE Healthcare Bio-Science |
| pMP590 | F. tularensis suicide vector | Kanr | 76 |
| pMP633 | Francisella-E. coli shuttle plasmid | Hygr | 76 |
| pST1010 | pBS::ΔclpP | Ampr | This study |
| pST1012 | pBS::ΔclpX | Ampr | This study |
| pST1100 | pBS::Δlon | Ampr | This study |
| pST1105 | pMP590::Δlon | Kanr | This study |
| pST1136 | pMP590::ΔclpP | Kanr | This study |
| pST1138 | pMP590::ΔclpX | Kanr | This study |
| pST1728 | pMP633::pTAC | Hygr | This study |
| pST1760 | pST1728::TIG-clpP | Hygr | This study |
| pST1864 | pST1728::pTIG-lon | Hygr | This study |
| pST1923 | pST1728::bgaB | Hygr | This study |
| pST1934 | pST1923::pTIG-bgaB | Hygr | This study |
| pST1936 | pST1923::pLON-bgaB | Hygr | This study |
| pST1938 | pST1923::pCLPB-bgaB | Hygr | This study |
| pST1940 | pST1923::pHUPB-bgaB | Hygr | This study |
| pST1956 | pST1923::pCLPP-bgaB | Hygr | This study |
| pST1958 | pST1923::pCLPX-bgaB | Hygr | This study |
| pST2700 | pGEX-2T derivate; a multiple-cloning site was added | Ampr | This study |
| pST3001 | pST2700::lon | Ampr | This study |
| pST3621 | pET28a derivate; an AscI site was added | Kanr | This study |
| pST3749 | pST2700::clpX | Ampr | This study |
| pST3750 | pST2700::clpP | Ampr | This study |
| pST4218 | pST3621::FTL1017 | Kanr | This study |
| pST4609 | pACYCDuet-1::FTL663 | Cmr | This study |
| pST4611 | pACYCDuet-1::FTL1957 | Cmr | This study |
| pST4635 | pBAD18::lon | Ampr | This study |
| pST4816 | pACYCDuet-1::FTL478 | Cmr | This study |
| pST4818 | pACYCDuet-1::FTL1017 | Cmr | This study |
| pST4819 | pACYCDuet-1::clpX | Cmr | This study |
| pST4861 | pEDL17::FTL663 | Hygr | This study |
| pST4863 | pEDL17::FTL1957 | Hygr | This study |
| pST4865 | pEDL17::FTL478 | Hygr | This study |
| pST4867 | pEDL17::FTL1017 | Hygr | This study |
| pST4869 | pEDL17::clpX | Hygr | This study |
| pST5007 | pACYCDuet-1::FTL578 | Cmr | This study |
| pST5009 | pACYCDuet-1::FTL1217 | Cmr | This study |
| pST5072 | pEDL17::FTL1217 | Hygr | This study |
| pST6927 | pACYCDuet-1::FTL40 | Cmr | This study |
| pST6928 | pACYCDuet-1::FTL398 | Cmr | This study |
| pST6930 | pACYCDuet-1::FTL687 | Cmr | This study |
| pST6931 | pACYCDuet-1::FTL891 | Cmr | This study |
| pST6935 | pACYCDuet-1::FTL1191 | Cmr | This study |
| pST6936 | pACYCDuet-1::FTL1228 | Cmr | This study |
| pST6937 | pACYCDuet-1::FTL1267 | Cmr | This study |
| pST6938 | pACYCDuet-1::FTL1358 | Cmr | This study |
| pST6939 | pACYCDuet-1::FTL1609 | Cmr | This study |
| pST6955 | pEDL17::FTL578 | Hygr | This study |
| pST6956 | pEDL17::FTL1228 | Hygr | This study |
| pST7043 | pACYCDuet-1::FTL87 | Cmr | This study |
| pST7044 | pACYCDuet-1::FTL593 | Cmr | This study |
| pST7045 | pACYCDuet-1::FTL595 | Cmr | This study |
| pST7046 | pACYCDuet-1::FTL599 | Cmr | This study |
| pST7048 | pACYCDuet-1::FTL1611 | Cmr | This study |
| pST7049 | pACYCDuet-1::FTL1724 | Cmr | This study |
Ampr, ampicillin resistance; Cmr, chloramphenicol resistance; Kanr, kanamycin resistance; Hygr, hygromycin resistance.
Isolation of bacterial RNA and reverse transcriptase PCR (RT-PCR).
Total RNA was isolated from F. tularensis LVS cultures using the SV total RNA isolation kit (Promega, Madison, WI) according to the supplier's instructions. LVS and its derivatives were cultured in MHB to an OD600 of ∼0.4 before being harvested for RNA purification. To remove residual DNA, RNA samples were treated with the DNA-free DNA treatment and removal kit (Ambion, Austin, TX). RNA concentration and integrity were determined by spectrophotometry and agarose gel electrophoresis, respectively.
Cotranscription of the F. tularensis genes was determined by RT-PCR essentially as described previously (43). The intergenic cDNAs were synthesized with the total RNA extract of LVS using the SuperScript III kit (Invitrogen, Carlsbad, CA) and primers Pr1050 (tig), Pr1426 (clpP), Pr1048 (clpX), Pr1203 (lon), and Pr1658 (hupB). The junction PCR products were generated with the above-described reverse transcription reactions and primer pairs Pr1049/Pr1050 (hyp-tig), Pr1298/Pr1426 (tig-clpP), Pr1425/Pr1048 (clpP-clpX), Pr1370/Pr1203 (clpX-lon), and Pr1002/Pr1658 (lon-hupB). Control reactions included amplifications of RNA lacking cDNA and chromosomal DNA.
Promoter mapping in the protease locus was performed using quantitative RT-PCR (qRT-PCR) with the total RNA extract of LVS and isogenic transposon mutants essentially as described previously (44, 45). The tig (tig::Tn, ST1284) and clpP (clpP::Tn, ST1292) transposon insertion mutants were generated in our previous study (8). Our unpublished data showed that the EZ-TN transposon possessed transcriptional termination activity and thereby exerted a polar effect on the gene downstream of the insertion site (J.-R. Zhang, unpublished observation). The target genes were amplified with primer pairs Pr11118/Pr11119 (tig), Pr1425/Pr1426 (clpP), Pr1427/Pr1428 (clpX), Pr1429/Pr1430 (lon), and Pr1812/Pr1813 (hupB). The data were normalized to the DNA helicase gene (FTL1656), which was amplified with primers Pr7867 and Pr7868 as described previously (46). The fold change of target gene transcription in each mutant is presented as mean percentage of the wild-type value ± standard error of the mean (SEM) (n = 3).
The transcriptional response to heat stress was determined with cultures of LVS or isogenic tig and clpP transposon mutants by qRT-PCR as described above. Briefly, LVS and its derivatives were grown in 5 ml MHB at 37°C to OD600 of 0.4 and split into two parts. One part of the culture was placed at 37°C, whereas the other was incubated at 44°C for 20 min before being harvested for RNA extraction, as described previously (46). The total RNA extracts were used to perform qRT-PCR as described above. The fold change of target gene transcription for the cultures incubated at 44°C is presented as a percentage of the value for the 37°C counterpart.
Transcription of the episomal Lon target genes in LVS or the isogenic Δlon mutant ST1120 was determined by qRT-PCR as described above. The Lon target genes were amplified with primer pairs Pr10246/Pr10247 (FTL578), Pr7861/Pr7862 (FTL663), Pr8151/Pr8152 (FTL1217), Pr10256/Pr10257 (FTL1228), Pr7863/Pr7864 (FTL1957), and Pr7865/Pr7866 (clpX). The data are presented as mean normalized fold expression relative to the value for the helicase gene ± SEM (n = 3).
Construction of F. tularensis LVS deletion mutants.
In-frame deletion in the coding region of lon, clpP, or clpX was done in F. tularensis LVS by allelic replacement and sacB-mediated counterselection as described previously (47). The up- and downstream sequences of each target gene were amplified by PCR from LVS genomic DNA with primer pairs Pr1235/Pr1238 and Pr1236/Pr1237 for lon, Pr1062/Pr1065 and Pr1063/Pr1064 for clpP, and Pr1058/Pr1061 and Pr1059/Pr1060 for clpX and linked by fusion PCR as described previously (45). Each fusion product was initially cloned into the XhoI (for clpP and clpX) or XhoI/NotI (for lon) sites of pBluescript II (SK−) (Stratagene, La Jolla, CA) and subcloned into the ApaI/BamHI (for clpP and clpX) or ApaI/NotI (for lon) sites of pMP590 (Table 1). The pMP590 derivatives were electroporated into LVS to generate kanamycin-resistant transformants, which were counterselected on chocolate agar plates containing 5% sucrose. The resulting kanamycin-sensitive/sucrose-resistant clones were tested for the loss of the target sequences by PCR amplification using the flanking primers (Pr1062/Pr1063 for clpP, Pr1058/Pr1059 for clpX, and Pr1235/Pr1236 for lon). The resultant clones were further characterized by Southern hybridization and DNA sequencing essentially as described previously (48). Genomic DNA from LVS or its derivatives was digested with HindIII and probed with a digoxigenin (DIG)-labeled PCR fragment representing the 3′ end of the tig coding sequence (a PCR product of primers Pr1298 and Pr1299).
Generation of complementation and transcriptional reporter plasmids.
The in trans complementation constructs were prepared by cloning the entire coding sequences of the LVS lon and clpP genes immediately downstream of the 505-bp tig promoter in plasmid pMP633 as described previously (47). This promoter was amplified from LVS genomic DNA with primers Pr1643 and Pr1644, linked to the 5′ end of the wild-type clpP or lon by fusion PCR (48), and cloned into the AvrII/KpnI site of pST1728 (see below). lon and clpP were amplified from LVS genomic DNA with primer pairs Pr1529/Pr1667 and Pr1645/Pr1642, respectively. The resulting plasmids pST1760 (clpP) and pST1864 (lon) were verified by DNA sequencing before being electroporated in the target LVS derivatives.
