

Abstract
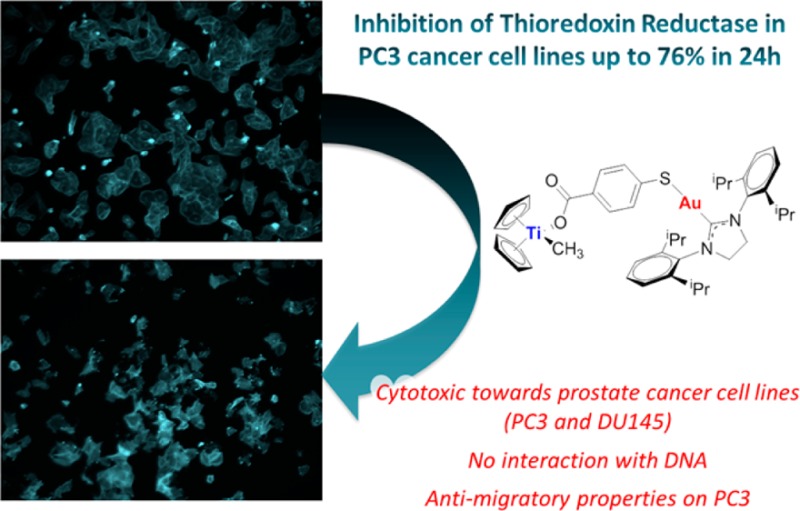
We report on the synthesis, characterization, and stability studies of new titanocene complexes containing a methyl group and a carboxylate ligand (mba = −OC(O)-p-C6H4-S−) bound to gold(I)–N-heterocyclic carbene fragments through the thiolate group: [(η5-C5H5)2TiMe(μ-mba)Au(NHC)]. The cytotoxicities of the heterometallic compounds along with those of novel monometallic gold–N-heterocyclic carbene precursors [(NHC)Au(mbaH)] have been evaluated against renal, prostate, colon, and breast cancer cell lines. The highest activity and selectivity and a synergistic effect of the resulting heterometallic species was found for the prostate and colon cancer cell lines. The colocalization of both titanium and gold metals (1:1 ratio) in PC3 prostate cancer cells was demonstrated for the selected compound 5a, indicating the robustness of the heterometallic compound in vitro. We describe here preliminary mechanistic data involving studies on the interaction of selected mono- and bimetallic compounds with plasmid (pBR322) used as a model nucleic acid and the inhibition of thioredoxin reductase in PC3 prostate cancer cells. The heterometallic compounds, which are highly apoptotic, exhibit strong antimigratory effects on the prostate cancer cell line PC3.
Introduction
The potential of heterometallic complexes as cancer chemotherapeutics has been recently highlighted.1 The improved activity of heteronuclear complexes as antitumor agents by incorporation of two different cytotoxic metals within the same molecule has been demonstrated. The beneficial influence may be due to a synergistic or cooperative effect. Bimetallic and trimetallic compounds with anticancer properties have been described. There have been reports on titanocenes incorporating Ru(II), Pt(II), and Pd(II) centers2,3 and a number of complexes containing ferrocene moieties and other metals.4 Bimetallic systems based on Ru(II)–Pt(II)5 or Ru(II)–Ir(III)6 have also been described. Ferrocenyl phosphanes were incorporated in the iminophosphorane skeleton of gold(III) and palladium(II) coordination complexes.4 Heterometallic compounds based on gold(I) fragments have been reported for titanocene,1,3,7,8 ruthenium(II),9−12 platinum(II),13 rhenium(I),14 and copper(II)10 derivatives.
We have reported on a number of titanocene–gold derivatives with potential as anticancer agents (zero-, first-, and second-generation derivatives 1–3 in Chart 1).1,3,8 We described cytotoxic species in which gold fragments coordinate to cyclopentadienyl–phosphane ligands that displayed a synergistic effect (such as 1 in Chart 1).3 In order to improve stability in physiological media and prevent the loss of cyclopentadienyl–gold fragments, first-generation derivatives (such as 2) were developed.8 While Ti–Cp hydrolysis still occurs at pH 7, the gold fragment remains linked to titanium by the carboxylate–phosphane ligand. These first-generation compounds showed excellent activity against renal cancer cell lines.8 In addition, the compounds were more selective toward cancerous cells and lacked systemic toxicity in mice models.
Chart 1. Potential Anticancer Titanocene–Gold Complexes Containing Different Linkers Described by Our Group1,3,8.
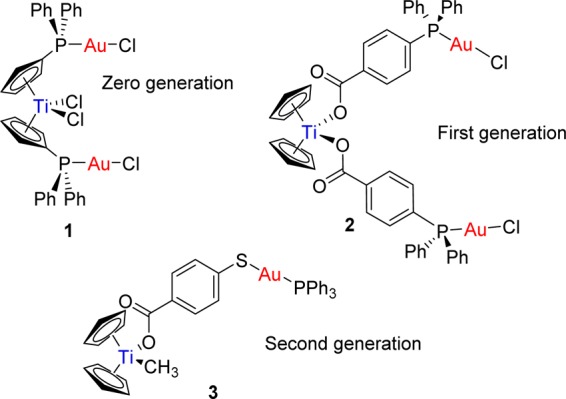
A further and successful modification was the introduction of a bifunctional ligand, mba (−OC(O)-p-C6H4-S–; derived from 4-mercaptobenzoic acid (H2mba)). We reported on compounds of the type [(η5-C5H5)2TiMe(μ-mba)Au(PR3)] (second generation such as 3 in Chart 1). Compound 3 was able to block renal cancer growth both in vitro and in vivo by pathway(s) that involve the inhibition of thioredoxin reductase and decreased expression of protein kinases known to drive cell migration.1 Preliminary evidence indicated that compound 3 may have appreciable anti-invasive properties. In addition, its robustness was demonstrated in cellular uptake experiments on Caki-1 cells by colocalization of Ti and Au metals in a 1:1 ratio.1
We aimed to exchange the gold(I)–phosphane fragments by gold(I)–N-heterocyclic carbene moieties to improve or modify the pharmacological profile of the previously reported heterometallic complexes [(η5-C5H5)2TiMe(μ-mba)Au(PR3)] (such as 3). NHC–gold(I) complexes are usually more stable than gold(I)–phosphane compounds, display strong antimitochondrial effects,15,16 and have demonstrated excellent inhibitory properties of certain enzymes such as thioredoxin reductase.15−23 They have also relevant anticancer effects in vitro15−25 (including some recent examples on heterometallic complexes9,10), and in vivo effects on melanoma have recently been described.26 We report here on the synthesis, characterization, and stability studies of the novel monometallic gold–N-heterocyclic carbene precursors [(NHC)Au(mbaH)] and heterometallic titanocene complexes of the type [(η5-C5H5)2TiMe(μ-mba)Au(NHC)]. We describe their in vitro activity against human renal, prostate, colon, and breast cancer cell lines and nontumorigenic human embryonic kidney cell lines HEK-293T. In addition, we present studies of cellular uptake, the effect of compounds on cell death, interactions with DNA, inhibitory effects on thioredoxin reductase in vitro, and antimigratory properties on PC3 prostate cancer cells.
Results and Discussion
Synthesis and Characterization
The synthesis of the new monometallic gold(I)–NHC compounds bearing the bifunctional ligand Hmba (Scheme 1) was carried out following the same strategy reported for the synthesis of species [Au(Hmba)(phosphane)].1
Scheme 1. Preparation of Monometallic Gold Complexes [Au(NHC)(Hmba)] (NHC = SIPr (4a), IPr (4b), IMes (4c), ICy (4d)).
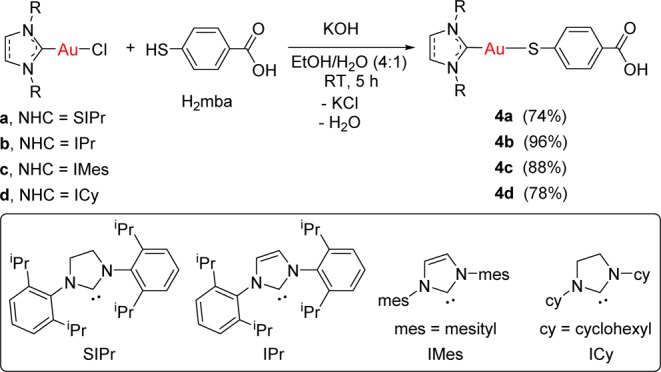
The thiol group on the bifunctional ligand H2mba (1 equiv) was deprotonated by reaction with 1 equiv of KOH for 20 min at room temperature. Subsequent addition of 1 equiv of the gold(I) N-heterocyclic carbene complexes a–d in situ, in a mixture of ethanol and water (4/1), led to the formation of the corresponding monometallic gold complexes [Au(NHC)(Hmba)] (NHC = SIPr (4a), IPr (4b), IMes (4c), ICy (4d)). Compounds 4a–d were isolated as pale orange solids in high yield and characterized by NMR and UV–vis spectroscopy, mass spectrometry, and elemental analysis (see the Experimental Section). The chemical shifts of the carbene carbons in the new derivatives (13C{1H} NMR spectra) appear at fields lower (∼10 ppm) than those reported for the parent chloro derivatives a–d,27,28 indicating a smaller electron-donating character of the Hmba ligand (σ donor) with respect to the chloride ligand (σ and π donor).
In the case of compound 4c, crystals suitable for X-ray diffraction (Figure 1) were obtained by layering n-pentane over a solution of compound 4c in tetrahydrofuran.
Figure 1.
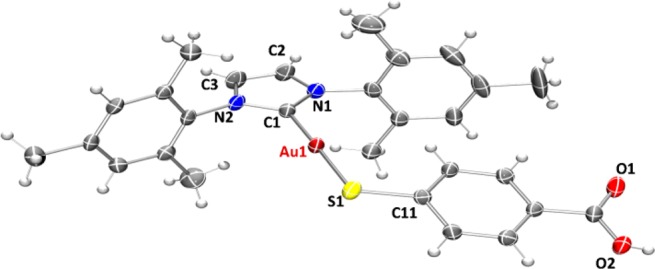
ORTEP view of the molecular structure of 4c showing the labeling scheme. The labels for hydrogen and some carbon atoms are omitted for clarity. A drawing of the molecular structure containing all labeled carbon atoms is provided in the Supporting Information.
