

Summary
Macrophages and dendritic cells (DCs) in murine spleen are essential for the maintenance of immune homeostasis by elimination of blood‐borne foreign particles and organisms. It has been reported that splenic DCs, especially CD8α + CD103+ DCs, are responsible for tolerance to apoptosis‐associated antigens. However, the molecular mechanism by which these DCs maintain immune homeostasis by blood‐borne apoptotic cell clearance remains elusive. Here, we found that the CCL22/CCR4 axis played a critical role in the maintenance of immune homeostasis during apoptotic cell clearance by splenic CD8α + CD103+ DCs. The present results revealed that systemic administration of apoptotic cells rapidly induced a large number of CCL22 and CCR4+ regulatory T (Treg) cells in the spleen of C57BL/6J mice. Further study demonstrated that CD8α + CD103+ DCs dominantly produce much higher CCL22 than CD8α + CD103− DCs. Moreover, the transient deletion of CD8α + CD103+ DCs caused a decrease in CCL22 levels together with CCR4+ Treg cell percentage. Subsequently, the levels of some pro‐inflammatory cytokines, such as interleukin‐17 and interferon‐γ in the spleen with the absence of CD8α + CD103+ DCs increased in response to the administration of apoptotic cells. Hence, intravenous injection of apoptotic cells induced a subsequent increase in CCL22 expression and CCR4+ Treg cells, which contribute to the maintenance of immune homeostasis at least partially by splenic CD8α + CD103+ DCs.
Keywords: CCL22, CCR4+ regulatory T cells, CD8α+CD103+ dendritic cells, spleen
Abbreviations
- CytC
cytochrome C
- DCs
dendritic cells
- MZ
marginal metallophilic zone
- NOD
non‐obese diabetic
- Treg cells
regulatory T cells
Introduction
In multicellular organisms, unnecessary or harmful cells are eliminated by apoptotic cell death.1 After undergoing apoptosis, cell corpses are rapidly recognized and phagocytosed by phagocytes, such as macrophages and dendritic cells (DCs). The phagocytes could engulf the dying cells via eat‐me signals, such as phosphatidylserine on dying cells and receptors such as T‐cell immunoglobulin mucin molecule (Tim4) and scavenger receptor type F family member 1 (Scarf1) for the eat‐me signals on phagocytes.2, 3 Inhibition or absence of these molecular functions in vivo causes failure of apoptotic cell clearance and subsequent high levels of autoantibodies, even autoimmune diseases, such as rheumatoid arthritis.4, 5 Apoptotic cell clearance by phagocytes is essential for maintenance of immune homeostasis under physiological conditions, and the impaired clearance of apoptotic cells is, at least partly, responsible for the aetiology of autoimmune diseases.
Macrophages and DCs play vital roles in engulfing the apoptotic cells through intravenous injection. The apoptotic cells injected intravenously can be swiftly phagocytosed by CD169+ macrophages, which are located in the splenic marginal metallophilic zone (MZ).6 Moreover, depletion of CD169+ macrophages will cause the failure of apoptotic cell‐mediated tolerance and accelerated diseases in mouse models of systemic lupus erythematosus and experimental autoimmune encephalomyelitis.6, 7 Splenic DCs rapidly engulf the blood‐borne apoptotic cells, especially in the absence of CD169+ macrophages. In addition, our previous data indicated that CD8α + CD103+ DCs in splenic MZ were responsible for blood‐borne apoptotic cell clearance.8 It has been revealed that CD8α + DCs could induce regulatory T (Treg) cell differentiation from naive CD4+ T‐cell precursors;9, 10, 11 however, the precise mechanisms of apoptotic cell‐mediated Treg accumulation by splenic CD8α + CD103+ DCs remain elusive.
The role of Treg cells in the maintenance of immune homeostasis by apoptotic cell clearance has been extensively explored.12, 13 The presentation of apoptotic cell‐associated antigens by antigen‐presenting cells in thymus or peripheral tissues leads to T‐cell anergy or deletion of self‐reactive T cells during normal tissue turnover.14, 15, 16 It has been revealed that the expansion of Treg cells is induced after apoptotic cell exposure in a transforming growth factor‐β (TGF‐β) ‐dependent manner. Moreover, expansion of Treg cells is involved in the protective effect of donor apoptotic splenocyte administration on occurrence of acute graft‐versus‐host disease.12, 17 However, the key cellular and molecular mechanisms by which the Treg cells play roles to promote apoptotic cell‐mediated immune homeostasis must still be clarified.
CC motif chemokines have been reported to mediate the recruitment of Treg cells to establish a suppressive microenvironment. In particular, CCL22‐mediated Treg cell recruitment is CCR4‐dependent and confers a long‐term protection from autoimmune diabetes in non‐obese diabetic (NOD) mice.18, 19 CCL22/CCR4‐mediated Treg cell accumulation in tumours leads to an adverse clinical outcome. Moreover, recent findings showed decreased levels of serum CCL22 in patients with multiple sclerosis and a rapid production of CCL22 in CD169+ macrophages after systemic challenge with apoptotic cells.20, 21 All of these data strongly promote us to hypothesize that CCL22 is involved in the maintenance of immune homeostasis during apoptotic cell clearance by CD8α + CD103+ DCs.
In the present study, we mainly investigated the role played by the CCL22/CCR4 axis in the maintenance of immune homeostasis during apoptotic cell clearance by splenic CD8α + CD103+ DCs. The results revealed that systemic administration of apoptotic cells rapidly induced a significant increase in CCL22 expression and the percentage of CCR4+ Treg cells in the spleen. Further study showed that splenic CD8α + CD103+ DCs dominantly produce higher levels of CCL22 than CD8α + CD103− DCs. Meanwhile, the transient deletion of CD8α + CD103+ DCs by cytochrome C (CytC) resulted in significantly lower CCL22 production and CCR4+ Treg cell percentage decrease. Subsequently, the expression of pro‐inflammatory cytokines, such as interleukin‐17 (IL‐17) and interferon‐γ (IFN‐γ), increased due to the deletion of CD8α + CD103+ DCs. These results demonstrate that CCL22 and CCR4+ Treg cells play a critical role in the maintenance of immune homeostasis for blood‐borne apoptotic cell clearance, at least partially, by splenic CD8α + CD103+ DCs.
Materials and methods
Mice
C57BL/6J mice were originally purchased from Vital River Laboratories (Beijing, China). All mice were housed at a constant temperature (24°) in a 12‐hr dark/12‐hr light‐cycle room in Shandong University Medical School Animal Care Facility, according to institutional guidelines. All mice used in the experiments were female 8‐ to 12‐week‐old animals unless otherwise noted. All experiments using mice described herein were approved by the Animal Care and Utilization Committee of Shandong University and were performed in accordance with applicable guidelines and regulations.
Apoptotic cell preparation and cell isolation
Thymocytes were obtained from C57BL/6J mice 5–6 weeks of age. Apoptosis of thymocytes was induced by 40 J/m2 UV radiation exposure using UV cross‐linker (UVP Inc., Upland, CA) followed by incubation at 37° in RPMI‐1640 with 10% fetal calf serum for 12 hr. The UV‐radiated cells were stained by using the Annexin V‐FITC Apoptosis Detection Kit (Beyotime, Haimen, China) for detection of the percentage of apoptotic cells. The indicated numbers of apoptotic cells in 200 μl of PBS were intravenously injected into mice through the tail vein. Spleens were obtained at various time‐points after the injection and flushed with 100 mU/ml collagenase D (Roche, Basel, Switzerland), teased apart with fine forceps, and digested with 400 mU/ml collagenase D for 15 min at 37°. After digestion, splenic DCs and T cells were enriched by MACS sorting with anti‐CD11c microbeads or anti‐CD4 microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany), respectively, for the following experiments.