Transcriptional reporter constructs were established in pMP633 as described previously (49). A starter plasmid was initially generated by cloning the E. coli pTAC promoter and multiple-cloning site (MCS) sequence of pGEX-2T (GE Healthcare Bio-Science, Piscataway, NJ) into the EcoRV site of pMP633, resulting in pST1728 (pMP633::pTAC). The insert was amplified with primers Pr1607 and Pr1608. To exclude the activity of the endogenous β-galactosidase, the Bacillus stearothermophilus β-galactosidase (bgaB) gene encoding a thermostable enzyme was used in this work. bgaB was amplified with primers Pr1732 and Pr1733 from plasmid pDL (49) and cloned into the KpnI/XhoI site of pST1728 (downstream of the pTAC promoter) as a transcriptional reporter, yielding plasmid pST1923 (pST1728::pTAC). The tig promoter reporter pST1934 (pST1923::pTIG-bgaB) was prepared by replacing the pTAC promoter with a 505-bp sequence upstream of the tig coding region (pTIG). pTIG was amplified from LVS genomic DNA with primers Pr1643 and Pr1690 and cloned into the AvrII/KpnI site of pST1923. Similar procedures were used to construct the reporter constructs for clpP (pST1956 with primers Pr1699 and Pr1700), clpX (pST1958 with primers Pr1701 and Pr1702), lon (pST1936 with primers Pr1370 and Pr1691), hupB (pST1940 with primers Pr1703 and Pr1704), and clpB (pST1938 with primers Pr1705 and Pr1706). The reporter plasmids were individually electroporated into strain LVS to generate the reporter strain for each target sequence. The β-galactosidase activity was determined with the LVS strain essentially as described previously (49). Briefly, each LVS reporter strain was grown to an OD600 of ∼0.4 in MHB supplemented with hygromycin (200 μg/ml) at 37°C and pelleted at 10,000 × g for 10 min at 4°C. Bacterial cells were lysed in Z buffer (100 mM Na2HPO4, 20 mM KCl, 2 mM MgSO4, 0.8 mg/ml cetyl trimethyl ammonium bromide, 0.4 mg/ml sodium deoxycholate, and 2.7 μl/ml β-mercaptoethanol) and incubated with 200 μl of the substrate (4 mg/ml of o-nitrophenyl-β-d-galactoside in Z buffer) at 55°C until sufficient color developed, and the reaction was stopped with 1 M sodium carbonate. The transcriptional activity of each reporter is expressed as the mean β-galactosidase activity ± SEM (n = 3) in Miller units.
Antibody production and immunoblotting.
Antisera against Francisella Lon, ClpP, ClpX, and FTL1017 were prepared with recombinant forms of these proteins in New Zealand White rabbits essentially as described previously (47). Lon, ClpP, and ClpX were expressed as glutathione S-transferase (GST) fusion proteins by amplifying their coding regions from LVS genomic DNA with primer pairs Pr3230/Pr3231 (lon), Pr5476/Pr5477 (clpP), and Pr5478/Pr5479 (clpX) and cloning each of the PCR products in the NotI/AscI site of pST2700, a modified pGEX-2T with additional cloning sites (Table 1). pST2700 was constructed by insertion of an MCS sequence into the BamHI/EcoRI site of pGEX-2T. The MCS fragment was generated by annealing reverse complementary oligonucleotides Pr3110 and Pr3111. His-tagged FTL1017 was similarly produced with primers Pr6434 and Pr6435 in pST3621, a pET28a (Novagen, Madison, WI, USA) derivative with additional AscI site between the XhoI and NotI sites of the original plasmid (Table 1). The resulting plasmids pST3001 (Lon), pST3750 (ClpP), pST3749 (ClpX), and pST4218 (FTL1017) were used to produce recombinant proteins by affinity chromatography with the glutathione-Sepharose 4 Fast Flow resins (Lon, ClpP, and ClpX) or Ni Sepharose 6 Fast Flow (FTL1017) resins (GE Healthcare Bio-Sciences) according to the supplier's instructions. The fusion proteins were visualized by SDS-PAGE, quantified with the bicinchoninic acid (BCA) assay kit (Solarbio, Beijing, China), and used to immunize rabbits for antiserum production. Immunoblot detection of F. tularensis proteins was carried out as described previously (47). The antisera and horseradish peroxidase (HRP)-conjugated secondary antibodies (ZSGB-Bio, Beijing, China) were used at dilutions of 1:5,000 and 1:10,000, respectively.
Mouse infection.
Experiments with F. tularensis LVS and its derivatives were carried out in female BALB/c mice (6 to 8 weeks old) as described previously (8). All animal experiments (including antibody production in rabbits) were in compliance with the guidelines of the Institutional Animal Care and Use Committee of Tsinghua University. The wild-type and mutant strains were individually diluted in phosphate-buffered saline (PBS) based on predetermined CFU values. Groups of six mice were each intranasally inoculated with ∼2.5 × 103 CFU of F. tularensis/mouse in a total volume of 40 μl and were sacrificed at days 0, 2, 4, and 7 postinfection. The lungs were aseptically removed and processed to determine the bacterial burden (CFU) as described previously (8). All of the mouse infection experiments were repeated at least twice. The bacterial burden levels in the representative experiments are presented as mean log CFU ± SEM (n = 6).
Macrophage infection.
Intracellular replication of F. tularensis LVS derivatives was assessed in the murine alveolar macrophage cell line MH-S and the human promyelomonocytic cell line THP-1 (ATCC, Manassas, VA) as described previously (47). MH-S and THP-1 cells were grown in RPMI 1640 medium supplemented with 10% (vol/vol) fetal bovine serum and 50 μM β-mercaptoethanol. MH-S cells were grown in 24-well plates (HyClone, Logan, UT) to approximately 80% confluence prior to infection. THP-1 cells were treated with phorbol myristate acetate (PMA) (200 ng/ml) for 48 h before being infected. Bacteria were grown overnight in MHB, pelleted by centrifugation, and resuspended to an OD600 of ∼0.2 in prewarmed medium without serum. After a brief rinse with medium, macrophages were infected at a multiplicity of infection (MOI) of 100 F. tularensis bacteria per macrophage (0 h). After 2 h of incubation at 37°C, the cells were washed with medium and incubated in gentamicin-containing medium (50 μg/ml) for 1 h at 37°C. The cells were rinsed three times with medium and subsequently cultured without antibiotic. The medium from the last washing was plated on antibiotic-free dishes to determine the killing efficacy. This treatment routinely resulted in complete eradication of the extracellular bacteria (data not shown). At 3, 24, and 48 h postinfection, the culture medium of infected macrophages was moved to sterile tubes before the cells were lysed in situ with prechilled sterile water, because there were substantial levels of bacteria in the medium at each of these time points. The medium and lysates from each well were mixed, diluted with sterile PBS, and spread on chocolate agar plates to enumerate CFU. Each infection experiment was repeated at least three times. The results of representative experiments are presented as mean log CFU/well ± SEM (n = 3).
Stress tolerance test.
The tolerance of F. tularensis to stress conditions was determined as described previously (34). Overnight cultures of the wild-type LVS and isogenic mutants were pelleted, washed once in PBS by centrifugation, and resuspended in PBS to an OD600 of 0.2 (∼108 CFU/ml). The suspensions were 10-fold serially diluted in PBS, and an aliquot of each dilution (10 μl) was spotted onto chocolate agar plates in the presence or absence of 1.5% NaCl. The LVS strains in each comparative group or on a plate were adjusted to a similar concentration of viable bacteria (CFU) and serially diluted in the same manner before being spotted. Plates were incubated for 5 to 7 days at 37°C or 39°C prior to photography. These conditions (39°C and 1.5% NaCl) were used because of minimal inhibition against growth of LVS; higher temperatures or NaCl concentrations typically resulted in a substantial loss of LVS growth in our pilot experiments (not shown).
MS.
For identification of the F. tularensis Lon substrates under heat stress, LVS and ST1120 were grown in 30 ml of MHB at 39°C to an OD600 of 0.8 with aeration. Cells were harvested by centrifugation at 3,500 rpm for 10 min at 4°C, resuspended in 1 ml of ice-cold Tri-Cl buffer (0.02 M Tris, 0.1 M NaCl, pH 8.0) supplemented with 1 mM phenylmethanesulfonyl fluoride (PMSF), and lysed by sonication. Protein concentrations of the lysates were determined with the BCA kit. The F. tularensis protein samples were processed for quantitative mass spectrometry (MS) essentially as described previously (50, 51). Briefly, total proteins (∼65 μg per sample) were separated in SDS-polyacrylamide gels, and gel slices were processed for in-gel digestion of proteins with trypsin (Promega, Fitchburg, WI) according to the instructions of the supplier. The peptides were extracted from the gel slices and labeled with isobaric tandem mass tag (TMT) 6-plex labeling reagent (Pierce, Rockford, IL, USA). Equal amounts of TMT-labeled proteins from different bacterial samples were combined and analyzed by liquid chromatography-tandem MS (LC-MS/MS). The MS/MS spectra from each LC-MS/MS run were searched against the F. tularensis LVS FASTA database downloaded from NCBI (release date, June 2013; 1,716 entries) using in-house Sequest HT Algorithm in Proteome Discoverer software (version 1.4) with the following parameters: peptide MS tolerance of 20 ppm, MS/MS tolerance of 20 milli-mass units (mmu), carbamidomethylation of Cys, TMT of lysine and N-terminal peptide as the fixed modification, and oxidation on Met as the variable modification. Peptide spectral matches (PSM) were validated using Percolator provided by Proteome Discoverer software based on q values at a 1% false-discovery rate (FDR). The false-discovery rate was also set to 0.01 for protein identifications. Relative protein quantification was performed using Proteome Discoverer software (version 1.4) according to the manufacturer's instructions on the reporter ion intensities per peptide. Proteins with at least two unique peptides were regarded as having confident identifications and were further quantified. Protein ratios were calculated as the median of all peptide hits belonging to a protein. Quantitative precision was expressed as protein ratio variability. A difference of 1.5-fold or more between proteins from two compared samples was initially considered to be significant. Other related parameters (e.g., number of unique peptides and sequence coverage rate) were also used for assessment of data reliability.
Protein degradation test.
Protein degradation by Lon was screened by coexpressing F. tularensis Lon and its putative substrate proteins in the Lon-deficient E. coli strain ER2566 (NEB) essentially as described previously (52). To produce a C-terminally His-tagged recombinant form of F. tularensis Lon, its entire coding region was amplified with primers Pr7259 and Pr7260 and cloned in the EcoRI/KpnI site of pBAD18 (53) under transcriptional control of the arabinose-inducible promoter (yielding plasmid pST4635). The other C-terminally His-tagged F. tularensis proteins were cloned in the NcoI/EcoRI site of pACYCDuet-1 (Novagen, Madison, WI, USA) behind an isopropyl-β-d-1-thiogalactopyranoside (IPTG)-inducible T7 promoter. The primers and resulting plasmids are listed in Table 2. Both pST4635 (F. tularensis Lon) and one of pACYCDuet-1 derivatives expressing other F. tularensis proteins were transformed into E. coli ER2566. At an OD600 of 0.3, arabinose or sterile water (negative control) was added to the cultures in LB broth to induce F. tularensis Lon for 2 h, followed by induction of the putative Lon substrates or control proteins with IPTG for 3 h at 16°C and 120 rpm (for FTL578, FTL663, and FTL1228) or 0.5 h at 37°C and 220 rpm (for the rest). New protein synthesis was blocked by adding spectinomycin (100 μg/ml) to the cultures as described previously (54). A fraction of the cultures (1 ml) was removed at the indicated time points, pelleted at 4°C, resuspended in 50 μl of ice-cold Tris-Cl buffer, lysed in 5× SDS-PAGE sample buffer (55), and analyzed by immunoblotting using anti-His6 monoclonal antibody (ZSGB-Bio) as described above. The protein half-life was calculated on the basis of the immunoblot signal for each protein as described previously (56). Immunoblot signals were quantified using Image Lab software (Bio-Rad, Hercules, CA, USA) according to the supplier's instructions.