The crystals of compound 4c were determined to be triclinic (space group P1̅) with Z = 4 formula units in the unit cell. The environment of the gold atoms is close to linear (C–Au–S 177.76(18)°) (Figure 1). A selection of structural parameters is given in Table 1. The individual monomeric units (Figure 1) show hydrogen bonds (∼1.83 Å) between the carboxylic groups of two neighboring units (Figure 2).
Table 1. Selected Structural Parameters of Complex 4c Obtained from X-ray Single-Crystal Diffraction Studiesa.
| Au(1)–C(1) | 1.995(6) | N(1)–C(2) | 1.381(8) |
| Au(1)–S(1) | 2.2790(17) | N(1)–C(21) | 1.453(7) |
| S(1)–C(11) | 1.746(6) | N(2)–C(3) | 1.382(7) |
| C(1)–N(1) | 1.353(7) | N(2)–C(31) | 1.456(7) |
| C(1)–N(2) | 1.341(7) | C(2)–C(3) | 1.335(9) |
| C(1)–Au(1)–S(1) | 177.76(18) | N(2)–C(1)–Au(1) | 129.8(4) |
| C(11)–S(1)–Au(1) | 105.92(19) | N(1)–C(1)–Au(1) | 126.0(4) |
| C(1)–N(1)–C(2) | 111.4(5) | C(1)–N(2)–C(3) | 111.1(5) |
| C(1)–N(1)–C(21) | 123.4(5) | C(1)–N(2)–C(31) | 124.8(4) |
| C(2)–N(1)–C(21) | 125.2(5) | C(3)–N(2)–C(31) | 124.1(5) |
| N(2)–C(1)–N(1) | 104.2(5) | C(3)–C(2)–N(1) | 106.2(5) |
Bond lengths are given in Å and angles in deg.
Figure 2.
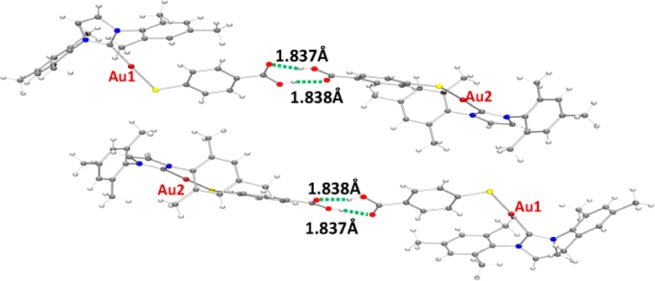
ORTEP view of the polymeric structure of compound 4c showing hydrogen bonds (green dotted line). The color code is as given in Figure 1.
Similar gold(I) polymeric compounds reported by us1 and others29 display additional Au–S interactions between the dimeric units which are not present in the structure of 4c. This fact might be related to the greater steric hindrance caused by the presence of a bulkier ligand such as the NHC ligands employed here.
The synthesis of the new heterometallic TiAu complexes is depicted in Scheme 2. The reaction of 1 equiv of each of the mononuclear gold(I) complexes 4a–d with 1 equiv of ([(η5-C5H5)2TiMe2] afforded the corresponding heterobimetallic complexes [([(η5-C5H5)2TiMe(μ-mba)Au(NHC)] (NHC = SIPr (5a), IPr (5b), IMes (5c), ICy (5d)), with concomitant elimination of 1 equiv of methane. Compounds 5a–d were obtained in moderate to high yields as air- and moisture-stable yellow solids. These compounds are less acidic than titanocene dichloride and soluble in DMSO/H2O, DMSO/PBS, or DMSO/media (1/99) mixtures at micromolar concentrations, which is relevant for subsequent biological testing. Moreover, they are more soluble at higher concentrations in DMSO/PBS mixtures than the previously described [(η5-C5H5)2TiMe(μ-mba)Au(PR3)] counterparts. Compounds 5a–d are stable as solids in air and at 5 °C for months and in CDCl3 solution for at least 3 days. They are stable in DMSO-d6 solution for weeks.
Scheme 2. Preparation of Heterometallic Titanocene–Gold Complexes [(η5-C5H5)2Ti(CH3){OC(O)-p-C6H4SAu(NHC)}] (NHC = SIPr (5a), IPr (5b), IMes (5c), ICy (5d)).
The structures of complexes 5a–d depicted in Scheme 1 have been proposed on the basis of NMR and UV–vis spectroscopy, mass spectrometry, and elemental analysis (see the Experimental Section). Moreover, IR experiments and DFT calculations were carried out in order to shed light on the coordination mode of the carboxylate groups. The differences found between the symmetric and antisymmetric stretching bands for the carboxylate groups in the solid state IR spectra (ranging from 210 to 351 cm–1) are greater than 200 cm–1, indicating a monodentate coordination mode.30,31 DFT calculations (e.g., Figure 3) also confirmed the monodentate nature of the carboxylate functionality. In the Supporting Information, data on different optimizations are provided. All calculations performed led to the species containing a monodentate carboxylate. Similar difference values (ranging from 200 to 328 cm–1) were found between the symmetric and antisymmetric stretching bands for the carboxylate groups in the IR calculated on the basis of the DFT studies.
Figure 3.
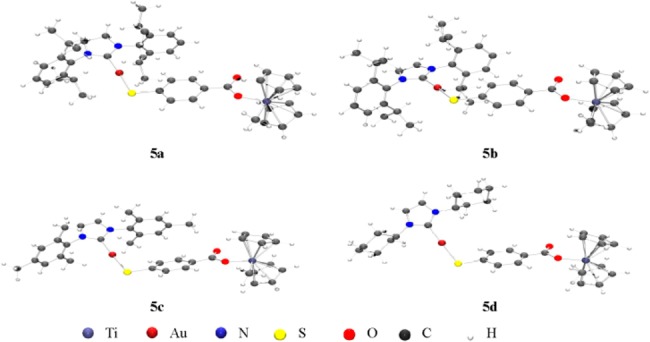
Optimized structures for heterometallic complexes 5a–d.
The stability of compounds 5a–d was evaluated by 1H NMR spectroscopy in DMSO-d6 and DMSO-d6/PBS-D2O (3/2) and by mass spectrometry over time (see the Supporting Information). NMR experiments were performed in DMSO-d6 and in DMSO-d6/PBS-D2O mixtures. The stability study of compounds 5a–d by 1H NMR in DMSO-d6 showed half-life values of 1, 3, 2, and 2 h, respectively: i.e., shorter than those for the corresponding phosphane derivatives1 (3, Chart 1). However, as shown in Table 2, compounds 5a–d exhibited longer half-lives in 3/2 of DMSO-d6/PBS-D2O mixtures. Titanocene dichloride is also known to hydrolyze with a higher rate in DMSO than in water.32 Mass spectrometry further supports the presence of species containing both titanium and gold in 1% DMSO/PBS solution after 24 h (see the Supporting Information).
Table 2. Half-Lives (h) of Heterobimetallic Compounds 5a–d in DMSO and in DMSO/PBS Mixtures by NMR.
| 5a | 5b | 5c | 5d | |
|---|---|---|---|---|
| 1H NMR, DMSO-d6 | 1 | 3 | 2 | 2 |
| 1H NMR, DMSO-d6 /PBS-D2O (3/2) | 24 | 24 | 24 | 48 |
Biological Activity
Assays of Cytotoxicity and Cell Death
The cytotoxicity of the heterometallic complexes [(η5-C5H5)2TiMe(μ-mba)Au(NHC)] (NHC = SIPr (5a), IPr (5b), IMes (5c), ICy (5d)), monometallic gold(I) complexes [Au(NHC)(Hmba)] (NHC = SIPr (4a), IPr (4b), IMes (4c), ICy (4d) in Scheme 1), and titanocene Y33 used as control was assayed by monitoring their ability to inhibit cell growth using the PrestoBlue Cell Viability assay (see the Experimental Section). The cytotoxic activity of the compounds was determined as described in the Experimental Section. In this assay, human cancer cell lines such as prostate PC3 and DU145, renal Caki-1, colon DLL1, triple negative breast MDA-MB-231, and nontumorigenic human embryonic kidney cell lines HEK-293T were incubated with the indicated compound for 72 h. The results are summarized in Table 3.
Table 3. IC50 Values (μM) in Human Cell Lines Determined with Heterometallic Ti–Au Compounds 5a–d, Monometallic Au Compounds 4a–d, and Titanocene Y as Controla.
| compound | PC3 | DU-145 | Caki-1 | DLD1 | MDA-MB-231 | HEK-293T |
|---|---|---|---|---|---|---|
| titanocene Y | 58.1 ± 11.2 | 55.2 ± 7.9 | 29.4 ± 4.2 | 56.2 ± 9.8 | 18.0 ± 3.6 | >200 |
| 5a | 9.8 ± 2.2 | 11.8 ± 3.0 | 21.0 ± 1.9 | 13.9 ± 1.7 | >100 | 58.8 ± 6.7 |
| 5b | 10.3 ± 2.8 | 18.9 ± 2.9 | 51.5 ± 3.7 | 30.4 ± 4.1 | >100 | >100 |
| 5c | 17.1 ± 2.9 | 13.76 ± 2.7 | 29.11 ± 4.1 | 19.9 ± 4.1 | >100 | 69.7 ± 9.9 |
| 5d | 11.8 ± 1.6 | 16.7 ± 2.0 | 42.9 ± 5.8 | 21.5 ± 2.0 | >100 | 77.1 ± 9.1 |
| 4a | 66.3 ± 6.4 | 74.8 ± 4.4 | 81.4 ± 2.9 | 78.2 ± 6 | >100 | >100 |
| 4b | 70.4 ± 6.8 | 60.9 ± 5.2 | 79.2 ± 11.7 | 82.6 ± 5.9 | >100 | >100 |
| 4c | 57.1 ± 5.1 | 67.6 ± 7.1 | 97.2 ± 8.6 | 73.1 ± 9.6 | >100 | 87.9 ± 6.4 |
| 4d | 65.1 ± 4.4 | 59.9 ± 4.7 | 88.9 ± 5.1 | 77.5 ± 8.1 | >100 | 97.2 ± 5.1 |
All compounds were dissolved in 1% of DMSO and diluted with water before addition to cell culture medium for a 72 h incubation period.