RNA isolation and quantitative real‐time RT‐PCR
Total RNA was extracted from control mice and apoptotic cells injected mice using an HP total RNA kit (Omega Bio‐Tek, Norcross, GA), according to the manufacturer's instructions. RNA isolation from MACS‐enriched splenic DCs, T cells and FACS‐sorted cells was performed using Trizol Reagent (Invitrogen, Carlsbad, CA). The total RNA was reverse transcribed into cDNA using the ReverTra Ace qPCR RT kit (Toyobo, Osaka, Japan) according to the manufacturer's instructions. One microlitre of cDNA was used for quantitative real‐time PCR performed with SYBR green PCR master mix (Toyobo) according to the manufacturer's instructions and specific primer pairs. The following primers were used for mouse: CCL22, CAGGCAGGTCTGGGTGAA (sense) and TAAAGGTGGCGTCGTTGG (antisense); CCL3, GGCATTCAGTTCCAGGTCAG (sense) and TCCCAGCCAGGTGTCATTT (antisense); IFN‐γ, CTGAGACAATGAACGCTACACA (sense) and TTTCTTCCACATCTATGCCACT (antisense); IL‐4, CGAGGTCACAGGAGAAGGG (sense) and CACCTTGGAAGCCCTACAGA (antisense); IL‐17, CTCCAGAAGGCCCTCAGACTAC (sense) and AGCTTTCCCTCCGCATTGACACAG (antisense); TGF‐β, GGACTCTCCACCTGCAAGAC (sense) and GACTGGCGAGCCTTAGTTTG (antisense); IL‐10, CCAAGCCTTATCGGAAATGA (sense) and TTTTCACAGGGGAGAAATCG (antisense); ROR‐γT, CTGGGATCCACTACGGGT (sense) and GCTGGTTCGGTCAATGGG (antisense); GATA‐3, CTCGGCCATTCGTACATGGAA (sense) and GGATACCTCTGCACCGTAGC (antisense); T‐bet, AGCAAGGACGGCGAATGTT (sense) and GGGTGGACATATAAGCGGTTC (antisense); Foxp3, GGCAGTTCAGGACGAGGG (sense) and GGTTCTTGTCAGAGGCA (antisense); β‐actin, TGCGTGACATCAAAGAGAAG (sense) and TCCATACCCAAGAAGGAAGG (antisense). The mRNA expression was calculated by the ▵▵C t‐method and depicted as relative expression of target genes to an endogenous reference gene actin in control groups.
Immunohistochemistry
Spleens were obtained from mice treated with or without apoptotic cell injection and frozen in Tissue Tek O.C.T. Compound (Sakura, Tokyo, Japan) at −80°. 5‐μm cryosections of the spleens were fixed in 4% paraformaldehyde and blocked with 10% normal goat sera for 45 min. Slides were incubated with FITC‐conjugated anti‐mouse‐CCL22 (Biolegend, San Diego, CA) at 4° overnight. The sections were counterstained with 4,6‐diamidino‐2‐phenylindole, mounted with FluorSave (Calbiochem, Darmstadt, Germany), and observed by fluorescence microscopy (Nikon, Tokyo, Japan) and analysed using NIS‐elements BR 3.2.
Western blotting
The proteins from splenocytes and splenic CD4+ T cells separated by MACS were extracted using cell lysis buffer (Beyotime) with PMSF (Beyotime). The protein concentration of the cell lysates was determined using a BCA Protein Assay Kit and equal amounts (15 μg) of protein were separated on 10% SDS–PAGE and then transferred onto PVDF membranes. Membranes were then blocked with 2% BSA in Tris‐buffered saline with Tween (TBST) containing 0·1% Tween‐20 for 1 hr. Membranes were probed overnight at 4° with the primary antibodies of rabbit monoclonal antibodies anti‐mouse Foxp3 (Sangon Biotech, Shanghai, China), or anti‐mouse β‐actin, followed by anti‐rabbit IgG antibodies (Sangon Biotech) conjugated with peroxidase for 1 hr at room temperature. After washing, the protein bands were visualized using the enhanced chemiluminescence method according to the manufacturer's instructions (KangWei Biotech, Beijing, China). Western blotting was performed at least three times for each sample.
Cell migration assay
The chemotactic ability of CCL22 in vitro was assessed by using a 5‐μm pore transwell system (Corning, Corning, NY). Then, 5 × 105 splenocytes or splenic CD4+ T cells from control or mice challenged with apoptotic cells were applied in the upper chambers of the transwell, and 500 μl of RPMI supplemented with or without 100–1000 pg/ml CCL22 (Peprotech, Rocky Hill, NJ) and/or anti‐CCL22 antibody was applied in the lower chambers to promote migration. After 5 hr, the migrated cells in the lower chambers were counted or stained with antibodies for CD4, CD25, FoxP3, or CCR4 for flow cytometry analysis as described below.
Flow cytometry
Splenocytes from control or apoptotic cell‐challenged mice were incubated with Fc blocker (clone 93; Biolegend) for 10 min at 4°, and then stained with antibodies for the indicated surface molecular. Anti‐CD4 (GK1.5), anti‐CD25 (3C‐7), anti‐CD8a (53‐6.7), anti‐CD11c (N418) and anti‐CCR4 (2G‐12), antibodies were purchased from Biolegend, anti‐CD11b (M1/70), anti‐CD103 (M290) were obtained from BD Biosciences (San Jose, CA). Intracellular staining for anti‐Foxp3 (MF‐14; Biolegend) was performed according to the manufacturer's instructions. Cells were acquired by FACS Aria 3 (BD Biosciences, San Jose, CA) and analysed by flowjo software version 8·8·7 (Tree Star, Ashland, OR).
To perform gene expression of CCL22 and CCL3in CD8α + CD103+ DCs, CD8α + CD103− DCs and CD8α − CD103− DCs, splenic DCs were enriched from C57BL/6J mice, stained with fluorescence labels for CD8α and CD103 antibodies and sorted by FACSAria3 (BD Biosciences). Quantitative PCR analysis was performed as described above.
ELISA
For analysis of CCL22 production, the splenocytes, MACS‐isolated splenic DCs and T cells from control mice and mice challenged with apoptotic cells or live cells were cultured in RPMI‐1640 with 10% fetal calf serum for 24 hr. The supernatants were harvested and used for detection of CCL22 production. The concentrations were measured by ELISA (Peprotech) according to the manufacturer's protocols.
Statistical analysis
Paired, two‐tailed Student t‐test was performed by using GraphPad Prism 5.0 software (GraphPad software, San Diego, CA) in all experiments. Data are presented as the mean ± SEM. A P‐value < 0·05 was considered significant.
Results
CCL22 expression of splenic DCs increased in mice receiving apoptotic cell injection
It has been reported that CCL22 plays an important role in Treg cell recruitment to protect against autoimmune diseases, such as NOD and multiple sclerosis.18, 19 Therefore, we first examined whether CCL22 levels changed after systemic administration of apoptotic cells using our system (Fig. 1a; see Supplementary material, Fig. S1). The results showed that mRNA expression of CCL22 in the spleen increased rapidly 6 hr after apoptotic thymocyte injection, which is supported by other similar results.21 Moreover, a 24‐fold increase of CCL22 mRNA was detected 12 hr after apoptotic cell injection, whereas apoptotic cells did not induce significant expression of CCL3 (Fig. 1a) a chemokine involved in activation and recruitment of lymphocytes during acute inflammation.22 In addition, we also detected CCL22 expression using immunofluorescence analysis with CCL22 antibody on the spleen frozen sections. The secretion of CCL22 in the spleen challenged with apoptotic cells was higher than that of controls (Fig. 1b).
Figure 1.