TABLE 2.
F. tularensis proteins tested for Lon degradation in Lon-deficient E. coli
| Gene | Protein function | Abundance ratio from MS dataa | Primer set, plasmidb | Degradationc | Reference(s)d |
|---|---|---|---|---|---|
| FTL663 | Hypothetical protein | 3.786 | Pr7261/Pr7262, pST4609 | Yes | 8 |
| FTL1957 | Heat shock protein | 2.447 | Pr7263/Pr7264, pST4611 | Yes | 46 |
| FTL578, ocd | Ornithine cyclodeaminase | 2.118 | Pr7755/Pr7756, pST5007 | Yes | 64 |
| FTL1217 | Hypothetical protein | 2.022 | Pr7751/Pr7752, pST5009 | Yes | |
| FTL1191, dnaK | Molecular chaperone DnaK | 1.409 | Pr10091/Pr10092, pST6935 | No | 64 |
| FTL1267 | Hypothetical protein | 1.350 | Pr10095/Pr10096, pST6937 | No | 7 |
| FTL891, tig | Trigger factor | 1.338 | Pr10083/Pr10084, pST6931 | No | 8 |
| FTL1609, flmK | Dolichyl-phosphate-mannose protein | 1.145 | Pr10099/Pr10100, pST6939 | No | 7 |
| FTL1228, sufD | SufS activator complex, SufD subunit | 1.059 | Pr10093/Pr10094, pST6936 | Yes | 6 |
| FTL893, clpX | ClpX | 1.028 | Pr7522/Pr7523, pST4819 | No | 8 |
| FTL1017, cmk | Cytidylate kinase | 1.011 | Pr7518/Pr7519, pST4818 | No | |
| FTL478, gcvH | Glycine cleavage system protein H | 1.000 | Pr7516/Pr7517, pST4816 | No | 7 |
| FTL398, purE | Phosphoribosylaminoimidazole carboxylase, catalytic subunit | 0.983 | Pr10077/Pr10078, pST6928 | No | 7 |
| FTL1358 | Cation efflux family protein | 0.957 | Pr10097/Pr10098, pST6938 | No | 7 |
| FTL1611, yfdH | Glycosyl transferase family protein | 0.932 | Pr9901/Pr9902, pST7048 | No | 7 |
| FTL1724 | Lipoprotein | 0.887 | Pr9903/Pr9904, pST7049 | No | 7 |
| FTL87 | Acetyltransferase | 0.881 | Pr9891/Pr9892, pST7043 | No | 77 |
| FTL40 | LysR family transcriptional regulator | 0.856 | Pr10075/Pr10076, pST6927 | No | 6 |
| FTL599, wbtG | Glycosyl transferase family protein | 0.838 | Pr9897/Pr9898 pST7046 | No | 7 |
| FTL687, emrA1 | HlyD family secretion protein | 0.814 | Pr10081/Pr10082 pST6930 | No | 7 |
| FTL593 | Galactosyl transferase | 0.797 | Pr9893/Pr9894 pST7044 | No | 78 |
| FTL595, wbtE | Galacturonosyl transferase | 0.794 | Pr9895/Pr9896 pST7045 | No | 7, 79 |
Ratio of abundance of the protein in the Δlon mutant to that in the wild-type strains.
Primer set used to amplify each target gene and pACYCDuet derivative containing the target gene.
Degradable or undegradable by the F. tularensis Lon protease in E. coli.
Previous characterization if available.
Protein degradation in LVS was performed virtually as described previously (54). The F. tularensis Lon substrates identified in the E. coli system were expressed as C-terminally His-tagged recombinant proteins from a tetracycline-inducible promoter in shuttle plasmid pEDL17 (57). The coding sequences of FTL478, FTL578, FTL663, FTL1017, FTL1217, FTL1228, FTL1957, and clpX were amplified from LVS genomic DNA with the following primer pairs, respectively: Pr7409/Pr7410, Pr10050/Pr10051, Pr7405/Pr7406, Pr7411/Pr7412, Pr7869/Pr7870, Pr10229/Pr10230, Pr7407/Pr7408, and Pr7631/Pr7632. The sequences were then cloned in the MluI/XmaI site of pEDL17 (replacing the original mOrgange2 gene), resulting in the following recombinant plasmids: pST4865 (FTL478), pST6955 (FTL578), pST4861 (FTL663), pST4867 (FTL1017), pST5072 (FTL1217), pST6956 (FTL1228), pST4863 (FTL1957), and pST4869 (clpX). The pEDL17 derivatives were individually electroporated into LVS derivatives. The resultant strains were cultivated to an OD600 of 0.6 in CDM broth before induction of target genes with 100 ng/ml anhydrotetracycline (ATc) (Clontech, Mountain View, CA, USA) for 4 h with aeration. A fraction of the cultures (1 ml) was removed and pelleted by centrifugation for detection of target proteins by immunoblot analysis using the anti-His6 antibody. For qRT-PCR, aliquots of the same samples were washed twice in ice-cold PBS by centrifugation and stored at −80°C until being used for total RNA extraction.
Statistical analysis.
Statistical differences between control and experimental groups in the macrophage and mouse infection experiments were determined using the Student t test in Microsoft Excel. Statistical significance is defined by a P value of <0.05 (*), <0.01 (**), or <0.001(***).
RESULTS
The genes in the F. tularensis protease locus are cotranscribed.
The gene sequence and organization of the protease locus are highly conserved among the four F. tularensis subspecies (Fig. 1A). The coding regions of tig, clpP, clpX, and lon are separated by only 25, 23, and 24 nucleotides, respectively, suggesting potential cotranscription of these genes. We tested this possibility by RT-PCR to amplify intergenic regions of the protease locus. As shown in Fig. 1B, RT-PCR yielded “gene junction” PCR products with predicted sizes and sequences. The specificity of the RT-PCR results was confirmed by the lack of detectable PCR products in the absence of the reverse transcriptase in the RT reactions. Consistently, no RT-PCR product was detected for the intergenic region between tig and the upstream gene FTL890 (encoding thymidine kinase). The RT-PCR analysis also indicated that hupB (encoding histone-like protein HU form B), the 3′ neighboring gene of lon, is cotranscribed with lon despite a relative large intergenic region (86 bp). Finally, a 1,241-bp lon coding sequence was successfully amplified under the same RT-PCR conditions (Fig. 1B, last panel). These results indicate that the four genes in the LVS protease locus are cotranscribed under the in vitro culture conditions.
We next tested possible promoter activity within the protease locus by using a β-galactosidase reporter (49). The sequence (∼400 bp) upstream of each gene in this locus was amplified and fused to the 5′ end of the promoterless B. stearothermophilus bgaB gene in the E. coli-Francisella shuttle vector pMP633. The reporter plasmids were electroporated into LVS to determine the transcriptional activity of each construct. Compared to the positive-control construct pST1938 containing the clpB promoter (260 Miller units), the tig reporter construct pST1934 produced a higher level of β-galactosidase activity (801 Miller units) (Fig. 1C), thus confirming the presence of a strong promoter upstream of the tig gene based on the RT-PCR result (Fig. 1B). Interestingly, the reporter constructs carrying the upstream sequences of lon (pST1936) and hupB (pST1940) also displayed β-galactosidase activity that was either comparable to (lon; 343 Miller units) or higher than (hupB; 647 Miller units) that of the clpB reporter control. This indicated that, besides the transcriptional read-through from the upstream genes, lon and hupB are also separately transcribed under these conditions. In sharp contrast to the reporter constructs of lon and hupB, the counterparts of clpP (pST1956) and clpX (pST1956) showed only residual levels of β-galactosidase activity (9 and 36 Miller units, respectively).
We also assessed the relative contributions of the tig and lon promoters to the transcription of the protease operon by qRT-PCR using two LVS mutants, each carrying an insertion of the EZ-TN transposon (8). Our preliminary data showed that the EZ-TN transposon possessed a transcriptional termination activity and thereby exerted polar effect on the downstream gene (Zhang, unpublished observation). We thus reasoned that insertion of this transposon in the 5′ region (e.g., tig and clpP) of the locus should block the expression of the downstream genes (e.g., clpX, lon, and hupB) if the genes in these genes are indeed cotranscribed. Consistent with the reporter experiment (Fig. 1C), the transcription of tig, clpP, and clpX was undetectable in the tig mutant, whereas tig expression was intact in the clpP mutant (Fig. 1D), thus verifying the cotranscription of these genes. There was still a low level of the lon mRNA in both the tig and clpP mutants, which was reduced by 56- and 66-fold compared with the level in the wild type. The transcript level of hupB was comparable between the wild type and mutants, indicating that the expression of hupB is independent of the protease locus, and the detected cotranscription with lon (Fig. 1B) was due to leaky transcription from the tig and/or lon promoter. The reporter and qRT-PCR results demonstrated that the tig promoter drives the transcription of tig, clpP, clpX, and lon but that lon is capable of independent transcription from its own promoter at a much lower capacity.
To determine whether the tig and lon promoters are regulated by stressful conditions, we determined the transcription of the protease operon under heat stress by qRT-PCR. Incubation of the mid-log-phase LVS culture at 44°C for 20 min significantly induced the expression of all four genes in the operon but not hupB (Fig. 1E). The transcripts of tig, clpP, clpX, and lon in the heat-shocked cells were increased by 1.5- to 2.5-fold compared with those in the bacteria incubated at 37°C. This result proved that the protease operon is upregulated by heat shock. This finding agrees with the presence of a putative RpoH-binding site in tig promoter sequence (46). Because the lon promoter also contains a putative RpoH-binding site (46), we tested whether its activity is also regulated by heat stress. The qRT-PCR result showed that the lon transcript was significantly increased by heat stress only in the wild type and not in the tig and clpP mutants (Fig. 1F), suggesting that the lon promoter is not regulated by heat stress. These experiments have demonstrated that the transcription of the tig, clpP, clpX, and lon genes is subject to regulation by heat stress.
Lon and ClpP contribute to the in vivo fitness of F. tularensis LVS.