The heterometallic compounds are considerably more toxic to the prostate cancer cell lines (PC3 and DU145) and the colon cancer cell line (DLD1) than titanocene Y. In addition, the heterometallic compounds 5a–d are more toxic in all the cell lines (excluding the triple negative breast cancer cell lines) than the monometallic gold compounds 4a–d on these cells. None of the heterometallic or monometallic gold compounds are toxic in the triple negative breast MDA-MB-231 cancer cell lines in concentrations lower than 100 μM, as opposed to titanocene Y. While the heterometallic compounds are toxic on renal Caki-1 cancer cell lines, the IC50 values are only comparable to those for titanocene Y for compounds 5a,c. These IC50 values are larger than those found for the first-generation titanocene–gold compounds previously described by us8 and larger than the IC50 value of the second-generation compounds of the type [(η5-C5H5)2TiMe(μ-mba)Au(PR3)]1 (especially 3). In terms of selectivity, the heterometallic compounds exhibit selectivity for the cancer cell lines (excluding the triple negative breast cancer cell line, MDA-MB-231), with compound 5b having a better selectivity in comparison to nontumorigenic human embryonic kidney cell lines HEK-293T. The new compounds display a better selectivity toward the HEK-293T cell line than the phosphane [(η5-C5H5)2TiMe(μ-mba)Au(PR3)] derivatives described before.
We did not find a strong correlation between the type of NHC ligand employed and the biological activity.
Lysates of PC3 cells treated with 5a were analyzed by inductively coupled plasma mass spectrometry (ICP-MS) to determine the colocalization and amounts of Au and Ti metals in these prostate cancer cell lines (see the Experimental Section for details). Lysate of untreated cells was employed as a control. It was observed that these cells have some basal levels of Au present (0.1 μg Au per mg cell protein). The cellular uptake of this compound increases with the increase in drug concentration in the media, indicating a dose-dependent uptake of compound 5a by PC3 cells (Figure 4).
Figure 4.

Cellular uptake of compound 5a in PC3 cells. The concentrations of compound 5a calculated on the basis of Ti and Au content in the cell lysates are similar, suggesting that the compound is robust and that both elements are colocalized in the cells.
Increasing the drug concentration from 10 to 20 μM resulted in a 2.6-fold increase in the cellular levels of compound 5a. More importantly, on correction for background levels of Au and Ti in cell lysates, the stoichiometric ratios of these elements were close to unity, suggesting that the compound remains stable in the intracellular environment after 72 h (or that at least uptake of both metals occurs concurrently).
We had found that the compound [(η5-C5H5)2TiMe(μ-mba)Au(PPh3)] (3) exerted cell death by inducing apoptosis.1 Titanocenes C, X, and Y are also known to induce apoptosis in different cancer cell lines.34 In order to gain some insight into the nature of the cytotoxicity of the new heterometallic compounds containing N-heterocyclic carbenes, we performed some experiments. The effect of titanocene Y, selected monometallic compounds 4a,b, and bimetallic 5a,b on necrosis and apoptosis on Caki-1 and PC3 was assessed by measuring protease activity using non-cell-permeable substrates and cell-permeable substrates and by measuring the total caspase-3 and -7 activities with the ApoTox-Glo triplex assay (see the Experimental Section). The effect of each treatment was determined by comparing treated and untreated cells after 72 h incubation. The results for each treatment were expressed as fold changes between nontreated (0.1% DMSO) and treated samples. ApoTox-Glo triplex assays were repeated twice (n = 2), and each repetition was run in quadruplicate.
The average of the four values was used for statistical calculations. The data (Figure 5) are presented as the mean value. It is important to note that, while all compounds are represented at once in graphs in Figure 5, the amounts of compound used to run the assays is different (according to their different IC50 values). The lower amounts used (20 μM) correspond to the new compounds 5a,b.
Figure 5.
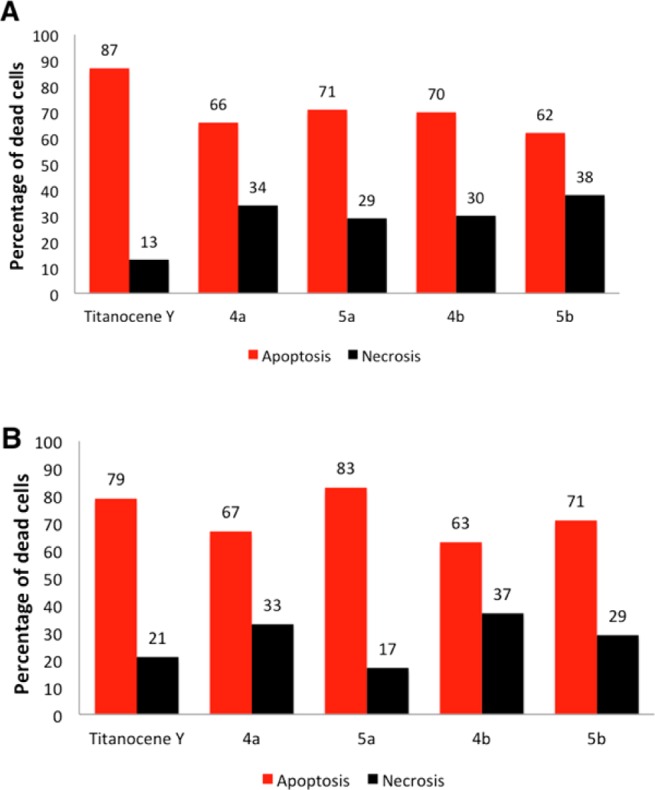
(A) Impairment of the viability of Caki-1 cells by compounds 4a (80 μM), 5a (20 μM), 4b (80 μM) 5b (50 μM), and titanocene Y (30 μM) by inducing apoptosis. (B) Impairment of the viability of PC3 cells by compounds 4a (60 μM), 5a (10 μM), 4b (70 μM), 5b (10 μM), and titanocene Y (30 μM) by inducing apoptosis.
From these data, it can be deduced that compounds 5a,b mainly induce apoptosis both in the renal Caki-1 and prostate PC3 cancer cell lines in a way similar to that for titanocene Y (highly apoptotic in vitro and in vivo34,35) and in the case of compound 5a in Caki-1 and both 5a and 5b in PC3 with a lower IC50 value. Titanocene Y was specially apoptotic on PC3 cancer cell lines. Monometallic compounds 4a,b were also mainly apoptotic, but their IC50 values are considerably higher than those of the new compounds 5a,b.
Migration Studies
In advanced tumors, increased cell migration is a hallmark of cancer cell invasion and metastasis.36,37 The effect of titanocenes on the migratory capability of cancer cells has been scarcely studied.38 We previously found that first-generation [(η5-C5H5)2Ti{OC(O)-p-C6H4–PPh2AuCl}2] (2)8 and second-generation [(η5-C5H5)2TiMe(μ-mba)Au(PPh3)] (3)1 titanocene–gold complexes displayed relevant antimigratory properties.1 We evaluated the anti-invasive properties of the most active heterometallic complexes [(η5-C5H5)2TiMe(μ-mba)Au(NHC)] (NHC = SIPr (5a), IPr (5b)) and titanocene Y, by using the same wound-healing scratch assay (Experimental Section) on prostate cancer PC3 cell lines. Twenty-four hours following a scratch through an in vitro confluent monolayer of prostate carcinoma PC3, cells treated with 15 μM titanocene Y invaded 69% of the scratch and cells treated with 5 μM of 5a or 5b invaded 42% or 33% of the scratch, respectively, while cells treated with 0.1% DMSO control invaded 88% of the scratch (Figure 6A). Figure 6B shows a comparison in terms of total reduction of migration among compounds 5a,b and titanocene Y. A similar assay was performed with gold monometallic compounds 4a,b (see Figure S60 in the Supporting Information), and it was found that under the same conditions (in vitro confluent monolayer of prostate carcinoma PC3 cells treated with 15 μM of the gold monometallic compounds) 4a,b invaded 73% and 80% of the scratch, respectively.
Figure 6.
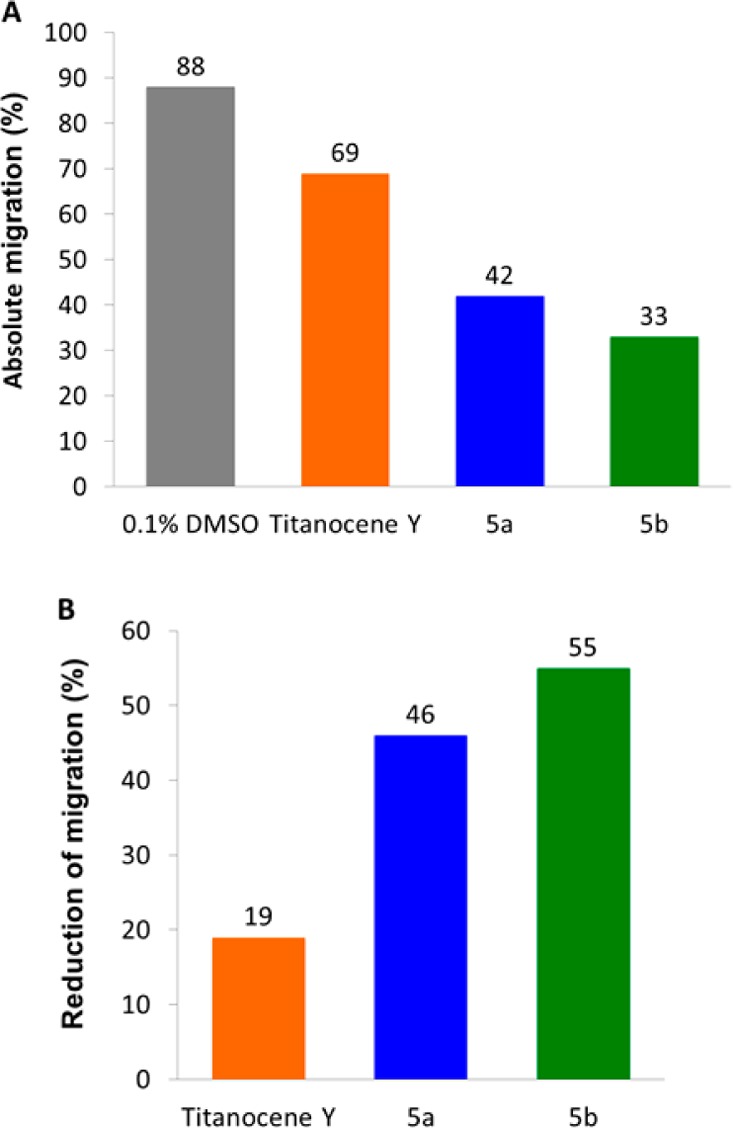
Cell migration in 5a or 5b treated PC3 cells. Migration of PC3 cells was assessed using a wound-healing assay following treatment with 15 μM titanocene Y or 5 μM of 5a or 5b incubated for 24 h (values normalized against 0.1% DMSO control): (A) absolute migration (%); (B) reduction of migration (%).