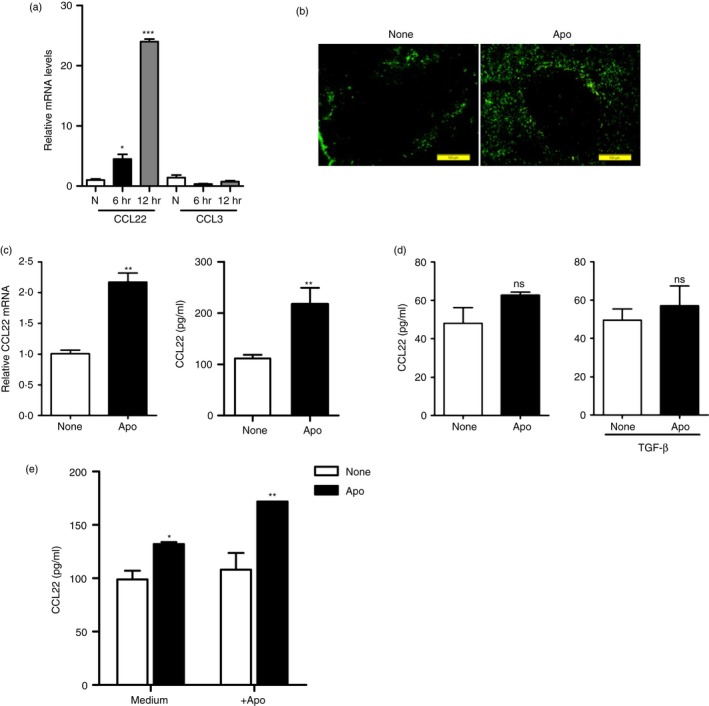
CCL22 secretion by splenic dendritic cells (DCs) increased in mice receiving injection of apoptotic cells. (a) C57BL/6J mice were randomly separated into three groups and intravenously injected PBS only (N) or with 1 × 107 apoptotic thymocytes. Total RNAs were isolated from splenocytes of control or apoptotic‐cell‐challenged mice 6 and 12 hr later. The mRNA expression levels of CCL22 and CCL3 at indicated times were detected using quantitative PCR analysis. *P < 0·05, ***P < 0·001 compared with control. (b) Immunohistochemistry analysis of CCL22 expression in spleen from control (None) and apoptotic‐cell‐challenged mice 12 hr later (Apo). Green fluorescence indicates CCL22‐positive signals. (c) Splenic CD11c+ DCs enriched from mice treated as described in (a) were enriched by MACS 12 hr later, and used for examining mRNA expression levels of CCL22 directly or cultured in vitro for CCL22 production detection using ELISA. **P < 0·01 compared with control. (d) Splenic CD4+ T cells enriched from mice treated as described in (a) were incubated with or without 5 ng/ml transforming growth factor‐β (TGF‐β) for 24 hr, and CCL22 production in supernatant was measured by ELISA. NS, no significant change. (e) Splenic CD11c+ cells enriched from mice with or without apoptotic cell injection as described in (a) were co‐cultured with or without apoptotic cells for 24 hr, and CCL22 production in supernatant was measured by ELISA. *P < 0·05, **P < 0·01 compared with control, respectively.
Splenic DCs have been reported to be a major source of macrophage‐derived chemokine CCL2223, and are responsible for phagocytosis of apoptotic cells.24, 25 For this reason, we next examined whether splenic DCs from apoptotic‐cell‐injected mice could secrete higher levels of CCL22 than naive DCs. Splenic CD11c+ DCs were isolated from apoptotic‐cell‐injected mice, and the mRNAs and culture supernatants were used to detect CCL22 expression. Quantitative PCR analysis showed that CCL22 mRNA in splenic DCs increased significantly due to apoptotic cell exposure than naive DCs (Fig. 1c). Also, the CCL22 protein in the supernatants of splenic DCs from apoptotic‐cell‐challenged mice was higher than in controls. However, the secretion of CCL22 by splenic CD4+ T cells from untreated mice was low and there was no significant change upon apoptotic cell administration even when stimulated with TGF‐β (Fig. 1d). Splenic DCs did not secrete higher CCL22 protein after live cell challenge (see Supplementary material, Fig. S2). To show the direct link between apoptotic cell phagocytosis, the co‐culture experiment of CD11c+ DCs and apoptotic cells was performed. The result showed (Fig. 1e) that the CD11c+ DCs from mice without apoptotic cell injection did not produce higher CCL22 levels when co‐cultured with apoptotic cells in vitro, but the CD11c+ DCs from mice receiving apoptotic cell injection produced higher CCL22 levels when co‐cultured with apoptotic cells. The result suggested that the microenvironment in mice receiving apoptotic cells is necessary for production of CCL22 after apoptotic cell injection. Taken together, it is indicated that splenic DCs produced higher levels of CCL22 in mice receiving apoptotic cell injection.
Splenic CD8α + CD103+ DCs dominantly secrete CCL22
Our previous data has shown that splenic CD8α + CD103+ DCs dominantly phagocytose blood‐borne apoptotic cells and induce immune tolerance to apoptotic cell‐associated antigens.8 Then, we detected the levels of CCL22 by splenic DCs with selective deletion of CD8α + CD103+ DCs by use of intravenous CytC injection. It has been reported that splenic CD8α + CD103+ DCs would be transiently deleted by use of CytC injection,8, 26 and our results (Fig. 2a; see Supplementary material, Fig. S3) also showed that CytC injection caused selective deletion of splenic CD8α + CD103+ DCs. Meanwhile, CCL22 production by total CD11c+ DCs was severely limited by the deletion of CD8α + CD103+ DCs in the spleen (Fig. 2b). In addition, the secretion of CCL22 did not increase in splenic DCs after CD8α + CD103+ DC deletion, even with apoptotic cell exposure (Fig. 2c). Similar limitation of CCL22 secretion was also observed using immunofluorescence histochemical analysis (Fig. 2d). Taken together, the results indicated that splenic CD8α + CD103+ DCs are important producers of CCL22 in the process of blood‐borne apoptotic cell clearance.
Figure 2.
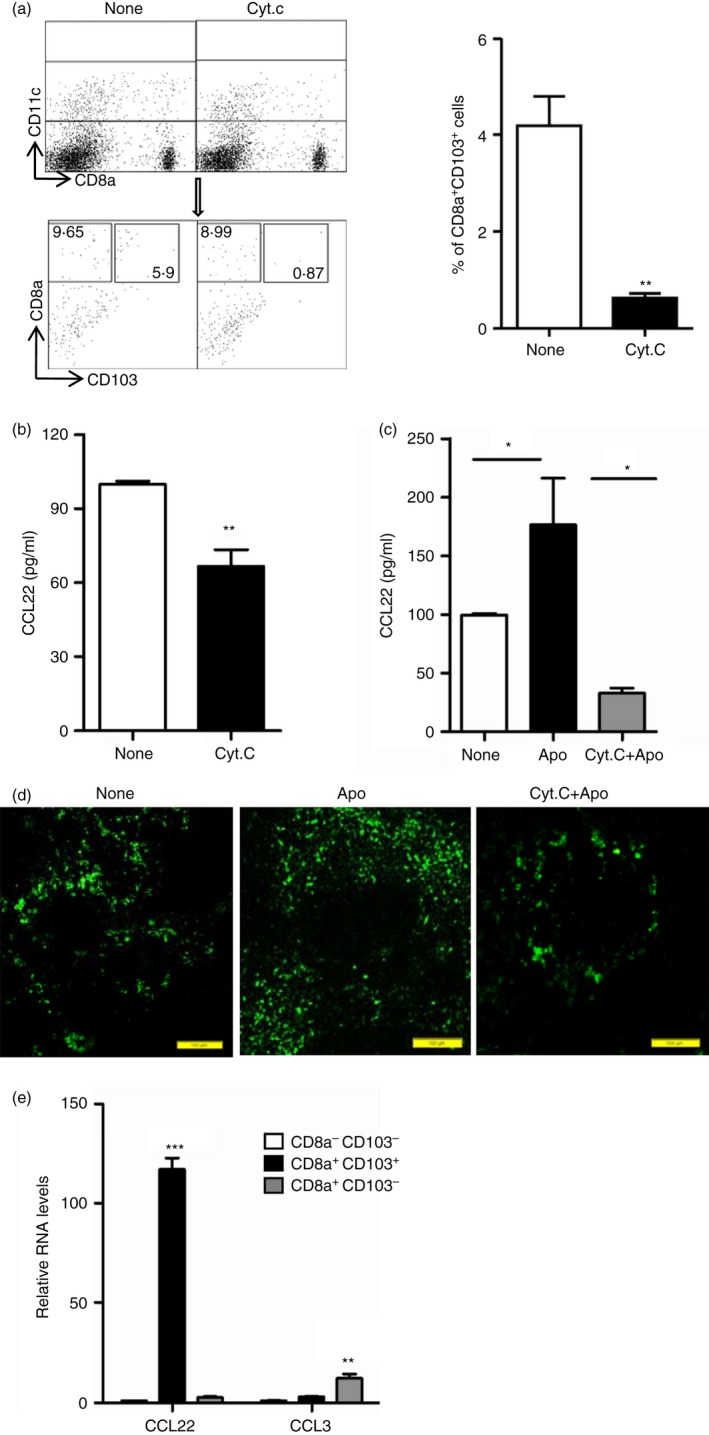
Splenic CD8α + CD103+ dendritic cells (DCs) dominantly secrete CCL22. (a) C57BL/6J mice were injected intravenously with 5 mg of Cytochrome c (CytC) or not (None). Twenty‐four hours later, splenocytes were isolated and stained with antibodies for CD11c, CD8α and CD103 for flow cytometry analysis, and the percentages of CD8α + CD103+ DCs in CD11c+ gated cells were compared. (b) Splenic CD11c+ DCs enriched from mice treated as described in (a) by MACS,were cultured in vitro for CCL22 production detection using ELISA. (c, d) Cytochrome c was injected intravenously into mice. Twenty‐four hours later, the mice were injected intravenously with 1 × 107 apoptotic thymocytes. Twelve hours later, splenic CD11c+ DCs were enriched by MACS from control (None), apoptotic cell injection only (Apo) and CytC with apoptotic‐cell‐injected (CytC+Apo) mice and cultured for CCL22 production detection using ELISA (c) and spleens from these three groups of mice were used for immunohistochemistry analysis of CCL22 expression (d). (e) Splenocytes from control mice (None) and apoptotic‐cell‐challenged mice (Apo) were stained with antibodies for CD11c, CD8α and CD103, and CD8α − CD103− DCs, CD8α + CD103− DCs and CD8α + CD103+ DCs in CD11c+ gated cells were sorted and used for examining the mRNA expression level of CCL22 and CCL3. *P < 0·05, **P < 0·01,***P < 0·001 compared with control, respectively.
