

Summary
MicroRNAs have been shown to play a role in B‐cell differentiation and activation. Here, we found miR‐182 to be highly induced in activated B cells. However, mice lacking miR‐182 have normal B‐cell and T‐cell development. Interestingly, mutant mice exhibited a defective antibody response at early time‐points in the immunization regimen when challenged with a T‐cell‐dependent antigen. Germinal centres were formed but the generation of extrafollicular plasma cells was defective in the spleens of immunized miR‐182‐deficient mice. Mutant mice were also not able to respond to a T‐cell‐independent type 2 antigen, which typically elicited an extrafollicular B‐cell response. Taken together, the data indicated that miR‐182 plays a critical role in driving extrafollicular B‐cell antibody responses.
Keywords: antibody response, extrafollicular B cells, germinal centres, miR‐182
Introduction
MicroRNAs (miRNAs) are short non‐coding RNAs of approximately 18–24 nucleotides in length that negatively regulate gene expression at the post‐transcriptional level.1 They act by either inducing the degradation of target mRNA or repressing the translation of mRNAs to proteins.2, 3, 4 MicroRNAs have been shown to play critical regulatory roles in a diverse set of normal biological processes during cellular differentiation, including neuronal patterning,5 lineage commitment in haematopoiesis,6 tissue homeostasis,7, 8, 9 apoptosis10, 11 and pathological progression, especially carcinogenesis.12, 13
The expression of miR‐182, a member of the miR‐183–miR‐96–miR‐182 cluster, is associated with many types of cancer, including prostate, lung and glioma.14, 15, 16 miR‐182 is highly expressed in the retina and hair cells of the inner ear.17, 18, 19 It is marginally expressed in immune cells but can be induced in activated splenic T and B cells.20, 21, 22 We and others have shown previously that miRNA played a crucial role in B‐cell development and differentiation.23, 24 In particular, miR‐181,25 miR‐150,26 miR‐15527 and miR‐17~9228 were shown to be required for normal B‐cell development and function. Initially, miR‐182 was shown to promote T‐cell‐mediated immune responses in gene knock‐down experiments.22 However, a recent study with miR‐182 knockout (KO) mice indicated that miR‐182 was dispensable for T‐cell development and activation.21 Hence, there remained controversy whether miR‐182 played a role in the immune system. In addition, a detailed study on the role of miR‐182 in B‐cell development and function is still lacking.
B cells can produce antibodies against foreign antigens with (thymus‐dependent, TD) or without (thymus‐independent, TI) T‐cell help.29 Upon recognition of the TD or TI antigen, B cells first differentiate into short‐lived plasma cells in the extrafollicular space, and this is critical in providing the first line of protection against the spread of pathogens. At later time‐points during the host response to T‐cell‐dependent antigens, germinal centres (GCs) are generated, where T‐cell–B‐cell interactions occur.30 GC B‐cell differentiation is aided by a subset of CD4+ T cells known as T‐follicular helper (Tfh) cells.31, 32
As miR‐182 was shown to be highly up‐regulated in activated B cells,20, 21 which might indicate that miR‐182 has a critical role to play during B‐cell terminal differentiation into GC B cells and plasma cells, we undertake to examine the role of miR‐182 in B‐cell differentiation, paying particular attention to the antibody response.
Materials and methods
Mouse immunization
The miR‐182 KO mice were obtained from Dr Naoharu Iwai17 and backcrossed from the mixed C57BL/6 and 129 background to wild‐type C57BL/6 background for five generations. Wild‐type B6 mice were maintained in‐house and used as controls. Mouse maintenance and usage in our animal facilities in the Biological Resource Centre are in compliance with guidelines issued by the National Advisory Committee on Laboratory Animal Research. Protocol (IACUC number 130857) was approved by the Institutional Animal Care and Use Committee (IACUC) of the Research Biological Resource Centre, Agency for Science, Technology. Measures were taken to minimize animal suffering.
To examine T‐cell‐dependent immune responses, sex‐matched 6‐ to 8‐week‐old mice were immunized intraperitoneally with 100 μg of alum‐precipitated NP38‐CGG (Biosearch Technologies, Petaluma, CA). Quantification of primary IgM and IgG1 antibody responses was carried out via ELISA using sera collected at 7, 14, 21 and 28 days after immunization. Mice were re‐challenged at day 30 with 50 μg of NP38‐CGG in PBS to examine the secondary immune response and IgM and IgG1 antibody titres were determined by ELISA using sera collected 5 days later. Mice were also immunized intraperitoneally with 50 μg of NP25‐Ficoll (Biosearch Technologies) to determine the T‐cell‐independent type II immune response. To measure NP‐specific IgM, IgG1 and IgG3 antibodies, sera were harvested at 4, 7, 14 and 21 days after NP25‐Ficoll immunizations.
Flow cytometry
Spleen, bone marrow, peritoneal cavity and Peyer's patches were extracted from mice and dissociated into single‐cell suspensions. The cells were then stained with mixtures of fluorochrome‐conjugated antibodies as follows: phycoerythrin (PE) ‐Cy7‐CD4, FITC‐CD8, peridinin chlorophyll protein‐conjugated T‐cell receptor‐β (PerCP‐TCR‐β), allophycocyanin (APC) ‐Cy7‐B220, APC‐Cy7‐CD19, PE‐Cy7‐CD38 (BioLegend, San Diego, CA), PerCP‐PD1, biotin‐CXCR5, FITC‐CD21, PE‐CD23, PE‐Fas (BD Biosciences, Franklin Lakes, NJ), PerCP‐IgM, FITC‐IgG1, PE‐IgD, FITC‐CD5, PE‐CD43 (eBiosciences, San Diego, CA). Streptavidin‐FITC was used to conjugate with biotin‐CXCR5. Dead cells were excluded using 4′, 6‐diamidino‐2‐phenyindole (DAPI) (Sigma‐Aldrich, St Louis, MO). Anti‐B220, Dump, anti‐IgG1 antibodies and NIP ((4‐hydroxy‐5‐iodo‐3‐nitrophenyl)acetyl) were used to visualize antigen‐specific IgG1 B cells. An R‐PE labelling kit‐SH (Dojindo Molecular Technologies, Rockville, MD) was used to generate PE‐conjugated NIP‐BSA (Biosearch Technologies). Dump for cell exclusion staining contained the following biotinylated antibodies, which were later conjugated with Streptavidin‐PerCP: anti‐CD3, anti‐Ter119, anti‐DX5, anti‐IgD, anti‐Th1.2, anti–Gr‐1, anti‐AA4.1, anti‐F4/80, anti‐NK1.1, anti‐CD138 and anti‐CD5 monoclonal antibodies.