We can conclude from this experiment that heterometallic compounds 5a,b possess antimigratory properties in PC3 cells in comparison to the control and are twice as powerful in inhibiting migration as titanocene Y. Recently enantiopure cyclopentadienyl Ti(IV) oximato compounds have been shown to affect the cell adhesion and migration of PC3 cancer cell lines.39 In this case the compounds (50 μM) showed a migration capability (62–72% of wound healing) significantly lower than that of control cells. The heterometallic complexes described here (5a,b) thus have strong antimigratory properties on PC3 cancer cell lines.
Interaction with Plasmid pBR322 DNA
We and others have previously found that titanocene–gold compounds interact weakly with calf thymus DNA or do not interact with plasmid pBR322 DNA, as is the case for many other gold compounds.1,3,7,8 Recent reports on titanocene dichloride and titanocene Y also indicate a weak interaction with DNA40,41 and the lack of suppression for DNA-processing enzymes.40 DNA interactions were tested with heterometallic compounds 5a−d or cisplatin by using plasmid (pBR322) DNA (Figure 7). This plasmid has two main forms: OC (open circular or relaxed form, form II) and CCC (covalently closed or supercoiled form, form I). Agarose gel electrophoresis assays were performed whereby decreased electrophoretic mobilities of both forms were taken as evidence of metal–DNA binding. Generally, the slower the mobility of supercoiled DNA (CCC, form I), the greater the DNA unwinding produced by the drug.42 For example, binding of cisplatin to plasmid DNA results in decreased mobility of the CCC form and increased mobility of the OC form. Treatment of plasmid DNA with increasing amounts of the new heterometallic compounds 5a–d did not affect the mobility of the faster-running supercoiled form (form I) even at the highest molar ratios (d). This result is in accordance with the lack of interaction shown by titanocene–gold compounds (zero, first, and second generation) and the lack of interaction displayed by monometallic gold compounds 4a–d (see Figure S59 in the Supporting Information).
Figure 7.
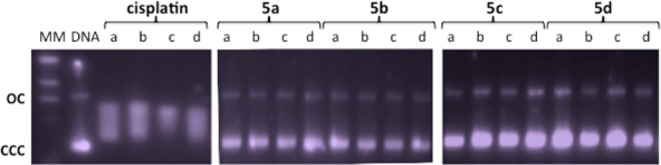
Electrophoresis mobility shift assays for cisplatin and heterometallic Ti–Au compounds 5a–d (see the Experimental Section for details). DNA refers to untreated plasmid pBR322. a, b, c, and d correspond to metal/DNA bp ratios of 0.25, 0.5, 1.0, and 2.0, respectively.
Inhibition of Thioredoxin Reductase in PC3 Cancer Cells
Many chemoresistant cancers produce changes in the cell antioxidant capacity. The overexpression of thioredoxin reductase (TrRx) is among the key defense and survival mechanisms of cisplatin-resistant cells. Thioredoxin reductase has become a potential target in cancer chemotherapy.43,44 We have reported on the inhibition of TrRx in Caki-1 cells by auranofin and the heterometallic titanocene–gold complex [(η5-C5H5)2TiMe(μ-mba)Au(PR3)] (3).1 Since Au–NHC compounds are known to inhibit TrRx, we measured the activity of thioredoxin reductase in PC3 prostate cancer cells, following incubation with monometallic compounds [Au(NHC)(Hmba)] (NHC = SIPr (4a), IPr (4b)) and bimetallic compounds [(η5-C5H5)2TiMe(μ-mba)Au(NHC)] (NHC = SIPr (5a), IPr (5b)). We found thioredoxin reductase activity to be lower in cells treated with 5 μM of 5a and 5b with observed inhibitions of 31% and 30%, respectively, after a 5 h incubation period (Figure 8). The inhibition was 61% (5a) and 76% (5b) after 24 h incubation. In the case of PC3 cells treated with 30 μM of monometallic gold compounds (4a,b) there was inhibition of thioredoxin reductase but to a lower extent (30% and 36% for 4a and 4b, respectively, after 24 h incubation). Surprisingly, we found that titanocene Y (15 μM) was also a strong TrRx inhibitor in PC3 cells (with a 57% or 80% reduction after 5 or 24 h of treatment, respectively, see Figure S61 in the Supporting Information). This experiment showed that the inhibition of TrRx is involved in the cell death mechanism of the new compounds and that the titanocene component has an influence on this target, although other cellular targets may not be excluded. In the past years, a number of other targets (such as gluthatione reductase, cysteine proteases such as cathepsins K and S, protein thyrosine phosphatases, glutathione peroxidase (GPx),44 iodothyronine deiodinase (ID),45 and IkB kinase) have been identified for gold(I) complexes.15 Helicases/topoisomerases and HIST1H4 core histones have been pointed out as targets of titanocene C,45 and we reported on the strong inhibitory effect of titanocene dichloride against PI3 protein kinases from a panel of 35 kinases of oncological interest.8
Figure 8.
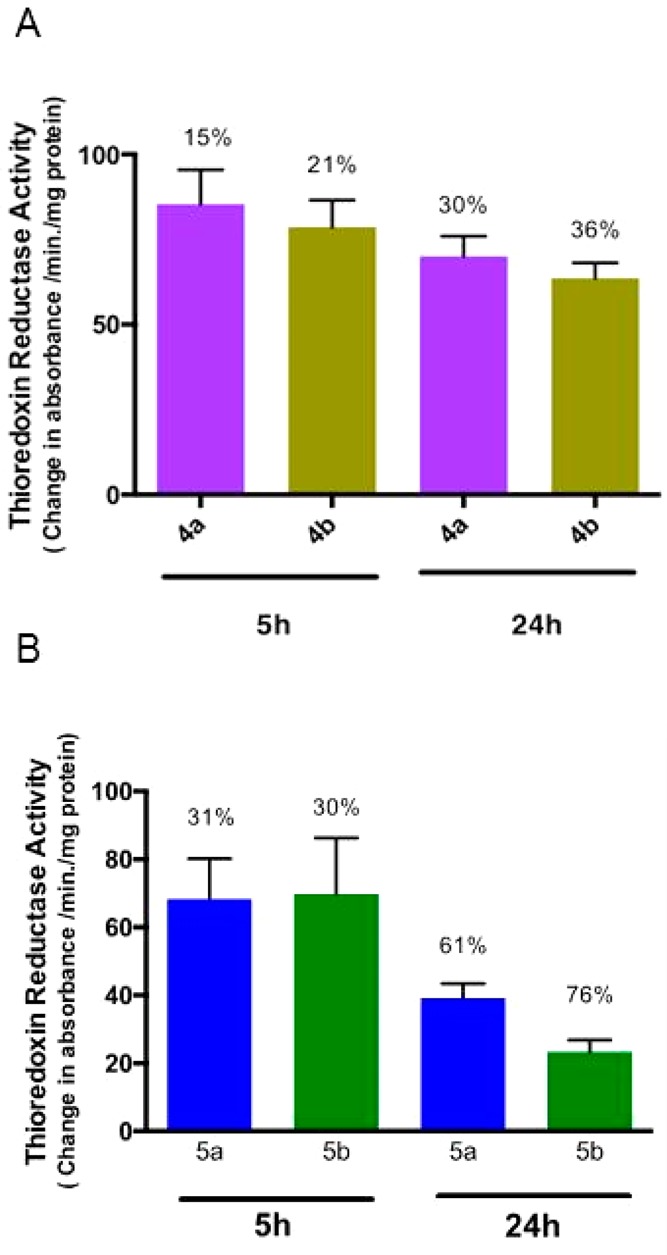
Thioredoxin reductase activity in 4a, 4b, 5a, or 5b treated PC3 cells: (A) activity of endogenous PC3 thioredoxin reductase from soluble whole cell lysates following incubation with 30 μM of 4a or 4b for 5 and 24 h (values normalized against DMSO control); (B) activity of endogenous PC3 thioredoxin reductase from soluble whole cell lysates following incubation with 5 μM of 5a or 5b for 5 h and 24 h (values normalized against DMSO control).
For titanocene–gold(I) heterometallic complexes, we have shown that the compound [(η5-C5H5)2TiMe(μ-mba)Au(PR3)] (3) not only inhibited TrRx in Caki-1 renal cancer cells with an IC50 value very similar to that of Auranofin but also was considerably more cytotoxic than auranofin in this cell line due to a more potent inhibition of the specific protein kinases AKT, p90-RSK, and MAPKAPK3 in vitro.1
All of these results warrant further studies on the mode of action of the new heterometallic compounds.
Conclusions
In conclusion, we have described the preparation of novel heterometallic titanocene–gold compounds incorporating gold(I)–N-heterocyclic carbene fragments. The exchange of the phosphane ligands by NHC ligands (L) in complexes of the type [(η5-C5H5)2TiMe(μ-mba)Au(L)] did result in lower IC50 values in renal Caki-1 cancer cell lines, although a significant activity and a considerably higher selectivity with respect to noncancerous cell lines was obtained in prostate and colon cancer cell lines for the new Ti–Au–NHC complexes. As for the analogous titanocene–gold compounds containing phosphanes, the new heterometallic carbene derivatives did not display a significant interaction with plasmid (pBR322) used as a model nucleic acid. Two selected compounds (5a,b) were found to be highly apoptotic and to inhibit TrRx in prostate PC3 cancer cell lines. These complexes also display strong antimigratory properties. The work presented here is the proof of concept that the substitution of PR3–gold(I) by NHC–gold(I) fragments in titanocene–gold complexes may afford derivatives with potential as cancer chemotherapeutics which will allow for further modification. With the NHC ligands described in this work we did not find a strong SAR correlation. Further optimization of the NHC ligands employed and more detailed mechanistic studies are needed in order to find candidates with improved pharmacological properties. These studies are currently under way in our laboratories.