To confirm whether CD8α + CD103+ DCs are the dominant DC subset to secrete CCL22 in the spleen, we further compared CCL22 expression in FACS‐sorted cell subsets: CD8α + CD103+ DCs, CD8α + CD103− DCs and CD8α − CD103− DCs by using FACSAria 3. It was shown that relatively larger amounts of CCL22 were expressed by CD8α + CD103+ DCs than by CD8α + CD103− DCs and CD8α + CD103− DCs. However, CD8α + CD103+ DCs express lower levels of CCL3 than CD8α + CD103− DCs (Fig. 2e). The results clearly demonstrated that CD8α + CD103+ DCs were the dominant source of CCL22 in the splenic DCs. Moreover, further analysis of the microarray data published in 2009 (see Supplementary material, Fig. S4; ref. 8) showed that CD8α + CD103+ DCs produced a higher CCL22 mRNA level than CD8α + CD103+ DCs. Therefore, these data indicated that apoptotic cell engulfment by CD8α + CD103+ DCs is directly responsible for CCL22 production.
Systemic administration of apoptotic cells induced a rapid increase in CCR4+ Treg cells
To understand whether up‐regulated CCL22 is related to establishment of the immunosuppressive microenvironment by intravenous apoptotic cell injection, we next confirmed the immunosuppressive environment established by systemic apoptotic cell administration. The results showed that the percentage of CD4+ CD25hi cells in CD4+ T cells or CD4+ FoxP3+ cells increased due to apoptotic cell challenge (Fig. 3a,b; see Supplementary material, Fig. S5). Both mRNA and protein expression of FoxP3 in splenic CD4+ T cells dramatically increased due to apoptotic cell exposure (Fig. 3b,c). Meanwhile, apoptotic cell injection resulted in fivefold and ninefold increases in the expression of suppressive factors IL‐10 and TGF‐β compared with baseline levels (Fig. 3c). The data indicated that the systemic administration of apoptotic cells induced a rapid increase in the percentage of Treg cells in the spleen.
Figure 3.
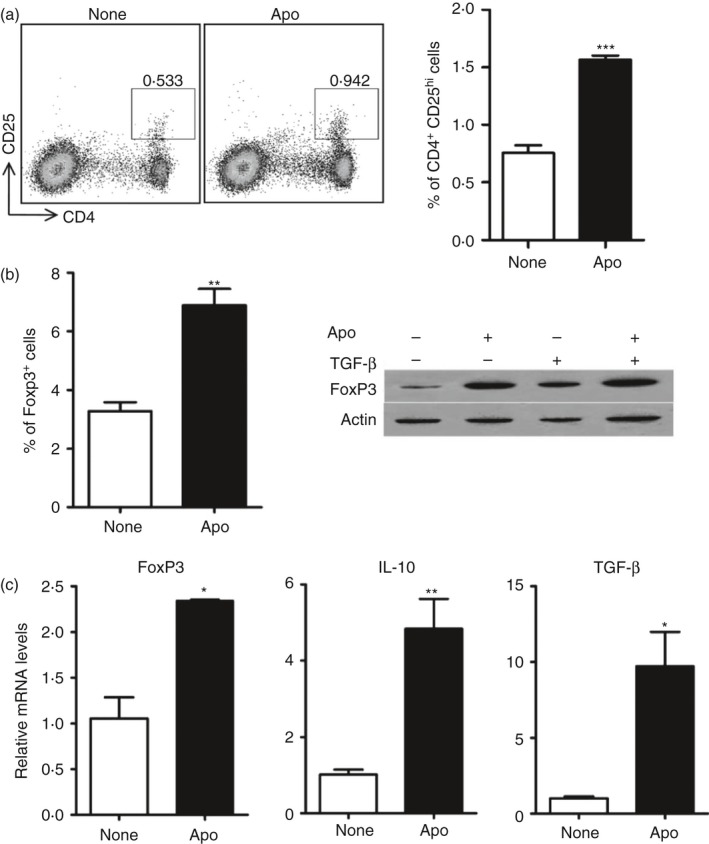
Systemic administration of apoptotic cells induced rapid increase of regulatory T (Treg) cell percentage in spleen. (a) C57BL/6J mice were intravenously injected with PBS only (None) or with 1 × 107 apoptotic cells (Apo). After 12 hr, splenocytes from control mice (None) and apoptotic‐cell‐challenged mice (Apo) were stained with antibodies for CD4 and CD25 and the percentage of CD4+ CD25hi was analysed by FACS. (b) Splenic CD4+ T cells from control and apoptotic‐cell‐challenged mice were used for detection of the percentage of the FoxP3+ cells in CD4+ T cells and compared using FACS analysis (left panel). A dot plot figure is shown in the Supplementary material (Fig. S2). The FoxP3 protein expression in splenic CD4+ T cells was also detected directly or after further culture in vitro in CD3‐bound plates with or without transforming growth factor‐β (TGF‐β) using western blotting analysis (right panel). (c) Splenic CD4+ T cells were enriched by MACS and used for examining mRNA expression levels of TGF‐β, FoxP3 and interleukin‐10 (IL‐10).*P < 0·05, **P < 0·01, ***P < 0.001 compared to control respectively.
It has been shown that Treg cells are accumulated by the CCL22/CCR4 axis.27 We hypothesized that the increased CCL22 was essential for CCR4+ Treg cell accumulation, because CD4+ FoxP3+ T cells increased after apoptotic cell injection (Fig. 3a). Hence, we detected CCR4 expression in different cell subsets in the spleen. The data showed that the percentage of CD4+ CCR4+ T cells increased significantly with intravenous apoptotic cell injection, whereas CD8+ CCR4+ T cells, CD11c+ CCR4+ DCs, and CD11b+ CCR4+ macrophages did not show significant changes (Fig. 4a). Moreover, the percentage of CCR4+ CD4+ CD25hi cells increased significantly in apoptotic‐cell‐challenged spleens compared with controls (Fig. 4b; see Supplementary material, Fig. S6).
Figure 4.