Multiple transposon insertion mutants of the LVS clpP, clpX, and lon genes were identified to be attenuated in a mouse lung infection model (8). We sought to confirm this finding with the unmarked ΔclpP, ΔclpX, and Δlon mutants of strain LVS. The clpP, clpX, and lon coding sequences were deleted by allelic replacement and counterselection. The resulting strains, ST1116 (ΔclpX), ST1118 (ΔclpP), and ST1120 (Δlon), were verified by PCR (Fig. 2A) and Southern blotting (Fig. 2B). The loss of ClpP, ClpX, or Lon in the unmarked mutants was confirmed by immunoblotting. The antisera raised with the recombinant forms of the F. tularensis proteins detected major protein bands in LVS, which were consistent with the predicted molecular sizes (in kDa) of ClpX (46.3), ClpP (22.2), and Lon (86.2) (Fig. 2C). In contrast, ClpP and ClpX were completely absent in the corresponding mutants (Fig. 2C). The Δlon mutant produced a truncated Lon of 44 kDa due to the in-frame deletion of the internal coding region. The missing ClpP and Lon proteins in the mutants were restored by in trans complementation with the wild-type clpP or lon of LVS. The presence of two additional bands (44 and 10 kDa) in the complemented ΔclpP mutant appeared to be due to overproduction of ClpP from the high-copy-number plasmid. No complementation was attempted with the ΔclpX mutant because it showed only marginal attenuation in animal infection or stress tolerance experiments (see below). We finally characterized the growth kinetics of these mutants in both nutritionally poor (CDM) and rich (MHB) media. The ΔclpP, ΔclpX, and Δlon mutants exhibited growth patterns comparable to that of the parent strain in CDM broth (Fig. 2D), although they grew slightly slower in the log phase in MHB medium (Fig. 2E). This result showed that the unmarked clpP, clpX, and lon deletion mutants did not suffer from a major growth defect under the in vitro culture conditions.
FIG 2.

Verification of unmarked deletions in clpX, clpP, and lon. (A) PCR detection of gene deletions. The protease locus was amplified from genomic DNA of LVS or isogenic clpX, clpP, and lon deletion mutants with the flanking region primers Pr1062/Pr1236. The molecular sizes of DNA markers are marked in kb. (B) Southern blot detection of gene deletions. Genomic DNA preparations of LVS and isogenic ΔclpX, ΔclpP, and Δlon mutants were digested with HindIII and separated by agarose gel electrophoresis. The DNA blot was probed with a PCR fragment (Pr1298/Pr1299) representing a 137-bp coding region of tig. (C) Detection of ClpX, ClpP, and Lon in LVS (WT) or isogenic clpX, clpP, and lon mutants. Cell lysates of each strain (10 μg of total proteins) were separated by SDS-PAGE and tested by immunoblotting with the specific rabbit antiserum for each protein. The sizes of protein standards are indicated at the left in kDa. (D) Growth kinetics of LVS and its derivatives in CDM. The optical densities at 600 nm of LVS (open circles) and the ΔclpX (filled circles), ΔclpP (filled triangles), and Δlon (filled squares) mutants were determined at the indicated time points. The values represent the means ± SEM for three independent cultures from one of the three independent experiments. (E) Growth kinetics of LVS and its derivatives in MHB.
To determine the contributions of ClpP, ClpX, and Lon to the infectivity of F. tularensis LVS, we compared the bacterial burdens in the lungs of the mice infected with LVS or isogenic protease gene mutants at four time points after intranasal inoculation. The results obtained from the mice sacrificed immediately after the infection procedure (time zero) indicated that all groups of mice received a similar level of viable bacteria (∼1.5 × 103 CFU) (Fig. 3). The Δlon mutant displayed significantly impaired growth at the later time points, with reduction in tissue bacterial burden by 5.6-fold on day 2, 13.5-fold on day 4, and 38.7-fold on day 7 (Fig. 3A). In contrast, the ΔclpP mutant showed much less attenuation throughout the testing period. While a marginal attenuation (0.7-fold reduction in tissue bacterial burden) was observed with the ΔclpP mutant-infected mice on day 2, there were small but statistically significant impairments in growth on day 4 (by 1.0-fold) and day 7 (by 1.7-fold) (Fig. 3B). The ΔclpX mutant showed even milder attenuation, with no (day 2) or modest (days 4 and 7) growth impairment. This result indicated that the Lon protease plays a more crucial role in the in vivo fitness of F. tularensis LVS than the ClpXP protease. It should also be mentioned that the levels of attenuation obtained with the unmarked lon, clpP, and clpX deletion mutants in this study are substantially lower than those with the corresponding transposon insertion mutants observed in our previous study (8). Potential reasons for this discrepancy are described in Discussion.
FIG 3.

Infectivities of the ΔclpX, ΔclpP, and Δlon mutants in BALB/c mice. BALB/c mice (n = 6) were infected with F. tularensis LVS (WT) or its derivatives via intranasal inoculation. Bacterial burdens of the lon (A), clpP (B), and clpX (C) mutants in the lung were determined at days 0, 2, 4, and 7 postinoculation. Each bar represents the mean log CFU ± SEM. All of the mouse infection experiments were repeated at least twice. The relative difference with each pair of LVS and isogenic mutant is indicated by fold change and a P value (*, <0.05; **, <0.01; ***, <0.001).
Lon and ClpP contribute to intramacrophage growth of F. tularensis LVS.
Based on the significant attenuation of the Δlon and ΔclpP mutants in mice (Fig. 3), we initially tested intramacrophage growth of these mutants in murine alveolar macrophage MH-S cells. At the end of the gentamicin treatment (3 h postinfection), all of the LVS mutants except for the ΔmglA strain had levels of intracellular bacteria similar to those the parent strain LVS (Fig. 4A). MglA is a transcriptional regulator required for intracellular growth of F. tularensis (58, 59). This result indicated that the ClpXP and Lon proteases are not essential for F. tularensis uptake or initial intracellular growth. However, macrophages infected with the ΔclpP mutant (ST1118) showed 38.0- and 15.5-fold reductions in intracellular growth at 24 and 48 h postinfection, respectively. Similarly, the Δlon strain ST1120 displayed significantly impaired intracellular growth at h 24 (4.8-fold) and 48 (46.3-fold). These defects were partially rescued by the clpP (pST1760) and lon (pST1764) complementation constructs in both the Δlon and ΔclpP mutants.
FIG 4.
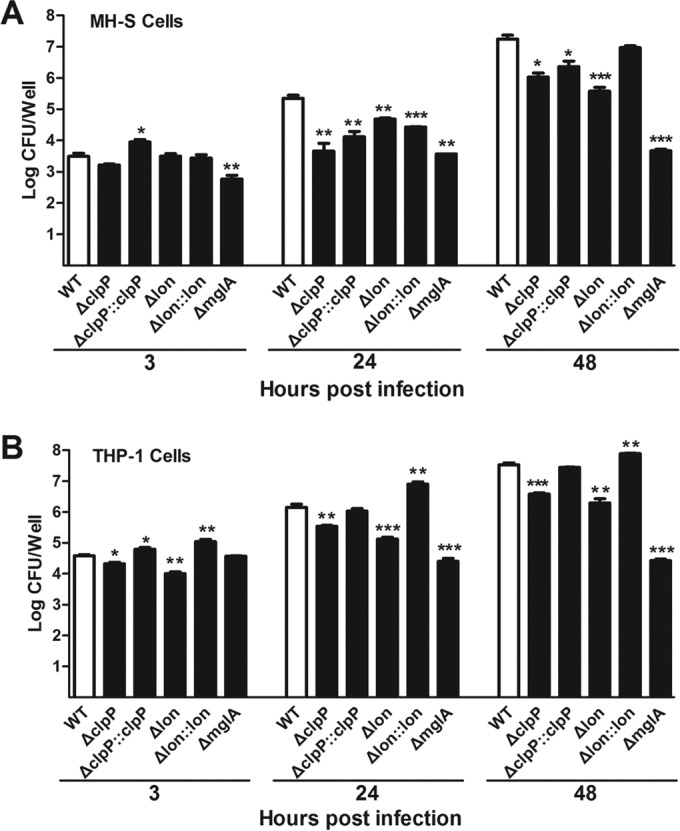
Intracellular growth of the lon and clpP mutants. MH-S cell (A) or THP-1 (B) cell monolayers (2.5 × 105 cells/well) were infected with F. tularensis LVS (WT), the isogenic ΔclpP and Δlon mutants, or complemented mutants; intracellular bacteria were enumerated at 3, 24, and 48 h postinfection. The data shown represent one of three independent experiments. Each bar represents the mean log CFU per well ± SEM (n = 3). Statistical analysis was performed only between LVS and each of the mutants.
We further sought to verify the phenotypes of the Δlon and ΔclpP mutants in human macrophage THP-1 cells. Although the overall attenuation levels were lower in the THP-1 cells than in the MH-S cells, both the Δlon and ΔclpP mutants showed significant defects in intracellular growth in the THP-1 cells at h 24 (4.4-fold for ΔclpP; 11.1-fold for Δlon) and 48 (9.0-fold for ΔclpP; 16.1-fold for Δlon) (Fig. 4B). These defects were almost completely alleviated by the lon and clpP complementation constructs. In both cell culture models, the Δlon mutant generally exhibited more severe impairment than the ΔclpP counterpart in multiple independent experiments (data not shown), suggesting nonredundant roles of each protease in enhancing F. tularensis intracellular growth. As a positive control, the mglA mutant showed minimal intracellular growth during the entire course of infection in both cell culture models, as reported previously (58). Lastly, consistent with the lung infection results (Fig. 3C), deleting clpX in LVS did not lead to significant alteration in the level of intracellular growth in either of the MH-S and THP-1 cell culture models (data not shown).
Lon and ClpP are required for F. tularensis LVS tolerance to stress conditions.
To understand the molecular mechanisms behind the contribution of the Lon and ClpP proteases to the F. tularensis fitness during intracellular infection of mammalian hosts, we initially tested the impact of clpP, clpX, or lon deletion on tolerance of LVS to heat stress. The ΔclpP (ST1116), ΔclpX (ST1118), and Δlon (ST1120) mutants showed slight but reproducible retardation in growth when they were cultivated at 37°C or 39°C in MHB (data not shown). At 37°C, growth on agar plates was comparable for these strains and the parent strain LVS (Fig. 5, top row). However, the growth of the ΔclpP and Δlon mutants on agar plates was reduced by >100-fold based on the CFU counts when the temperature was shifted to 39°C (Fig. 5). The growth defects of the ΔclpP and Δlon mutants under heat stress were completely restored by in trans complementation with the wild-type clpP or lon gene, respectively (Fig. 5). In contrast, the elevated temperature did not significantly alter the growth of the ΔclpX mutant (Fig. 5, middle row).
FIG 5.