Experimental Section
Chemistry
Synthesis and Characterization: General Procedure
Imidazolium salts (SiPr·HCl,46 IPr·HCl,46 IMes·HCl,47 ICy·HCl48), [AuCl(tht)],49 [AuCl(NHC)] (NHC = SIPr (1),24 IPr (2),24 IMes (3),28 ICy (4)28), and titanocene Y33 were prepared as previously reported. Cp2TiCl2 and H[AuCl4] were purchased from Strem Chemicals. Tetrahydrothiophene was purchased from Sigma-Aldrich. All purchased reactants were used without further purification. Reaction solvents were purchased anhydrous from Fisher Scientific (ACS grade) and purified by use of a PureSolv purification unit from Innovative Technology, Inc. Deuterated solvents were purchased from Cambridge Isotope Laboratories, Inc., kept over molecular sieves (3 Å, beads, 4–8 mesh), and degassed by the freeze–pump–thaw method. NMR spectra were recorded using a Bruker AV400 (1H NMR at 400 MHz, 13C{1H} NMR at 100.6 MHz). Chemical shifts (δ) are given in ppm using CDCl3 as the solvent, unless otherwise stated. 1H and 13C NMR resonances were measured relative to solvent peaks considering tetramethylsilane at 0 ppm. Coupling constants J are given in hertz. IR spectra (4000–250 cm–1) were recorded on a Nicolet 6700 Fourier transform infrared spectrophotometer on samples in the solid state (ATR accessory). Elemental analyses were performed on a PerkinElmer 2400 CHNS/O series II analyzer by Atlantic Microlab Inc. (US). Mass spectra (electrospray ionization, ESI-high resolution) were performed on a Waters Q-Tof Ultima instrument. The theoretical isotopic distributions have been calculated using enviPat Web 2.0.1. Stability studies were performed in a Cary 100 Bio UV–visible spectrophotometer. The pH was measured in an OAKTON pH conductivity meter in 5 × 10–5 M 1/99 DMSO/H2O solutions.
[Au(Hmba)(NHC)] (4a–d)
H2mba (0.154 g, 1 mmol) was added to a solution of KOH (0.056 g, 1 mmol) in 20 mL of ethanol (16 mL) and water (4 mL) and stirred for 20 min at room temperature. Afterward, the corresponding [Au(NHC)Cl] (1 mmol) was added to the solution and the mixture was stirred for 5 h. The solvents were then removed under reduced pressure, and the residue was washed with water (3 × 2 mL) and then diethyl ether (3 × 3 mL) to afford compounds 4a–d as white powdery solids.
4a (NHC = SIPr): 74% yield (0.548 g). Anal. Calcd for C34H43AuN2O2S (740.75): C, 55.13; H, 5.85; N, 3.78; S, 4.33. Found: C, 55.03; H, 5.73; N, 3.88; S, 4.06. 1H NMR (CDCl3): δ 1.36 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 1.41 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 3.12 (m, 4H, CH(CH3)2), 4.09 (s, 4H, CH2-imidazole), 6.74 (d, 3JHH = 8.3 Hz, 2H, ArH), 7.30 (d, 3JHH = 7.8 Hz, 4H, ArH), 7.42 (d, 3JHH = 8.3 Hz, 2H, ArH), 7.52 (t, 3JHH = 7.8 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 24.57 (s, CH(CH3)2), 25.41 (s, CH(CH3)2), 29.40 (s, CH(CH3)2), 53.99 (s, CH2-imidazole), 122.44 (s, 2-C6H3), 125.02 (s, 3-C6H3), 129.36 (s, 3-C6H4), 130.41 (s, 4-C6H3), 131.50 (s, 2-C6H4), 134.35 (s, 1-C6H3), 147.28 (s, 4-C6H4), 154.69 (s, 1-C6H4), 171.04 (s, C = O), 205.68 (s, C-carbene). IR (cm–1): 2962 m,br (OH), 2360 m, 1671 s, 1585 s (νasym CO2), 1338 s, 1289 vs (νsym CO2), 1175 m, 1086 m, 759 m. MS (ESI+) [m/z]: 847.36 (100%) [M]+.
4b (NHC = IPr): 96% yield (0.709 g). Anal. Calcd for C34H41AuN2O2S (738.73): C, 55.28; H, 5.59; N, 3.79; S, 4.34. Found: C, 55.32; H, 5.64; N, 3.77; S, 4.10. 1H NMR (CDCl3): δ 1.24 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 1.34 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 2.62 (m, 4H, CH(CH3)2), 6.88 (d, 3JHH = 8.3 Hz, 2H, ArH), 7.22 (s, 2H, CH-imidazole), 7.35 (d, 3JHH = 7.8 Hz, 4H, ArH), 7.46 (d, 3JHH = 8.3 Hz, 2H, ArH), 7.59 (t, 3JHH = 7.8 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 24.44 (s, CH(CH3)2), 24.79 (s, CH(CH3)2), 29.23 (s, CH(CH3)2), 122.53 (s, 2-C6H3), 123.31 (s, CH-imidazole), 124.65 (s, 3-C6H3), 129.39 (s, 3-C6H4), 131.11 (s, 4-C6H3), 131.58 (s, 2-C6H4), 134.34 (s, 1-C6H3), 146.23 (s, 4-C6H4), 154.98 (s, 1-C6H4), 171.27 (s, C=O), 186.36 (s, C-carbene). IR (cm–1): 2958 m,br (OH), 2361 w, 1677 s, 1585 s (νasym CO2), 1285 m (νsym CO2), 1176 m, 1088 m, 759 m.
4c (NHC = IMes): 88% yield (0.576 g). Anal. Calcd for C28H29AuN2O2S (654.57): C, 51.38; H, 4.47; N, 4.28; S, 4.90. Found: C, 51.41; H, 4.45; N, 4.25; S, 4.99. 1H NMR (CDCl3): δ 2.15 (s, CH3, 12H), 2.41 (s, 6H, CH3), 7.02 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.06 (s, 4H, ArH), 7.14 (s, 2H, CH-imidazole), 7.50 (d, 3JHH = 8.4 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 18.14 (s, CH3), 21.64 (s, CH3), 122.38 (s, CH-imidazole), 122.67 (s, 4-C6H2), 129.38 (s, 2-C6H4), 129.81 (s, 3-C6H2), 135.06 (s, 2-C6H2), 135.32 (s, 1-C6H2), 140.27 (s, 4-C6H4), 154.68 (s, 1-C6H4), 170.40 (s, C=O), 184.56 (s, C-carbene). IR (cm–1): 2948 m,br (OH), 1668 vs, 1583 s (νasym CO2), 1411 m, 1283 vs (νsym CO2), 1170 m, 1087 m, 767 m.
4d (NHC = ICy): 78% yield (0.454 g). Anal. Calcd for C22H29AuN2O2S (582.51): C, 45.36; H, 5.02; N, 4.81; S, 5.50. Found: C, 45.20; H, 5.04; N, 4.73; S, 5.45. 1H NMR (CDCl3): δ 1.21–2.16 (m, 20H C6H11), 4.61 (tt, 2H, 3JHH = 12.1, 3.8 Hz C6H11), 6.97 (s, 2H, CH-imidazole), 7.72 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.78 (d, 3JHH = 8.4 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 25.47 (s, 4-C6H11), 25.79 (s, 3-C6H11), 34.57 (s, 2-C6H11), 61.27 (s, 1-C6H11), 117.35 (s, CH-imidazole), 123.36 (s, 4-C6H4), 129.84 (s, 2-C6H4), 132.28 (s, 3-C6H4), 154.46 (s, 1-C6H4), 170.60 (s, C=O), 179.90 (s, C-carbene). IR (cm–1): 2932 m,br (OH), 2357 w, 1677 s, 1581 s (νasym CO2), 1417 s, 1287 vs (νsym CO2), 1083 m, 766 m.
[(η5-C5H5)2TiMe(μ-mba)Au(NHC)] (5a–d)
The corresponding monometallic gold complex 4a–d (0.41 mmol) was dissolved in tetrahydrofuran (15 mL) and added via cannula over a solution of Cp2TiMe2 (0.084 g, 0.41 mmol) in toluene (5 mL) to give rise to a bright orange solution that was stirred for 1.5 h at room temperature. The solution was filtered off, and the solvents were then removed under reduced pressure to afford an oily solid that was washed with a dichloromethane/diethyl ether/hexane mixture (2/6/2) (3 × 5 mL). The heterometallic complexes were then isolated as orange solids.
5a (NHC = SIPr): 62% yield (0.237 g). Anal. Calcd for C45H55AuN2O2STi·H2O (950.84): C, 56.84; H, 6.04; N, 2.95; S, 3.37. Found: C, 57.15; H, 6.15; N, 2.62; S, 3.02. 1H NMR (CDCl3): δ 0.99 (s, 3H, Ti-CH3), 1.35 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 1.41 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2), 3.11 (m, 4H, CH(CH3)2), 4.08 (s, 4H, CH-imidazole), 6.19 (s, 10H, Cp), 6.64 (d, 3JHH = 8.5 Hz, 2H, ArH), 7.05 (d, 3JHH = 8.5 Hz, ArH, 2H), 7.28 (d, 3JHH = 7.8 Hz, ArH, 4H), 7.50 (t, 2H, 3JHH = 7.7 Hz, ArH). 13C{1H} NMR (CDCl3): δ 24.24 (s, CH(CH3)2), 25.06 (s, CH(CH3)2), 29.05 (s, CH(CH3)2), 43.73 (s, Ti-CH3, 53.63 (s, CH-imidazole), 114.62 (s, Cp), 125.00 (s, 3-C6H3), 127.21 (s, 2-C6H3), 129.19, (s, 3-C6H4), 130.37 (s, 4-C6H3), 131.17 (s, 2-C6H4), 134.41 (s, 1-C6H3), 147.24 (s, 4-C6H4), 150.87 (s, 1-C6H4), 172.35 (s, C=O), 206.00 (s, C-carbene). IR (cm–1): 2959 m (Cp), 1585 s (νasym CO2), 1493 s, 1275 vs (νsym CO2), 1169 m (Cp), 1085 m (Cp). pH 5.92.