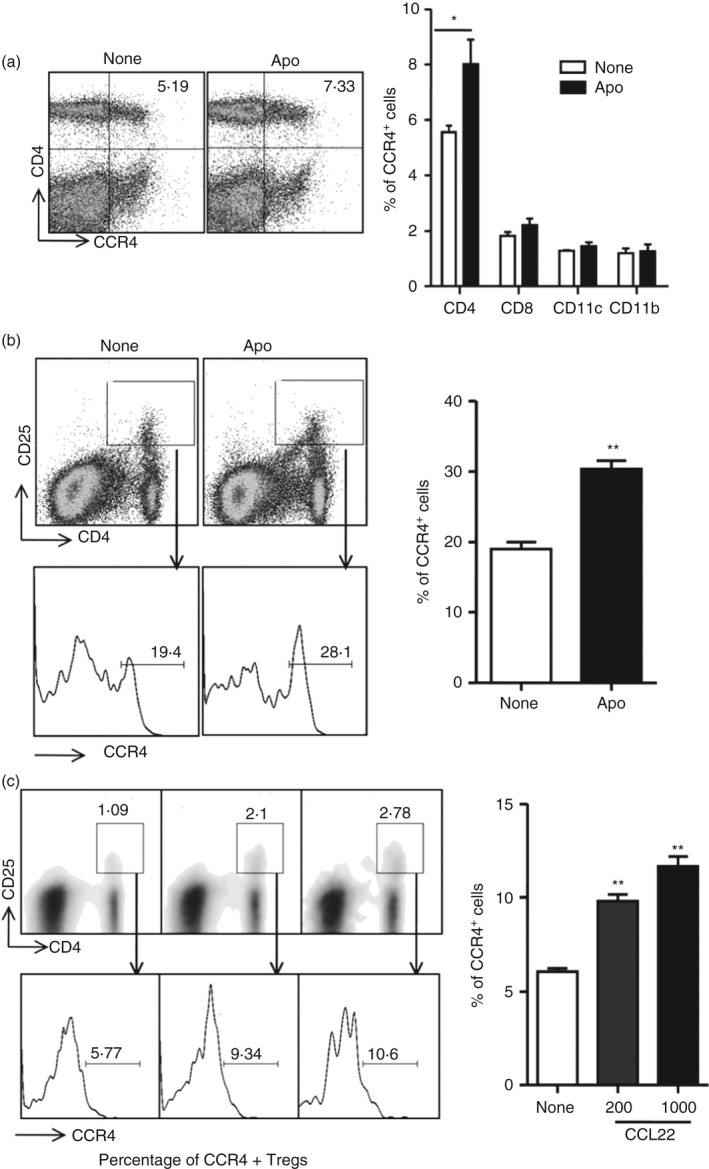
Rapid CCR4+ regulatory T (Treg) cell accumulation in spleen in response to apoptotic cell injection. (a) Splenocytes from control (None) and apoptotic‐cell‐challenged mice (Apo) were stained with antibodies for CD4, CD8, CD11c, CD11b together with antibody for CCR4, respectively, and the percentages of CD4+ CCR4+ cells, CD8+ CCR4+ cells, CD11c+ CCR4+ cells and CD11b+ CCR4+ cells were analysed, respectively, by FACS. (b) Splenocytes from control (None) and apoptotic‐cell‐challenged (Apo) mice were also stained with antibodies for CD4, CD25 and CCR4 for flow cytometry analysis, and the percentage of CCR4 in CD4+ CD25hi were compared. (c) Splenocytes from apoptotic‐cell‐challenged mice were applied in the upper chamber of the transwell, supplemented or not supplemented with CCL22. After 3 hr, the migrated cells in the lower chamber were stained with antibodies for CD4, CD25 and CCR4 for flow cytometry analysis, and the percentages of CCR4 in CD4+ CD25hi were compared. *P < 0·05, **P < 0·01 compared with control, respectively.
To investigate whether the increased CCL22 induces Treg cell migration, we also performed cell migration assays ex vivo. First, we demonstrated that CCL22 indeed regulated splenocyte migration in a CCL22 dose‐dependent manner and many more splenocytes challenged by apoptotic cells migrated than those in controls (data not shown). We next detected whether CCL22 could recruit CCR4+ Treg cells from the apoptotic‐cell‐challenged spleens. The results in Fig. 4(c) revealed that apoptotic cell‐driven CCR4+ Treg cells migrated in the presence of CCL22, and the migration was dose‐dependent. Moreover, it has been showed that the CCL22‐dependent migration of CCR4+ Treg cells could be blocked by using anti‐CCL22 antibody to neutralize CCL22 (see Supplementary material, Fig. S7). Taken together, these findings strongly indicated that the systemic administration of apoptotic cells induced a rapid increase in CCR4+ Treg cells, and CCL22 was an important factor involved in CCR4+ Treg cell accumulation in apoptotic cell‐challenged spleens.
Failure of CCR4+ Treg cells increased by apoptotic cell challenge in the spleen with deletion of CD8α + CD103+ DCs
Apoptotic cell clearance by phagocytes is essential for the maintenance of immune homeostasis under physiological conditions. Hence, we next investigated whether the impaired CCL22 secretion by splenic DCs in the absence of CD8α + CD103+ DCs (Fig. 2c,d) resulted in failure of maintenance of immune homeostasis. FACS analysis showed the percentage of CD4+ CD25hi increased 12 hr after apoptotic cell injection, which was not detected in the absence of CD8α + CD103+ DCs (Fig. 5a). In addition, the injection of CytC did not affect the percentage of CD4+ FoxP3+ in the spleen (see Supplementary material, Fig. S3). We also detected the FoxP3 protein in CD4+ T cells by Western blotting analysis and the results showed that FoxP3 levels were up‐regulated by apoptotic cell administration. However, the increase of FoxP3 expression was not observed in the absence of CD8α + CD103+ DCs (Fig. 5b).
Figure 5.
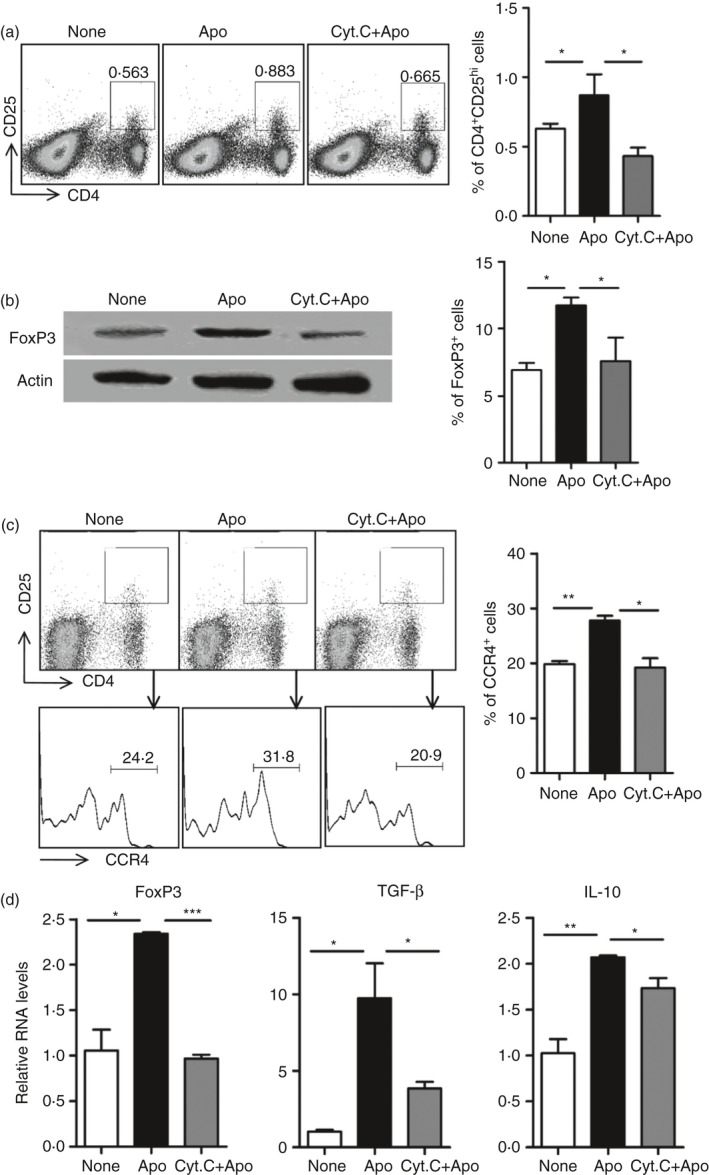
Failure to increase CCR4+ regulatory T (Treg) cell percentage by apoptotic cell challenge in spleen with deletion of CD8α + CD103+ dendritic cells (DCs). (a) Splenocytes from mice described as in Fig. 2(c) were stained with antibodies for CD4 and CD25 and the percentage of CD4+ CD25hi were analysed by FACS. (b) Splenic CD4+ T cells enriched from mice described as in Fig. 2(c) were used for detection of FoxP3 protein expression using Western blotting analysis detection directly (left panel). Percentage of the FoxP3+ cells in CD4+ T cells was also compared using FACS analysis (right panel). (c) Splenocytes were further stained with antibodies for CD4, CD25 and CCR4, and the percentage of CCR4 in CD4+ CD25hi was analysed by FACS. (d) Splenic CD4+ T cells enriched from mice described as in Fig. 2(c) were investigated using quantitative PCR analysis of mRNA expression levels of FoxP3, transforming growth factor‐β (TGF‐β) and interleukin‐10 (IL‐10). *P < 0·05, **P < 0·01, ***P < 0·001 compared with control, respectively.