For intracellular staining of Bcl‐6 and interleukin‐21 (IL‐21), cells were first labelled for surface markers, incubated with BD Cytofix/Cytoperm for 15 min, washed with BD Perm/Wash buffer, incubated with BD Cytoperm Plus for 10 min, washed, and incubated with BD Cytofix/Cytoperm for 5 min. Cells were then labelled with antibodies to PE‐bcl‐6, APC‐IL‐21 (eBioscience).
All antibodies were diluted 100 times with FACS buffer (PBS containing 5% fetal bovine serum). Samples were analysed on an LSRII cytometer (BD Biosciences) and data were further analysed with flowjo software (TreeStar, Ashland, OR). Fluorescence compensation was set to correct for spill‐over. For gating strategy used in flow cytometry analysis, cells were first gated for singlets (FSC‐W versus FSC‐A) and lymphocytes (SSC‐A versus FSC‐A). The lymphocyte gate was further analysed for their uptake of the Live/Dead DAPI stain to determine live versus dead cells and their expression of CD4 and CD19/B220 to differentiate T or B cells for further gating as shown in the figures.
ELISA and ELISPOT
4‐Hydroxy‐3‐nitrophenylacytyl (NP)‐specific IgM, IgG1 and IgG3 antibodies were determined by ELISA, and IgG1 antibody‐secreting cell (ASC) formation were detected by ELISPOT, following previously published protocols.33
For ELISA, 384‐well flat‐bottomed NUNC plates were coated overnight with NP14‐BSA (5 μg/ml), washed and blocked for 2 hr with 2% BSA in PBS (blocking buffer) at room temperature. Sera samples were serially diluted with blocking buffer and incubated for 2 hr at room temperature. After washing thoroughly, biotinylated anti‐IgM and anti‐IgG1 antibodies (1 : 1000 dilution in blocking buffer) were added into each well for 1 hr and followed by incubation with streptavidin‐horseradish peroxidase. 3,3′,5,5′‐Tetramethyl benzidine thiobarbituric acid substrate was used to assess bound secondary antibodies and data were collected using a Tecan microplate reader (Tecan, Männedorf, Switzerland).
For ELISPOT assay, multiScreen filter plates (Millipore, Billerica, MA) were coated with 50 μg/ml NP14‐BSA in PBS at 4° overnight, and blocked with 2% BSA in PBS at 37° for 2 hr. Splenocytes or bone marrow cells were added to each well and incubated in a humidified incubator containing 5% CO2. After a 2‐hr incubation, the filter plates were washed with PBS containing 0·1% Tween‐20 (Sigma‐Aldrich) twice and then incubated with streptavidin–alkaline phosphatase (AKP)‐conjugated anti‐IgG1 antibodies. AKP activity was developed using BCIP/NBT‐plus substrate (Mabtech, Nacka Strand, Sweden).
B‐cell isolation and real‐time RT‐PCR analyses
Negative selection by CD43 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany) were used to isolate B cells from spleens of wild‐type and miR‐182 KO mice. To induce plasmablast differentiation in vitro, purified B cells were re‐suspended at 2 × 106 cells/ml and incubated for 1–3 days in the presence of anti‐IgM F(ab′)2 alone (10 μg/ml), anti‐CD40 alone (1 μg/ml), anti‐IgM F(ab′)2 (10 μg/ml) and anti‐CD40 (1 μg/ml) antibodies, or lipopolysaccharide alone (20 μg/ml).
Total RNA was isolated using TRIZOL Reagent (Invitrogen, Carlsbad, CA). TaqMan Gene Expression Assays (Applied Biosystems, Foster City, CA) was used for real‐time RT‐PCR on an ABI Prism7500 instrument (Applied Biosystems). The miR‐182 predesigned TaqMan Gene Expression Assays [mmu‐miR‐182 (ID 002599)] were used in real‐time RT‐PCR, and normalized to U6 snRNA expression. Data analysis was performed using the comparative Ct (threshold cycle) method.
Confocal microscopy
Spleens sections were harvested, embedded in Tissue‐Tek and ‘snap‐frozen’ in dry ice and stored at −80°. Cryostat sections (8 μm in thickness) were prepared, air‐dried and fixed in ice‐cold acetone for 15 min. Sections were blocked with 5% rat serum and 2% BSA in PBS and stained with PE‐anti‐IgG1 and FITC‐anti‐IgD antibodies (BD Biosciences PharMingen, San Diego, CA). Antibodies were diluted (1 : 50) in PBS containing 5% rat serum. Sections were further mounted with CYTOSEAL 60 (Electron Microscopy Sciences, Hatfield, PA) and analysed with an Olympus FV1000 microscope (Olympus, Tokyo, Japan) using a × 20 objective, and the images were acquired with olympus fluoview Version 2.1 software.
Statistics
Two‐tailed Student's t‐test was used for statistical analyses using prism (GraphPad). P‐values < 0·05 were considered significant.