5b (NHC = IPr): 68% yield (0.259 g). Anal. Calcd for C45H53AuN2O2STi·H2O (948.83): C, 56.96; H, 5.84; N, 2.95; S, 3.38. Found: C, 57.41; H, 5.73; N, 2.94; S, 3.39. 1H NMR (CDCl3): δ 0.99 (s, 3H, Ti-CH3), 1.23 (d, 3JHH = 6.9 Hz, 12H, CH(CH3)2), 1.33 (d, 3JHH = 6.8 Hz, 12H, CH(CH3)2,), 2.61 (m, 4H, CH(CH3)2), 6.20 (s, 10H, Cp), 6.78 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.09 (d, 3JHH = 8.3 Hz, 2H, ArH), 7.21 (s, 2H, CH-imidazole), 7.33 (d, 3JHH = 7.8 Hz, 4H, ArH), 7.58 (t, 3JHH = 7.8 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 24.47 (s, CH(CH3)2), 24.79 (s, CH(CH3)2), 29.23 (s, CH(CH3)2), 44.07 (s, Ti-CH3, 114.28 (s, Cp), 123.24 (s, CH-imidazole), 124.63 (s, 3-C6H3), 127.25 (s, 2-C6H3), 129.22 (s, 3-C6H4), 131.07 (s, 4-C6H3), 131.26 (s, 2-C6H4), 134.40 (s, 1-C6H3), 146.21 (s, 4-C6H4), 151.13 (s, 1-C6H4), 172.36 (s, C=O), 186.77 (s, C-carbene). IR (cm–1): 2959 m (Cp), 2361 w, 1636 m, 1584 s (νasym CO2), 1469 m, 1286 vs (νsym CO2), 1169 m (Cp), 1085 m (Cp). pH 6.16.
5c (NHC = IMes): 81% yield (0.281 g). Anal. Calcd for C39H41AuN2O2STi (846.65): C, 55.33; H, 4.88; N, 3.31; S, 3.79. Found: C, 55.22; H, 4.92; N, 3.19; S, 3.58. 1H NMR (CDCl3): δ 0.98 (s, 3H, Ti-CH3), 2.14 (s, 12H, CH3), 2.41 (s, 6H, CH3), 6.19 (s, 10H, Cp), 6.94 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.04 (s, 4H, ArH), 7.13 (s, 2H, CH-imidazole), 7.15 (d, 3JHH = 8.4 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 18.15 (s, CH3), 21.63 (s, CH3), 44.28 (s, Ti-CH3), 114.28 (s, Cp), 122.31 (s, CH-imidazole), 122.49 (s, 4-C6H2), 129.23 (s, 2-C6H4), 129.39 (s, 3-C6H2), 131.58 (s, 2-C6H2), 135.27 (s, 1-C6H2), 140.19 (s, 4-C6H4), 150.66 (s, 1-C6H4), 172.28 (s, C=O), 184.88 (s, C-carbene). IR (cm–1): 2954 m (Cp), 1631 w, 1584 m (νasym CO2), 1286 vs (νsym CO2), 1168 m (Cp), 1085 m (Cp). pH 6.15.
5d (NHC = ICy): 79% yield (0.250 g). Anal. Calcd for C33H41AuN2O2STi·H2O (792.60): C, 50.01; H, 5.47; N, 3.53; S, 4.05. Found: C, 50.01; H, 5.31; N, 3.45; S, 4.03. 1H NMR (CDCl3): δ 0.96 (s, 3H, Ti-CH3) 1.21–2.16 (m, 20H, C6H11), 4.61 (m, 2H, C6H11), 6.19 (s, 10H, Cp), 6.96 (s, 2H, CH-imidazole), 7.39 (d, 3JHH = 8.4 Hz, 2H, ArH), 7.62 (d, 3JHH = 8.4 Hz, 2H, ArH). 13C{1H} NMR (CDCl3): δ 25.50 (s, 4-C6H11), 25.79 (s, 3-C6H11), 34.54 (s, 2-C6H11), 44.20 (s, Ti-CH3), 61.20 (s, 1-C6H11), 114.64 (s, Cp) 117.28 (s, CH-imidazole), 128.21 (s, 4-C6H4), 129.67 (s, 2-C6H4), 132.06 (s, 3-C6H4), 150.47 (s, 1-C6H4), 172.27 (s, C=O), 180.19 (s, C-carbene). IR (cm–1): 2931 m (Cp), 1636 m, 1584 m (νasym CO2), 1285 vs (νsym CO2), 1168 m (Cp), 1084 m (Cp). pH 5.82.
X-ray Crystallography
Suitable single crystals of compound 4c were obtained by layering pentane over a solution of tetrahydrofuran. Details of the crystallographic data and a complete list of selected structural parameters are given in Tables S1 and S2 in the Supporting Information, respectively. The crystal was mounted on a glass fiber, and the diffraction measurements were performed with a Nonius Kappa CCD area-detector diffractometer with Mo Kα radiation (λ = 0.71073 Å). The structure were solved by direct methods and refined by least-squares techniques on weighted F2 values for all reflections (SHELXTL, 6.14). All non-hydrogen atoms were assigned anisotropic displacement parameters and refined without positional constraints. All hydrogen atoms were calculated with a riding model. Complex neutral-atom scattering factors were used. The program SQUEEZE, a part of the Platon2 package of crystallographic software, was used to calculate the solvent disorder area and remove its contribution to the overall intensity data. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. The assigned deposition number at the Cambridge Crystallographic Data Centre for compound 4c is 1438894.
DFT Calculations
The calculations have been performed using the hybrid density functional method B3LYP,50,51 as implemented in Gaussian09.52 Geometries were optimized with the 6-311G(d) basis set for the P and S elements, the 6-31G(d,p) basis set for the C, N, P, S, and H elements, and the SDD pseudopotential for the titanium, iron, and gold metal centers.53,54 Frequency calculations have been done at the same level of theory as the geometry optimizations to confirm the nature of the stationary points.
Biology
Interactions of the New Compounds with Plasmid pBR322 (Gel Electrophoresis Mobility Shift Assay)
Ten microliter aliquots of pBR322 plasmid DNA (20 μg/mL) in buffer (5 mM Tris/HCl, 50 mM NaClO4, pH 7.39) were incubated with different concentrations of the compounds (4a–d, 5a–d, and cisplatin as control) (in the range 0.25 and 4.0 metal complex to DNA bp) at 37 °C for 20 h in the dark. Samples of free DNA and cisplatin-DNA were prepared as controls. After the incubation period, the samples were loaded onto the 1% agarose gel. The samples were separated by electrophoresis for 1.5 h at 80 V in Tris-acetate/EDTA buffer (TAE). Afterward, the gel was stained for 30 min with a solution of GelRed nucleic acid stain.
Cell Culture and PrestoBlue Cell Viability Assay for Caki-1, DLD-1, PC3, DU145, MDA-MB 231, and HEK-293T Cells
Human renal clear-cell carcinoma Caki-1, human breast adenocarcinoma cells MDA-MB-231, human prostate adenocarcinoma cells PC3, human prostate carcinoma cells DU145, and the human colorectal adenocarcinoma cells DLD-1 in comparison with healthy human embryonic kidney cells HEK-293T were used to study the cytotoxic activity of bimetallic carbenes 5a–d and their monometallic controls 4a–d. The cells were all obtained from the American Type Culture Collection (ATCC) (Manassas, VA). All of the cells were grown adherently. The Caki-1, PC3, Du145, and DLD-1 cells were cultured in Roswell Park Memorial Institute (RPMI-1640) (Mediatech Inc., Manassas, VA) medium, while MDA-MB-231 and HEK-293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Mediatech Inc., Manassas, VA); all media were supplemented with 10% fetal bovine serum (FBS, Life Technologies, Grand Island, NY), 1% Minimum Essential Media (MEM) nonessential amino acids (NEAA, Mediatech), and 1% penicillin–streptomycin (Pen Strep, Mediatech). All cells were cultured at 37 °C under 5% CO2 and 95% air in a humidified incubator. For evaluation of cell viability, cells were seeded at a concentration of 5 × 103 cells/well in 90 μL of DMEM or RPMI without phenol red and without antibiotics, supplemented with 10% FBS and 2 mM l-glutamine into tissue culture grade 96-well flat bottom microplates (BioLite Microwell Plate, Fisher Scientific, Waltham, MA) and grown for 24 h at 37 °C under 5% CO2 and 95% air in a humidified incubator. All compounds were dissolved in DMSO and diluted to 1% in media before addition to cell culture medium. The intermediate dilutions of the compounds were added to the wells (10 μL) to obtain concentrations of 1, 10, and 100 μM, 0.1% DMSO was used as control, and the cells were incubated for 72 h. PrestoBlue was used to quantitatively measure variations in cell viability of treated cells. Following 72 h drug exposure, 11 μL of per well of 10× PrestoBlue (Life Technologies, Carlsbad, CA) labeling mixture was added to the cells at a final concentration of 1× and incubated for 1.5 h at 37 °C under 5% CO2 and 95% air in a humidified incubator. The optical absorbance of each well in a 96-well plate was quantified using a 16 BioTek ELx 808 absorbance microplate reader (BioTek Winooski, VT) set at 570 nm excitation and 600 nm emission wavelength. The percentage of surviving cells was calculated from the ratio of absorbance of treated to untreated cells. The IC50 (μM) value was calculated as the concentration reducing the proliferation of the cells by 50% and is presented as a mean (±S.EM) of at least two independent experiments each with triplicate measurements.
Stability of Compound 5a in Vitro and Colocalization of Ti/Au Metals in PC3 Cells
PC3 cells were incubated with 10 and 20 μM compound 5a for 72 h. Postincubation, the cells were washed twice with cold PBS and lysed with cell lysis buffer comprising of 1% (v/v) Triton-X-100, 25 mM HEPES, 100 mM NaCl, 1 mM EDTA, 10% (v/v) glycerol, and protease and phosphatase inhibitors. Lysates from untreated cells incubated with media supplemented with DMSO for the same duration were used as controls. Gold and titanium contents in the cell lysates were determined using ICP-MS. One hundred microliter portions of lysates were transferred into a glass vials, and 1 mL of concentrated acid mix (comprised of 75% of 16 N nitric acid and 25% of 12 N hydrochloric acid) was added. The mixture was heated at 90 °C for 5 h. After cooling, the samples were diluted with water and 40 ppb of indium internal standard was added and analyzed in a Thermo Scientific XSERIES 2 ICP-MS with ESI PC3 Peltier cooled spray chamber with an SC-FAST injection loop and SC-4 autosampler. All of the elements were analyzed using He/H2 collision-reaction mode. A standard curve (0, 1, 5, 10, and 20 μM) of compound 5a was processed similarly to determine the linearity of extraction efficiency of Au and Ti. The protein contents of the cell lysates were determined using a bicinchoninic acid based protein assay kit (Thermo Scientific). The final levels of either Ti or Au were normalized to the cellular protein levels.