Furthermore, the additional study using FACS showed that the percentage of CCR4+ Treg cells in the spleen increased upon apoptotic cell injection, but this was not observed in the absence of CD8α + CD103+ DCs (Fig. 5c; see Supplementary material, Fig. S8). The mRNA expressions of FoxP3, TGF‐β and IL‐10 in CD4+ T cells were also detected, and their expressions were much higher in the apoptotic‐cell‐challenged CD4+ T cells than in controls, and they were down‐regulated in the absence of CD8α + CD103+ DCs (Fig. 5d). All the data indicated that the absence of CD8α + CD103+ DCs resulted in a failure of CCR4+ Treg cells increased by apoptotic cell administration in the spleen.
Higher pro‐inflammatory cytokine response to apoptotic cell administration in the absence of CD8α + CD103+ DCs
Impaired apoptotic cell clearance would cause activation of self‐reactive immune cells in some specific gene‐deficient mouse models. Hence, we detected the mRNA expression of the pro‐inflammatory cytokines IFN‐γ, IL‐4, IL‐17 in CD4+ T cells to investigate the T‐cell response to apoptotic cell administration in the spleen with the absence of CD8α + CD103+ DCs. All of these cytokines decreased dramatically upon apoptotic cell injection; however, they were up‐regulated in the absence of CD8α + CD103+ DCs (Fig. 6a). We further detected the expression of the transcription factors T‐bet, GATA‐3 and ROR‐γT mediating differentiation of T helper types 1, 2 and 17, respectively. The results showed that the expression of ROR‐γT was down‐regulated in the environment mediated by apoptotic cell administration, and it was up‐regulated in the absence of CD8α + CD103+ DCs (Fig. 6b). In particular, the expressions of GATA‐3 and ROR‐γT in CD4+ T cells isolated from the spleen with the absence of CD8α + CD103+ DCs were much higher than those in apoptotic‐cell‐challenged CD4+ T cells, although no significant changes detected in T‐bet. Taken together, the maintenance of immune homeostasis mediated by apoptotic cell clearance would be severely destroyed, at least partially resulted from the failure of CCR4+ Treg cell induction in apoptotic‐cell‐challenged spleens in the absence of CD8α + CD103+ DCs.
Figure 6.
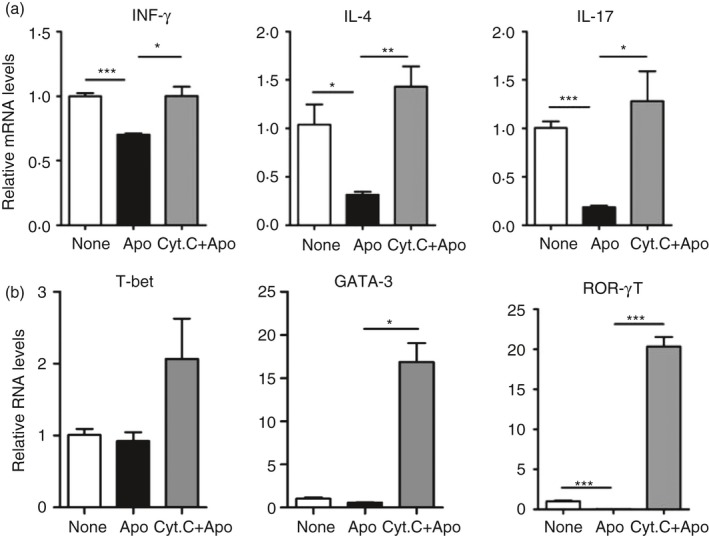
Higher pro‐inflammatory cytokine response to apoptotic cell administration in the absence of CD8α + CD103+ dendritic cells (DCs). Splenic CD4+ T cells enriched from mice described as in Fig. 2(c) were performed using quantitative PCR analysis of mRNA expression levels of interferon‐γ (IFN‐γ), interleukin‐17 (IL‐17) and IL‐4 (a) and T‐Bet,GATA‐3 and ROR‐γT (b). *P < 0·05, **P < 0·01, ***P < 0·001 compared with control, respectively.
Discussion
Several previous studies have reported that phagocytes such as macrophages and DCs play important roles in the maintenance of immune homeostasis mediated by apoptotic cell clearance to prevent autoimmune diseases.4, 6, 13 Most studies have revealed some mechanisms focusing on the steps of the engulfment and clearance of apoptotic cells by phagocytes. However, more evidence is still needed to clarify how these phagocytes, especially the DC subset, that are responsible for apoptotic cell clearance mediate immune homeostasis by engulfing apoptotic cells.28, 29, 30, 31 The current results reported some new findings about how splenic CD8α + CD103+ DCs, a subset responsible for apoptotic cell clearance, maintain immune homeostasis by engulfing apoptotic cells. First, the levels of CCL22 and the percentage of CCR4+ Treg cells increased rapidly in the spleen upon systemic apoptotic cell administration. Second, selective depletion of CD8α + CD103+ DCs caused the failure of the increases in CCL22 levels and CCR4+ Treg percentage in response to apoptotic cell administration. Moreover, the expression of pro‐inflammatory cytokines, such as IL‐17 and IFN‐γ in splenic CD4+ T cells increased significantly when challenged by apoptotic cells with the deletion of CD8α + CD103+ DCs.
CCL22, a member of the CC chemokine families, is mainly produced by monocyte‐derived macrophages and DCs upon stimulation with microbial products.32, 33 A recent study has reported that the mean serum levels of CCL22 in newly diagnosed patients with multiple sclerosis were significantly lower than those in treated patients with multiple sclerosis and healthy people,20 suggesting that CCL22 serves as a biomarker for autoimmune diseases. However, the roles of CCL22 in the regulation of immune homeostasis by apoptotic cell clearance are poorly defined, although impairment of apoptotic cell phagocytosis causes autoimmune diseases.34, 35 Here, we first examined how CCL22 increased rapidly in response to apoptotic cell administration, which was limited by deletion of CD8α + CD103+ DCs. Also, the CD8α + CD103+ DCs dominantly expressed relatively higher levels of CCL22 than other splenic DC subsets. Splenic CD8α + CD103+ DCs were a critical subset responsible for tolerance induction by apoptotic cell clearance, which has been clearly reported by Tanaka's team.8 CD8α + CD103+ DCs are a subpopulation of splenic DCs and mainly locate in the splenic MZ, an area capable of constantly screening blood for foreign particles and organisms.8, 36 It is suggested that CCL22 would be a critical factor for splenic CD8α + CD103+ DCs to mediate immune homeostasis by apoptotic cell clearance.