Results
Induction of miR‐182 expression during B‐cell activation
Previous work had shown the abundance of miR‐182 in cancer cells, where it played a role in promoting their proliferation.34 However, the role of miR‐182 in the immune system has been relatively unexplored. Studies showed that miR‐182 expression was largely undetectable in naive B cells, but highly expressed in activated B cells including those that were in vitro stimulated or found in the germinal centre.20, 21 To determine if miR‐182 is indeed induced upon B‐cell activation, we purified B cells and stimulated them with either anti‐IgM, or anti‐CD40 antibodies, or both, or LPS, and analysed the expression of miR‐182 by quantitative RT‐PCR. We showed that miR‐182 was highly induced in activated B cells (Fig. 1). At 3 days after activation, the expression of miR‐182 was much higher (30‐fold to 250‐fold) in activated B cells compared with naive B cells in all stimulation conditions tested. Interestingly, the induction of miR‐182 was higher in samples stimulated by anti‐IgM than in those stimulated by anti‐CD40 antibodies. As control, the expression of miR‐182 was undetectable in B cells obtained from miR‐182 KO mice. Taken together, we confirmed the up‐regulation of miR‐182 expression in activated B cells, which suggested that it could play a role in B‐cell function.
Figure 1.
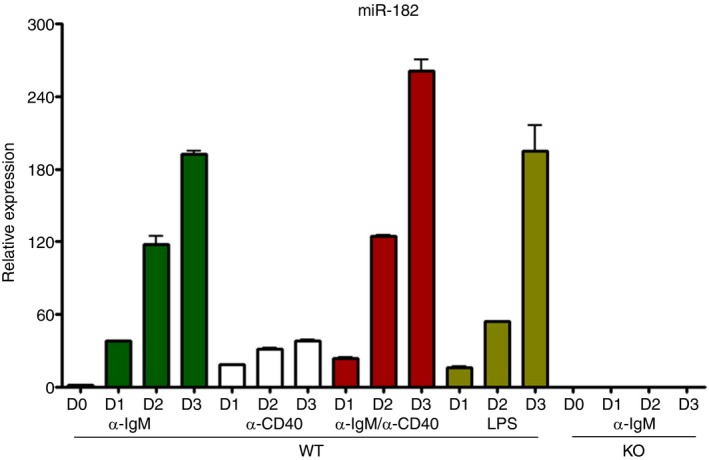
Profiling of miR‐182 expression in stimulated B cells. Real‐time RT‐PCR analyses indicate that the microRNA miR‐182 is preferentially expressed in activated B cells. Purified wild‐type and miR‐182 knockout (KO) B cells were stimulated with anti‐IgM (F(ab′)2) alone (10 μg/ml), anti‐CD40 alone (1 μg/ml), anti‐IgM (F(ab′)2) (10 μg/ml) and anti‐CD40 (1 μg/ml) antibodies, or lipopolysaccharide (LPS) alone (20 μg/ml) for 1–3 days. Cells were harvested and miR‐182 expression was determined by real‐time RT‐PCR, normalized to U6 snRNA expression. Data shown represent averages with standard deviations from three independent samples.
miR‐182 KO mice have normal B‐ and T‐cell development
To examine the role of miR‐182 in B‐cell activation, we first assessed if B‐cell development would be perturbed in miR‐182 KO mice. As shown in Fig. 2(a), miR‐182 KO mice have intact B‐cell lymphopoiesis in the bone marrow with normal populations of B220low IgM− pro/pre‐B cells, B220low IgM+ immature B and B220+ IgM+ circulating mature B cells. Follicular (CD23+ CD21+) and marginal zone (CD23− CD21++) B‐cell subsets were also found to be unperturbed in the spleens of mutant mice (Fig. 2b). In the peritoneal cavity, B‐1a (CD5+ CD43+), B‐1b (CD5− CD43+) and B‐2 (CD5− CD43−) cell populations were also comparable between wild‐type (WT) and miR‐182 KO mice (Fig. 2c). In addition, miR‐182 KO mice also have normal T‐cell populations in the thymus with normal generation of CD4 and CD8 single‐positive (CD4+ CD8− or CD4− CD8+) and CD4+ CD8+ double‐positive thymocytes (Fig. 2d). Taken together, the data indicated that miR‐182 KO mice have normal B‐cell and T‐cell development and therefore could be used to study the role of miR‐182 in B‐cell activation and terminal differentiation.
Figure 2.
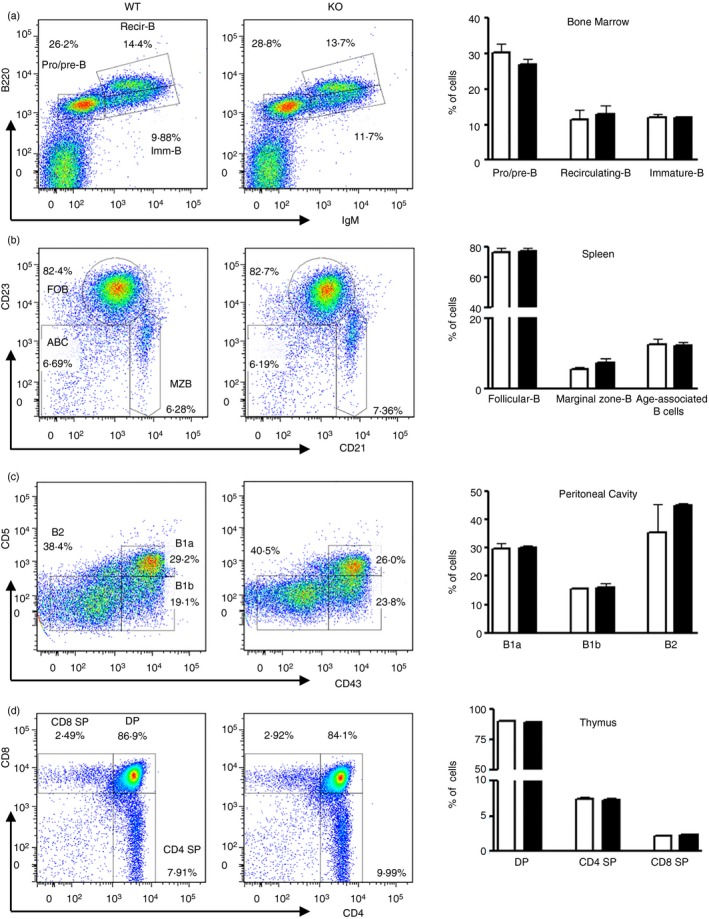
Examination of B‐ and T‐cell populations in wild‐type and miR‐182 knockout (KO) mice. Flow cytometric analyses of B‐cell populations in the bone marrow (a), spleen (b) and peritoneal cavity (c), and T‐cell subsets in the thymus (d) of wild‐type and miR‐182 KO mice. (a) Bone marrow cell suspension was stained for B220 and IgM expression and numbers adjacent to gated areas indicate per cent of total bone marrow cells. (b) Gated splenic CD19+ cells were further interrogated for their expression of CD23 and CD21 to determine populations of CD23+ CD21+ follicular B, CD23− CD21++ marginal zone B, and a mature B‐cell subset termed age‐associated B cells that are CD23− CD21−. Numbers shown are per cent of CD19+ B cells. (c) CD5 and CD43 expression were used to differentiate cell populations in the peritoneal cavity, namely CD5+ CD43+ B‐1a, CD5− CD43+ B‐1b, and CD5− CD43− B‐2 cells. Numbers shown are per cent of CD19+ cells. (d) Thymocytes were stained for CD8 and CD4 expression and numbers adjacent to gated areas indicate per cent of live cells. Frequencies of each subset of cells in wild‐type and miR‐182 KO mice are shown. Data shown represent averages with standard deviations from three independent experiments.