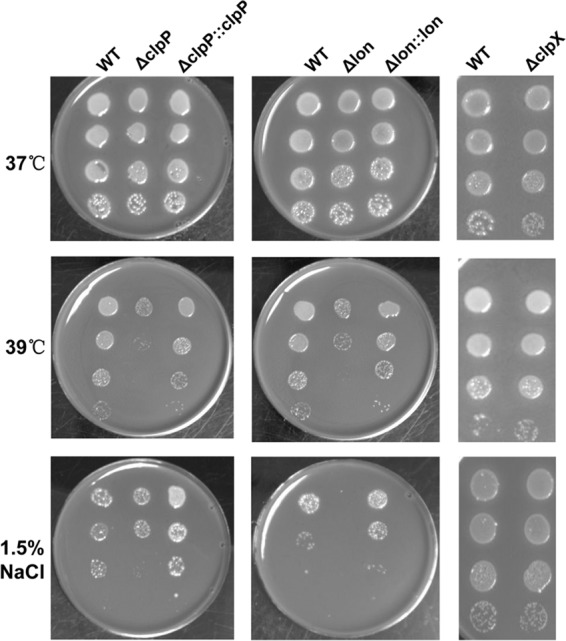
Growth of the lon, clpP, and clpX mutants under stress conditions. Overnight cultures of LVS (WT) and its derivatives were adjusted to an OD600 of 0.2 in PBS and diluted 10-, 100-, 1,000-, and 10,000-fold in PBS. A chocolate agar plate was spotted with 10 μl of each dilution and incubated at 37°C, at 39°C, or in the presence of 1.5% NaCl.
We also tested the impact of clpP, clpX, or lon deletion on F. tularensis fitness under high-salt conditions. In a pattern similar to that for the heat stress conditions, adding 1.5% NaCl to the agar medium slowed the growth of the ΔclpP and Δlon mutants by >100-fold at 37°C compared with that of the wild type, but the ΔclpX strain grew similarly in the presence of 1.5% NaCl (Fig. 5, bottom row). The growth defects of the ΔclpP and Δlon mutants under heat stress and in the presence of 1.5% NaCl were completely alleviated by in trans complementation with lon and clpP, respectively (Fig. 5). These data demonstrated that ClpP and Lon but not ClpX are important for F. tularensis fitness under stressful conditions.
Identification of the Francisella Lon substrates under heat stress.
Because the Δlon and ΔclpP mutants showed a clear growth defect at 39°C (Fig. 5), we reasoned that the these mutants had suffered from abnormal accumulation of the Lon and ClpP substrates under the heat shock condition, which are proteolytically removed in the parent strain LVS. This condition was thus used to identify the substrates of the Lon protease by determining the proteomic profiles from three sets of LVS and Δlon (ST1120) samples (three biological replicas). Each set was derived from a pair of independently prepared cultures of LVS and ST1120. Equal amounts of total proteins (∼65 μg per sample) from each of 6 samples (3 per strain) were processed by SDS-PAGE, digested with trypsin, and differentially labeled with a unique TMT tag before being pooled for LC-MS/MS analysis as described in Materials and Methods. A total of 1,099 F. tularensis proteins were identified from the combined data of the LVS and ST1120 triplicate samples, representing 64.0% of the entire proteome (1,716 proteins) predicted from the LVS genome (accession number NC_007880). The complete list of the proteins identified in this analysis is shown in Table S2 in the supplemental material. Comparative analysis revealed 29 proteins that were significantly more abundant in the Δlon mutant than in LVS (see Table S3 in the supplemental material), referred to as putative Lon substrates here.
To identify authentic substrates of the F. tularensis Lon protease and test the reliability of the MS data, we performed protein degradation tests in E. coli with a total of 22 F. tularensis proteins identified in the MS analysis, including four putative Lon substrates, three additional proteins with mutant/wild-type ratios slightly below the cutoff values (1.3 to 1.50), and 15 proteins with even lower values (<1.3) (Table 2). All of these proteins, except for FTL1217 and FTL1957, have been implicated to be necessary for F. tularensis pathogenesis in previous studies (see the references in Table 2). FTL1217 was included in the analysis because it shares 45.2% amino acid sequence identity with FTL663, which had the highest protein ratio value between the lon mutant and wild-type strains. FTL1957 was selected on the basis of its protein sequence homology with heat shock chaperone IbpA of E. coli, a known substrate of the Lon protease (15). lon and each target gene were coexpressed with an arabinose (for lon)- or IPTG (for target proteins)-inducible promoter as His-tagged recombinant proteins from two different plasmids in E. coli ER2566, which lacks the endogenous lon gene. Proteolytic degradation of the target proteins was assessed by monitoring the stability of the target proteins in the presence or absence of the F. tularensis Lon protease. To minimize the impact of the newly synthesized proteins in the assay, spectinomycin was added to the reaction mixtures at the beginning of each experiment to block translation.
In the presence of F. tularensis Lon, five proteins (FTL578, FTL663, FTL1217, FTL1228, and FTL1957) were rapidly diminished after nascent protein synthesis was blocked by spectinomycin (Fig. 6A). In the absence of F. tularensis Lon, these proteins were stable (Fig. 6B), indicating that these proteins are authentic substrates of the F. tularensis Lon protease. FTL1217 was reduced even in the absence of Lon, suggesting that it was also susceptible to degradation by an endogenous E. coli protease(s). In sharp contrast, the levels of the remaining 17 proteins were not obviously altered by the presence of Lon during the entire course of the test (80 min), as exemplified by FTL478, FTL1017, and ClpX (Fig. 6A and B, Table 2, and data not shown). The result suggested that these proteins are not F. tularensis Lon substrates. All of the four proteins (FTL578, FTL663, FTL1217, and FTL1957) selected from the list of overrepresented proteins (see Table S3 in the supplemental material) were degraded by the Lon protease. FTL1228 was the only Lon substrate identified in this test from the 18 proteins that had mutant/wild-type ratios below the cutoff value (Table 2). This analysis has thus demonstrated that phenotypic enrichment (heat stress in this case) of the Lon substrates coupled with the quantitative proteomic and protein degradation analyses can be an effective approach to identify the substrates of the Lon protease.
FIG 6.

Degradation of the F. tularensis proteins by Lon in E. coli ER2566 (Lon−). (A) Detection of protein stability in the presence of the F. tularensis Lon protease. The Lon-deficient E. coli strain ER2566 carrying two expression plasmids for the coproduction of the F. tularensis Lon protease and its putative Lon substrate were treated with a combination of arabinose (for Lon) and IPTG (for the target protein). After adding spectinomycin (0-min time point), the cells were harvested 80 min later (80-min time point) to determine the amount of each target protein by immunoblotting using the anti-His6 antibody. The sizes of protein standards are indicated at the left in kDa. (B) Detection of protein stability in the absence of the F. tularensis Lon protease, as for panel A but with no induction for Lon protease production. (C) Assessment of protein half-life in the presence of the F. tularensis Lon protease, as for panel A with additional sampling time points.
We further determined the degradation kinetics of the five Francisella Lon substrates in E. coli ER2566. The amount of each protein was evaluated at various time points after addition of spectinomycin. All of the five proteins were rapidly degraded by Francisella Lon protease; the half-lives of FTL578, FTL663, FTL1217, FTL1228, and FTL1957 were 6 ± 1, 8 ± 1, 8 ± 1, 5 ± 1, and 8 ± 1 min, respectively (Fig. 6C). This result is similar to the degradation kinetics of SoxS (half life = 2 min) and SulA (half life = 1.2 min), two known Lon substrates of E. coli (60, 61). In sharp contrast, FTL478, FTL1017, and ClpX were highly stable, with a half-life of longer than 80 min. This experiment provides further evidence that FTL578, FTL663, FTL1217, FTL1228, and FTL1957 of F. tularensis LVS are authentic Lon substrates.
Characterization of the Lon substrates in F. tularensis LVS.
We attempted to characterize the 5 substrates of the F. tularensis Lon protease in LVS by a strategy similar to that we used in E. coli. The coding regions of FTL578, FTL663, FTL1217, FTL1228, and FTL1957 were cloned in the E. coli-F. tularensis shuttle plasmid pEDL17 to express His-tagged proteins in a tetracycline-inducible manner. However, the original plan to visualize degradation of the five proteins in LVS was unsuccessful. All of the 5 proteins were barely detectable in LVS by immunoblotting after the corresponding pEDL17 derivatives were transformed in LVS and induced by ATc (data not shown). We then used an alternative approach by comparing the relative levels of target proteins in LVS and ST1120 (Δlon mutant). As a stable protein control, ClpX was detectable in both LVS and ST1120 after ATc induction (Fig. 7A), thus validating the protein expression system in LVS. Although FTL578 (Fig. 7B), FTL663 (Fig. 7C), and FTL1957 (Fig. 7D) were barely detectable by immunoblotting in LVS derivatives containing the corresponding pEDL17 constructs after induction with ATc, they were readily detectable in ST1120 (Δlon), suggesting that these proteins were specifically degraded by the Lon protease in the parent strain LVS. To determine whether these Lon substrates are also degradable by the ClpP protease, we performed a similar protein expression experiment in a clpP-deficient mutant of LVS (ST1118). The immunoblotting result showed that FTL578 (Fig. 7B) and FTL663 (Fig. 7C) were barely detectable in both LVS and ST1118, but the levels of FTL1957 were comparable in the lon and clpP mutants (Fig. 7D, lanes 4 and 6). Quantification of the immunoblotting signals showed that strains ST1118 (ΔclpP) and ST1120 (Δlon) produced approximately 4.6-fold more FTL1957 than the wild type. This result suggested that FTL578 and FTL663 are unique substrates of Lon but that FTL1957 is degradable by both Lon and ClpP.
FIG 7.
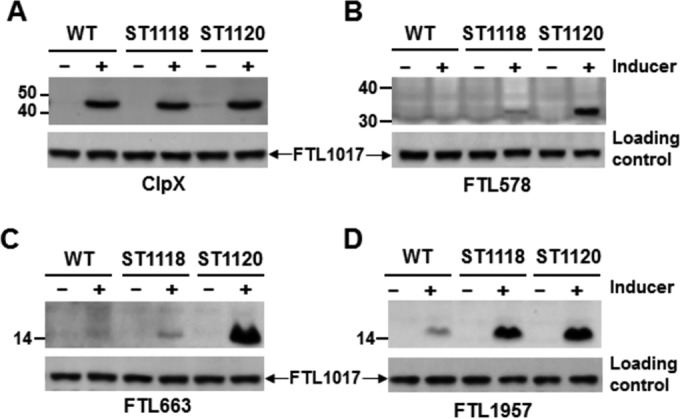
Stability of the Lon substrate proteins in LVS and its protease-deficient mutants. Each pEDL17 derivative containing the coding region of clpX (A), FTL578 (B), FTL663 (C), or FTL1957 (D) behind a tetracycline-inducible promoter was transformed in LVS (WT), ST1118 (ΔclpP), or ST1120 (Δlon). A cell lysate of each resulting strain was prepared from the culture grown in the absence (−) or presence (+) of the inducer. The recombinant protein from the episomal copy of each target gene was detected by immunoblotting as described in Materials and Methods. The protein encoded by endogenous (chromosomal) FTL1017 was detected with an antiserum as a loading control.