It has been revealed that CCL22 is an important chemotactic factor for attracting Treg cells expressing high levels of the chemokine receptor CCR4.37 Injection of double‐stranded adeno‐associated virus encoding CCL22 recruits endogenous Treg cells to the islets and protects NOD mice against autoimmune diabetes.19 Similarly, our data also showed a rapid increase of CCR4+ Treg cells in response to apoptotic cell administration together with the increased level of CCL22. However, the increase of CCR4+ Treg cells was not observed in the absence of CD8α + CD103+ DCs. Moreover, the selective deletion of CD8α + CD103+ DCs resulted in the expression of pro‐inflammatory cytokines, such as IL‐17 and IFN‐γ, increased significantly, which may be caused at least partially by impairment of apoptotic cell clearance. The data suggested that the CCL22/CCR4 axis contributed to immune homeostasis regulated by CD8α + CD103+ DCs engulfing apoptotic cells. In‐vivo‐generated antigen‐specific Treg cells has been shown to have therapeutic effects on experimental autoimmune encephalomyelitis and NOD in mice, which attracts much attention for its possible clinical applications to treat autoimmune diseases.38 Further study on the mechanism how the CCL22/CCR4 axis is involved in the pathogenesis and treatment of autoimmune diseases should be carried out.
CD103+ DCs in several tissues, especially in intestine, lung, pancreas and mesenteric lymph nodes, have been reported to induce peripheral tolerance.11, 39, 40, 41 Moreover, recent research showed that human peripheral blood mononuclear cell‐derived CD103+ DCs and retinoic‐acid‐primed CD103+ DCs also have the ability to promote the differentiation of Treg cells.42, 43 Taken together, peripheral CD103+ DCs are the critical regulators of immune homeostasis. In addition, CCL22 may be used as a candidate to study the molecular mechanisms involved in tolerance induction by peripheral CD103+ DCs.
Intravenously injected apoptotic cells are initially accumulated in the MZ. CD8α + CD103+ DCs in MZ form a critical subset together with CD169+ macrophages for the uptake of circulating apoptotic‐cell‐associated antigens. The increase of CCL22 levels and Treg cell percentage in response to apoptotic cell administration strongly supported how the CCL22/CCR4 axis plays a critical role in the maintenance of the immunosuppressive microenvironment.21 Mazzini et al. reported that CX3CR1+ macrophages captured soluble food antigens from the gut lumen and transferred them to CD103+ DCs to induce oral tolerance, suggesting that intestinal macrophages and DCs close the gap on tolerance.44, 45 From this point of view, our present data also supported the possibility that there is similar gap junction transfer of apoptotic‐cell‐associated antigens between CD8α + CD103+ DCs and CD169+ macrophages, inducing a suppressive microenvironment in the spleen, because these two subsets are the main phagocytes in the spleen responsible for clearance of blood‐borne apoptotic cells.
In summary, our results suggested that the CCL22/CCR4 axis is a critical regulator for the maintenance of immune homeostasis in apoptotic cell phagocytosis under physiological conditions by splenic CD8α + CD103+ DCs. Systemic injection of apoptotic cells induced a subsequent increase in CCL22 expression and the splenic CCR4+ Treg cell percentage, which were not observed due to the selective depletion of CD8α + CD103+ DCs in the spleen. Considering CCL22 is involved in some autoimmune diseases, this report will provide important clues for clinical medicines. Therefore, further study is needed for understanding the roles of CD8α + CD103+ DCs–CCL22 axis in autoimmune diseases.
Authors' contributions
We would like to thank Prof. Lifen Gao and Zhiyan Liu at Shandong University for immunohistochemistry technique support and Ms Limei Wang at Shandong University for cell sorting. HS, HX, WD, YY and LQ performed experiments. HS, HX, LX analysed data, and QC, HS and HX wrote the paper.
Disclosures
All authors declare that no conflicts of interest.
Supporting information
Figure S1. Analysis of apoptotic thymocytes upon UV exposure.
Figure S2. CCL22 levels did not increase in CD11c+ dendritic cell from mice receiving live cells injection.
Figure S3. Analysis of percentage of CD4+, CD8+, CD11c+, CD11b+ cells and regulatory T cells in the spleen due to Cytochrome C injection.
Figure S4. Analysis of the microarray data published on JI, 2009 by Qiu et al.
Figure S5. Analysis of FoxP3+ cells in CD4+ T cells upon apoptotic cell injection.
Figure S6. Analysis of percentage of CD4+ CD25hi cells due to apoptotic cell administration.
Figure S7. CCL22 drives the migration of regulatory T cells.
Figure S8. Analysis of FoxP3+ cells in CD4+ T cells in absence of CD8a+ CD103+ upon apoptotic cells injection.
Acknowledgements
The present work was supported by the project grant from The National Natural Science Foundation of China (81202306); China Postdoctoral Science Foundation (201252M1343, 2013T60674) and Shandong Postdoctoral Science Foundation (201201005).
References
- 1. Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell 2010; 140:619–30. [DOI] [PubMed] [Google Scholar]
- 2. Ravichandran KS. Beginnings of a good apoptotic meal: the find‐me and eat‐me signaling pathways. Immunity 2011; 35:445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ramirez‐Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ et al The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol 2013; 14:917–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Miyanishi M, Segawa K, Nagata S. Synergistic effect of Tim4 and MFG‐E8 null mutations on the development of autoimmunity. Int Immunol 2012; 24:551–9. [DOI] [PubMed] [Google Scholar]
- 5. Hanayama R, Miyasaka K, Nakaya M, Nagata S. MFG‐E8‐dependent clearance of apoptotic cells, and autoimmunity caused by its failure. Curr Dir Autoimmun 2006; 9:162–72. [DOI] [PubMed] [Google Scholar]
- 6. Miyake Y, Asano K, Kaise H, Uemura M, Nakayama M, Tanaka M. Critical role of macrophages in the marginal zone in the suppression of immune responses to apoptotic cell‐associated antigens. J Clin Invest 2007; 117:2268–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McGaha TL, Chen Y, Ravishankar B, van Rooijen N, Karlsson MC. Marginal zone macrophages suppress innate and adaptive immunity to apoptotic cells in the spleen. Blood 2011; 117:5403–12. [DOI] [PubMed] [Google Scholar]
- 8. Qiu CH, Miyake Y, Kaise H, Kitamura H, Ohara O, Tanaka M. Novel subset of CD8α + dendritic cells localized in the marginal zone is responsible for tolerance to cell‐associated antigens. J Immunol 2009; 182:4127–36. [DOI] [PubMed] [Google Scholar]
- 9. Liu K, Iyoda T, Saternus M, Kimura Y, Inaba K, Steinman RM. Immune tolerance after delivery of dying cells to dendritic cells in situ . J Exp Med 2002; 196:1091–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dudziak D, Kamphorst AO, Heidkamp GF, Buchholz VR, Trumpfheller C, Yamazaki S et al Differential antigen processing by dendritic cell subsets in vivo . Science 2007; 315:107–11. [DOI] [PubMed] [Google Scholar]
- 11. Desch AN, Randolph GJ, Murphy K, Gautier EL, Kedl RM, Lahoud MH et al CD103+ pulmonary dendritic cells preferentially acquire and present apoptotic cell‐associated antigen. J Exp Med 2011; 208:1789–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kleinclauss F, Perruche S, Masson E, de Carvalho Bittencourt M, Biichle S, Remy‐Martin JP et al Intravenous apoptotic spleen cell infusion induces a TGF‐β‐dependent regulatory T‐cell expansion. Cell Death Differ 2006; 13:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol 2014; 14:166–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T cell repertoire. Nature 1999; 402:255–62. [DOI] [PubMed] [Google Scholar]