miR‐182 deficiency does not perturb the expansion of Tfh and GC B cells
To determine if there is a role for miR‐182 in B‐cell activation, we first examined the immune cells found in the Peyer's patches of WT and miR‐182 KO mice. Peyer's patches are sites of chronic immune responses, with persistent GC reactions. Our flow cytometry analyses indicated that miR‐182 KO mice were able to generate comparable fractions of CD4+ TCRβ + PD‐1high CXCR5+ Tfh cells (Fig. 3a,b) compared with WT controls. Tfh cell differentiation is driven by the master transcription factor Bcl‐6.35 Interestingly, Tfh cells from both WT and miR‐182 KO mice express equivalent amounts of Bcl‐6, further suggesting that the Tfh cells in miR‐182 KO mice were not compromised (Fig. 3c). Other than Tfh cells, GC also contains antigen‐activated B cells (GC B cells, B220+ CD38−Fas+). Again, GC B cells were found to be present in comparable fractions in WT and miR‐182 KO mice (Fig. 3d,e). These data suggest that although miR‐182 is highly induced during B‐cell activation, it might not play any role in GC B‐cell differentiation in the Peyer's patches.
Figure 3.
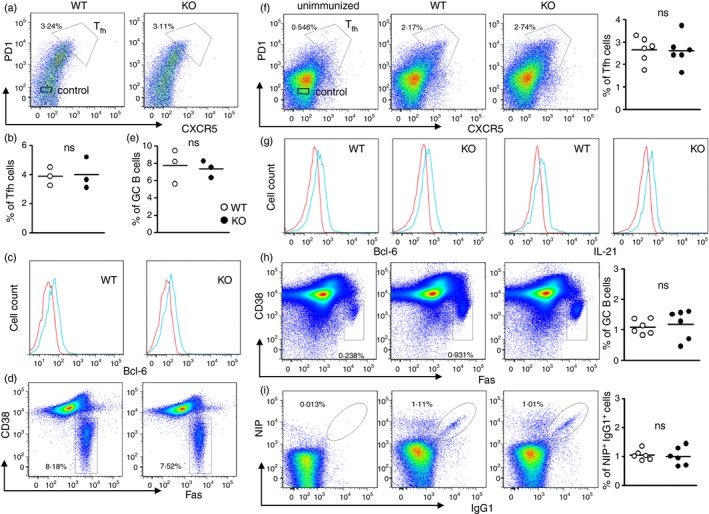
Analyses of T follicular helper (Tfh) cells and germinal centre (GC) B cells in miR‐182 knockout (KO) mice. (a) Flow cytometric analysis of Tfh cells in the Peyer's patches of unchallenged wild‐type and miR‐182 KO mice. CD4+ TCR β + cells were examined for PD1 and CXCR5 expression and CD4+ TCR β + CXCR5+ PD‐1+ Tfh cells were shown. Numbers indicate per cent of CD4+ TCR β + cells. (b) Tfh cell frequency in wild‐type and miR‐182 KO mice. (c) Histogram profile of Bcl‐6 expression in Tfh cells (blue histogram) and non‐Tfh cells (red histogram). (d) Flow cytometric analyses of the Peyer's patches of wild‐type and miR‐182 KO mice depicting activated B‐cell populations (B220+ Fas+ CD38−), and numbers indicate per cent of total Peyer's patches cells. Numbers indicate per cent of B220+ cells. (e) Frequency of GC B cells in wild‐type and miR‐182 KO mice (on the same panel as B). (f) Wild‐type and miR‐182 KO mice were analysed 10 days after challenge with NP 38‐CGG. Flow cytometry analysis and quantification of the percentage of Tfh cells in the spleens of immunized wild‐type and miR‐182 KO mice. Numbers shown are per cent of CD4+ TCR β + cells. (g) Histogram profile of Bcl‐6 and interleukin‐21 (IL‐21) expression in Tfh cells (blue histogram) and non‐Tfh cells (red histogram). (h) Flow cytometry analysis and quantification of the percentage of B220+ Fas+ CD38− GC B cells in the spleens of immunized wild‐type and miR‐182 KO mice. Numbers shown are per cent of B220+ cells. (i) FACS analysis of antigen‐specific NIP + IgG1+ B cells from B220+Dump− cells in the spleens of immunized wild‐type and miR‐182 KO mice. Numbers shown are per cent of antigen‐specific NIP + IgG1+ B cells from the B220+ Dump− gate. Frequencies of antigen‐specific NIP + IgG1+ B cells in immunized wild‐type and miR‐182 KO spleens.