The above-described episomal protein expression approach failed to produce detectable levels of FTL1217and FTL1228 even in ST1120 (Δlon) (data not shown). One of the possibilities to explain this outcome is inactive transcription from the episomal FTL1217 and FTL1228 constructs. We tested this hypothesis by comparing the mRNA levels of each target gene between the LVS and ST1120 backgrounds by qRT-PCR. As predicted from the protein level of ClpX (Fig. 7A), clpX was transcribed at similar levels in LVS and ST1120 (Fig. 8A). Similar results were observed for the transcripts for FTL578, FTL663, and FTL1957 (Fig. 8B to D), further confirming that the differences in the amounts of the FTL578, FTL663, and FTL1957 proteins between the two strain backgrounds were due to the absence of the Lon protease in the Δlon mutant. The transcription of FTL1217 (Fig. 8E) and FTL1228 (Fig. 8F) from the episomal constructs in LVS was enhanced to a similar degree by ATc induction, demonstrating that the absence of these two proteins in LVS was not due to failure at the transcription level. For an unknown reason(s), the mRNA levels of both FTL1217 and FTL1228 were substantially lower in the Δlon mutant for both the endogenous (Fig. 8E and F, open bars) and episomal (Fig. 8E and F, filled bars) copies of these genes. This result might have contributed to the undetectable production of both the proteins in the Δlon mutant (ST1120). In the context of the protein degradation result from the E. coli system (Fig. 6), the protein stability experiments in F. tularensis further confirmed that FTL578, FTL663, and FTL1957 are Lon substrates in LVS and very likely in virulent F. tularensis subspecies because of the high conservation of the protease locus and these proteins among strains of different F. tularensis subspecies.
FIG 8.

Transcription of the genes encoding the F. tularensis Lon substrates in LVS and its isogenic Δlon mutant. The parent strain (WT) or its Δlon mutant (ST1120) containing an episomal tetracycline-inducible copy of the target gene (on the pEDL17 shuttle plasmid) was grown to an OD600 of 0.6 in CDM in the presence of hygromycin (200 μg/ml) at 37°C before being treated with the tetracycline analog ATc for 4 h. The total RNA samples were purified from each pair of LVS and ST1120 carrying the same expression construct of clpX (A), FTL578 (B), FTL663 (C), FTL1957 (D), FTL1217 (E), or FTL1228 (F). The mRNA level of each target gene in the presence (filled bars) or absence (open bars) of ATc was quantified by qRT-PCR, normalized on the basis of the helicase gene, and presented as the mean level ± SEM (n = 3).
DISCUSSION
This work represents the first systematic study of the F. tularensis protease locus. Our initial experiments demonstrated that the Lon and ClpP proteases encoded by the F. tularensis protease locus are important for bacterial fitness in mammalian hosts by using unmarked deletion mutants of LVS. This finding is consistent with the impaired growth of LVS in cultured macrophages and under stressful conditions. Our high-throughput quantitative proteomic approach identified many putative F. tularensis Lon substrates. Further analysis of these putative Lon substrates by protein degradation testing in E. coli revealed five novel Lon substrates. Three (FTL578, FTL663, and FTL1957) of these Lon substrates were rapidly degraded by the Lon protease in the F. tularensis setting, because they were undetectable (FTL578 and FTL663) or barely detectable (FTL1957) in the wild-type LVS even when they were overexpressed. Identification of these Lon substrates has provided important clues for a more complete understanding of the Lon protease in the F. tularensis stress response and pathogenesis.
The contributions of the five Lon substrates identified in this work to the physiology and pathogenesis of F. tularensis are largely undefined, although the substrates are present in all four subspecies of this species. FTL578 is a putative ornithine cyclodeaminase (Ocd), which catalyzes the conversion of l-ornithine to l-proline in several plant and soil bacteria (62, 63). An FTL578 transposon mutant of F. novicida U112 exhibited reduced growth in macrophages (64), but its activity as an ornithine cyclodeaminase remains to be defined in F. tularensis. FTL1228 is homologous to the Fe-S cluster assembly protein (SufD). In E. coli, SufD forms the Fe-S cluster biosynthesis complex with SufB and SufC during oxidative stress (65). All of the fully sequenced F. tularensis genomes possess homologs of sufBCD as an apparent operon. The sufD mutant of F. novicida U112 is attenuated in the inhalation infection model of mice (6). Neither Ocd nor SufD has been previously shown as a substrate protein of the Lon protease in any bacteria. FTL663, FTL1217, and FTL1957 appear to be heat shock proteins, because FTL663 and FTL1957 are induced by elevated temperature in LVS (46). FTL1217 shares 45.2% amino acid sequence identity with FTL663. An FTL663 transposon insertion mutant of LVS was attenuated in the mouse lung infection model (8). FTL1957 is homologous to the members of the small heat shock protein family, such as E. coli IbpA and IbpB. IbpA and IbpB are involved in keeping client proteins in a refoldable state under stress conditions (66–68). Both IbpA and IbpB are also substrates of the Lon protease (15). Degradation of Ibps by Lon in E. coli keeps these proteins at levels appropriate to their detrimental binding to functional proteins and allows refolding of Ibp-bound client proteins (15). The Lon protease in F. tularensis may target FTL1957 and other small heat shock proteins for similar reasons under heat stress.
Our results indicated that the Lon and ClpP proteases of F. tularensis regulate the homeostasis of different proteins and functional pathways. First, there are clear phenotypic differences between the lon and clpP mutants of LVS. The animal infection trial indicated that the Δlon mutant is substantially more attenuated than the isogenic clpP mutant in the mouse lung infection model. The same trend is true in the intramacrophage growth and tolerance to 1.5% NaCl of the lon and clpP mutants, although both mutants showed similar levels of deficiency in growth under heat stress. These results consistently showed that Lon is more instrumental in F. tularensis-host interactions but that both the Lon and ClpP proteases are similarly important in the stress response of F. tularensis. Second, Lon and ClpP target different substrates. The protein stability experiment in LVS showed that ClpP does not appear to degrade two of the Lon substrates (FTL578 and FTL663), but another Lon substrate (FTL1957) was degraded by ClpP. These lines of information imply that Lon and ClpP regulate homeostasis of different F. tularensis proteins, although certain proteins are degraded by both proteases. Lastly, functional partition between Lon and ClpP is also supported by the observation that clpP is transcribed only from the common promoter of the protease locus but the transcription of lon is additionally driven by its own promoter, in addition to that from the tig promoter. Because the protease locus is extremely conserved in gene sequence and organization among different subspecies of F. tularensis, the data from this study thus provide highly valuable information for a comprehensive understanding of protein homeostasis and pathogenesis in F. tularensis.
The discrepancy between the results obtained with the unmarked deletion and transposon mutants of clpX may be explained by the polar effect of the transposon insertion on the transcription of the downstream genes. Because clpX is cotranscribed with two downstream genes, lon and hupB (Fig. 1B), the attenuation in the in vivo fitness observed with the Tn5-based EZ::TN transposon mutant could be due to the transcriptional blockage of lon and/or hupB (8). The lack of phenotypic changes with the clpX deletion mutant in in vivo infectivity and stress tolerance is reminiscent of similar observations made with the ClpX and ClpA in E. coli. ClpP is necessary for degradation of the hybrid protein CRAG, but this activity occurs in a clpA clpB clpX triple mutant (69). This phenomenon is thought to be mediated by the overlapping functions of the chaperones (e.g., trigger factor, GroEL, and GroES) (69, 70). It is possible that these chaperones play redundant roles with ClpX in facilitating protein degradation by ClpP in F. tularensis. Alternatively, F. tularensis ClpP may functionally couple with an uncharacterized ATPase partner(s), like its counterparts in other bacteria. ClpX appears to be a major ATPase partner of the ClpP protease, given the fact that clpX is cotranscribed with clpP in the protease locus; F. tularensis does not appear to possess homologs of E. coli ClpA or the other ATPases that couple with ClpP in other bacteria (71). However, there is not evidence to rule out an unknown ATPase(s) that may act as a functional ATPase partner for ClpP in the absence of ClpX.
To our knowledge, this represents the first study to identify the Lon substrates at a genome level by a high-throughput approach. Previous studies have identified many ClpP substrates in E. coli (37), C. crescentus (28), S. aureus (38, 39), and Mycobacterium spp. (40), but there is no similar report of searching for Lon substrates in any bacteria. Our approach takes advantage of Lon substrate enrichment in the F. tularensis Δlon mutant under the heat stress condition, the high-throughput capacity of the quantitative proteomic technology, and convenient tools for testing protein degradation by exogenous Lon protease in E. coli. This screen led to the identification of five substrates of the F. tularensis Lon protease. In this context, our experience in this work has indicated that characterizing Lon substrates in the native host bacteria can be challenging because of the difficulty in producing the substrate proteins to workable levels. As an example, wild-type F. tularensis LVS did not produce four of the five Lon substrates to a detectable amount even when they were overexpressed by a well-characterized tetracycline-inducible promoter from a multicopy plasmid; two of the overexpressed Lon substrates remained undetectable even in the Δlon mutant of LVS. As a result, we were able to characterize only three of the five F. tularensis Lon substrates (FTL578, FTL663, and FTL1957) because they were expressed to detectable levels in the Δlon mutant of LVS. Because the non-Lon substrate proteins, as exemplified by ClpX, were readily produced by the same episomal system in both the wild-type LVS and its Δlon mutant, it appears that these Lon substrates were tightly restricted in the native host. The two undetectable Lon substrates (FTL1217 and FTL1228) in the Δlon mutant may be degraded by other proteases, such as ClpP and HslU. This observation indicates additional technical difficulties in direct screening of Lon substrates in the native host bacteria. This makes the approach used to identify Lon substrates in this work appealing for the identification of the Lon substrates in other bacteria, particularly those whose genetic manipulation is not readily achievable.
Homologs of the bacterial Lon proteases are also present in the mitochondrial matrix of humans (LONP1), mice (Lon), and many other eukaryotic organisms (72). Lon has been implicated in controlling mitochondrial matrix protein quality, regulating aerobic respiratory function, degrading oxidized proteins, and maintaining mitochondrial DNA (mtDNA) nucleoid integrity (73). Dysfunction of human LONP1 has been associated with ageing, cancer, and CODAS syndrome, a multisystem developmental disorder in humans (73, 74). LONP1 has been proposed as an anticancer target (10). In part because Lon is absent in Saccharomyces cerevisiae, the best-characterized eukaryotic model organism, its substrates and functions are much less understood than those of the bacterial counterparts. Only a few putative substrates of human LONP1 have been described (10). In this context, the approach we developed in this study may also be applicable to systematic identification of the Lon substrates in humans and other eukaryotic organisms.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to A. Sjostedt for the mglA mutant, J. Beckwith for pBAD18, and T. H. Kawula for pEDL17.
Footnotes
Supplemental material for this article may be found at http://dx.doi.org/10.1128/IAI.00076-16.