Cell Death for Titanocene Y, 4a,b, and 5a,b
For apoptosis, viability, and necrosis assays, the Caki-1 and PC3 cells were seeded in 96-well opaque-walled tissue culture plates with clear bottoms (Thermo Scientific Nunc; Somerset, NJ) at an initial density of 5 × 104 red and without antibiotics, supplemented with 10% FBS and 2 mM l-glutamine. Following 24 h incubation, Caki-1 cells were treated with 30 μM of titanocene Y, 80 μM of compounds 4a and 4b, and and 10 μM of 5a or 5b for 72 h, while PC3 cells were treated with 30 μM of titanocene Y, 60 μM of 4a, 70 μM of 4b, and 10 μM of 5a or 5b for 72 h. The cells were then assayed using the ApoTox-Glo triplex assay (Promega GmbH, High-Tech-Park, Mannheim, Germany). Twenty microliter portions of viability/cytotoxicity reagent containing both glycylphenylalanyl-aminofluorocoumarin (GF-AFC) and bis-alanylalanyl-phenylalanyl-rhodamine 110 (bis-AAF-R110) substrates were added to each well, and they were briefly mixed by orbital shaking at 200 rpm for 30 s and then incubated at 37 °C for 2 h. Fluorescence was measured at 400 nm for excitation/505 nm for emission (viability) and 485 nm for excitation/520 nm for emission (cytotoxicity/necrosis) using a BioTek Fluorescence Microplate Reader (BioTek U.S., Winooski, VT)). Next, 100 μL of Caspase-Glo 3/7 reagents was added to each well, and the samples were briefly mixed by orbital shaking at 200 rpm for 30 s and then incubated at room temperature for 1 h. Luminescence was measured for 1 s and is proportional to the amount of caspase activity present (BioTek U.S., Winooski, VT). The results for each treatment were expressed as fold change between nontreated (0.1% DMSO) and treated samples. ApoTox-Glo triplex assays were repeated twice (n = 2), and each repetition was run in quadruplicate. The average of the four values was used for statistical calculations. The data are presented as the mean values.
In Vitro Migration Assay (Wound Healing Assay)
For the assessment of cell migration, confluent PC3 cells maintained in standard medium were wounded with a plastic micropipette tip (tip 20–200 μL). After washing, the medium was replaced by fresh medium containing 5 μM of either 5a or 5b, 15 μM of titanocene Y, and 15 μM of 4a or 4b or 0.1% DMSO solution (control). Photographs of the wounded area were taken after 0 and 24 h using phase-contrast microscopy. For evaluation of wound closure, four randomly selected points along each wound area were marked and the horizontal distance of migrating cells from the initial wound was measured (Labomed TCM400 Inverted Phase Microscope Series, equipped with a digital camera (Fisher Scientific Moticam 10). The assays were done twice, and for each trial two images were analyzed per time point.
Method for Thioredoxin Reductase Activity Assay
Whole cell lysates was assayed using PC3 cells treated in vitro with 5 μM of 5a or 5b, 15 μM of titanocene Y, 30 μM of 4a or 4b, or 0.1% DMSO solution (control). After 5 or 24 h of treatment, cells were washed three times in PBS and lysed by douncing using scrappers and sheer force through a syringe with a 34 gauge in assay buffer (Abcam Thioredoxin Reductase Assay kit, ab83463) with 1 mM Protease Inhibitor Cocktail (Abcam, ab65621). The lysates were centrifuged at 10000 rcf for 15 min at 4 °C to isolate insoluble material. The total protein concentrations of soluble lysates were measured using the Bradford assay. The soluble lysates were incubated for 20 min in assay buffer or assay buffer with a proprietary thioredoxin reductase specific inhibitor before adding a specific substrate, DTNB (5,5′-dithiobis(2-nitrobenzoic acid)), and measuring the activity at 1 min intervals for 30 min using a BioTek Fluorescence Microplate Reader (BioTek U.S., Winooski, VT) at λ 412 nm. Lysates were tested in duplicate. TrxR activity was calculated on the basis of the linear amount of TNB (2-nitro-5-thiobenzoic acid) produced per minute per milligram of total protein and adjusted for background activity from enzymes other than TrxR in the lysates.
Acknowledgments
We thank the National Cancer Institute (NCI) for grant 1SC1CA182844 (M.C.). M.C. is very grateful to Mr. Leonard Tow and the Tow Foundation for a Tow Professorship 2015-2017. O.N. acknowledges the Dalton Division of the Royal Society of Chemistry for a travel bursary that facilitated this collaboration. We thank Prof. Swayam Prabha and Dr. Buddhadev Layek from the Center for Translational Drug Delivery at the University of Minnesota for performing the ICP-MS analysis (cellular uptake for compound 5a) in the PC3 cells.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.organomet.6b00051.
NMR, IR and UV–vis spectra of all new compounds, MS ESI+ spectra of all compounds and theoretical isotopic distributions of relevant peaks, DFT calculations for all new compounds, crystallographic data for 4c, interaction of monometallic gold compounds 4a–d with plasmid pBR322 DNA, migration assays with compounds 4a,c, and inhibition of thioredoxin reductase (TrxR) studies of titanocene Y at 5 and 24 h (PDF)
Cartesian coordinates for calculated structures (XYZ)
Crystallographic data for 4c (CIF)
Author Contributions
# These authors contributed equally to the work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Fernández-Gallardo J.; Elie B. T.; Sadhukha T.; Prabha S.; Sanaú M.; Rotenberg S. A.; Ramos J. W.; Contel M. Chem. Sci. 2015, 6, 5269–5283. 10.1039/C5SC01753J. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Contel M.; Fernández-Gallardo J.; Elie B. T.; Ramos J. W. U.S. Pat. Appl. 9,315,531, 2016, 2015.
- Pelletier F.; Comte V.; Massard A.; Wenzel M.; Toulot S.; Richard P.; Picquet M.; Le Gendre P.; Zava O.; Edafe F.; Casini A.; Dyson P. J. J. Med. Chem. 2010, 53, 6923–6933. 10.1021/jm1004804. [DOI] [PubMed] [Google Scholar]
- González-Pantoja J. F.; Stern M.; Jarzecki A. A.; Royo E.; Robles-Escajeda E.; Varela-Ramirez A.; Aguilera R. J.; Contel M. Inorg. Chem. 2011, 50, 11099–11110. 10.1021/ic201647h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lease N.; Vasilevski V.; Carreira M.; de Almeida A.; Sanaú M.; Hirva P.; Casini A.; Contel M. J. Med. Chem. 2013, 56, 5806–5818. 10.1021/jm4007615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson C. M.; Taylor I. R.; Tibbetts M. F.; Philpott J.; Hu Y.; Tanski J. M. Inorg. Chem. 2012, 51, 12917–1294. and references therein 10.1021/ic301981s. [DOI] [PubMed] [Google Scholar]
- Tripathy S. K.; De U.; Dehury N.; Pal S.; Kim H. S.; Patra S. Dalton Trans. 2014, 43, 14546–14549. 10.1039/C4DT01033G. [DOI] [PubMed] [Google Scholar]
- Wenzel M.; Bertrand B.; Eymin M.-J.; Comte V.; Harvey J. A.; Richard P.; Groessl M.; Zava O.; Amrouche H.; Harvey P. D.; Le Gendre P.; Picquet M.; Casini A. Inorg. Chem. 2011, 50, 9472–9480. 10.1021/ic201155y. [DOI] [PubMed] [Google Scholar]
- Fernandez-Gallardo J.; Elie B. T.; Sulzmaier F.; Sanaú M.; Ramos J. W.; Contel M. Organometallics 2014, 33, 6669–6681. 10.1021/om500965k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boselli L.; Carraz M.; Mazeres S.; Paloque L.; Gonzalez G.; Benoit-Vical F.; Valentin A.; Hemmert C.; Gornitzka H. Organometallics 2015, 34, 1046–1055. 10.1021/om501158m. [DOI] [Google Scholar]
- Bertrand B.; Citta A.; Franken I. L.; Picquet M.; Folda A.; Scalcon V.; Rigobello M. P.; Le Gendre P.; Casini A.; Bodio E. JBIC, J. Biol. Inorg. Chem. 2015, 20, 1005–1020. 10.1007/s00775-015-1283-1. [DOI] [PubMed] [Google Scholar]
- Massai L.; Fernandez-Gallardo J.; Guerri A.; Arcangeli A.; Pillozzi S.; Contel M.; Messori L. Dalton Trans. 2015, 44, 11067–11076. 10.1039/C5DT01614B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelosevic H.; Guzei I. A.; Spencer L. C.; Persson T.; Kriel F. H.; Hewer R.; Nell M. J.; Gut J.; van Resburg C. E. J.; Rosenthal P.; Coates J.; Darkwa J.; Elmroth S. K. C. J. Organomet. Chem. 2012, 720, 52–59. 10.1016/j.jorganchem.2012.08.034. [DOI] [Google Scholar]
- Wenzel M.; Bigaeva E.; Richard P.; Le Gendre P.; Picquet M.; Casini A.; Bodio E. J. Inorg. Biochem. 2014, 141, 10–16. 10.1016/j.jinorgbio.2014.07.011. [DOI] [PubMed] [Google Scholar]
- Fernandez-Moreira V.; Marzo I.; Gimeno C. Chem. Sci. 2014, 5, 4434–4446. 10.1039/C4SC01684J. [DOI] [Google Scholar]