To confirm this observation, we immunized miR‐182 KO mice with T‐cell‐dependent antigen NP38‐CGG in alum to examine the role of miR‐182 in B‐cell terminal differentiation. At 10 days post‐immunization, we isolated splenocytes from mutant mice and compared their composition of Tfh and GC B cells to those found in WT mice. Consistent with the data obtained from Fig 3(a–e), we found comparable CD4+ TCRβ + PD‐1high CXCR5+ Tfh cells in WT and mutant mice (Fig. 3f). Enumeration of Tfh cells confirmed that these cells were generated in normal fractions in miR‐182 KO mice. Tfh cells are characterized by their expressions of Bcl‐6 and IL‐21.35 Hence we compared the respective expression levels of Bcl‐6 and IL‐21 in Tfh cells of WT and KO mice. Both WT and miR‐182 KO mice displayed increased expression of Bcl‐6 and IL‐21 in their Tfh populations compared with PD‐1− CXCR5− non‐Tfh cells (Fig. 3g). Furthermore, B220+ CD38− Fas+ GC B cells were also found to be present in normal fractions in the spleens of miR‐182 KO mice (Fig. 3h). Further examination of class‐switched antigen‐binding NIP+ IgG1+ GC B cells corroborated that these cells were generated in normal fractions in miR‐182 KO mice (Fig. 3i). Taken together, the data indicated that miR‐182 was not involved in the generation of Tfh and GC B cells.
miR‐182 deficiency impairs early T‐cell‐dependent immune response
Germinal centres play critical roles during adaptive immune responses and support antibody class‐switching and plasma cell differentiation.30 Our data (Fig 3) indicated that the loss of miR‐182 did not affect the GC reaction. To examine if miR‐182 deficiency would affect the antibody response, we examined antigen‐specific antibodies in the sera of immunized WT and miR‐182 KO mice. The TD antigen NP38‐CGG elicits mainly IgM and IgG1 antibody responses.36 Hence, we focused on NP‐specific IgM and IgG1 antibody titres at various time‐points (days) post‐immunization. Interestingly, miR‐182 deficiency led to defective generation of antigen‐specific IgM and IgG1 antibodies at early time‐points (days 7 and 14) after primary immunization in miR‐182 KO mice (Fig. 4a). The ELISA data suggested that miR‐182 is required for the early generation of antibodies against T‐cell‐dependent antigens.
Figure 4.
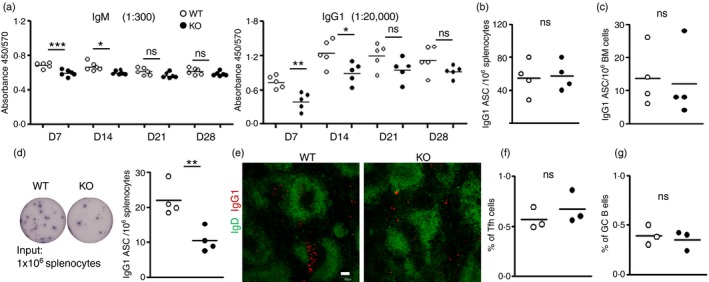
Impaired early T‐cell‐dependent antibody response in miR‐182 knockout (KO) mice. (a) Serum IgM and IgG1 antigen‐specific antibody levels in wild‐type (empty circles) and miR‐182 KO (filled circles) mice determined via ELISA at indicated time‐points after NP 38‐CGG immunization. Sera were diluted 300 and 20 000 times, respectively, to measure NP‐specific IgM and IgG1 antibodies. Data indicated are representative of two independent experiments and each experiment comprised at least five mice per group. (b, c) ELISPOT analyses of antigen‐binding IgG1 secreting cells in the spleen (b) and bone marrow (c) of wild‐type and miR‐182 KO mice 10 days after primary immunization. (d) ELISPOT analyses and quantification of antigen specific IgG1 antibody‐secreting cells (ASCs) in the spleens of wild‐type and miR‐182 KO mice at day 5 after primary challenge. (e) Splenic tissue section analyses of NP 38‐CGG‐challenged wild‐type and miR‐182 KO mice at day 5 post‐immunization by confocal microscopy. Anti‐IgD (green, to visualize B‐cell follicles) and anti‐IgG1 (red, to visualize IgG1 ASC) antibodies were used in the staining. White bar, 100 μm. Image indicated representative from three spleens analysed per group. Quantification of flow cytometry analysis of the percentage of follicular helper T (Tfh) cells (f) and germinal centre (GC) B cells (g) in the spleens of immunized wild‐type and miR‐182 KO mice. Numbers shown are per cent of CD4+ TCR β + and B220+ cells, respectively. Each circle stands for data from one mouse examined. *P < 0·05; **P < 0·01; ***P < 0·001.
Following immunization with a T‐cell‐dependent antigen, B cells start proliferating and differentiate into antibody‐secreting plasma cells and this process either occurs in the extrafollicular compartments or in the germinal centres of secondary lymphoid organs. Since NIP+ IgG1+ B cells were observed in GC upon immunization (Fig. 3i), and yet there were reduced serum IgM and IgG1 antibodies at days 7 and 14 (Fig. 4a) in immunized miR‐182 KO mice, we wondered if plasma cells or ASC would be generated in normal fractions in miR‐182 KO mice upon antigen challenge. Plasma cells were thought to arise a few days after antigen challenge. Hence, we examined the generation of antigen‐specific IgG1 ASC during early time‐points in the immunization regime. Interestingly, comparable numbers of NP‐specific IgG1 ASC were detected by ELISPOT in the spleen (Fig. 4b) and bone marrow (Fig. 4c) of miR‐182 KO mice compared with control mice at day 10 post‐immunization. Given the impaired serum antibody generation at early time‐points in the immunization regimen, we decided to measure the number of NP‐specific IgG1 ASC at day 5 post‐immunization. At this time‐point in the immunization process, the ASC present arose mainly from an extrafollicular B‐cell response. The number of ASC was found to be significantly reduced in the spleens of miR‐182 KO mice at this early time‐point (Fig. 4d), which might account for the observed reduction of serum antibodies early in the immunization regimen. Next, we further confirmed this finding by confocal microscopy. Substantial numbers of IgG1 ASC were found to cluster in the extrafollicular regions of the spleen in immunized WT mice but these cells were drastically reduced in numbers in the KO mice at day 5 post‐immunization (Fig. 4e). At this time‐point in the antigen challenge, both WT and miR‐182 KO mice started to initiate the GC reaction with the generation of comparable fractions of Tfh cells (Fig. 4f) and GC B cells (Fig. 4g). These data together suggest that mice lacking miR‐182 are impaired in the generation of early short‐lived extrafollicular plasma cells, which helped to explain the impaired antibody responses seen at early time‐points during the immunization process.