REFERENCES
- 1.Oyston PC, Sjostedt A, Titball RW. 2004. Tularaemia: bioterrorism defence renews interest in Francisella tularensis. Nat Rev Microbiol 2:967–978. doi: 10.1038/nrmicro1045. [DOI] [PubMed] [Google Scholar]
- 2.Olsufjev NG, Meshcheryakova IS. 1982. Infraspecific taxonomy of tularemia agent Francisella tularensis McCoy et Chapin. J Hyg Epidemiol Microbiol Immunol 26:291–299. [PubMed] [Google Scholar]
- 3.Feldman KA, Enscore RE, Lathrop SL, Matyas BT, McGuill M, Schriefer ME, Stiles-Enos D, Dennis DT, Petersen LR, Hayes EB. 2001. An outbreak of primary pneumonic tularemia on Martha's Vineyard. N Engl J Med 345:1601–1606. doi: 10.1056/NEJMoa011374. [DOI] [PubMed] [Google Scholar]
- 4.Anthony LS, Kongshavn PA. 1987. Experimental murine tularemia caused by Francisella tularensis, live vaccine strain: a model of acquired cellular resistance. Microb Pathog 2:3–14. doi: 10.1016/0882-4010(87)90110-0. [DOI] [PubMed] [Google Scholar]
- 5.Eigelsbach HT, Downs CM. 1961. Prophylactic effectiveness of live and killed tularemia vaccines. I. Production of vaccine and evaluation in the white mouse and guinea pig. J Immunol 87:415–425. [PubMed] [Google Scholar]
- 6.Kraemer PS, Mitchell A, Pelletier MR, Gallagher LA, Wasnick M, Rohmer L, Brittnacher MJ, Manoil C, Skerett SJ, Salama NR. 2009. Genome-wide screen in Francisella novicida for genes required for pulmonary and systemic infection in mice. Infect Immun 77:232–244. doi: 10.1128/IAI.00978-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weiss DS, Brotcke A, Henry T, Margolis JJ, Chan K, Monack DM. 2007. In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A 104:6037–6042. doi: 10.1073/pnas.0609675104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Su J, Yang J, Zhao D, Kawula TH, Banas JA, Zhang J-R. 2007. Genome-wide identification of Francisella tularensis virulence determinants. Infect Immun 75:3089–3101. doi: 10.1128/IAI.01865-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sauer RT, Baker TA. 2011. AAA+ proteases: ATP-fueled machines of protein destruction. Annu Rev Biochem 80:587–612. doi: 10.1146/annurev-biochem-060408-172623. [DOI] [PubMed] [Google Scholar]
- 10.Goard CA, Schimmer AD. 2014. Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene 33:2690–2699. doi: 10.1038/onc.2013.228. [DOI] [PubMed] [Google Scholar]
- 11.Gottesman S. 2003. Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19:565–587. doi: 10.1146/annurev.cellbio.19.110701.153228. [DOI] [PubMed] [Google Scholar]
- 12.Torres-Cabassa A, Gottesman S, Frederick RD, Dolph PJ, Coplin DL. 1987. Control of extracellular polysaccharide synthesis in Erwinia stewartii and Escherichia coli K-12: a common regulatory function. J Bacteriol 169:4525–4531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mukherjee S, Bree AC, Liu J, Patrick JE, Chien P, Kearns DB. 2015. Adaptor-mediated Lon proteolysis restricts Bacillus subtilis hyperflagellation. Proc Natl Acad Sci U S A 112:250–255. doi: 10.1073/pnas.1417419112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jaskolska M, Gerdes K. 2015. CRP-dependent positive autoregulation and proteolytic degradation regulate competence activator Sxy of Escherichia coli. Mol Microbiol 95:833–845. doi: 10.1111/mmi.12901. [DOI] [PubMed] [Google Scholar]
- 15.Bissonnette SA, Rivera-Rivera I, Sauer RT, Baker TA. 2010. The IbpA and IbpB small heat-shock proteins are substrates of the AAA+ Lon protease. Mol Microbiol 75:1539–1549. doi: 10.1111/j.1365-2958.2010.07070.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leslie DJ, Heinen C, Schramm FD, Thuring M, Aakre CD, Murray SM, Laub MT, Jonas K. 2015. Nutritional control of DNA replication initiation through the proteolysis and regulated translation of DnaA. PLoS Genet 11:e1005342. doi: 10.1371/journal.pgen.1005342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jonas K, Liu J, Chien P, Laub MT. 2013. Proteotoxic stress induces a cell-cycle arrest by stimulating Lon to degrade the replication initiator DnaA. Cell 154:623–636. doi: 10.1016/j.cell.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ricci V, Blair JM, Piddock LJ. 2014. RamA, which controls expression of the MDR efflux pump AcrAB-TolC, is regulated by the Lon protease. J Antimicrob Chemother 69:643–650. doi: 10.1093/jac/dkt432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ingmer H, Brondsted L. 2009. Proteases in bacterial pathogenesis. Res Microbiol 160:704–710. doi: 10.1016/j.resmic.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 20.Robertson GT, Kovach ME, Allen CA, Ficht TA, Roop RM 2nd. 2000. The Brucella abortus Lon functions as a generalized stress response protease and is required for wild-type virulence in BALB/c mice. Mol Microbiol 35:577–588. [DOI] [PubMed] [Google Scholar]
- 21.Breidenstein EB, Janot L, Strehmel J, Fernandez L, Taylor PK, Kukavica-Ibrulj I, Gellatly SL, Levesque RC, Overhage J, Hancock RE. 2012. The Lon protease is essential for full virulence in Pseudomonas aeruginosa. PLoS One 7:e49123. doi: 10.1371/journal.pone.0049123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takaya A, Suzuki M, Matsui H, Tomoyasu T, Sashinami H, Nakane A, Yamamoto T. 2003. Lon, a stress-induced ATP-dependent protease, is critically important for systemic Salmonella enterica serovar Typhimurium infection of mice. Infect Immun 71:690–696. doi: 10.1128/IAI.71.2.690-696.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takaya A, Tomoyasu T, Tokumitsu A, Morioka M, Yamamoto T. 2002. The ATP-dependent lon protease of Salmonella enterica serovar Typhimurium regulates invasion and expression of genes carried on Salmonella pathogenicity island 1. J Bacteriol 184:224–232. doi: 10.1128/JB.184.1.224-232.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herbst K, Bujara M, Heroven AK, Opitz W, Weichert M, Zimmermann A, Dersch P. 2009. Intrinsic thermal sensing controls proteolysis of Yersinia virulence regulator RovA. PLoS Pathog 5:e1000435. doi: 10.1371/journal.ppat.1000435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kruger E, Zuhlke D, Witt E, Ludwig H, Hecker M. 2001. Clp-mediated proteolysis in Gram-positive bacteria is autoregulated by the stability of a repressor. EMBO J 20:852–863. doi: 10.1093/emboj/20.4.852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chastanet A, Prudhomme M, Claverys JP, Msadek T. 2001. Regulation of Streptococcus pneumoniae clp genes and their role in competence development and stress survival. J Bacteriol 183:7295–7307. doi: 10.1128/JB.183.24.7295-7307.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Camberg JL, Hoskins JR, Wickner S. 2009. ClpXP protease degrades the cytoskeletal protein, FtsZ, and modulates FtsZ polymer dynamics. Proc Natl Acad Sci U S A 106:10614–10619. doi: 10.1073/pnas.0904886106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhat NH, Vass RH, Stoddard PR, Shin DK, Chien P. 2013. Identification of ClpP substrates in Caulobacter crescentus reveals a role for regulated proteolysis in bacterial development. Mol Microbiol 88:1083–1092. doi: 10.1111/mmi.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jenal U, Fuchs T. 1998. An essential protease involved in bacterial cell-cycle control. EMBO J 17:5658–5669. doi: 10.1093/emboj/17.19.5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Turgay K, Hahn J, Burghoorn J, Dubnau D. 1998. Competence in Bacillus subtilis is controlled by regulated proteolysis of a transcription factor. EMBO J 17:6730–6738. doi: 10.1093/emboj/17.22.6730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roy AB, Petrova OE, Sauer K. 2012. The phosphodiesterase DipA (PA5017) is essential for Pseudomonas aeruginosa biofilm dispersion. J Bacteriol 194:2904–2915. doi: 10.1128/JB.05346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Loughlin MF, Arandhara V, Okolie C, Aldsworth TG, Jenks PJ. 2009. Helicobacter pylori mutants defective in the clpP ATP-dependent protease and the chaperone clpA display reduced macrophage and murine survival. Microb Pathog 46:53–57. doi: 10.1016/j.micpath.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 33.Raju RM, Unnikrishnan M, Rubin DH, Krishnamoorthy V, Kandror O, Akopian TN, Goldberg AL, Rubin EJ. 2012. Mycobacterium tuberculosis ClpP1 and ClpP2 function together in protein degradation and are required for viability in vitro and during infection. PLoS Pathog 8:e1002511. doi: 10.1371/journal.ppat.1002511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Frees D, Qazi SN, Hill PJ, Ingmer H. 2003. Alternative roles of ClpX and ClpP in Staphylococcus aureus stress tolerance and virulence. Mol Microbiol 48:1565–1578. doi: 10.1046/j.1365-2958.2003.03524.x. [DOI] [PubMed] [Google Scholar]
- 35.Brotz-Oesterhelt H, Beyer D, Kroll HP, Endermann R, Ladel C, Schroeder W, Hinzen B, Raddatz S, Paulsen H, Henninger K, Bandow JE, Sahl HG, Labischinski H. 2005. Dysregulation of bacterial proteolytic machinery by a new class of antibiotics. Nat Med 11:1082–1087. doi: 10.1038/nm1306. [DOI] [PubMed] [Google Scholar]
- 36.Conlon BP, Nakayasu ES, Fleck LE, LaFleur MD, Isabella VM, Coleman K, Leonard SN, Smith RD, Adkins JN, Lewis K. 2013. Activated ClpP kills persisters and eradicates a chronic biofilm infection. Nature 503:365–370. doi: 10.1038/nature12790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flynn JM, Neher SB, Kim YI, Sauer RT, Baker TA. 2003. Proteomic discovery of cellular substrates of the ClpXP protease reveals five classes of ClpX-recognition signals. Mol Cell 11:671–683. doi: 10.1016/S1097-2765(03)00060-1. [DOI] [PubMed] [Google Scholar]
- 38.Feng J, Michalik S, Varming AN, Andersen JH, Albrecht D, Jelsbak L, Krieger S, Ohlsen K, Hecker M, Gerth U, Ingmer H, Frees D. 2013. Trapping and proteomic identification of cellular substrates of the ClpP protease in Staphylococcus aureus. J Proteome Res 12:547–558. doi: 10.1021/pr300394r. [DOI] [PubMed] [Google Scholar]