- 15. Kyewski B, Klein L. A central role for central tolerance. Annu Rev Immunol 2006; 24:571–606. [DOI] [PubMed] [Google Scholar]
- 16. Kurts C. Cross‐presentation: inducing CD8 T cell immunity and tolerance. J Mol Med 2000; 78:326–32. [DOI] [PubMed] [Google Scholar]
- 17. Mevorach D, Zuckerman T, Reiner I, Shimoni A, Samuel S, Nagler A et al Single infusion of donor mononuclear early apoptotic cells as prophylaxis for graft‐versus‐host disease in myeloablative HLA‐matched allogeneic bone marrow transplantation: a phase I/IIa clinical trial. Biol Blood Marrow Transplant 2014; 20:58–65. [DOI] [PubMed] [Google Scholar]
- 18. Lee I, Wang L, Wells AD, Dorf ME, Ozkaynak E, Hancock WW. Recruitment of Foxp3+ T regulatory cells mediating allograft tolerance depends on the CCR4 chemokine receptor. J Exp Med 2005; 201:1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Montane J, Bischoff L, Soukhatcheva G, Dai DL, Hardenberg G, Levings MK et al Prevention of murine autoimmune diabetes byCCL22‐mediated Tregs recruitment to the pancreatic islets. J Clin Invest 2011; 121:3024–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jafarzadeh A, Ebrahimi HA, Bagherzadeh S, Zarkesh F, Iranmanesh F, Najafzadeh A et al Lower serum levels of Th2‐related chemokine CCL22 in women patients with multiple sclerosis: a comparison between patients and healthy women. Inflammation 2014; 37:604–10. [DOI] [PubMed] [Google Scholar]
- 21. Ravishankar B, Shinde R, Liu H, Chaudhary K, Bradley J, Lemos HP et al Marginal zone CD169+ macrophages coordinate apoptotic cell‐driven cellular recruitment and tolerance. Proc Natl Acad Sci USA 2014; 111:4215–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Heinrichs D, Berres M, Nellen A, Fischer P, Scholten D, Trautwein C et al The chemokine CCL3 promotes experimental liver fibrosis in mice. PLoS ONE 2013; 8:e66106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vulcano MAC, Stoppacciaro A, Bagnati R, D'Amico G, Struyf S, Transidico P et al Dendritic cells as a major source of macrophage‐derived chemokine/CCL22 in vitro and in vivo . Eur J Immunol 2001; 31:812–22. [DOI] [PubMed] [Google Scholar]
- 24. Iyoda T, Shimoyama S, Liu K, Omatsu Y, Akiyama Y, Maeda Y et al The CD8+ dendritic cell subset selectively endocytoses dying cells in culture and in vivo . J Exp Med 2002; 195:1289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakayama M, Akiba H, Takeda K, Kojima Y, Hashiguchi M, Azuma M et al Tim‐3 mediates phagocytosis of apoptotic cells and cross‐presentation. Blood 2009; 113:3821–30. [DOI] [PubMed] [Google Scholar]
- 26. Lin ML, Zhan Y, Proietto AI, Prato S, Wu L, Heath WR et al Selective suicide of cross‐presenting CD8+ dendritic cells by cytochrome c injection shows functional heterogeneity within this subset. Proc Natl Acad Sci USA 2008; 105:3029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Iellem A, Mariani M, Lang R, Recalde H, Panina‐Bordignon P, Sinigaglia F et al Unique chemotactic response profile and specific expression of chemokine receptors CCR4 and CCR8 by CD4+CD25+ regulatory T cells. J Exp Med 2001; 194:847–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S. Caspase‐mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 2014; 344:1164–8. [DOI] [PubMed] [Google Scholar]
- 29. Murakami Y, Tian L, Voss OH, Margulies DH, Krzewski K, Coligan JE. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ 2014; 21:1746–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tyurin VA, Balasubramanian K, Winnica D, Tyurina YY, Vikulina AS, He RR et al Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat‐me’ signals: cleavage and inhibition of phagocytosis by Lp‐PLA2. Cell Death Differ 2014; 21:825–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prabagar MG, Do Y, Ryu S, Park JY, Choi HJ, Choi WS et al SIGN‐R1, a C‐type lectin, enhances apoptotic cell clearance through the complement deposition pathway by interacting with C1q in the spleen. Cell Death Differ 2013; 20:535–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yamashita U, Kuroda E. Regulation of macrophage‐derived chemokine (MDC, CCL22) production. Crit Rev Immunol 2002; 22:105–14. [PubMed] [Google Scholar]
- 33. Layseca‐Espinosa E, Korniotis S, Montandon R, Gras C, Bouillié M, Gonzalez‐Amaro R et al CCL22‐producing CD8α‐ myeloid dendritic cells mediate regulatory T cell recruitment in response to G‐CSF treatment. J Immunol 2013; 191:2266–72. [DOI] [PubMed] [Google Scholar]
- 34. Nagata S. Apoptosis and autoimmune diseases. Ann N Y Acad Sci 2010; 1209:10–6. [DOI] [PubMed] [Google Scholar]
- 35. Szondy Z, Garabuczi E, Joos G, Tsay GJ, Sarang Z. Impaired clearance of apoptotic cells in chronic inflammatory diseases: therapeutic implications. Front Immunol 2014; 5:354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Idoyaga J, Suda N, Suda K, Park CG, Steinman RM. Antibody to Langerin/CD207 localizes large numbers of CD8α + dendritic cells to the marginal zone of mouse spleen. Proc Natl Acad Sci USA 2009; 106:1524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yoshie O, Matsushima K. CCR4 and its ligands: from bench to bedside. Int Immunol 2015; 27:11–20. [DOI] [PubMed] [Google Scholar]
- 38. Kasagi S, Zhang P, Che L, Abbatiello B, Maruyama T, Nakatsukasa H et al In vivo‐generated antigen‐specific regulatory T cells treat autoimmunity without compromising antibacterial immune response. Sci Transl Med 2014; 6:241ra78. [DOI] [PubMed] [Google Scholar]
- 39. Yamazaki S, Morita A. Dendritic cells in the periphery control antigen‐specific natural and induced regulatory T cells. Front Immunol 2013; 4:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Welzen‐Coppens JM, van Helden‐Meeuwsen CG, Leenen PJ, Drexhage HA, Versnel MA. Reduced numbers of dendritic cells with a tolerogenic phenotype in the prediabetic pancreas of NOD mice. J Leukoc Biol 2012; 92:1207–13. [DOI] [PubMed] [Google Scholar]
- 41. Huang G, Wang Y, Chi H. Control of T cell fates and immune tolerance by p38α signaling in mucosal CD103+ dendritic cells. J Immunol 2013; 191:650–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim MJ, Jeong EK, Kwon EY, Joo JY, Lee JY, Choi J. Human CD103+ dendritic cells promote the differentiation of Porphyromonas gingivalis heat shock protein peptide‐specific regulatory T cells. J Periodontal Implant Sci 2014; 44:235–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bakdash G, Vogelpoel LT, van Capel TM, Kapsenberg ML, de Jong EC. Retinoic acid primes human dendritic cells to induce gut‐homing, IL‐10‐producing regulatory T cells. Mucosal Immunol 2015; 8:265–78. [DOI] [PubMed] [Google Scholar]
- 44. Mazzini E, Massimiliano L, Penna G, Rescigno M. Oral tolerance can be established via gap junction transfer of fed antigens from CX3CR1+ macrophages to CD103+ dendritic cells. Immunity 2014; 40:248–61. [DOI] [PubMed] [Google Scholar]
- 45. Shakhar G, Kolesnikov M. Intestinal macrophages and DCs close the gap on tolerance. Immunity 2014; 40:171–3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Analysis of apoptotic thymocytes upon UV exposure.
Figure S2. CCL22 levels did not increase in CD11c+ dendritic cell from mice receiving live cells injection.
Figure S3. Analysis of percentage of CD4+, CD8+, CD11c+, CD11b+ cells and regulatory T cells in the spleen due to Cytochrome C injection.
Figure S4. Analysis of the microarray data published on JI, 2009 by Qiu et al.
Figure S5. Analysis of FoxP3+ cells in CD4+ T cells upon apoptotic cell injection.
Figure S6. Analysis of percentage of CD4+ CD25hi cells due to apoptotic cell administration.
Figure S7. CCL22 drives the migration of regulatory T cells.
Figure S8. Analysis of FoxP3+ cells in CD4+ T cells in absence of CD8a+ CD103+ upon apoptotic cells injection.