To assess whether miR‐182 deficiency would affect the ability of miR‐182 KO mice to mount a rapid successful secondary immune response upon re‐encounter with the same antigen, we re‐challenged the mice at 30 days after the primary immunization. As depicted in Fig. 5, both WT and miR‐182 KO mice were able to elicit a significant recall IgM and IgG1 response upon re‐encountering the same antigen. These data suggest that miR‐182 KO mice harbour an intact immune memory compartment.
Figure 5.
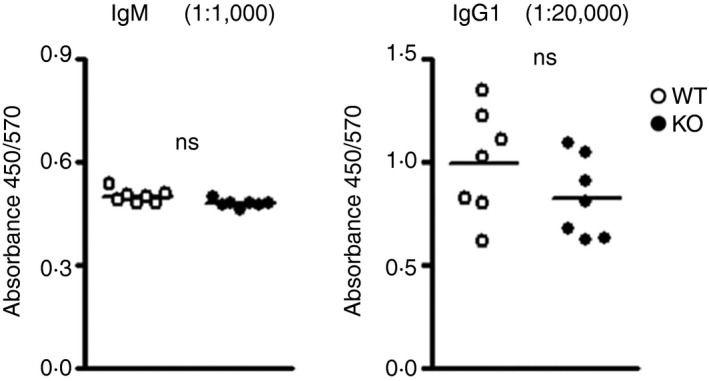
Examination of antibody recalled response in miR‐182 knockout (KO) mice. Mice previously challenged with alum‐precipitated NP 38‐CGG were further immunized with soluble NP 38‐CGG 30 days later. Serially diluted sera samples were examined using ELISA to determine antigen‐binding NP + IgM (1: 1000 dilution) and IgG1 (1: 20 000 dilution) titres in wild‐type (empty circles) and miR‐182 KO (filled circles) mice 5 days after re‐challenge.
Defective T‐cell‐independent type 2 immune response in miR‐182 KO mice
The defective extrafollicular immune response observed in miR‐182 KO mice during the T‐cell‐dependent challenge prompted us to further characterize the T‐cell‐independent immune response in these mice. The T‐cell‐independent response predominantly involved extrafollicular B‐cell responses when antigen‐activated B cells differentiate into plasma cells and produce antibodies without T‐cell help. We immunized WT and miR‐182 KO mice intraperitoneally with the T‐cell‐independent type II antigen (TI‐II), NP25‐Ficoll, and measured sera immunoglobulin titres at days 4, 7, 14 and 21 after antigenic challenge. As seen in Fig 6(a), WT mice were able to rapidly produce a substantial amount of IgM antibodies from day 4 and to generate class‐switched IgG1 and IgG3 antibodies from day 7 upon immunization with NP25‐Ficoll. However, miR‐182 KO mice were impaired in mounting an immune response to NP25‐Ficoll with drastic reduction in the production of NP‐specific IgM, IgG1 and IgG3 antibodies throughout the immunization period. Hence, the lack of miR‐182 leads to defective TI‐II humoral immune responses.
Figure 6.
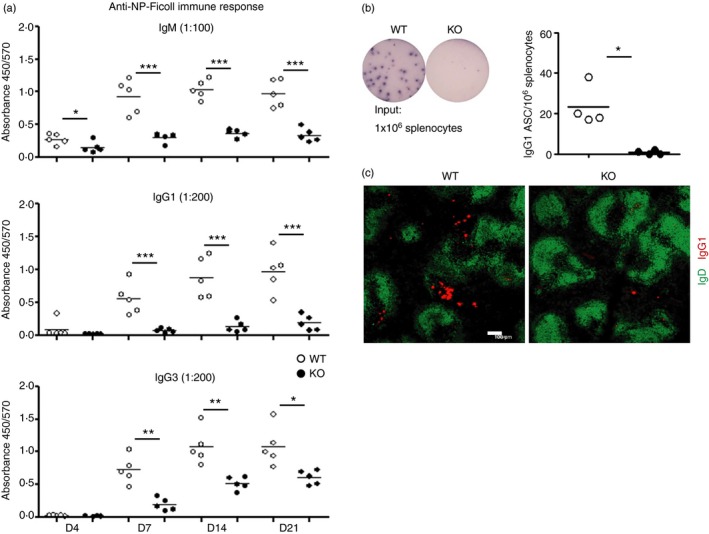
Impaired T‐cell‐independent antibody response in miR‐182 knockout (KO) mice. (a) Mice were immunized with NP 25‐Ficoll, and sera antigen‐specific antibodies in wild‐type (empty circles) and miR‐182 KO (filled circles) mice were measured via ELISA at the indicated days upon challenge. Sera were diluted 100‐, 200‐ and 200‐fold, respectively, to determine IgM, IgG1 and IgG3 isotype concentrations. Data indicated are representative of two independent experiments and each experiment comprised of at least five mice per group. *P < 0·05; **P < 0·01; ***P < 0·001. (b) ELISPOT analyses and enumeration of antigen specific IgG1 antibody‐secreting cells (ASC) are shown in the spleens of wild‐type and miR‐182 KO mice at day 5 post immunization. Each circle stands for data from one mouse examined, *P < 0·05. (c) Splenic tissue section analyses of NP 25‐Ficoll‐challenged wild‐type and miR‐182 KO mice at day 5 post‐immunization by confocal microscopy. Anti‐IgD (green) and anti‐IgG1 (red) antibodies were used in the staining. White bar, 100 μm. Image indicated representative of three spleens analysed per group.