- 39.Graham JW, Lei MG, Lee CY. 2013. Trapping and identification of cellular substrates of the Staphylococcus aureus ClpC chaperone. J Bacteriol 195:4506–4516. doi: 10.1128/JB.00758-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Raju RM, Jedrychowski MP, Wei JR, Pinkham JT, Park AS, O'Brien K, Rehren G, Schnappinger D, Gygi SP, Rubin EJ. 2014. Post-translational regulation via Clp protease is critical for survival of Mycobacterium tuberculosis. PLoS Pathog 10:e1003994. doi: 10.1371/journal.ppat.1003994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carroll P, Faray-Kele MC, Parish T. 2011. Identifying vulnerable pathways in Mycobacterium tuberculosis by using a knockdown approach. Appl Environ Microbiol 77:5040–5043. doi: 10.1128/AEM.02880-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chamberlain RE. 1965. Evaluation of live tularemia vaccine prepared in a chemically defined medium. Appl Microbiol 13:232–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng K, Blick RJ, Liu W, Hansen EJ. 2006. Identification of Francisella tularensis genes affected by iron limitation. Infect Immun 74:4224–4236. doi: 10.1128/IAI.01975-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, Brouwer D, Nair V, Fischer ER, Wicke L, Curda AJ, Kupko JJ III, Martens C, Crane DD, Bosio CM, Porcella SF, Celli J. 2009. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol 11:1128–1150. doi: 10.1111/j.1462-5822.2009.01316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wen Z, Sertil O, Cheng Y, Zhang S, Liu X, Wang WC, Zhang JR. 2015. Sequence elements upstream of the core promoter are necessary for full transcription of the capsule gene operon in Streptococcus pneumoniae strain D39. Infect Immun 83:1957–1972. doi: 10.1128/IAI.02944-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grall N, Livny J, Waldor M, Barel M, Charbit A, Meibom KL. 2009. Pivotal role of the Francisella tularensis heat-shock sigma factor RpoH. Microbiology 155:2560–2572. doi: 10.1099/mic.0.029058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su J, Asare R, Yang J, Nair MK, Mazurkiewicz JE, Abu-Kwaik Y, Zhang JR. 2011. The capBCA locus is required for intracellular growth of Francisella tularensis LVS. Front Microbiol 2:83. doi: 10.3389/fmicb.2011.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu L, Ma Y, Zhang JR. 2006. Streptococcus pneumoniae recruits complement factor H through the amino terminus of CbpA. J Biol Chem 281:15464–15474. doi: 10.1074/jbc.M602404200. [DOI] [PubMed] [Google Scholar]
- 49.Yuan G, Wong SL. 1995. Regulation of groE expression in Bacillus subtilis: the involvement of the sigma A-like promoter and the roles of the inverted repeat sequence (CIRCE). J Bacteriol 177:5427–5433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Weekes MP, Tomasec P, Huttlin EL, Fielding CA, Nusinow D, Stanton RJ, Wang EC, Aicheler R, Murrell I, Wilkinson GW, Lehner PJ, Gygi SP. 2014. Quantitative temporal viromics: an approach to investigate host-pathogen interaction. Cell 157:1460–1472. doi: 10.1016/j.cell.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin LX, Huo Y, Zheng ZG, Jiang XY, Deng HY, Chen YL, Lian QQ, Ge RS, Deng HT. 2014. Down-regulation of Ras-related protein Rab 5C-dependent endocytosis and glycolysis in cisplatin-resistant ovarian cancer cell lines. Mol Cell Proteomics 13:3138–3151. doi: 10.1074/mcp.M113.033217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wohlever ML, Nager AR, Baker TA, Sauer RT. 2013. Engineering fluorescent protein substrates for the AAA+ Lon protease. Protein Eng Des Sel 26:299–305. doi: 10.1093/protein/gzs105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Choy JS, Aung LL, Karzai AW. 2007. Lon protease degrades transfer-messenger RNA-tagged proteins. J Bacteriol 189:6564–6571. doi: 10.1128/JB.00860-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sambrook J, Fritsch EF, Maniatis T. 1989. Molecular cloning: a laboratory manual, 2nd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 56.Shah IM, Wolf RE Jr. 2006. Sequence requirements for Lon-dependent degradation of the Escherichia coli transcription activator SoxS: identification of the SoxS residues critical to proteolysis and specific inhibition of in vitro degradation by a peptide comprised of the N-terminal 21 amino acid residues. J Mol Biol 357:718–731. doi: 10.1016/j.jmb.2005.12.088. [DOI] [PubMed] [Google Scholar]
- 57.LoVullo ED, Miller CN, Pavelka MS Jr, Kawula TH. 2012. TetR-based gene regulation systems for Francisella tularensis. Appl Environ Microbiol 78:6883–6889. doi: 10.1128/AEM.01679-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baron GS, Nano FE. 1998. MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol Microbiol 29:247–259. doi: 10.1046/j.1365-2958.1998.00926.x. [DOI] [PubMed] [Google Scholar]
- 59.Lauriano CM, Barker JR, Yoon SS, Nano FE, Arulanandam BP, Hassett DJ, Klose KE. 2004. MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc Natl Acad Sci U S A 101:4246–4249. doi: 10.1073/pnas.0307690101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mizusawa S, Gottesman S. 1983. Protein degradation in Escherichia coli: the lon gene controls the stability of sulA protein. Proc Natl Acad Sci U S A 80:358–362. doi: 10.1073/pnas.80.2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Griffith KL, Shah IM, Wolf RE Jr. 2004. Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol Microbiol 51:1801–1816. doi: 10.1046/j.1365-2958.2003.03952.x. [DOI] [PubMed] [Google Scholar]
- 62.Goodman JL, Wang S, Alam S, Ruzicka FJ, Frey PA, Wedekind JE. 2004. Ornithine cyclodeaminase: structure, mechanism of action, and implications for the mu-crystallin family. Biochemistry 43:13883–13891. doi: 10.1021/bi048207i. [DOI] [PubMed] [Google Scholar]
- 63.Cho K, Fuqua C, Martin BS, Winans SC. 1996. Identification of Agrobacterium tumefaciens genes that direct the complete catabolism of octopine. J Bacteriol 178:1872–1880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tempel R, Lai XH, Crosa L, Kozlowicz B, Heffron F. 2006. Attenuated Francisella novicida transposon mutants protect mice against wild-type challenge. Infect Immun 74:5095–5105. doi: 10.1128/IAI.00598-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Boyd ES, Thomas KM, Dai Y, Boyd JM, Outten FW. 2014. Interplay between oxygen and Fe-S cluster biogenesis: insights from the Suf pathway. Biochemistry 53:5834–5847. doi: 10.1021/bi500488r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Matuszewska M, Kuczynska-Wisnik D, Laskowska E, Liberek K. 2005. The small heat shock protein IbpA of Escherichia coli cooperates with IbpB in stabilization of thermally aggregated proteins in a disaggregation competent state. J Biol Chem 280:12292–12298. doi: 10.1074/jbc.M412706200. [DOI] [PubMed] [Google Scholar]
- 67.Ratajczak E, Zietkiewicz S, Liberek K. 2009. Distinct activities of Escherichia coli small heat shock proteins IbpA and IbpB promote efficient protein disaggregation. J Mol Biol 386:178–189. doi: 10.1016/j.jmb.2008.12.009. [DOI] [PubMed] [Google Scholar]
- 68.Matuszewska E, Kwiatkowska J, Kuczynska-Wisnik D, Laskowska E. 2008. Escherichia coli heat-shock proteins IbpA/B are involved in resistance to oxidative stress induced by copper. Microbiology 154:1739–1747. doi: 10.1099/mic.0.2007/014696-0. [DOI] [PubMed] [Google Scholar]
- 69.Kandror O, Sherman M, Rhode M, Goldberg AL. 1995. Trigger factor is involved in GroEL-dependent protein degradation in Escherichia coli and promotes binding of GroEL to unfolded proteins. EMBO J 14:6021–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kandror O, Sherman M, Goldberg A. 1999. Rapid degradation of an abnormal protein in Escherichia coli proceeds through repeated cycles of association with GroEL. J Biol Chem 274:37743–37749. doi: 10.1074/jbc.274.53.37743. [DOI] [PubMed] [Google Scholar]
- 71.Robertson GT, Ng WL, Gilmour R, Winkler ME. 2003. Essentiality of clpX, but not clpP, clpL, clpC, or clpE, in Streptococcus pneumoniae R6. J Bacteriol 185:2961–2966. doi: 10.1128/JB.185.9.2961-2966.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gur E. 2013. The Lon AAA+ protease. Subcell Biochem 66:35–51. doi: 10.1007/978-94-007-5940-4_2. [DOI] [PubMed] [Google Scholar]
- 73.Pinti M, Gibellini L, Liu Y, Xu S, Lu B, Cossarizza A. 2015. Mitochondrial Lon protease at the crossroads of oxidative stress, ageing and cancer. Cell Mol Life Sci 72:4807–4824. doi: 10.1007/s00018-015-2039-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strauss KA, Jinks RN, Puffenberger EG, Venkatesh S, Singh K, Cheng I, Mikita N, Thilagavathi J, Lee J, Sarafianos S, Benkert A, Koehler A, Zhu A, Trovillion V, McGlincy M, Morlet T, Deardorff M, Innes AM, Prasad C, Chudley AE, Lee IN, Suzuki CK. 2015. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am J Hum Genet 96:121–135. doi: 10.1016/j.ajhg.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vonkavaara M, Telepnev MV, Ryden P, Sjostedt A, Stoven S. 2008. Drosophila melanogaster as a model for elucidating the pathogenicity of Francisella tularensis. Cell Microbiol 10:1327–1338. doi: 10.1111/j.1462-5822.2008.01129.x. [DOI] [PubMed] [Google Scholar]
- 76.LoVullo ED, Sherrill LA, Perez LL, Pavelka MS Jr. 2006. Genetic tools for highly pathogenic Francisella tularensis subsp. tularensis. Microbiology 152:3425–3435. doi: 10.1099/mic.0.29121-0. [DOI] [PubMed] [Google Scholar]
- 77.Margolis JJ, El-Etr S, Joubert LM, Moore E, Robison R, Rasley A, Spormann AM, Monack DM. 2010. Contributions of Francisella tularensis subsp. novicida chitinases and Sec secretion system to biofilm formation on chitin. Appl Environ Microbiol 76:596–608. doi: 10.1128/AEM.02037-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Siddaramappa S, Challacombe JF, Petersen JM, Pillai S, Hogg G, Kuske CR. 2011. Common ancestry and novel genetic traits of Francisella novicida-like isolates from North America and Australia as revealed by comparative genomic analyses. Appl Environ Microbiol 77:5110–5122. doi: 10.1128/AEM.00337-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomas RM, Titball RW, Oyston PC, Griffin K, Waters E, Hitchen PG, Michell SL, Grice ID, Wilson JC, Prior JL. 2007. The immunologically distinct O antigens from Francisella tularensis subspecies tularensis and Francisella novicida are both virulence determinants and protective antigens. Infect Immun 75:371–378. doi: 10.1128/IAI.01241-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.