- Zou T.; Lum C. T.; Lok C.-N.; Zhang J.-J.; Che C.-M. Chem. Soc. Rev. 2015, 44, 8786–8801. and references therein 10.1039/C5CS00132C. [DOI] [PubMed] [Google Scholar]
- Bertrand B.; Casini A. Dalton Trans. 2014, 43, 4209–4219. and references therein 10.1039/C3DT52524D. [DOI] [PubMed] [Google Scholar]
- Oehninger L.; Ott I. Dalton Trans. 2013, 42, 3269–3284. and references therein 10.1039/C2DT32617E. [DOI] [PubMed] [Google Scholar]
- Liu W.; Gust R. Chem. Soc. Rev. 2013, 42, 755–773. and references therein 10.1039/C2CS35314H. [DOI] [PubMed] [Google Scholar]
- Barnard P. J.; Berners-Price S. J. Coord. Chem. Rev. 2007, 251, 1889–1902. and references therein 10.1016/j.ccr.2007.04.006. [DOI] [Google Scholar]
- Rubbiani R.; Can S.; Kitanovic I.; Alborzinia H.; Stefanopoulou M.; Kokoschka M.; Monchgesang S.; Sheldrick W. S.; Wolf S.; Ott I. J. Med. Chem. 2011, 54, 8646–8657. 10.1021/jm201220n. [DOI] [PubMed] [Google Scholar]
- Schuh E.; Pfluger C.; Citta A.; Folda A.; Rigobello M. P.; Bindoli A.; Casini A.; Mohr F. J. Med. Chem. 2012, 55, 5518–5528. and references therein 10.1021/jm300428v. [DOI] [PubMed] [Google Scholar]
- Rubbiani R.; Schuh E.; Meyer A.; Lemke J.; Wimberg J.; Metzler-Nolte N.; Meyer F.; Mohr F.; Ott I. MedChemComm 2013, 4, 942–948. 10.1039/c3md00076a. [DOI] [Google Scholar]
- Rubbiani R.; Salassa L.; de Almeida A.; Casini A.; Ott I. ChemMedChem 2014, 9, 1205–1210. 10.1002/cmdc.201400056. [DOI] [PubMed] [Google Scholar]
- Serebryanskaya T. V.; Zolotarev A. A.; Ott I. MedChemComm 2015, 6, 1186–1189. 10.1039/C5MD00185D. [DOI] [Google Scholar]
- Some selected recent examples:; a Rodrigues M.; Russo L.; Aguilo E.; Rodriguez L.; Ott I.; Perez-Garcia L. RSC Adv. 2016, 6, 2202–2209. 10.1039/C5RA21621D. [DOI] [Google Scholar]; b Maftei C. V.; Fodor E.; Jones P. G.; Freytag M.; Franz M. H.; Kelter G.; Fiebig H.-H.; Tamm M.; Neda I. Eur. J. Med. Chem. 2015, 101, 431–441. 10.1016/j.ejmech.2015.06.053. [DOI] [PubMed] [Google Scholar]; c Holenya P.; Can S.; Rubbiani R.; Alborzinia H.; Junger A.; Cheng X.; Ott I.; Wolf S. Metallomics 2014, 6, 1591–1601. 10.1039/C4MT00075G. [DOI] [PubMed] [Google Scholar]; d Baron M.; Bellemin-Laponnaz S.; Tubaro C.; Basato M.; Bogialli S.; Dolmella A. J. Inorg. Biochem. 2014, 141, 94–102. 10.1016/j.jinorgbio.2014.08.013. [DOI] [PubMed] [Google Scholar]; e Bertrand B.; Stefan L.; Pirrotta M.; Monchaud D.; Bodio E.; Richard P.; Le Gendre P.; Wardeman E.; de Jager M. H.; Groothuis G. M. M.; Picquet M.; Casini A. Inorg. Chem. 2014, 53, 2296–2303. 10.1021/ic403011h. [DOI] [PubMed] [Google Scholar]; f Messori L.; Marchetti L.; Massai L.; Scaletti F.; Guerri A.; Landini I.; Nobili S.; Perrone G.; Mini E.; Leoni E.; Pasquali M.; Gabianni C. Inorg. Chem. 2014, 53, 2396–2403. 10.1021/ic401731a. [DOI] [PubMed] [Google Scholar]; g Gutierrez A.; Gimeno M. C.; Marzo I.; Metzler-Nolte N. Eur. J. Inorg. Chem. 2014, 2014, 2512–2519. 10.1002/ejic.201402103. [DOI] [Google Scholar]; h Rana B. K.; Nandy A.; Bertolasi V.; Bielawski C. W.; Das Saha K.; Dinda J. Organometallics 2014, 33, 2544–2548. 10.1021/om500118x. [DOI] [Google Scholar]; i Boselli L.; Ader I.; Carraz M.; Hemmert C.; Cuvillier O.; Gornitzka H. Eur. J. Med. Chem. 2014, 85, 87–94. 10.1016/j.ejmech.2014.07.086. [DOI] [PubMed] [Google Scholar]; j Sun R. W.-Y.; Zhang M.; Li D.; Zhang Z.-F.; Cai H.; Li M.; Xian Y.-J.; Ng S. W.; Wong A.S.-T. Chem. Eur. J. 2015, 21, 18534–18538. 10.1002/chem.201503656. [DOI] [PubMed] [Google Scholar]
- a Zou T.; Lum C. T.; Lok C.-N.; To W.-P.; Low K.-H.; Che C.-M. Angew. Chem., Int. Ed. 2014, 53, 5810–5814. 10.1002/anie.201400142. [DOI] [PubMed] [Google Scholar]; b Nandy A.; Dey S. K.; Das S.; Munda R. N.; Dinda J.; Das Saha K. Mol. Cancer 2014, 13, 57. 10.1186/1476-4598-13-57. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Muenzner J. K.; Biersack B.; Kalie H.; Andronache I. C.; Kaps L.; Schuppan D.; Sasse F.; Schobert R. ChemMedChem 2014, 9, 1195–1204. 10.1002/cmdc.201400049. [DOI] [PubMed] [Google Scholar]
- Visbal R.; Laguna A.; Gimeno M. C. Chem. Commun. 2013, 49, 5642–5644. 10.1039/c3cc42919a. [DOI] [PubMed] [Google Scholar]
- de Fremont P.; Scott N. M.; Stevens E. D.; Nolan S. P. Organometallics 2005, 24, 2411–2418. 10.1021/om050111c. [DOI] [Google Scholar]
- Wilton-Ely J. D. E. T.; Schier A.; Mitzel N. W.; Schmidbaur H. J. Chem. Soc.; Dalton Trans. 2001, 7, 1058–1062. 10.1039/b100113m. [DOI] [Google Scholar]
- Deacon G. B.; Phillips R. J. Coord. Chem. Rev. 1980, 33, 227–250. 10.1016/S0010-8545(00)80455-5. [DOI] [Google Scholar]
- Martínez D.; Motevalli M.; Watkinson M. Dalton Trans. 2010, 39, 446–455. 10.1039/B913865J. [DOI] [PubMed] [Google Scholar]
- a Chen X.; Zhou L. J. Mol. Struct.: THEOCHEM 2010, 940, 45–49. 10.1016/j.theochem.2009.10.007. [DOI] [Google Scholar]; b Olszewski U.; Hamilton G. Anti-Cancer Agents Med. Chem. 2010, 10, 302–311. and references therein 10.2174/187152010791162261. [DOI] [PubMed] [Google Scholar]
- Sweeney N. J.; Mendoza O.; Muller-Bunz H.; Pampillon C.; Rehmann F. -J. K.; Strohfeldt K.; Tacke M. J. Organomet. Chem. 2005, 690, 4537–4544. 10.1016/j.jorganchem.2005.06.039. [DOI] [Google Scholar]
- Fichtner I.; Behrens D.; Claffey J.; Deally A.; Gleeson B.; Patil S.; Weber H.; Tacke M. Letters in Drug Design & Discovery 2011, 8, 302–307. 10.2174/157018011794839367. [DOI] [Google Scholar]
- Walther W.; Fichtner I.; Deally A.; Hogan M.; Tacke M. Letters in Drug Design & Discovery 2013, 10, 375–381. 10.2174/1570180811310050002. [DOI] [Google Scholar]
- Moutasim K. A.; Nystrom M. L.; Thomas G. J. Methods Mol. Biol. 2011, 731, 333–343. 10.1007/978-1-61779-080-5_27. [DOI] [PubMed] [Google Scholar]
- Hulkower K. I.; Herber R. L. Pharmaceutics 2011, 3, 107–124. 10.3390/pharmaceutics3010107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gmeiner A.; Effenberger-Neidnicht K.; Zoldakova M.; Schobert R. Appl. Organomet. Chem. 2011, 25, 117–120. 10.1002/aoc.1725. [DOI] [Google Scholar]
- De la Cueva-Alique I.; Muñoz-Moreno L.; Benebdelouahab Y.; Elie B. T.; El Amrani M. A.; Mosquera M. E. G.; Contel M.; Bajo A. M.; Cuenca T.; Royo E. J. Inorg. Biochem. 2016, 156, 22–34. 10.1016/j.jinorgbio.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravera M.; Gabano E.; Baracco S.; Osella D. Inorg. Chim. Acta 2009, 362, 1303–1306. and references therein 10.1016/j.ica.2008.06.022. [DOI] [Google Scholar]
- Lally G.; Deally A.; Hackenberg F.; Quinn S. J.; Tacke M. Letters in Drug Design & Discovery 2013, 10, 675–682. 10.2174/15701808113100890027. [DOI] [Google Scholar]
- Fox K.Drug-DNA Interaction Protocols; Humana Press: Totowa, NJ, 1997; Methods in Molecular Biology. 10.1385/089603447X [DOI] [Google Scholar]
- Lim H.-V.; Hong S.; Jin W.; Lim S.; Kim S.-J.; Kang H.-J.; Park E.-H.; Ahn K.; Lim C.-J. Exp. Mol. Med. 2005, 37, 497–506. 10.1038/emm.2005.62. [DOI] [PubMed] [Google Scholar]
- Marzano C.; Gandin V.; Folda A.; Scutari G.; Bindoli A.; Rigobello A. P. Free Radical Biol. Med. 2007, 42, 872–881. 10.1016/j.freeradbiomed.2006.12.021. [DOI] [PubMed] [Google Scholar]
- Olszewski U.; Claffey J.; Hogan M.; Tacke M.; Zeillinger R.; Bednarski P. J.; Hamilton G. Invest. New Drugs 2011, 29, 607–614. 10.1007/s10637-010-9395-5. [DOI] [PubMed] [Google Scholar]
- Arduengo A. J. III; Krafczyk R.; Schmutzler R. Tetrahedron 1999, 55, 14523–14534. 10.1016/S0040-4020(99)00927-8. [DOI] [Google Scholar]
- Huang J.; Nolan S. P. J. Am. Chem. Soc. 1999, 121, 9889–9890. 10.1021/ja991703n. [DOI] [Google Scholar]
- Mistryukov E. A. Mendeleev Commun. 2006, 16, 258–259. 10.1070/MC2006v016n05ABEH002347. [DOI] [Google Scholar]
- Uson R.; Laguna A.; Laguna M. Inorg. Synth. 1989, 26, 85–91. 10.1002/9780470132579.ch17. [DOI] [Google Scholar]
- Becke A. D. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- Becke A. D. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M. J.; Heyd J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam N. J.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, Revision C.2; Gaussian Inc., Wallingford, CT, 2009.
- Dolg M.; Wedig U.; Stoll H.; Preuss H. J. Chem. Phys. 1987, 86, 866–872. 10.1063/1.452288. [DOI] [Google Scholar]
- Andrae D.; Haussermann U.; Dolg M.; Stoll H.; Preuss H. Theoretica Chimica Acta 1990, 77, 123–141. 10.1007/BF01114537. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.