Given the observation of a defective serum antibody response, we examined the formation of NP‐specific IgG1 ASC at day 5 post‐immunization. Consistently with our observation during the early time‐points of the TD immune response, IgG1 ASC were also found to be reduced in the spleens of miR‐182 KO mice at this time‐point upon NP25‐Ficoll challenge (Fig. 6b), which accounted for the reduced serum antibody responses seen in these mice. Histological studies using spleen tissue sections obtained from day 5 post‐immunized mice further corroborated the absence of IgG1 ASC in the spleens of KO mice compared with WT controls (Fig. 6c). These data together suggest that miR‐182‐deficient B cells are impaired in their generation of short‐lived extrafollicular plasma cells and so led to a defective antibody response to TI‐II antigen.
Discussion
Previous profiling studies have revealed differentiated expression of miRNAs during different stages of B‐cell development. A number of miRNAs have also been implicated in B‐cell activation.37 Specifically, miR‐155 was shown to be important for GC and post‐GC plasma cell responses38, 39 and our laboratory had also elucidated a role for miR‐17~92 in plasma cell function.28 Hence, we embarked on this study to examine the role of miR‐182 in the humoral immune response given that miR‐182 has been found to be highly up‐regulated in activated B cells. We found that miR‐182 KO mice have intact B‐cell and T‐cell development and could generate comparable Tfh and GC B‐cell populations upon challenge with a T‐cell‐dependent antigen. The miR‐182 KO mice were also able to generate normal recalled antibody responses upon re‐challenge with the same antigen. Hence, the data suggest that miR‐182 might be dispensable for B‐cell activation during T‐cell‐dependent immune challenge. However, when we examined more closely the kinetics of the primary T‐cell‐dependent immune response in vivo, we found that miR‐182 KO mice exhibited defective generation of antigen‐specific antibodies at an early time‐point in the immunization scheme. This finding suggested that the generation of extrafollicular plasma cells could be defective in miR‐182 KO mice. In addition, miR‐182 KO mice were also found to be unable to produce antibodies to a TI‐II antigen, which typically elicits an extrafollicular antibody response, corroborating our earlier findings. Taken together, our data suggest that miR‐182 is required for the extrafollicular B‐cell antibody responses.
While our work was in progress, Pucella et al. published that miR‐182‐deficiency had minimal impact on B‐cell biology and that there was a lack of correlation between miR‐182 expression and function in B cells.21 The group reported normal B‐cell development and GC responses. These findings were consistent with ours. However, in their study, the authors measured the antibody response at one time‐point in the secondary challenge, which mainly interrogated the recalled memory immune response in the miR‐182 KO mice. They did not examine in detail the primary antibody response and in particular, the early time‐points of the immune response to a TD antigen. Furthermore, the authors also did not examine the antibody response to TI‐II antigen. Hence our study not only confirms the normal B‐cell development data seen in the Pucella et al. study but substantially advances the field by revealing that miR‐182 plays an important role in regulating the extrafollicular antibody response. Without miR‐182, there was a delay in the production of antigen‐specific antibodies at early time‐points in a TD antigenic challenge and a complete impairment of antibody response in a TI type II antigenic challenge.
It would be interesting to identify the targets of miR‐182 in the modulation of the extrafollicular B‐cell responses. So far, we have examined a few candidates using an in vitro culture system of plasma cell generation but none seems to be responsible for the phenotype. However, the in vitro culture system might not reflect the in vivo situation and so could hamper our understanding of the role of miR‐182 in B‐cell biology. Nevertheless, we will carry out future experiments to elucidate the real targets of miR‐182 in activated B cells that are responsible for the defective extrafollicular response. Future work is needed to help us understand how miR‐182 affects the short‐lived extrafollicular plasma cells. For example, does miR‐182 affect the generation, differentiation or survival of the extrafollicular plasma cells?
Competing interests statement
The authors declare no competing interests.
Acknowledgements
We thank all members from the Immunology group in the Bioprocessing Technology Institute for insightful suggestions and discussions. This work was funded by the Biomedical Research Council of the Singapore Agency for Science Technology and Research.
References
- 1. Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nat Rev Genet 2009; 10:94–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol 2014; 15:509–24. [DOI] [PubMed] [Google Scholar]
- 3. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet 2011; 12:99–110. [DOI] [PubMed] [Google Scholar]
- 4. Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell 2009; 136:215–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Johnston RJ, Hobert O. A microRNA controlling left/right neuronal asymmetry in Caenorhabditis elegans. Nature 2003; 426:845–9. [DOI] [PubMed] [Google Scholar]
- 6. Chen C‐Z, Li L, Lodish HF, Bartel DP. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004; 303:83–6. [DOI] [PubMed] [Google Scholar]
- 7. Cui Q, Yu Z, Purisima EO, Wang E. Principles of microRNA regulation of a human cellular signaling network. Mol Syst Biol 2006; 2, doi: 10.1038/msb4100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Plaisance V, Abderrahmani A, Perret‐Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA‐9 controls the expression of Granuphilin/Slp4 and the secretory response of insulin‐producing cells. J Biol Chem 2006; 281:26932–42. [DOI] [PubMed] [Google Scholar]
- 9. Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, et al Requirement of bic/microRNA‐155 for normal immune function. Science 2007; 316:608–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Baehrecke EH. miRNAs: micro managers of programmed cell death. Curr Biol 2003; 13:R473–5. [DOI] [PubMed] [Google Scholar]
- 11. Cimmino A, Calin GA, Fabbri M, Iorio MV, Ferracin M, Shimizu M, et al miR‐15 and miR‐16 induce apoptosis by targeting BCL2. Proc Natl Acad Sci USA 2005; 102:13944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schickel R, Boyerinas B, Park SM, Peter ME. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008; 27:5959–74. [DOI] [PubMed] [Google Scholar]
- 13. Lu J, Getz G, Miska EA, Alvarez‐Saavedra E, Lamb J, Peck D, et al MicroRNA expression profiles classify human cancers. Nature 2005; 435:834–8. [DOI] [PubMed] [Google Scholar]
- 14. Schaefer A, Jung M, Mollenkopf H‐J, Wagner I, Stephan C, Jentzmik F, et al Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int J Cancer 2010; 126:1166–76. [DOI] [PubMed] [Google Scholar]
- 15. Benjamin H, Lebanony D, Rosenwald S, Cohen L, Gibori H, Barabash N, et al A diagnostic assay based on microRNA expression accurately identifies malignant pleural mesothelioma. J Mol Diagn 2010; 12:771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jiang L, Mao P, Song L, Wu J, Huang J, Lin C, et al miR‐182 as a prognostic marker for glioma progression and patient survival. Am J Pathol 2010; 177:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jin Z‐B, Hirokawa G, Gui L, Takahashi R, Osakada F, Hiura Y, et al Targeted deletion of miR‐182, an abundant retinal microRNA. Molecular Vision 2009; 15:523–33. [PMC free article] [PubMed] [Google Scholar]
- 18. Friedman LM, Dror AA, Mor E, Tenne T, Toren G, Satoh T, et al MicroRNAs are essential for development and function of inner ear hair cells in vertebrates. Proc Natl Acad Sci U S A 2009; 106:7915–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sacheli R, Nguyen L, Borgs L, Vandenbosch R, Bodson M, Lefebvre P, et al Expression patterns of miR‐96, miR‐182 and miR‐183 in the developing inner ear. Gene Expr Patterns 2009; 9:364–70. [DOI] [PubMed] [Google Scholar]
- 20. Kuchen S, Resch W, Yamane A, Kuo N, Li Z, Chakraborty T, et al Regulation of microRNA expression and abundance during lymphopoiesis. Immunity 2010; 32:828–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pucella JN, Yen W‐F, Kim MV, van der Veeken J, Socci ND, Naito Y, et al miR‐182 is largely dispensable for adaptive immunity: lack of correlation between expression and function. J Immunol 2015; 194:2635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stittrich A‐B, Haftmann C, Sgouroudis E, Kuhl AA, Hegazy AN, Panse I, et al The microRNA miR‐182 is induced by IL‐2 and promotes clonal expansion of activated helper T lymphocytes. Nat Immunol 2010; 11:1057–62. [DOI] [PubMed] [Google Scholar]
- 23. Xu S, Guo K, Zeng Q, Huo J, Lam K‐P. The RNase III enzyme Dicer is essential for germinal center B‐cell formation. Blood 2011; 119:767–76. [DOI] [PubMed] [Google Scholar]
- 24. de Yébenes VG, Bartolomé‐Izquierdo N, Ramiro AR. Regulation of B‐cell development and function by microRNAs. Immunol Rev 2013; 253:25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. de Yébenes VG, Belver L, Pisano DG, González S, Villasante A, Croce C, et al miR‐181b negatively regulates activation‐induced cytidine deaminase in B cells. J Exp Med 2008; 205:2199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Xiao C, Calado DP, Galler G, Thai T‐H, Patterson HC, Wang J, et al MiR‐150 controls B cell differentiation by targeting the transcription factor c‐Myb. Cell 2007; 131:146–59. [DOI] [PubMed] [Google Scholar]
- 27. Calame K. MicroRNA‐155 function in B Cells. Immunity 2007; 27:825–7. [DOI] [PubMed] [Google Scholar]
- 28. Xu S, Ou X, Huo J, Lim K, Huang Y, Chee S, et al Mir‐17‐92 regulates bone marrow homing of plasma cells and production of immunoglobulin G2c. Nat Commun 2015; 6, doi: 10.1038/ncomms7764. [DOI] [PubMed] [Google Scholar]
- 29. Liu Y‐J. Sites of B lymphocyte selection, activation, and tolerance in spleen. J Exp Med 1997; 186:625–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Klein U, Dalla‐Favera R. Germinal centres: role in B‐cell physiology and malignancy. Nat Rev Immunol 2008; 8:22–33. [DOI] [PubMed] [Google Scholar]
- 31. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol 2011; 29:621–63. [DOI] [PubMed] [Google Scholar]
- 32. Nurieva RI, Chung Y. Understanding the development and function of T follicular helper cells. Cell Mol Immunol 2010; 7:190–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Y‐F, Xu S, Ou X, Lam K‐P. Shp1 signalling is required to establish the long‐lived bone marrow plasma cell pool. Nat Commun 2014; 5, doi: 10.1038/ncomms5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chi J, Ballabio E, Chen X‐H, Kušec R, Taylor S, Hay D, et al MicroRNA expression in multiple myeloma is associated with genetic subtype, isotype and survival. Biology Direct 2011; 6:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ma CS, Deenick EK, Batten M, Tangye SG. The origins, function, and regulation of T follicular helper cells. J Exp Med 2012; 209:1241–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Allen D, Cumano A, Dildrop R, Kocks C, Rajewsky K, Rajewsky N, et al Timing, genetic requirements and functional consequences of somatic hypermutation during B‐cell development. Immunol Rev 1987; 96:5–22. [DOI] [PubMed] [Google Scholar]
- 37. Davidson‐Moncada J, Papavasiliou FN, Tam W. MicroRNAs of the immune system: roles in inflammation and cancer. Ann N Y Acad Sci 2010; 1183:183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thai T‐H, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, et al Regulation of the germinal center response by microRNA‐155. Science 2007; 316:604–8. [DOI] [PubMed] [Google Scholar]
- 39. Vigorito E, Perks KL, Abreu‐Goodger C, Bunting S, Xiang Z, Kohlhaas S, et al microRNA‐155 regulates the generation of immunoglobulin class‐switched plasma cells. Immunity 2007; 27:847–59. [DOI] [PMC free article] [PubMed] [Google Scholar]